
Deletion Mutants of Francisella Phagosomal Transporters FptA and FptF Are Highly Attenuated for Virulence and Are Protective Against Lethal Intranasal Francisella LVS Challenge in a Murine Model of Respiratory Tularemia

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
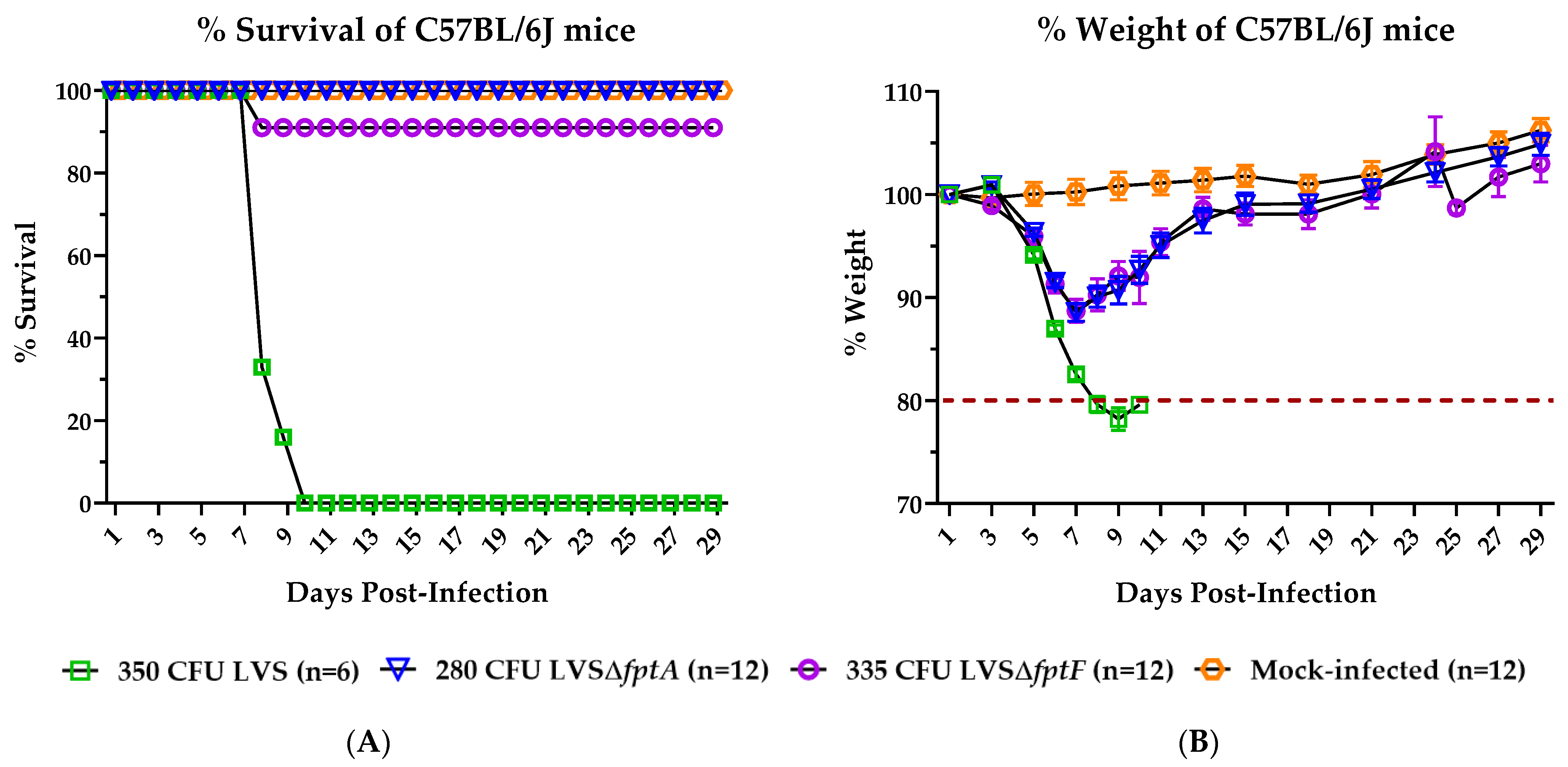
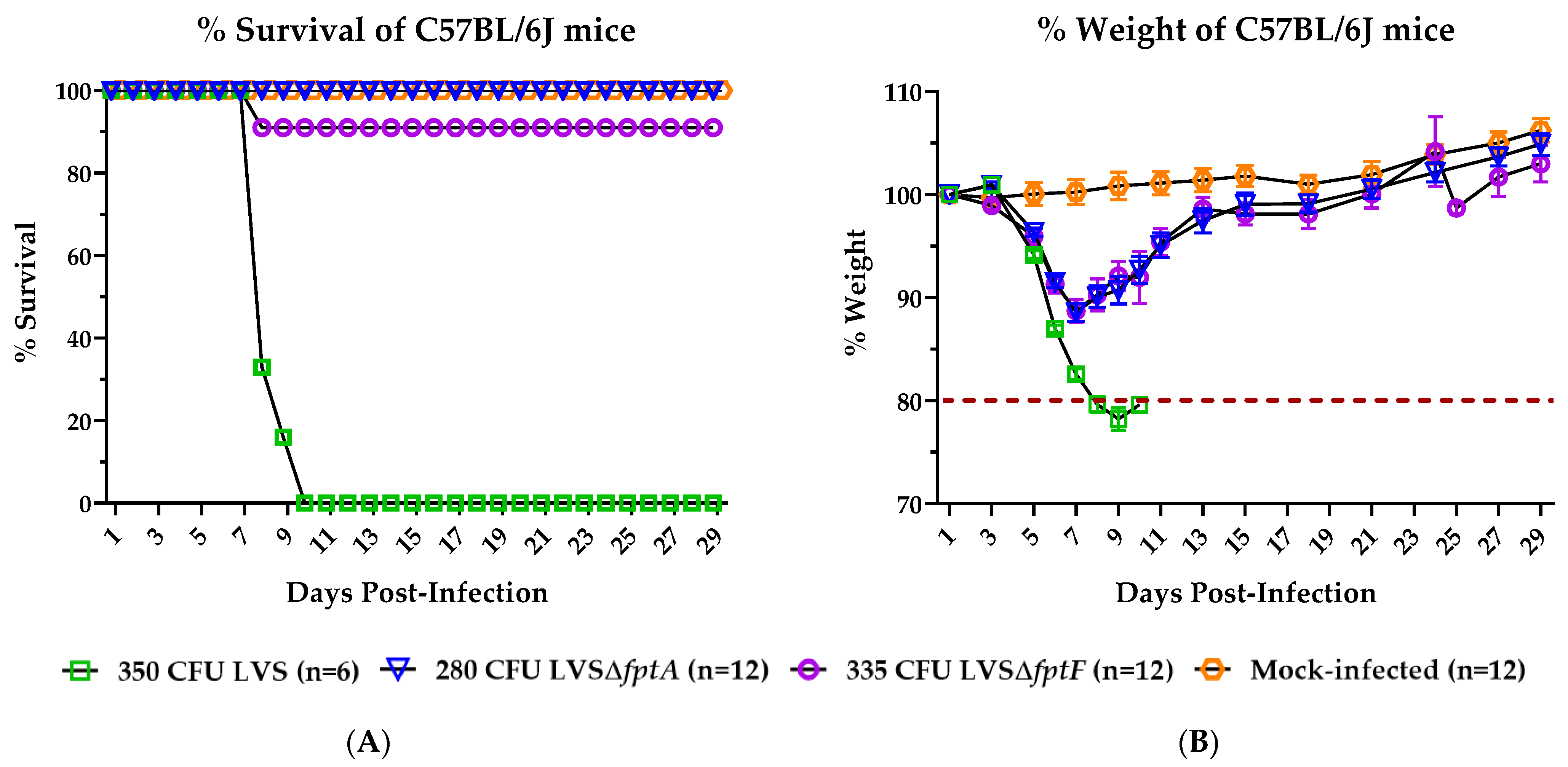
2.1. fpt Mutants Are Attenuated in the C57BL/6J Murine Model of Respiratory Tularemia Compared to Parental LVS
2.2. Inoculation with LVSΔfptA or LVSΔfptF Confers Protection against Lethal Challenge with Parental LVS in the C57BL/6J Murine Model of Respiratory Tularemia
2.3. fpt Mutants Have Reduced Bacterial Organ Burdens in the Lungs, Livers, and Spleens of C57BL/6J Mice versus Mice Infected with Parental LVS
