
Endosymbiont Capture, a Repeated Process of Endosymbiont Transfer with Replacement in Trypanosomatids Angomonas spp.

 , , , ,
, , , ,  and
and 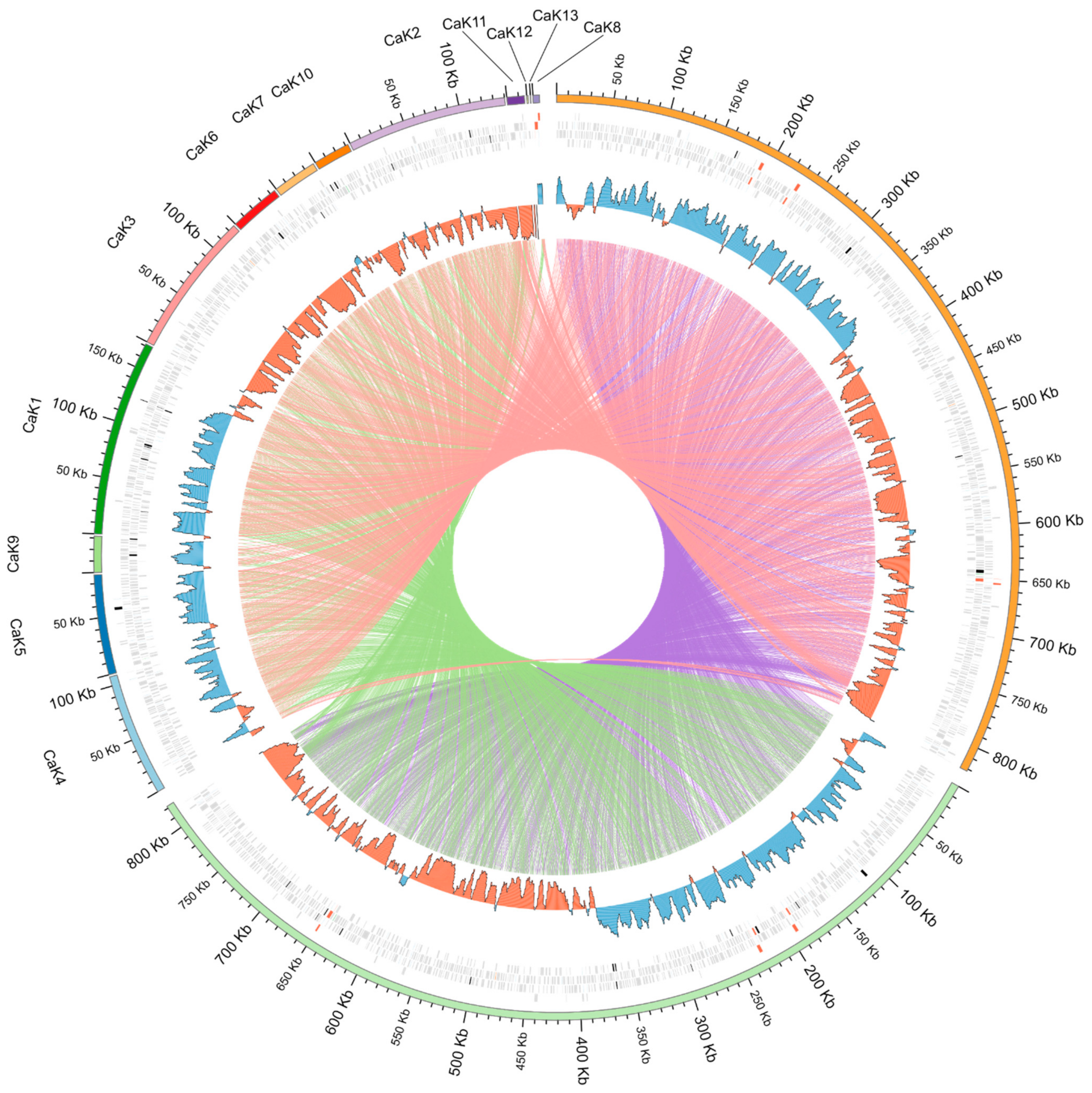{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Genomic Sequences
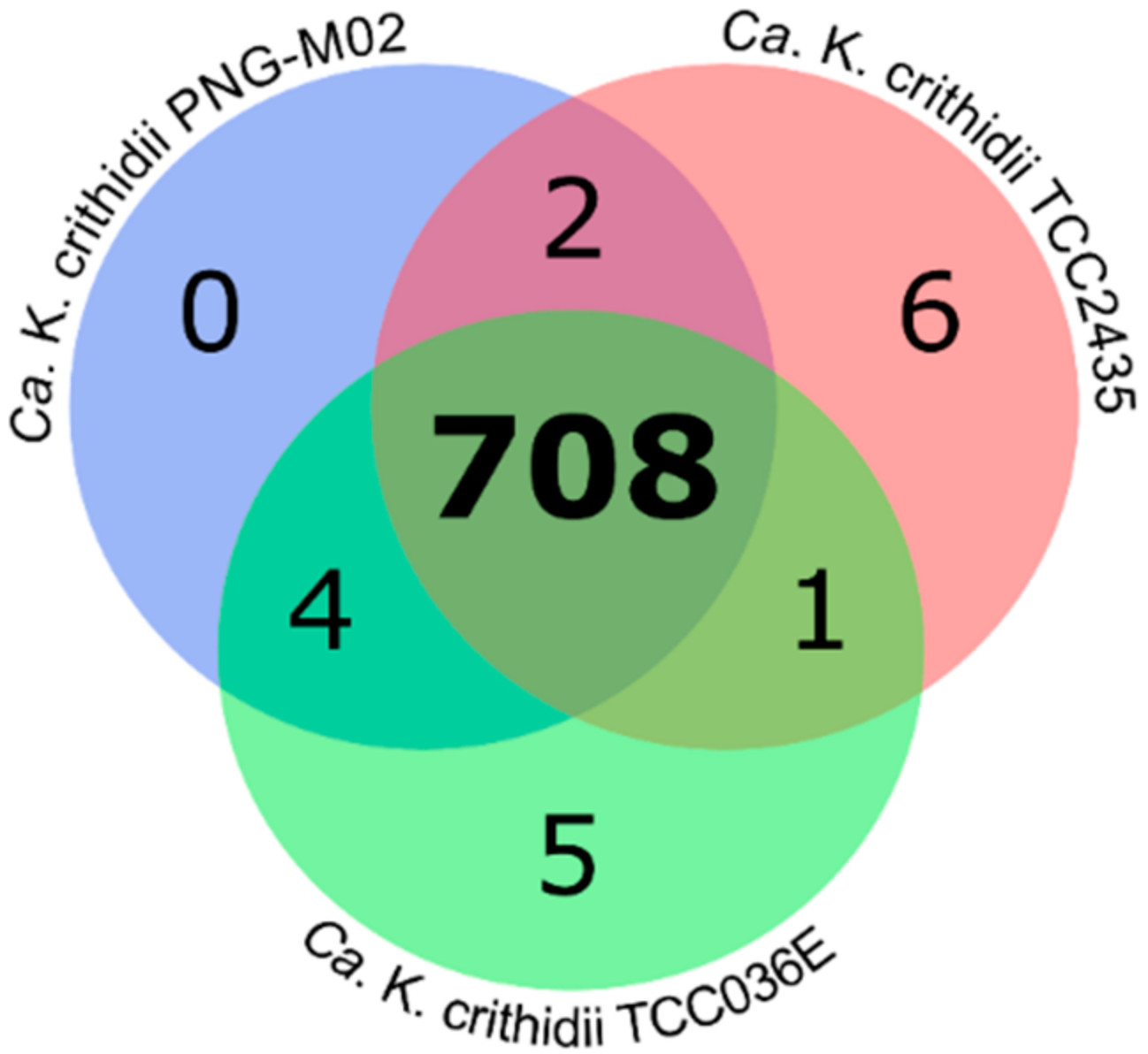
2.2. Analysis of Orthologous Groups (OGs) of Proteins
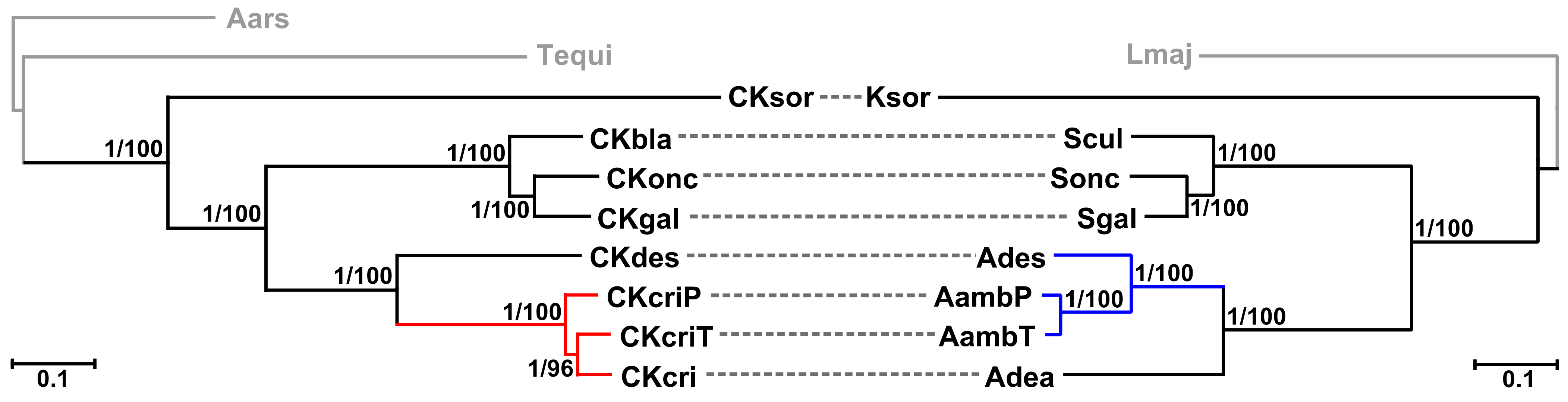
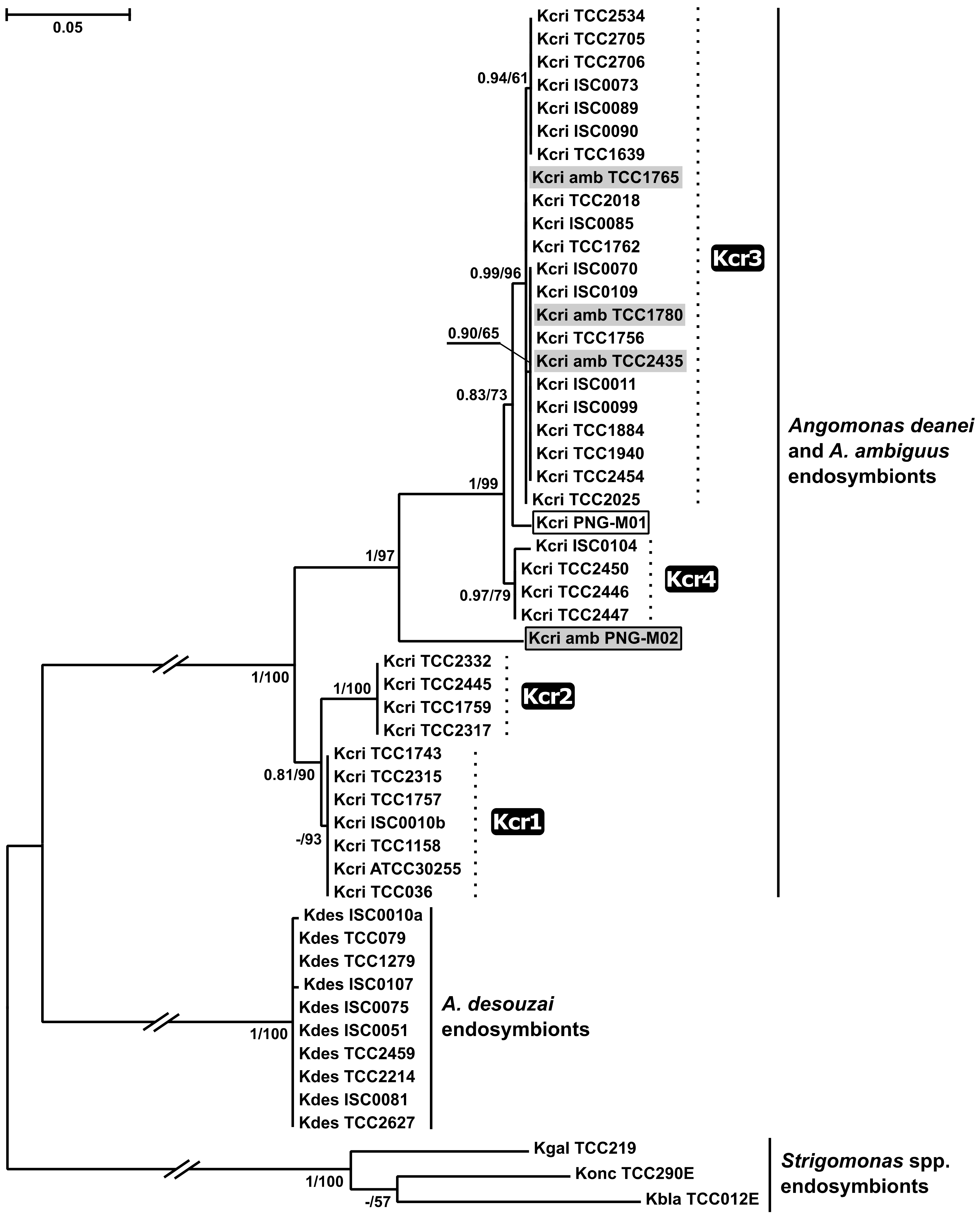
2.3. Phylogenetic Analyses
3. Discussion
4. Materials and Methods
4.1. Trypanosomatid Strains: Origin and Cultivation
4.2. Genome Sequencing, Assembly, and Annotation
4.3. Synteny Analysis of Bacterial Genomes
4.4. Phylogenomic Analyses
4.5. Amplification and Phylogenetic Analysis of GAPDH Gene
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kostygov, A.Y.; Karnkowska, A.; Votýpka, J.; Tashyreva, D.; Maciszewski, K.; Yurchenko, V.; Lukeš, J. Euglenozoa: Taxonomy, diversity and ecology, symbioses and viruses. Open Biol. 2021, 11, 200407. [Google Scholar] [CrossRef]
- Lukeš, J.; Butenko, A.; Hashimi, H.; Maslov, D.A.; Votýpka, J.; Yurchenko, V. Trypanosomatids are much more than just trypanosomes: Clues from the expanded family tree. Trends Parasitol. 2018, 34, 466–480. [Google Scholar] [CrossRef]
- Butenko, A.; Hammond, M.; Field, M.C.; Ginger, M.L.; Yurchenko, V.; Lukeš, J. Reductionist pathways for parasitism in euglenozoans? Expanded datasets provide new insights. Trends Parasitol. 2021, 37, 100–116. [Google Scholar] [CrossRef] [PubMed]
- Maslov, D.A.; Opperdoes, F.R.; Kostygov, A.Y.; Hashimi, H.; Lukeš, J.; Yurchenko, V. Recent advances in trypanosomatid research: Genome organization, expression, metabolism, taxonomy and evolution. Parasitology 2019, 146, 1–27. [Google Scholar] [CrossRef]
