
Sow Contact Is a Major Driver in the Development of the Nasal Microbiota of Piglets




Abstract
1. Introduction
2. Results
2.1. Sample Collection and Sequencing
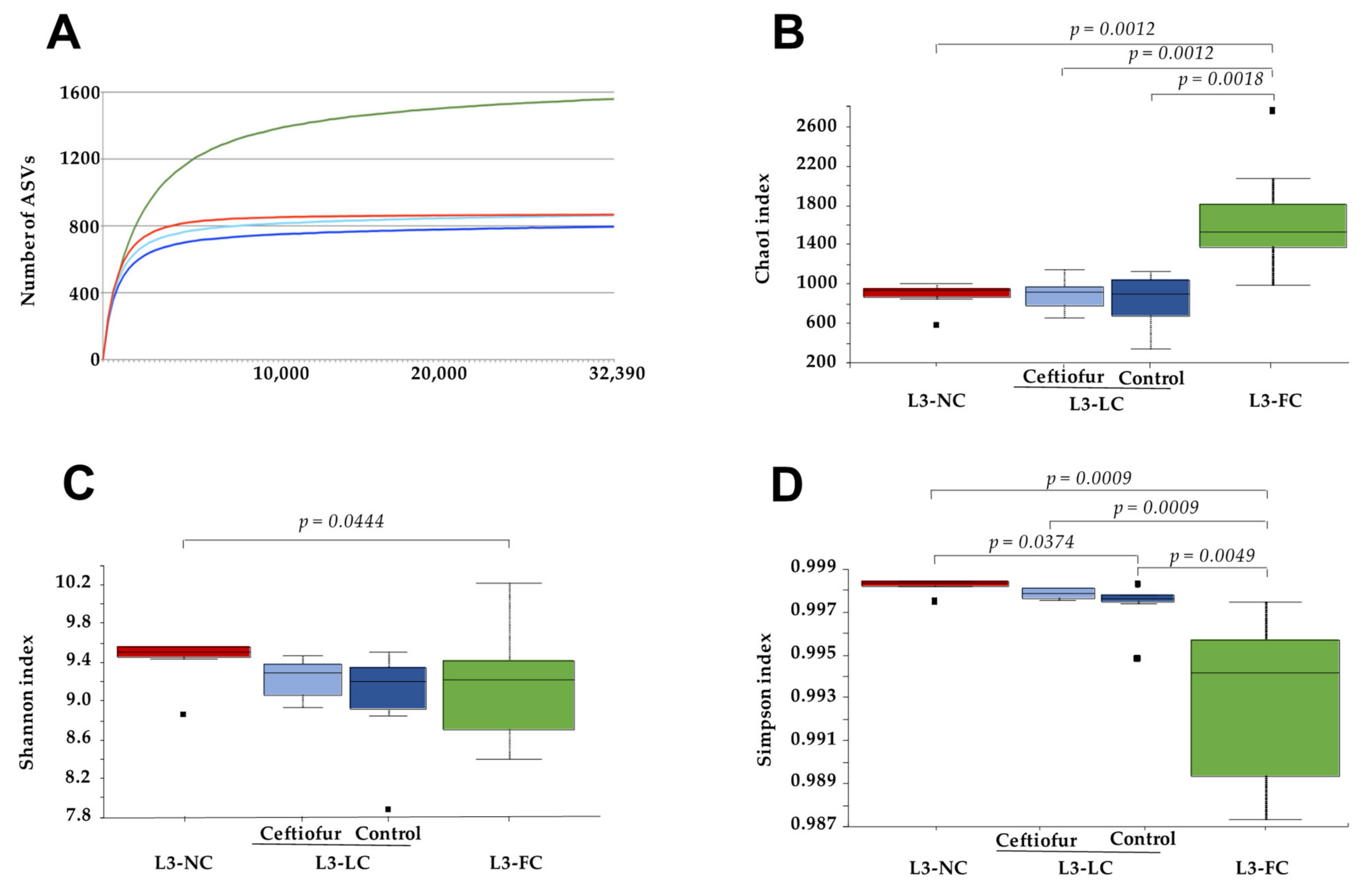
2.2. Alpha Diversity of the Nasal Microbiota of Piglets with Different Degree of Sow Contact
2.3. Nasal Microbiota Composition of Piglets Differed Depending on the Degree of Contact with Sows
2.4. Comparison of the Nasal Microbiota Core from Animals with Variable Sow Contact
2.5. Sow–Piglet Contact Had a Stronger Impact on the Nasal Microbiota of BSL3 Piglets Than the Environment
3. Discussion
4. Materials and Methods
4.1. Samples Included in the Study
4.2. DNA Extraction and 16S rRNA Gene Sequencing
4.3. Microbiota In Silico Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pickard, J.M.; Zeng, M.Y.; Caruso, R.; Núñez, G. Gut Microbiota: Role in Pathogen Colonization, Immune Responses and Inflammatory Disease. Immunol. Rev. 2017, 279, 70–89. [Google Scholar] [CrossRef]
- Barko, P.C.; McMichael, M.A.; Swanson, K.S.; Williams, D.A. The Gastrointestinal Microbiome: A Review. J. Vet. Intern. Med. 2018, 32, 9–25. [Google Scholar] [CrossRef]
- Lazar, V.; Ditu, L.-M.; Pircalabioru, G.G.; Gheorghe, I.; Curutiu, C.; Holban, A.M.; Picu, A.; Petcu, L.; Chifiriuc, M.C. Aspects of Gut Microbiota and Immune System Interactions in Infectious Diseases, Immunopathology, and Cancer. Front. Immunol. 2018, 9, 1830. [Google Scholar] [CrossRef]
- Rawls, M.; Ellis, A.K. The microbiome of the nose. Ann. Allergy Asthma Immunol. 2019, 122, 17–24. [Google Scholar] [CrossRef]