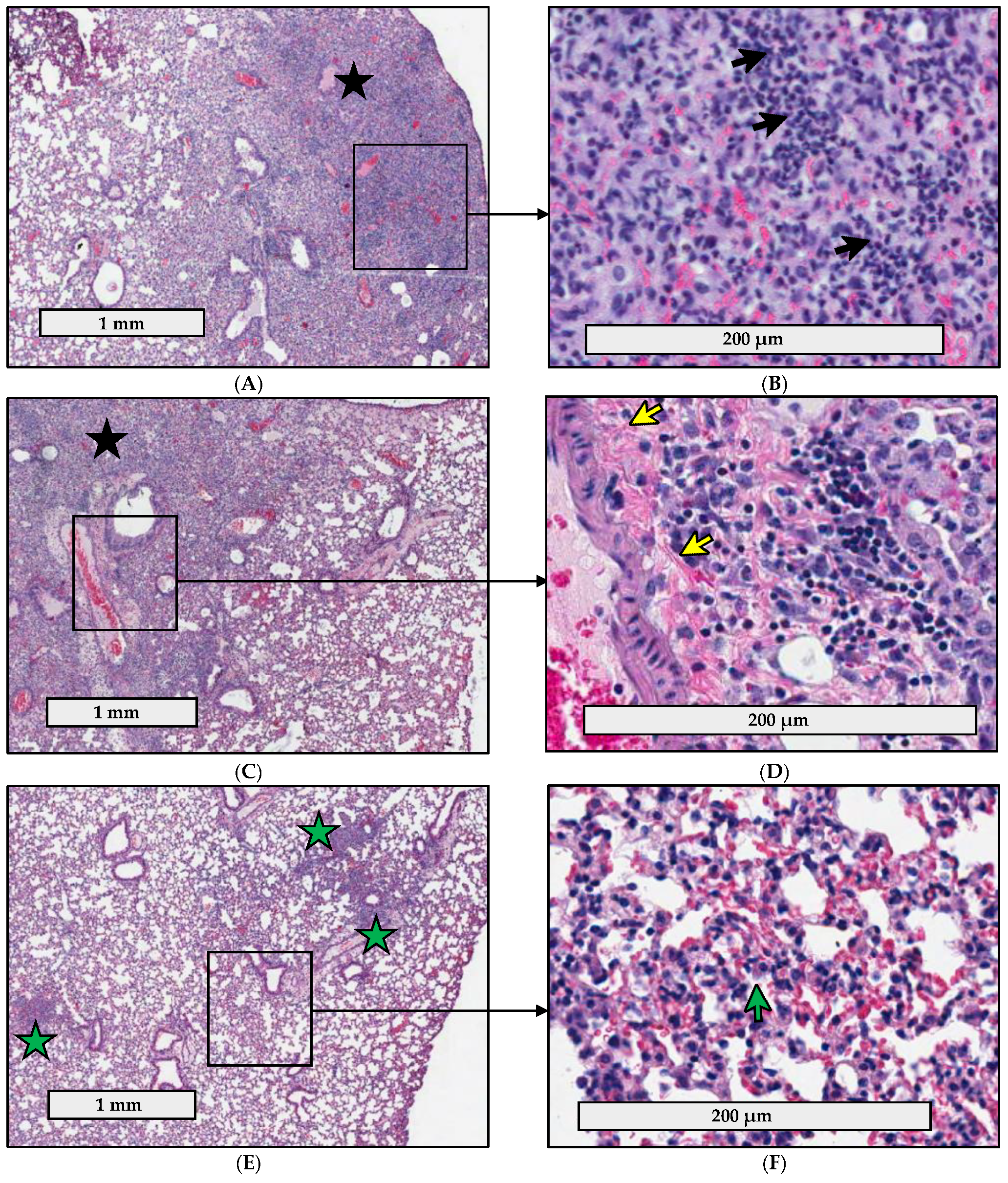
2.4. Pathology Is Less Severe in the Lungs of C57BL/6J Mice Infected with fpt Mutants than in the Lungs of Mice Infected with Parental LVS
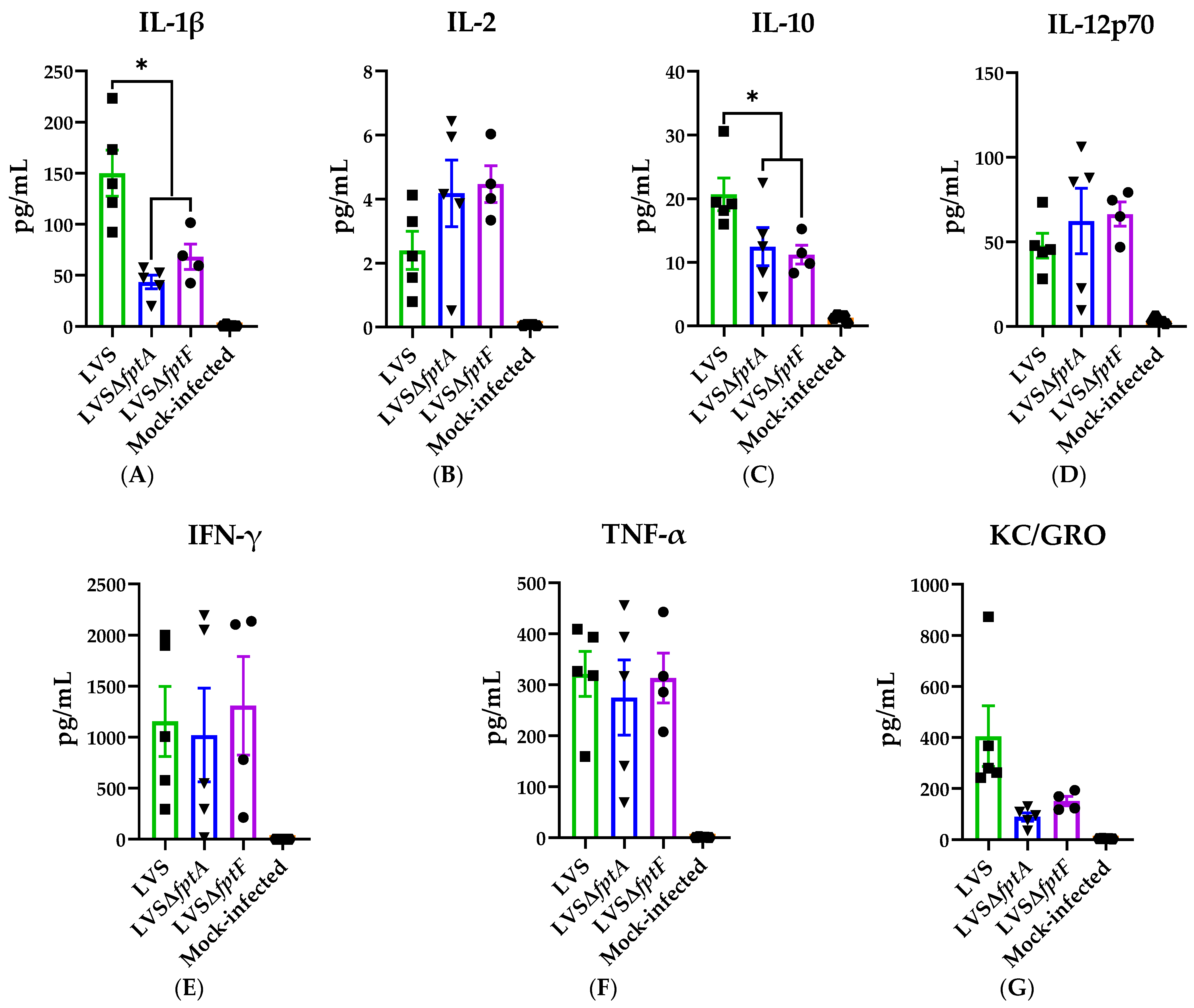
2.5. fpt Mutants Induce Altered Proinflammatory Cytokine Responses in the Bronchoalveolar Lavage Fluid of C57BL/6J Mice Compared to Parental LVS
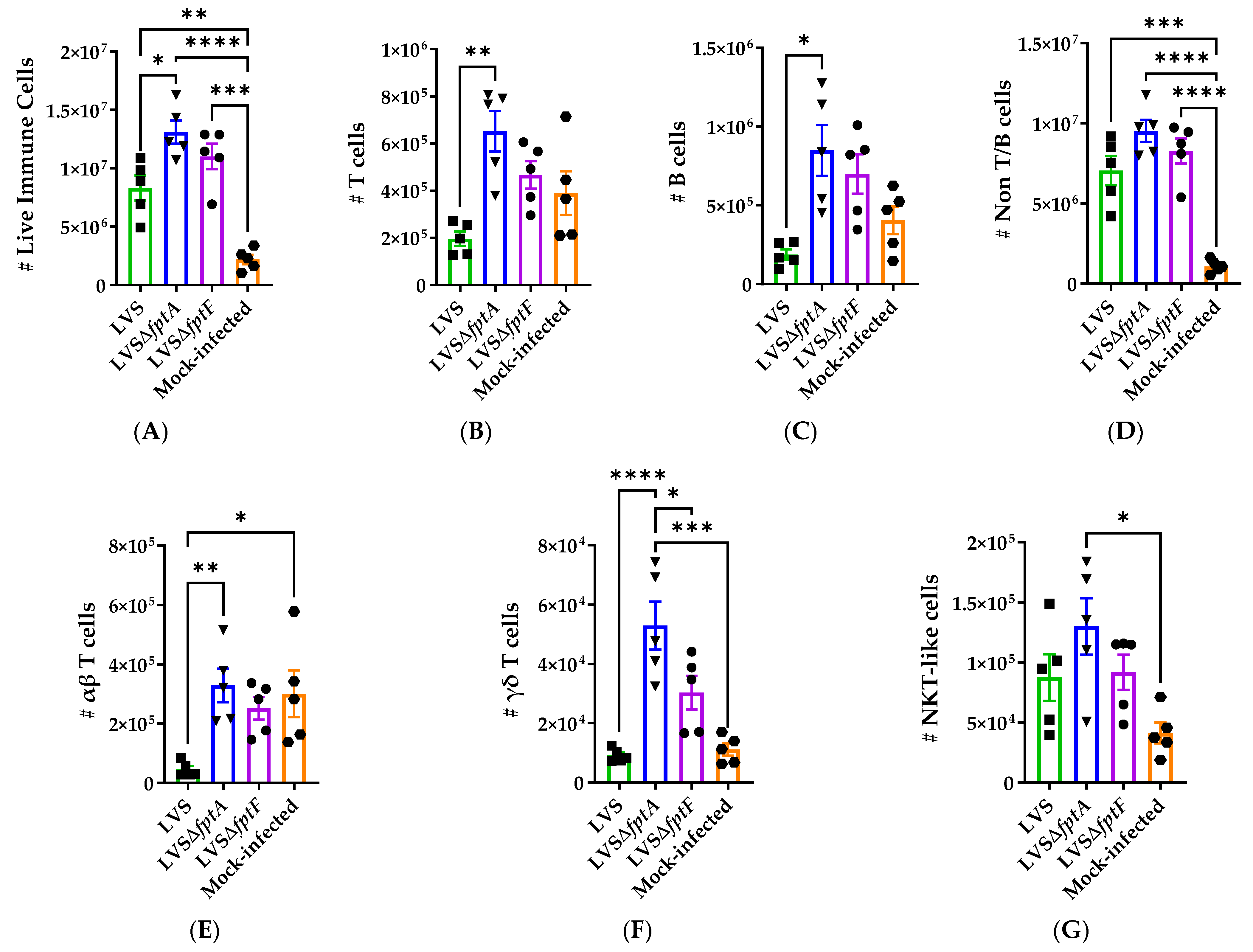
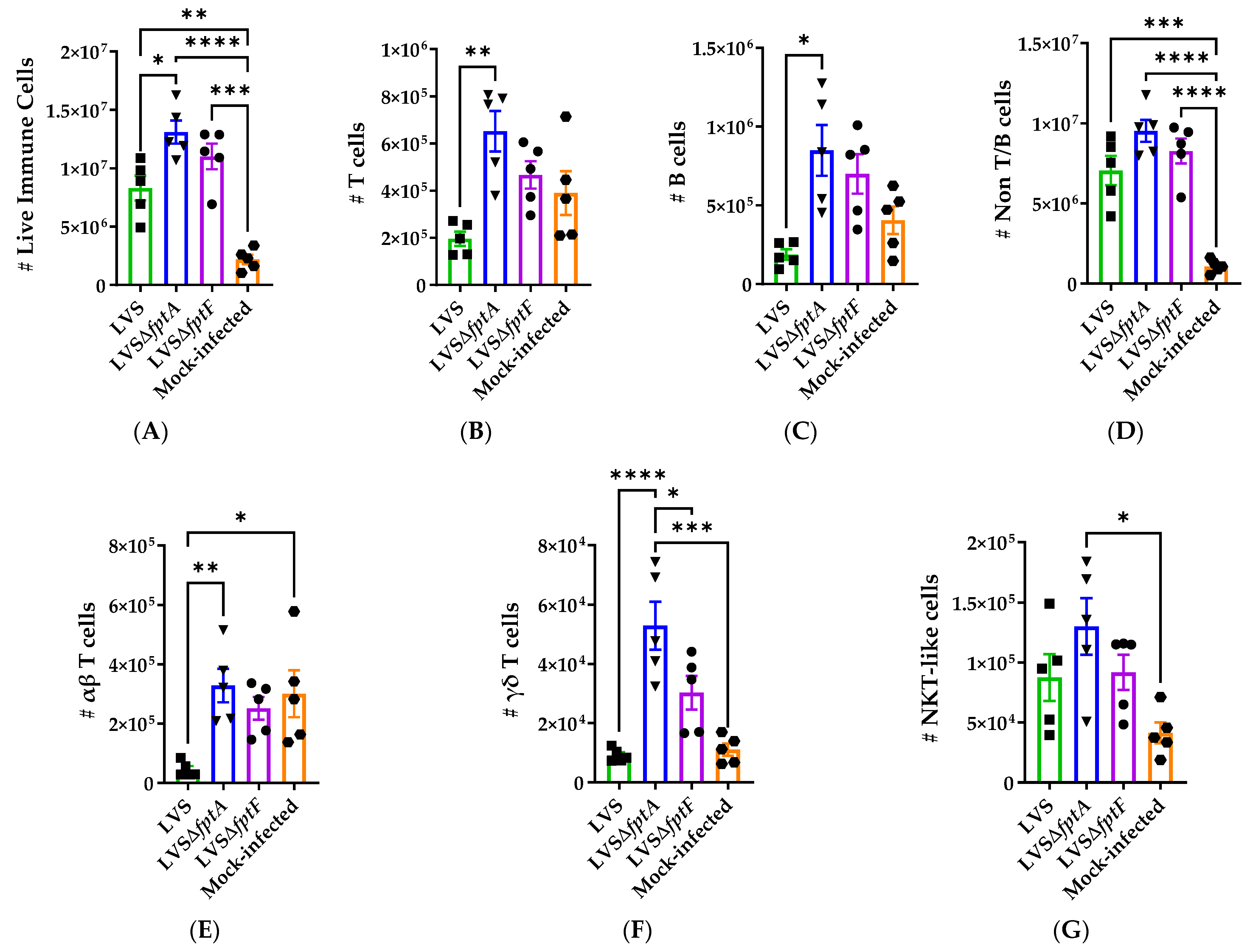
2.6. fpt Mutants Induce Altered Host Immune Responses in the Lungs of C57BL/6J Mice Compared to Parental LVS
3. Discussion
4. Materials and Methods
4.1. Bacteria and Growth Conditions
4.2. Mice
4.3. Attenuation of fpt Mutant Strains in Mice
4.4. Protective Capacity of fpt Mutant Strains in Mice
4.5. Quantification of Bacterial Organ Burdens in Mice
4.6. Histopathology Analysis of Infected Mouse Lungs
4.7. Quantification of Secreted Cytokines in Murine Bronchoalveolar Lavage Fluid
4.8. Characterization of the Immune Response in Infected Mouse Lungs
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Steiner, D.J.; Furuya, Y.; Metzger, D.W. Host-Pathogen Interactions and Immune Evasion Strategies in Francisella tularensis Pathogenicity. Infect. Drug Resist. 2014, 7, 239–251. [Google Scholar] [CrossRef] [Green Version]
- Centers for Disease Control and Prevention: Tularemia. Available online: https://www.cdc.gov/tularemia/statistics/index.html (accessed on 4 April 2021).