- Ganyukova, A.I.; Frolov, A.O.; Malysheva, M.N.; Spodareva, V.V.; Yurchenko, V.; Kostygov, A.Y. A novel endosymbiont-containing trypanosomatid Phytomonas borealis sp. n. from the predatory bug Picromerus bidens (Heteroptera: Pentatomidae). Folia Parasitol. 2020, 67, 4. [Google Scholar] [CrossRef] [PubMed]
- Kostygov, A.Y.; Dobáková, E.; Grybchuk-Ieremenko, A.; Váhala, D.; Maslov, D.A.; Votýpka, J.; Lukeš, J.; Yurchenko, V. Novel trypanosomatid-bacterium association: Evolution of endosymbiosis in action. mBio 2016, 7, e01985-15. [Google Scholar] [CrossRef]
- Teixeira, M.M.; Borghesan, T.C.; Ferreira, R.C.; Santos, M.A.; Takata, C.S.; Campaner, M.; Nunes, V.L.; Milder, R.V.; de Souza, W.; Camargo, E.P. Phylogenetic validation of the genera Angomonas and Strigomonas of trypanosomatids harboring bacterial endosymbionts with the description of new species of trypanosomatids and of proteobacterial symbionts. Protist 2011, 162, 503–524. [Google Scholar] [CrossRef] [PubMed]
- Catta-Preta, C.M.; Brum, F.L.; da Silva, C.C.; Zuma, A.A.; Elias, M.C.; de Souza, W.; Schenkman, S.; Motta, M.C. Endosymbiosis in trypanosomatid protozoa: The bacterium division is controlled during the host cell cycle. Front. Microbiol. 2015, 6, 520. [Google Scholar] [CrossRef] [PubMed]
- Kostygov, A.Y.; Butenko, A.; Nenarokova, A.; Tashyreva, D.; Flegontov, P.; Lukeš, J.; Yurchenko, V. Genome of Ca. pandoraea novymonadis, an endosymbiotic bacterium of the trypanosomatid Novymonas esmeraldas. Front. Microbiol. 2017, 8, 1940. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.C.; Alves, J.M.; Serrano, M.G.; Buck, G.A.; Vasconcelos, A.T.; Sagot, M.F.; Teixeira, M.M.; Camargo, E.P.; Motta, M.C. Biosynthesis of vitamins and cofactors in bacterium-harbouring trypanosomatids depends on the symbiotic association as revealed by genomic analyses. PLoS ONE 2013, 8, e79786. [Google Scholar] [CrossRef]