- Fouhse, J.M.; Zijlstra, R.T.; Willing, B.P. The Role of Gut Microbiota in the Health and Disease of Pigs. Anim. Front. 2016, 6, 30–36. [Google Scholar] [CrossRef]
- Niederwerder, M.C. Role of the Microbiome in Swine Respiratory Disease. Vet. Microbiol. 2017, 209, 97–106. [Google Scholar] [CrossRef]
- Nowland, T.; Plush, K.; Barton, M.; Kirkwood, R. Development and Function of the Intestinal Microbiome and Potential Implications for Pig Production. Animals 2019, 9, 76. [Google Scholar] [CrossRef] [PubMed]
- Lloyd-Price, J.; Abu-Ali, G.; Huttenhower, C. The Healthy Human Microbiome. Genome Med. 2016, 8, 51. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Ryan, C.A.; Boyaval, P.; Dempsey, E.M.; Ross, R.P.; Stanton, C. Maternal Vertical Transmission Affecting Early-Life Microbiota Development. Trends Microbiol. 2020, 28, 28–45. [Google Scholar] [CrossRef]
- Meat—Food and Agriculture Organization of the United Nations. 2020. Available online: http://www.fao.org/3/ca8861en/Meat.pdf (accessed on 26 March 2021).
- Cheng, G.; Hao, H.; Xie, S.; Wang, X.; Dai, M.; Huang, L.; Yuan, Z. Antibiotic Alternatives: The Substitution of Antibiotics in Animal Husbandry? Front. Microbiol. 2014, 5, 217. [Google Scholar] [CrossRef] [PubMed]
- Guidelines for the Prudent Use of Antimicrobials in Veterinary Medicine. Official Journal of the European Union (2015/C 299/04). Available online: https://ec.europa.eu/health/sites/health/files/antimicrobial_resistance/docs/2015_prudent_use_guidelines_en.pdf (accessed on 26 March 2021).