- Dennis, D.T.; Inglesby, T.V.; Henderson, D.A.; Bartlett, J.G.; Ascher, M.S.; Eitzen, E.; Fine, A.D.; Friedlander, A.M.; Hauer, J.; Layton, M.; et al. Tularemia as a Biological Weapon: Medical and Public Health Management. J. Am. Med. Assoc. 2001, 285, 2763–2773. [Google Scholar] [CrossRef] [PubMed]
- Feldman, K.A.; Enscore, R.E.; Lathrop, S.L.; Matyas, B.T.; McGuill, M.; Schriefer, M.E.; Stiles-Enos, D.; Dennis, D.T.; Petersen, L.R.; Hayes, E.B. An Outbreak of Primary Pneumonic Tularemia on Martha’s Vineyard. N. Engl. J. Med. 2001, 345, 1601–1606. [Google Scholar] [CrossRef]
- Gill, V.; Cunha, B.A. Tularemia Pneumonia. Semin. Respir. Infect. 1997, 12, 61–67. [Google Scholar] [PubMed]
- Hornick, R.B.; Eigelsbach, H.T. Aerogenic Immunization of Man with Live Tularemia Vaccine. Bacteriol. Rev. 1966, 30, 532–538. [Google Scholar] [CrossRef] [PubMed]
- McCrumb, F.R. Aerosol Infection of Man with Pasteurella tularensis. Bacteriol. Rev. 1961, 25, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Oyston, P.C.; Sjostedt, A.; Titball, R.W. Tularaemia: Bioterrorism Defence Renews Interest in Francisella tularensis. Nat. Rev. Microbiol. 2004, 2, 967–978. [Google Scholar] [CrossRef]
- Sjostedt, A. Tularemia: History, Epidemiology, Pathogen Physiology, and Clinical Manifestations. Ann. N. Y. Acad. Sci. 2007, 1105, 1–29. [Google Scholar] [CrossRef]
- Keim, P.; Johansson, A.; Wagner, D.M. Molecular Epidemiology, Evolution, and Ecology of Francisella. Ann. N. Y. Acad. Sci. 2007, 1105, 30–66. [Google Scholar] [CrossRef]
- Jia, Q.; Horwitz, M.A. Live Attenuated Tularemia Vaccines for Protection Against Respiratory Challenge with Virulent F. tularensis subsp. tularensis. Front. Cell. Infect. Microbiol. 2018, 8, 154. [Google Scholar] [CrossRef] [PubMed]
- Marohn, M.E.; Barry, E.M. Live Attenuated Tularemia Vaccines: Recent Developments and Future Goals. Vaccine 2013, 31, 3485–3491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulligan, M.J.; Stapleton, J.T.; Keitel, W.A.; Frey, S.E.; Chen, W.H.; Rouphael, N.; Edupuganti, S.; Beck, A.; Winokur, P.L.; El Sahly, H.M.; et al. Tularemia Vaccine: Safety, Reactogenicity, “Take” Skin Reactions, and Antibody Responses Following Vaccination with a New Lot of the Francisella tularensis Live Vaccine Strain—A Phase 2 Randomized Clinical Trial. Vaccine 2017, 35, 4730–4737. [Google Scholar] [CrossRef] [PubMed]
- Roberts, L.M.; Powell, D.A.; Frelinger, J.A. Adaptive Immunity to Francisella tularensis and Considerations for Vaccine Development. Front. Cell. Infect. Microbiol. 2018, 8, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sunagar, R.; Kumar, S.; Franz, B.J.; Gosselin, E.J. Tularemia Vaccine Development: Paralysis or Progress? Vaccine (Auckl.) 2016, 6, 9–23. [Google Scholar] [CrossRef] [Green Version]
- Isherwood, K.E.; Titball, R.W.; Davies, D.H.; Felgner, P.L.; Morrow, W.J. Vaccination Strategies for Francisella tularensis. Adv. Drug Deliv. Rev. 2005, 57, 1403–1414. [Google Scholar] [CrossRef] [PubMed]
- Wayne Conlan, J.; Oyston, P.C. Vaccines Against Francisella tularensis. Ann. N. Y. Acad. Sci. 2007, 1105, 325–350. [Google Scholar] [CrossRef]
- Tigertt, W.D. Soviet Viable Pasteurella tularensis vaccines: A Review of Selected Articles. Bacteriol. Rev. 1962, 26, 354–373. [Google Scholar] [CrossRef]
- Saslaw, S.; Eigelsbach, H.T.; Prior, J.A.; Wilson, H.E.; Carhart, S. Tularemia Vaccine Study. II. Respiratory Challenge. Arch. Intern. Med. 1961, 107, 702–714. [Google Scholar] [CrossRef]
- Saslaw, S.; Eigelsbach, H.T.; Wilson, H.E.; Prior, J.A.; Carhart, S. Tularemia Vaccine Study. I. Intracutaneous Challenge. Arch. Intern. Med. 1961, 107. [Google Scholar] [CrossRef]
- Burke, D.S. Immunization Against Tularemia: Analysis of the Effectiveness of Live Francisella tularensis Vaccine in Prevention of Laboratory-Acquired Tularemia. J. Infect. Dis. 1977, 135, 55–60. [Google Scholar] [CrossRef]
- Pasetti, M.F.; Cuberos, L.; Horn, T.L.; Shearer, J.D.; Matthews, S.J.; House, R.V.; Sztein, M.B. An Improved Francisella tularensis Live Vaccine Strain (LVS) is Well Tolerated and Highly Immunogenic when Administered to Rabbits in Escalating Doses Using Various Immunization Routes. Vaccine 2008, 26, 1773–1785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oyston, P.C.; Quarry, J.E. Tularemia Vaccine: Past, Present and Future. Antonie Van Leeuwenhoek 2005, 87, 277–281. [Google Scholar] [CrossRef]
- Twine, S.M.; Petit, M.D.; Fulton, K.M.; House, R.V.; Conlan, J.W. Immunoproteomics Analysis of the Murine Antibody Response to Vaccination with an Improved Francisella tularensis Live Vaccine Strain (LVS). PLoS ONE 2010, 5, e10000. [Google Scholar] [CrossRef] [PubMed]
- El Sahly, H.M.; Atmar, R.L.; Patel, S.M.; Wells, J.M.; Cate, T.; Ho, M.; Guo, K.; Pasetti, M.F.; Lewis, D.E.; Sztein, M.B.; et al. Safety, Reactogenicity and Immunogenicity of Francisella tularensis Live Vaccine Strain in Humans. Vaccine 2009, 27, 4905–4911. [Google Scholar] [CrossRef] [Green Version]
- Stundick, M.V.; Albrecht, M.T.; Houchens, C.R.; Smith, A.P.; Dreier, T.M.; Larsen, J.C. Animal Models for Francisella tularensis and Burkholderia Species: Scientific and Regulatory Gaps Toward Approval of Antibiotics Under the FDA Animal Rule. Vet. Pathol. 2013, 50, 877–892. [Google Scholar] [CrossRef]
- Rohmer, L.; Brittnacher, M.; Svensson, K.; Buckley, D.; Haugen, E.; Zhou, Y.; Chang, J.; Levy, R.; Hayden, H.; Forsman, M.; et al. Potential Source of Francisella tularensis Live Vaccine Strain Attenuation Determined by Genome Comparison. Infect. Immun. 2006, 74, 6895–6906. [Google Scholar] [CrossRef] [Green Version]