- Alves, J.M.; Klein, C.C.; da Silva, F.M.; Costa-Martins, A.G.; Serrano, M.G.; Buck, G.A.; Vasconcelos, A.T.; Sagot, M.F.; Teixeira, M.M.; Motta, M.C.; et al. Endosymbiosis in trypanosomatids: The genomic cooperation between bacterium and host in the synthesis of essential amino acids is heavily influenced by multiple horizontal gene transfers. BMC Evol. Biol. 2013, 13, 190. [Google Scholar] [CrossRef]
- Alves, J.M.; Voegtly, L.; Matveyev, A.V.; Lara, A.M.; da Silva, F.M.; Serrano, M.G.; Buck, G.A.; Teixeira, M.M.; Camargo, E.P. Identification and phylogenetic analysis of heme synthesis genes in trypanosomatids and their bacterial endosymbionts. PLoS ONE 2011, 6, e23518. [Google Scholar] [CrossRef] [PubMed]
- Votýpka, J.; Kostygov, A.Y.; Kraeva, N.; Grybchuk-Ieremenko, A.; Tesařová, M.; Grybchuk, D.; Lukeš, J.; Yurchenko, V. Kentomonas gen. n., a new genus of endosymbiont-containing trypanosomatids of Strigomonadinae subfam. n. Protist 2014, 165, 825–838. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Maslov, D.A.; Chang, K.P. Monophyletic origin of beta-division proteobacterial endosymbionts and their coevolution with insect trypanosomatid protozoa Blastocrithidia culicis and Crithidia spp. Proc. Natl. Acad. Sci. USA 1994, 91, 8437–8441. [Google Scholar] [CrossRef]
- Borghesan, T.C.; Campaner, M.; Matsumoto, T.E.; Espinosa, O.A.; Razafindranaivo, V.; Paiva, F.; Carranza, J.C.; Añez, N.; Neves, L.; Teixeira, M.M.G.; et al. Genetic diversity and phylogenetic relationships of coevolving symbiont-harboring insect trypanosomatids, and their Neotropical dispersal by invader African blowflies (Calliphoridae). Front. Microbiol. 2018, 9, 131. [Google Scholar] [CrossRef] [PubMed]