- Correa-Fiz, F.; Gonçalves dos Santos, J.M.; Illas, F.; Aragon, V. Antimicrobial Removal on Piglets Promotes Health and Higher Bacterial Diversity in the Nasal Microbiota. Sci. Rep. 2019, 9, 6545. [Google Scholar] [CrossRef]
- Guevarra, R.B.; Lee, J.H.; Lee, S.H.; Seok, M.-J.; Kim, D.W.; Kang, B.N.; Johnson, T.J.; Isaacson, R.E.; Kim, H.B. Piglet Gut Microbial Shifts Early in Life: Causes and Effects. J. Anim. Sci. Biotechnol. 2019, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Pirolo, M.; Espinosa-Gongora, C.; Bogaert, D.; Guardabassi, L. The Porcine Respiratory Microbiome: Recent Insights and Future Challenges. Anim. Microbiome 2021, 3, 1–13. [Google Scholar] [CrossRef]
- Jiang, L.; Feng, C.; Tao, S.; Li, N.; Zuo, B.; Han, D.; Wang, J. Maternal Imprinting of the Neonatal Microbiota Colonization in Intrauterine Growth Restricted Piglets: A Review. J. Anim. Sci. Biotechnol. 2019, 10, 1–8. [Google Scholar] [CrossRef]
- Sakwinska, O.; Foata, F.; Berger, B.; Brüssow, H.; Combremont, S.; Mercenier, A.; Dogra, S.; Soh, S.-E.; Yen, J.C.K.; Heong, G.Y.S.; et al. Does the Maternal Vaginal Microbiota Play a Role in Seeding the Microbiota of Neonatal Gut and Nose? Benef. Microbes 2017, 8, 763–778. [Google Scholar] [CrossRef]
- Correa-Fiz, F.; Fraile, L.; Aragon, V. Piglet Nasal Microbiota at Weaning May Influence the Development of Glässer’s Disease during the Rearing Period. BMC Genom. 2016, 17, 404. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.M.; Crenshaw, J.D.; Polo, J. The Biological Stress of Early Weaned Piglets. J. Anim. Sci. Biotechnol. 2013, 4, 19. [Google Scholar] [CrossRef] [PubMed]
- Baekbo, P.; Kristensen, C.S.; Larsen, L.E. Porcine Circovirus Diseases: A Review of PMWS: Porcine Circovirus Diseases. Transbound. Emerg. Dis. 2012, 59, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Jayaraman, B.; Nyachoti, C.M. Husbandry Practices and Gut Health Outcomes in Weaned Piglets: A Review. Anim. Nutr. 2017, 3, 205–211. [Google Scholar] [CrossRef]
- Blanco-Fuertes, M.; Correa-Fiz, F.; Fraile, L.; Sibila, M.; Aragon, V. Altered Nasal Microbiota Composition Associated with Development of Polyserositis by Mycoplasma hyorhinis. Pathogens 2021, 10, 603. [Google Scholar] [CrossRef] [PubMed]
- Murase, K.; Watanabe, T.; Arai, S.; Kim, H.; Tohya, M.; Ishida-Kuroki, K.; Võ, T.H.; Nguyễn, T.P.B.; Nakagawa, I.; Osawa, R.; et al. Characterization of Pig Saliva as the Major Natural Habitat of Streptococcus suis by Analyzing Oral, Fecal, Vaginal, and Environmental Microbiota. PLoS ONE 2019, 14, e0215983. [Google Scholar] [CrossRef] [PubMed]
- Pena Cortes, L.C.; LeVeque, R.M.; Funk, J.; Marsh, T.L.; Mulks, M.H. Development of the Tonsillar Microbiome in Pigs from Newborn through Weaning. BMC Microbiol. 2018, 18, 35. [Google Scholar] [CrossRef]
- Wang, M.; Radlowski, E.C.; Monaco, M.H.; Fahey, J.G.C.; Gaskins, H.R.; Donovan, S.M. Mode of Delivery and Early Nutrition Modulate Microbial Colonization and Fermentation Products in Neonatal Piglets. J. Nutr. 2013, 143, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Larivie, G. Salmonella Shedding Status of the Sow Affects the Microbiota of Their Piglets at Weaning. J. Appl. Microbiol. 2019, 126, 411–423. [Google Scholar] [CrossRef] [PubMed]