- Kotloff, K.L.; Noriega, F.; Losonsky, G.A.; Sztein, M.B.; Wasserman, S.S.; Nataro, J.P.; Levine, M.M. Safety, Immunogenicity, and Transmissibility in Humans of CVD 1203, a Live Oral Shigella flexneri 2a Vaccine Candidate Attenuated by Deletions in aroA and virG. Infect. Immun. 1996, 64, 4542–4548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tacket, C.O.; Hone, D.M.; Losonsky, G.A.; Guers, L.; Edelman, R.; Levine, M.M. Clinical Acceptability and Immunogenicity of CVD 908 Salmonella typhi Vaccine Strain. Vaccine 1992, 10, 443–446. [Google Scholar] [CrossRef]
- Elkins, K.L.; Cowley, S.C.; Bosio, C.M. Innate and Adaptive Immune Responses to an Intracellular Bacterium, Francisella tularensis Live Vaccine Strain. Microbes Infect. 2003, 5, 135–142. [Google Scholar] [CrossRef]
- Tarnvik, A. Nature of Protective Immunity to Francisella tularensis. Rev. Infect. Dis. 1989, 11, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Sauer, J.D.; Bachman, M.A.; Swanson, M.S. The Phagosomal Transporter A Couples Threonine Acquisition to Differentiation and Replication of Legionella pneumophila in Macrophages. Proc. Natl. Acad. Sci. USA 2005, 102, 9924–9929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, V.S.; Shlykov, M.A.; Castillo, R.; Sun, E.I.; Saier, M.H., Jr. The Major Facilitator Superfamily (MFS) Revisited. FEBS J. 2012, 279, 2022–2035. [Google Scholar] [CrossRef] [PubMed]
- Marohn, M.E.; Santiago, A.E.; Shirey, K.A.; Lipsky, M.; Vogel, S.N.; Barry, E.M. Members of the Francisella tularensis Phagosomal Transporter Subfamily of Major Facilitator Superfamily Transporters are Critical for Pathogenesis. Infect. Immun. 2012, 80, 2390–2401. [Google Scholar] [CrossRef] [Green Version]
- Gesbert, G.; Ramond, E.; Tros, F.; Dairou, J.; Frapy, E.; Barel, M.; Charbit, A. Importance of Branched-Chain Amino Acid Utilization in Francisella Intracellular Adaptation. Infect. Immun. 2015, 83, 173–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gesbert, G.; Ramond, E.; Rigard, M.; Frapy, E.; Dupuis, M.; Dubail, I.; Barel, M.; Henry, T.; Meibom, K.; Charbit, A. Asparagine Assimilation is Critical for Intracellular Replication and Dissemination of Francisella. Cell. Microbiol. 2014, 16, 434–449. [Google Scholar] [CrossRef]
- Balzano, P.M.; Cunningham, A.L.; Grassel, C.; Barry, E.M. Deletion of the Major Facilitator Superfamily Transporter fptB Alters Host Cell Interactions and Attenuates Virulence of Type A Francisella tularensis. Infect. Immun. 2018, 86. [Google Scholar] [CrossRef] [Green Version]
- Conlan, J.W.; Chen, W.; Bosio, C.M.; Cowley, S.C.; Elkins, K.L. Infection of Mice with Francisella as an Immunological Model. Curr. Protoc. Immunol. 2011, 93, 19.14.1–19.14.16. [Google Scholar] [CrossRef] [Green Version]
- Baskerville, A.; Hambleton, P. Pathogenesis and Pathology of Respiratory Tularaemia in the Rabbit. Br. J. Exp. Pathol. 1976, 57, 339–347. [Google Scholar]
- Hall, J.D.; Woolard, M.D.; Gunn, B.M.; Craven, R.R.; Taft-Benz, S.; Frelinger, J.A.; Kawula, T.H. Infected-Host-Cell Repertoire and Cellular Response in the Lung Following Inhalation of Francisella tularensis Schu S4, LVS, or U112. Infect. Immun. 2008, 76, 5843–5852. [Google Scholar] [CrossRef] [Green Version]
- Conlan, J.W.; KuoLee, R.; Shen, H.; Webb, A. Different Host Defences are Required to Protect Mice from Primary Systemic vs. Pulmonary Infection with the Facultative Intracellular Bacterial Pathogen, Francisella tularensis LVS. Microb. Pathog. 2002, 32, 127–134. [Google Scholar] [CrossRef]
- Conlan, J.W.; Chen, W.; Shen, H.; Webb, A.; KuoLee, R. Experimental Tularemia in Mice Challenged by Aerosol or Intradermally with Virulent Strains of Francisella tularensis: Bacteriologic and Histopathologic Studies. Microb. Pathog. 2003, 34, 239–248. [Google Scholar] [CrossRef]
- Wickstrum, J.R.; Bokhari, S.M.; Fischer, J.L.; Pinson, D.M.; Yeh, H.W.; Horvat, R.T.; Parmely, M.J. Francisella tularensis Induces Extensive Caspase-3 Activation and Apoptotic Cell Death in the Tissues of Infected Mice. Infect. Immun. 2009, 77, 4827–4836. [Google Scholar] [CrossRef] [Green Version]
- Cole, L.E.; Elkins, K.L.; Michalek, S.M.; Qureshi, N.; Eaton, L.J.; Rallabhandi, P.; Cuesta, N.; Vogel, S.N. Immunologic Consequences of Francisella tularensis Live Vaccine Strain Infection: Role of the Innate Immune Response in Infection and Immunity. J. Immunol. 2006, 176, 6888–6899. [Google Scholar] [CrossRef] [Green Version]
- Mares, C.A.; Ojeda, S.S.; Morris, E.G.; Li, Q.; Teale, J.M. Initial Delay in the Immune Response to Francisella tularensis is Followed by Hypercytokinemia Characteristic of Severe Sepsis and Correlating with Upregulation and Release of Damage-Associated Molecular Patterns. Infect. Immun. 2008, 76, 3001–3010. [Google Scholar] [CrossRef] [Green Version]
- Mares, C.A.; Sharma, J.; Ojeda, S.S.; Li, Q.; Campos, J.A.; Morris, E.G.; Coalson, J.J.; Teale, J.M. Attenuated Response of Aged Mice to Respiratory Francisella novicida is Characterized by Reduced Cell Death and Absence of Subsequent Hypercytokinemia. PLoS ONE 2010, 5, e14088. [Google Scholar] [CrossRef] [Green Version]
- Putzova, D.; Senitkova, I.; Stulik, J. Tularemia Vaccines. Folia Microbiol. 2016, 61, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Conlan, J.W. Tularemia Vaccines: Recent Developments and Remaining Hurdles. Future Microbiol. 2011, 6, 391–405. [Google Scholar] [CrossRef] [PubMed]
- Barry, E.M.; Cole, L.E.; Santiago, A.E. Vaccines Against Tularemia. Hum. Vaccines 2009, 5, 832–838. [Google Scholar] [CrossRef]
- Mann, B.J.; Ark, N.M. Rationally Designed Tularemia Vaccines. Expert Rev. Vaccines 2009, 8, 877–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, Q.; Lee, B.Y.; Bowen, R.; Dillon, B.J.; Som, S.M.; Horwitz, M.A. A Francisella tularensis Live Vaccine Strain (LVS) Mutant with a Deletion in capB, Encoding a Putative Capsular Biosynthesis Protein, is Significantly More Attenuated than LVS yet Induces Potent Protective Immunity in Mice Against F. tularensis Challenge. Infect. Immun. 2010, 78, 4341–4355. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.H.; Pinkham, J.T.; Heninger, S.J.; Chalabaev, S.; Kasper, D.L. Genetic Modification of the O-Polysaccharide of Francisella tularensis Results in an Avirulent Live Attenuated Vaccine. J. Infect. Dis. 2012, 205, 1056–1065. [Google Scholar] [CrossRef]