- Alves, J.M.; Serrano, M.G.; Maia da Silva, F.; Voegtly, L.J.; Matveyev, A.V.; Teixeira, M.M.; Camargo, E.P.; Buck, G.A. Genome evolution and phylogenomic analysis of Candidatus kinetoplastibacterium, the betaproteobacterial endosymbionts of Strigomonas and Angomonas. Genome Biol. Evol. 2013, 5, 338–350. [Google Scholar] [CrossRef]
- Motta, M.C.; Martins, A.C.; de Souza, S.S.; Catta-Preta, C.M.; Silva, R.; Klein, C.C.; de Almeida, L.G.; de Lima Cunha, O.; Ciapina, L.P.; Brocchi, M.; et al. Predicting the proteins of Angomonas deanei, Strigomonas culicis and their respective endosymbionts reveals new aspects of the trypanosomatidae family. PLoS ONE 2013, 8, e60209. [Google Scholar] [CrossRef]
- Silva, F.M.; Kostygov, A.Y.; Spodareva, V.V.; Butenko, A.; Tossou, R.; Lukeš, J.; Yurchenko, V.; Alves, J.M.P. The reduced genome of Candidatus Kinetoplastibacterium sorsogonicusi, the endosymbiont of Kentomonas sorsogonicus (Trypanosomatidae): Loss of the haem-synthesis pathway. Parasitology 2018, 145, 1287–1293. [Google Scholar] [CrossRef]
- Martinez-Cano, D.J.; Reyes-Prieto, M.; Martinez-Romero, E.; Partida-Martinez, L.P.; Latorre, A.; Moya, A.; Delaye, L. Evolution of small prokaryotic genomes. Front. Microbiol. 2014, 5, 742. [Google Scholar] [CrossRef]
- Wernegreen, J.J. Endosymbiont evolution: Predictions from theory and surprises from genomes. Ann. N. Y. Acad. Sci. 2015, 1360, 16–35. [Google Scholar] [CrossRef]
- Wernegreen, J.J. Endosymbiosis. Curr. Biol. 2012, 22, R555–R561. [Google Scholar] [CrossRef]
- McCutcheon, J.P.; Boyd, B.M.; Dale, C. The life of an insect endosymbiont from the cradle to the grave. Curr. Biol. 2019, 29, R485–R495. [Google Scholar] [CrossRef] [PubMed]