- Blanco, I.; Galina-Pantoja, L.; Oliveira, S.; Pijoan, C.; Sánchez, C.; Canals, A. Comparison between Haemophilus parasuis Infection in Colostrums-Deprived and Sow-Reared Piglets. Vet. Microbiol. 2004, 103, 21–27. [Google Scholar] [CrossRef]
- Opriessnig, T.; Gerber, P.F.; Halbur, P.G. Refinement of a Colostrum-Deprived Pig Model for Infectious Disease Research. MethodsX 2018, 5, 403–413. [Google Scholar] [CrossRef]
- Vahle, J.L.; Haynes, J.S.; Andrews, J.J. Interaction of Haemophilus parasuis with Nasal and Tracheal Mucosa Following Intranasal Inoculation of Cesarean Derived Colostrum Deprived (CDCD) swine. Can. J. Vet. Res. 1997, 61, 200–206. [Google Scholar]
- Weiss, S.; Xu, Z.Z.; Peddada, S.; Amir, A.; Bittinger, K.; Gonzalez, A.; Lozupone, C.; Zaneveld, J.R.; Vázquez-Baeza, Y.; Birmingham, A.; et al. Normalization and Microbial Differential Abundance Strategies Depend upon Data Characteristics. Microbiome 2017, 5, 27. [Google Scholar] [CrossRef] [PubMed]
- Correa-Fiz, F.; Blanco-Fuertes, M.; Navas, M.J.; Lacasta, A.; Bishop, R.P.; Githaka, N.; Onzere, C.; Le Potier, M.-F.; Almagro-Delgado, V.; Martinez, J.; et al. Comparative Analysis of the Fecal Microbiota from Different Species of Domesticated and Wild Suids. Sci. Rep. 2019, 9, 13616. [Google Scholar] [CrossRef] [PubMed]
- Lowe, B.A.; Marsh, T.L.; Isaacs-Cosgrove, N.; Kirkwood, R.N.; Kiupel, M.; Mulks, M.H. Microbial Communities in the Tonsils of Healthy Pigs. Vet. Microbiol. 2011, 147, 346–357. [Google Scholar] [CrossRef]
- De Arriba, L.M.; Lopez-Serrano, S.; Galofre-Mila, N.; Aragon, V. Characterisation of Bergeyella Spp. Isolated from the Nasal Cavities of Piglets. Vet. J. 2018, 234, 1–6. [Google Scholar] [CrossRef]
- Maes, D.; Sibila, M.; Kuhnert, P.; Segalés, J.; Haesebrouck, F.; Pieters, M. Update on Mycoplasma hyopneumoniae Infections in Pigs: Knowledge Gaps for Improved Disease Control. Transbound. Emerg. Dis. 2018, 65, 110–124. [Google Scholar] [CrossRef] [PubMed]
- Vacca, M.; Celano, G.; Calabrese, F.M.; Portincasa, P.; Gobbetti, M.; De Angelis, M. The Controversial Role of Human Gut Lachnospiraceae. Microorganisms 2020, 8, 573. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, G.E.; Metzler-Zebeli, B.U.; Lawlor, P.G. Impact of Intestinal Microbiota on Growth and Feed Efficiency in Pigs: A Review. Microorganisms 2020, 8, 1886. [Google Scholar] [CrossRef] [PubMed]
- Amat, S.; Lantz, H.; Munyaka, P.M.; Willing, B.P. Prevotella in Pigs: The Positive and Negative Associations with Production and Health. Microorganisms 2020, 8, 1584. [Google Scholar] [CrossRef] [PubMed]
- Ormerod, K.L.; Wood, D.L.A.; Lachner, N.; Gellatly, S.L.; Daly, J.N.; Parsons, J.D.; Dal’Molin, C.G.O.; Palfreyman, R.W.; Nielsen, L.K.; Cooper, M.A.; et al. Genomic Characterization of the Uncultured Bacteroidales Family S24-7 Inhabiting the Guts of Homeothermic Animals. Microbiome 2016, 4, 36. [Google Scholar] [CrossRef]