- Bakshi, C.S.; Malik, M.; Mahawar, M.; Kirimanjeswara, G.S.; Hazlett, K.R.; Palmer, L.E.; Furie, M.B.; Singh, R.; Melendez, J.A.; Sellati, T.J.; et al. An Improved Vaccine for Prevention of Respiratory Tularemia Caused by Francisella tularensis SchuS4 Strain. Vaccine 2008, 26, 5276–5288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karki, H.S.; Ham, J.H. The Roles of the Shikimate Pathway Genes, aroA and aroB, in Virulence, Growth and UV Tolerance of Burkholderia glumae Strain 411gr-6. Mol. Plant Pathol. 2014, 15, 940–947. [Google Scholar] [CrossRef] [PubMed]
- Avitia-Dominguez, C.; Sierra-Campos, E.; Salas-Pacheco, J.M.; Najera, H.; Rojo-Dominguez, A.; Cisneros-Martinez, J.; Tellez-Valencia, A. Inhibition and Biochemical Characterization of Methicillin-Resistant Staphylococcus aureus Shikimate Dehydrogenase: An in Silico and Kinetic Study. Molecules 2014, 19, 4491–4509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charles, I.G.; Lamb, H.K.; Pickard, D.; Dougan, G.; Hawkins, A.R. Isolation, Characterization and Nucleotide Sequences of the aroC Genes Encoding Chorismate Synthase from Salmonella typhi and Escherichia coli. J. Gen. Microbiol. 1990, 136, 353–358. [Google Scholar] [CrossRef] [Green Version]
- Hoiseth, S.K.; Stocker, B.A. Aromatic-Dependent Salmonella typhimurium are Non-Virulent and Effective as Live Vaccines. Nature 1981, 291, 238–239. [Google Scholar] [CrossRef] [PubMed]
- Hone, D.M.; Harris, A.M.; Chatfield, S.; Dougan, G.; Levine, M.M. Construction of Genetically Defined Double aro Mutants of Salmonella typhi. Vaccine 1991, 9, 810–816. [Google Scholar] [CrossRef]
- Peek, J.; Castiglione, G.; Shi, T.; Christendat, D. Isolation and Molecular Characterization of the Shikimate Dehydrogenase Domain from the Toxoplasma gondii AROM Complex. Mol. Biochem. Parasitol. 2014, 194, 16–19. [Google Scholar] [CrossRef]
- Cunningham, A.L.; Mann, B.J.; Qin, A.; Santiago, A.E.; Grassel, C.; Lipsky, M.; Vogel, S.N.; Barry, E.M. Characterization of Schu S4 aro Mutants as Live Attenuated Tularemia Vaccine Candidates. Virulence 2020, 11, 283–294. [Google Scholar] [CrossRef] [Green Version]
- Reed, D.S.; Smith, L.P.; Cole, K.S.; Santiago, A.E.; Mann, B.J.; Barry, E.M. Live Attenuated Mutants of Francisella tularensis Protect Rabbits Against Aerosol Challenge with a Virulent Type A Strain. Infect. Immun. 2014, 82, 2098–2105. [Google Scholar] [CrossRef] [Green Version]
- Santiago, A.E.; Mann, B.J.; Qin, A.; Cunningham, A.L.; Cole, L.E.; Grassel, C.; Vogel, S.N.; Levine, M.M.; Barry, E.M. Characterization of Francisella tularensis Schu S4 Defined Mutants as Live-Attenuated Vaccine Candidates. Pathog. Dis. 2015, 73, ftv036. [Google Scholar] [CrossRef]
- Pechous, R.; Celli, J.; Penoske, R.; Hayes, S.F.; Frank, D.W.; Zahrt, T.C. Construction and Characterization of an Attenuated Purine Auxotroph in a Francisella tularensis Live Vaccine Strain. Infect. Immun. 2006, 74, 4452–4461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pechous, R.D.; McCarthy, T.R.; Mohapatra, N.P.; Soni, S.; Penoske, R.M.; Salzman, N.H.; Frank, D.W.; Gunn, J.S.; Zahrt, T.C. A Francisella tularensis Schu S4 Purine Auxotroph is Highly Attenuated in Mice but Offers Limited Protection Against Homologous Intranasal Challenge. PLoS ONE 2008, 3, e2487. [Google Scholar] [CrossRef] [PubMed]
- Ireland, P.M.; LeButt, H.; Thomas, R.M.; Oyston, P.C.F. A Francisella tularensis SCHU S4 Mutant Deficient in Gamma-Glutamyltransferase Activity Induces Protective Immunity: Characterization of an Attenuated Vaccine Candidate. Microbiology 2011, 157, 3172–3179. [Google Scholar] [CrossRef] [Green Version]
- Twine, S.; Bystrom, M.; Chen, W.; Forsman, M.; Golovliov, I.; Johansson, A.; Kelly, J.; Lindgren, H.; Svensson, K.; Zingmark, C.; et al. A Mutant of Francisella tularensis Strain SCHU S4 Lacking the Ability to Express a 58-Kilodalton Protein is Attenuated for Virulence and is an Effective Live Vaccine. Infect. Immun. 2005, 73, 8345–8352. [Google Scholar] [CrossRef] [Green Version]
- Ramakrishnan, G.; Sen, B.; Johnson, R. Paralogous Outer Membrane Proteins Mediate Uptake of Different Forms of Iron and Synergistically Govern Virulence in Francisella tularensis tularensis. J. Biol. Chem. 2012, 287, 25191–25202. [Google Scholar] [CrossRef] [Green Version]
- Lindgren, M.; Tancred, L.; Golovliov, I.; Conlan, W.; Twine, S.M.; Sjostedt, A. Identification of Mechanisms for Attenuation of the FSC043 Mutant of Francisella tularensis SCHU S4. Infect. Immun. 2014, 82, 3622–3635. [Google Scholar] [CrossRef] [Green Version]
- Fortier, A.H.; Slayter, M.V.; Ziemba, R.; Meltzer, M.S.; Nacy, C.A. Live Vaccine Strain of Francisella tularensis: Infection and Immunity in Mice. Infect. Immun. 1991, 59, 2922–2928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurtz, S.L.; Foreman, O.; Bosio, C.M.; Anver, M.R.; Elkins, K.L. Interleukin-6 is Essential for Primary Resistance to Francisella tularensis Live Vaccine Strain Infection. Infect. Immun. 2013, 81, 585–597. [Google Scholar] [CrossRef] [Green Version]
- Metzger, D.W.; Bakshi, C.S.; Kirimanjeswara, G. Mucosal Immunopathogenesis of Francisella tularensis. Ann. N. Y. Acad. Sci. 2007, 1105, 266–283. [Google Scholar] [CrossRef]
- Miller, M.A.; Stabenow, J.M.; Parvathareddy, J.; Wodowski, A.J.; Fabrizio, T.P.; Bina, X.R.; Zalduondo, L.; Bina, J.E. Visualization of Murine Intranasal Dosing Efficiency Using Luminescent Francisella tularensis: Effect of Instillation Volume and Form of Anesthesia. PLoS ONE 2012, 7, e31359. [Google Scholar] [CrossRef] [Green Version]