- Boscaro, V.; Fokin, S.I.; Petroni, G.; Verni, F.; Keeling, P.J.; Vannini, C. Symbiont replacement between bacteria of different classes reveals additional layers of complexity in the evolution of symbiosis in the ciliate Euplotes. Protist 2018, 169, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Vannini, C.; Ferrantini, F.; Ristori, A.; Verni, F.; Petroni, G. Betaproteobacterial symbionts of the ciliate Euplotes: Origin and tangled evolutionary path of an obligate microbial association. Environ. Microbiol. 2012, 14, 2553–2563. [Google Scholar] [CrossRef]
- Bennett, G.M.; Moran, N.A. Heritable symbiosis: The advantages and perils of an evolutionary rabbit hole. Proc. Natl. Acad. Sci. USA 2015, 112, 10169–10176. [Google Scholar] [CrossRef]
- Toews, D.P.; Brelsford, A. The biogeography of mitochondrial and nuclear discordance in animals. Mol. Ecol. 2012, 21, 3907–3930. [Google Scholar] [CrossRef]
- Tsitrone, A.; Kirkpatrick, M.; Levin, D.A. A model for chloroplast capture. Evolution 2003, 57, 1776–1782. [Google Scholar] [CrossRef]
- Harrison, R.G.; Larson, E.L. Hybridization, introgression, and the nature of species boundaries. J. Hered. 2014, 105 (Suppl. 1), 795–809. [Google Scholar] [CrossRef]
- Petit, R.J.; Excoffier, L. Gene flow and species delimitation. Trends Ecol. Evol. 2009, 24, 386–393. [Google Scholar] [CrossRef]
- Du, F.K.; Petit, R.J.; Liu, J.Q. More introgression with less gene flow: Chloroplast vs. mitochondrial DNA in the Picea asperata complex in China, and comparison with other conifers. Mol. Ecol. 2009, 18, 1396–1407. [Google Scholar] [CrossRef]
- Týč, J.; Votýpka, J.; Klepetková, H.; Šuláková, H.; Jirků, M.; Lukeš, J. Growing diversity of trypanosomatid parasites of flies (Diptera: Brachcera): Frequent cosmopolitism and moderate host specificity. Mol. Phylogenet. Evol. 2013, 69, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Ganyukova, A.I.; Malysheva, M.N.; Frolov, A.O. Life cycle, ultrastructure and host-parasite relationships of Angomonas deanei (Kinetoplastea: Trypanosomatidae) in the blowfly Lucilia sericata (Diptera: Calliphoridae). Protistology 2020, 14, 204–218. [Google Scholar] [CrossRef]