- Visweswaraiah, A.; Novotny, L.A.; Hjemdahl-Monsen, E.J.; Bakaletz, L.O.; Thanavala, Y. Tracking the Tissue Distribution of Marker Dye Following Intranasal Delivery in Mice and Chinchillas: A Multifactorial Analysis of Parameters Affecting Nasal Retention. Vaccine 2002, 20, 3209–3220. [Google Scholar] [CrossRef]
- Bosio, C.M.; Dow, S.W. Francisella tularensis Induces Aberrant Activation of Pulmonary Dendritic Cells. J. Immunol. 2005, 175, 6792–6801. [Google Scholar] [CrossRef] [Green Version]
- Ben Nasr, A.; Haithcoat, J.; Masterson, J.E.; Gunn, J.S.; Eaves-Pyles, T.; Klimpel, G.R. Critical Role for Serum Opsonins and Complement Receptors CR3 (CD11b/CD18) and CR4 (CD11c/CD18) in Phagocytosis of Francisella tularensis by Human Dendritic Cells (DC): Uptake of Francisella Leads to Activation of Immature DC and Intracellular Survival of the Bacteria. J. Leukoc. Biol. 2006, 80, 774–786. [Google Scholar] [CrossRef]
- Hall, J.D.; Craven, R.R.; Fuller, J.R.; Pickles, R.J.; Kawula, T.H. Francisella tularensis Replicates within Alveolar Type II Epithelial Cells in vitro and in vivo Following Inhalation. Infect. Immun. 2007, 75, 1034–1039. [Google Scholar] [CrossRef] [Green Version]
- Sharma, J.; Li, Q.; Mishra, B.B.; Pena, C.; Teale, J.M. Lethal Pulmonary Infection with Francisella novicida is Associated with Severe Sepsis. J. Leukoc. Biol. 2009, 86, 491–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, J.; Mares, C.A.; Li, Q.; Morris, E.G.; Teale, J.M. Features of Sepsis Caused by Pulmonary Infection with Francisella tularensis Type A Strain. Microb. Pathog. 2011, 51, 39–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, A.; Mann, B.J. Identification of Transposon Insertion Mutants of Francisella tularensis tularensis Strain Schu S4 Deficient in Intracellular Replication in the Hepatic Cell Line HepG2. BMC Microbiol. 2006, 6, 69. [Google Scholar] [CrossRef] [Green Version]
- Periasamy, S.; Singh, A.; Sahay, B.; Rahman, T.; Feustel, P.J.; Pham, G.H.; Gosselin, E.J.; Sellati, T.J. Development of Tolerogenic Dendritic Cells and Regulatory T Cells Favors Exponential Bacterial Growth and Survival During Early Respiratory Tularemia. J. Leukoc. Biol. 2011, 90, 493–507. [Google Scholar] [CrossRef] [Green Version]
- Malik, M.; Bakshi, C.S.; McCabe, K.; Catlett, S.V.; Shah, A.; Singh, R.; Jackson, P.L.; Gaggar, A.; Metzger, D.W.; Melendez, J.A.; et al. Matrix Metalloproteinase 9 Activity Enhances Host Susceptibility to Pulmonary Infection with Type A and B Strains of Francisella tularensis. J. Immunol. 2007, 178, 1013–1020. [Google Scholar] [CrossRef] [Green Version]
- Periasamy, S.; Avram, D.; McCabe, A.; MacNamara, K.C.; Sellati, T.J.; Harton, J.A. An Immature Myeloid/Myeloid-Suppressor Cell Response Associated with Necrotizing Inflammation Mediates Lethal Pulmonary Tularemia. PLoS Pathog. 2016, 12, e1005517. [Google Scholar] [CrossRef] [Green Version]
- Silverman, M.S.; Greenman, V.; McKee, A.E.; Hadley, K.; Hodge, F.A.; Burriss, C. Cellular Response of Mice to Infection with Pasteurella tularensis (Live Vaccine Strain) Following Continuous Exposure to Low Dose Gamma Radiation. J. Infect. Dis. 1969, 120, 366–371. [Google Scholar] [CrossRef]
- Slight, S.R.; Monin, L.; Gopal, R.; Avery, L.; Davis, M.; Cleveland, H.; Oury, T.D.; Rangel-Moreno, J.; Khader, S.A. IL-10 Restrains IL-17 to Limit Lung Pathology Characteristics Following Pulmonary Infection with Francisella tularensis Live Vaccine Strain. Am. J. Pathol. 2013, 183, 1397–1404. [Google Scholar] [CrossRef] [Green Version]
- Conlan, J.W.; Zhao, X.; Harris, G.; Shen, H.; Bolanowski, M.; Rietz, C.; Sjostedt, A.; Chen, W. Molecular Immunology of Experimental Primary Tularemia in Mice Infected by Respiratory or Intradermal Routes with Type A Francisella tularensis. Mol. Immunol. 2008, 45, 2962–2969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anthony, L.S.; Ghadirian, E.; Nestel, F.P.; Kongshavn, P.A. The Requirement for Gamma Interferon in Resistance of Mice to Experimental Tularemia. Microb. Pathog. 1989, 7, 421–428. [Google Scholar] [CrossRef]
- Elkins, K.L.; Rhinehart-Jones, T.R.; Culkin, S.J.; Yee, D.; Winegar, R.K. Minimal Requirements for Murine Resistance to Infection with Francisella tularensis LVS. Infect. Immun. 1996, 64, 3288–3293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elkins, K.L.; Rhinehart-Jones, T.; Nacy, C.A.; Winegar, R.K.; Fortier, A.H. T-Cell-Independent Resistance to Infection and Generation of Immunity to Francisella tularensis. Infect. Immun. 1993, 61, 823–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leiby, D.A.; Fortier, A.H.; Crawford, R.M.; Schreiber, R.D.; Nacy, C.A. In vivo Modulation of the Murine Immune Response to Francisella tularensis LVS by Administration of Anticytokine Antibodies. Infect. Immun. 1992, 60, 84–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, L.M.; Davies, J.S.; Sempowski, G.D.; Frelinger, J.A. IFN-Gamma, but not IL-17A, is Required for Survival During Secondary Pulmonary Francisella tularensis Live Vaccine Stain Infection. Vaccine 2014, 32, 3595–3603. [Google Scholar] [CrossRef] [Green Version]
- Collazo, C.M.; Sher, A.; Meierovics, A.I.; Elkins, K.L. Myeloid Differentiation Factor-88 (MyD88) is Essential for Control of Primary in vivo Francisella tularensis LVS Infection, but not for Control of Intra-Macrophage Bacterial Replication. Microbes Infect. 2006, 8, 779–790. [Google Scholar] [CrossRef]
- Sjostedt, A.; North, R.J.; Conlan, J.W. The Requirement of Tumour Necrosis Factor-Alpha and Interferon-Gamma for the Expression of Protective Immunity to Secondary Murine Tularaemia Depends on the Size of the Challenge Inoculum. Microbiology 1996, 142 Pt 6, 1369–1374. [Google Scholar] [CrossRef] [Green Version]