- Carvalho, A.L.M. Estudos sobre a posição sistemática, a biologia e a transmissão de tripanosomatídeos encontrados em Zelus leucogrammus (Perty, 1834) (Hemiptera, Reduviidae). Rev. Pathol. Trop. 1973, 2, 223–274. [Google Scholar]
- Frolov, A.O.; Kostygov, A.Y.; Yurchenko, V. Development of monoxenous trypanosomatids and phytomonads in insects. Trends Parasitol. 2021, 37, 538–551. [Google Scholar] [CrossRef]
- Stegemann, S.; Keuthe, M.; Greiner, S.; Bock, R. Horizontal transfer of chloroplast genomes between plant species. Proc. Natl. Acad. Sci. USA 2012, 109, 2434–2438. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Antipov, D.; Korobeynikov, A.; McLean, J.S.; Pevzner, P.A. hybridSPAdes: An algorithm for hybrid assembly of short and long reads. Bioinformatics 2016, 32, 1009–1015. [Google Scholar] [CrossRef]
- Steinbiss, S.; Silva-Franco, F.; Brunk, B.; Foth, B.; Hertz-Fowler, C.; Berriman, M.; Otto, T.D. Companion: A web server for annotation and analysis of parasite genomes. Nucleic Acids Res. 2016, 44, W29–W34. [Google Scholar] [CrossRef]
- Delcher, A.L.; Bratke, K.A.; Powers, E.C.; Salzberg, S.L. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 2007, 23, 673–679. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Hunt, M.; De Silva, N.; Otto, T.D.; Parkhill, J.; Keane, J.A.; Harris, S.R. Circlator: Automated circularization of genome assemblies using long sequencing reads. Genome Biol. 2015, 16, 294. [Google Scholar] [CrossRef]
- Darling, A.E.; Mau, B.; Perna, N.T. ProgressiveMauve: Multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 2010, 5, e11147. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef]
- Kuck, P.; Longo, G.C. FASconCAT-G: Extensive functions for multiple sequence alignment preparations concerning phylogenetic studies. Front. Zool. 2014, 11, 81. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Hohna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skalický, T.; Alves, J.M.P.; Morais, A.C.; Režnarová, J.; Butenko, A.; Lukeš, J.; Serrano, M.G.; Buck, G.A.; Teixeira, M.M.G.; Camargo, E.P.; et al. Endosymbiont Capture, a Repeated Process of Endosymbiont Transfer with Replacement in Trypanosomatids Angomonas spp. Pathogens 2021, 10, 702. https://doi.org/10.3390/pathogens10060702
Skalický T, Alves JMP, Morais AC, Režnarová J, Butenko A, Lukeš J, Serrano MG, Buck GA, Teixeira MMG, Camargo EP, et al. Endosymbiont Capture, a Repeated Process of Endosymbiont Transfer with Replacement in Trypanosomatids Angomonas spp. Pathogens. 2021; 10(6):702. https://doi.org/10.3390/pathogens10060702
Chicago/Turabian StyleSkalický, Tomáš, João M. P. Alves, Anderson C. Morais, Jana Režnarová, Anzhelika Butenko, Julius Lukeš, Myrna G. Serrano, Gregory A. Buck, Marta M. G. Teixeira, Erney P. Camargo, and et al. 2021. "Endosymbiont Capture, a Repeated Process of Endosymbiont Transfer with Replacement in Trypanosomatids Angomonas spp." Pathogens 10, no. 6: 702. https://doi.org/10.3390/pathogens10060702
APA StyleSkalický, T., Alves, J. M. P., Morais, A. C., Režnarová, J., Butenko, A., Lukeš, J., Serrano, M. G., Buck, G. A., Teixeira, M. M. G., Camargo, E. P., Sanders, M., Cotton, J. A., Yurchenko, V., & Kostygov, A. Y. (2021). Endosymbiont Capture, a Repeated Process of Endosymbiont Transfer with Replacement in Trypanosomatids Angomonas spp. Pathogens, 10(6), 702. https://doi.org/10.3390/pathogens10060702