- Collazo, C.M.; Meierovics, A.I.; De Pascalis, R.; Wu, T.H.; Lyons, C.R.; Elkins, K.L. T Cells from Lungs and Livers of Francisella tularensis-Immune Mice Control the Growth of Intracellular Bacteria. Infect. Immun. 2009, 77, 2010–2021. [Google Scholar] [CrossRef] [Green Version]
- Del Barrio, L.; Sahoo, M.; Lantier, L.; Reynolds, J.M.; Ceballos-Olvera, I.; Re, F. Production of Anti-LPS IgM by B1a B Cells Depends on IL-1beta and is Protective against Lung Infection with Francisella tularensis LVS. PLoS Pathog. 2015, 11, e1004706. [Google Scholar] [CrossRef]
- Jayakar, H.R.; Parvathareddy, J.; Fitzpatrick, E.A.; Bina, X.R.; Bina, J.E.; Re, F.; Emery, F.D.; Miller, M.A. A galU Mutant of Francisella tularensis is Attenuated for Virulence in a Murine Pulmonary Model of Tularemia. BMC Microbiol. 2011, 11, 179. [Google Scholar] [CrossRef] [Green Version]
- Belkaid, Y.; Oldenhove, G. Tuning Microenvironments: Induction of Regulatory T Cells by Dendritic Cells. Immunity 2008, 29, 362–371. [Google Scholar] [CrossRef] [Green Version]
- Lin, A.; Lore, K. Granulocytes: New Members of the Antigen-Presenting Cell Family. Front. Immunol. 2017, 8, 1781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radsak, M.; Iking-Konert, C.; Stegmaier, S.; Andrassy, K.; Hansch, G.M. Polymorphonuclear Neutrophils as Accessory Cells for T-Cell Activation: Major Histocompatibility Complex Class II Restricted Antigen-Dependent Induction of T-Cell Proliferation. Immunology 2000, 101, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Vono, M.; Lin, A.; Norrby-Teglund, A.; Koup, R.A.; Liang, F.; Lore, K. Neutrophils Acquire the Capacity for Antigen Presentation to Memory CD4(+) T Cells in vitro and ex vivo. Blood 2017, 129, 1991–2001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Culkin, S.J.; Rhinehart-Jones, T.; Elkins, K.L. A Novel Role for B Cells in Early Protective Immunity to an Intracellular Pathogen, Francisella tularensis Strain LVS. J. Immunol. 1997, 158, 3277–3284. [Google Scholar]
- Liu, Y.J. Dendritic Cell Subsets and Lineages, and Their Functions in Innate and Adaptive Immunity. Cell 2001, 106, 259–262. [Google Scholar] [CrossRef] [Green Version]
- Lopez, M.C.; Duckett, N.S.; Baron, S.D.; Metzger, D.W. Early Activation of NK Cells After Lung Infection with the Intracellular Bacterium, Francisella tularensis LVS. Cell. Immunol. 2004, 232, 75–85. [Google Scholar] [CrossRef]
- Maniar, A.; Zhang, X.; Lin, W.; Gastman, B.R.; Pauza, C.D.; Strome, S.E.; Chapoval, A.I. Human GammaDelta T Lymphocytes Induce Robust NK Cell-Mediated Antitumor Cytotoxicity Through CD137 Engagement. Blood 2010, 116, 1726–1733. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Vaccination Dose (i.n.) on Day 1 (CFU) | Survival Post-Vaccination on Day 28 | LD50 | Survival Post-Challenge with ~500 CFU LVS (i.n.) on Day 29 1 | Vaccine Efficacy 2 | Sterilizing Immunity 3 |
|---|---|---|---|---|---|---|
| PBS | - | 24/24 | 1/24 | - | - | |
| LVS | 60 | 1/6 | <60 CFU | 1/1 | 100% | Yes |
| 121 | 0/6 | - | - | - | ||
| 175 | 1/6 | 1/1 | 100% | Yes | ||
| 242 | 0/6 | - | - | - | ||
| 350 | 0/6 | - | - | - | ||
| 483 | 0/6 | - | - | - | ||
| 633 | 0/6 | - | - | - | ||
| LVSΔfptA | ~280 | 12/12 | ~1400 CFU | 12/12 | 100% | Yes |
| ~560 | 10/12 | 10/10 | 100% | - | ||
| ~1120 | 5/11 4 | 5/5 | 100% | - | ||
| ~1680 | 7/11 5 | 7/7 | 100% | Yes | ||
| 3250 | 0/4 | - | - | - | ||
| 32,500 | 0/4 | - | - | - | ||
| LVSΔfptF | ~335 | 11/12 | ~2010–5167 CFU | 11/11 | 100% | Yes |
| ~670 | 9/9 6 | 9/9 | 100% | - | ||
| ~1340 | 11/12 | 11/11 | 100% | - | ||
| ~2010 | 8/12 | 8/8 | 100% | Yes | ||
| 5167 | 0/4 | - | - | - | ||
| 51,667 | 0/4 | - | - | - |
| Strain | LVS | LVSΔfptA | LVSΔfptF | Mock-Infected |
|---|---|---|---|---|
| (1a) Global Extent of Inflammation | 1.75 | 1.5 | 1.25 | 0.0 |
| (1b) Surface Area of Alveolar Destruction | 1.5 | 1.25 | 0.25 | 0.0 |
| (1c) Interstitial Involvement | 0.75 | 0.75 | 0.5 | 0.0 |
| (1d) Foci of Inflammation | 1.0 | 0.75 | 0.5 | 0.0 |
| (2a) Neutrophils | 1.0 | 0.5 | 0.0 | 0.0 |
| (2b) Macrophages | 0.75 | 0.75 | 0.5 | 0.0 |
| (2c) Lymphocytes | 1.0 | 0.75 | 0.75 | 0.0 |
| (3a) Fibrin Deposition | 0.75 | 0.75 | 0.0 | 0.0 |
| (3b) Hyperplasia of Type II Pneumocytes | 1.0 | 0.75 | 0.75 | 0.0 |
| Cummulative Score | 9.5 | 7.75 | 4.5 | 0.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hobbs, B.E.; Matson, C.A.; Theofilou, V.I.; Webb, T.J.; Younis, R.H.; Barry, E.M. Deletion Mutants of Francisella Phagosomal Transporters FptA and FptF Are Highly Attenuated for Virulence and Are Protective Against Lethal Intranasal Francisella LVS Challenge in a Murine Model of Respiratory Tularemia. Pathogens 2021, 10, 799. https://doi.org/10.3390/pathogens10070799
Hobbs BE, Matson CA, Theofilou VI, Webb TJ, Younis RH, Barry EM. Deletion Mutants of Francisella Phagosomal Transporters FptA and FptF Are Highly Attenuated for Virulence and Are Protective Against Lethal Intranasal Francisella LVS Challenge in a Murine Model of Respiratory Tularemia. Pathogens. 2021; 10(7):799. https://doi.org/10.3390/pathogens10070799
Chicago/Turabian StyleHobbs, Brandi E., Courtney A. Matson, Vasileios I. Theofilou, Tonya J. Webb, Rania H. Younis, and Eileen M. Barry. 2021. "Deletion Mutants of Francisella Phagosomal Transporters FptA and FptF Are Highly Attenuated for Virulence and Are Protective Against Lethal Intranasal Francisella LVS Challenge in a Murine Model of Respiratory Tularemia" Pathogens 10, no. 7: 799. https://doi.org/10.3390/pathogens10070799
APA StyleHobbs, B. E., Matson, C. A., Theofilou, V. I., Webb, T. J., Younis, R. H., & Barry, E. M. (2021). Deletion Mutants of Francisella Phagosomal Transporters FptA and FptF Are Highly Attenuated for Virulence and Are Protective Against Lethal Intranasal Francisella LVS Challenge in a Murine Model of Respiratory Tularemia. Pathogens, 10(7), 799. https://doi.org/10.3390/pathogens10070799