- Wexler, H.M. Bacteroides: The Good, the Bad, and the Nitty-Gritty. CMR 2007, 20, 593–621. [Google Scholar] [CrossRef]
- Mortensen, M.S.; Rasmussen, M.A.; Stokholm, J.; Brejnrod, A.D.; Balle, C.; Thorsen, J.; Krogfelt, K.A.; Bisgaard, H.; Sørensen, S.J. Modeling Transfer of Vaginal Microbiota from Mother to Infant in Early Life. eLife 2021, 10, e57051. [Google Scholar] [CrossRef] [PubMed]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science Using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods. 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A Versatile Open Source Tool for Metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- McDonald, D. An Improved Greengenes Taxonomy with Explicit Ranks for Ecological and Evolutionary Analyses of Bacteria and Archaea. ISME J. 2012, 6, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. Blast+: Architecture and Applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Lane, D.J. 16s/23s rRna Sequencing. Nucleic Acid Techniques in Bacterial Systematics; John Wiley and Sons: New York, NY, USA, 1991; pp. 115–175. [Google Scholar]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. Fasttree 2–Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Chao, A. Nonparametric Estimation of the Number of Classes in a Population. Scand. J. Stat. 1984, 4, 265–270. [Google Scholar]
- Shannon, C.; Weaver, W. The Mathematical Theory of Communication. Bell Syst. Tech. J. 1948, 27, 379–423. [Google Scholar] [CrossRef]
- Simpson, E.H. Measurement of Diversity. Nature 1949, 163, 688. [Google Scholar] [CrossRef]
- Kruskal, W.H.; Wallis, W.A. Use of Ranks in One-Criterion Variance Analysis. J. Am. Stat. Assoc. 1952, 47, 583–621. [Google Scholar] [CrossRef]
- Halko, N.; Martinsson, P.G.; Shkolnisky, Y.; Tygert, M. An Algorithm for the Principal Component Analysis of Large Data Sets. SIAM J. Sci. Comput. 2010, 33, 2580–2594. [Google Scholar] [CrossRef]
- Legendre, P.; Legendre, L. Numerical Ecology, 3rd ed.; Elsevier: Amsterdam, The Netherlands, 2012; p. 499. [Google Scholar]
- Vázquez-Baeza, Y.; Pirrung, M.; Gonzalez, A.; Knight, R. EMPeror: A Tool for Visualizing High-Throughput Microbial Community Data. GigaScience 2013, 2, 16. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, C.; Knight, R. UniFrac: A New Phylogenetic Method for Comparing Microbial Communities. AEM 2005, 71, 8228–8235. [Google Scholar] [CrossRef]
- Lozupone, C.A.; Hamady, M.; Kelley, S.T.; Knight, R. Quantitative and Qualitative β Diversity Measures Lead to Different Insights into Factors That Structure Microbial Communities. AEM 2007, 73, 1576–1585. [Google Scholar] [CrossRef]
- Anderson, M.J. A New Method for Non-Parametric Multivariate Analysis of Variance. Austral Ecol. 2001, 26, 32–46. [Google Scholar]
- Jari Oksanen, F.; Blanchet, G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Peter, R.; Minchin, R.B.; O’Hara, G.; Simpson, L.; et al. Vegan: Community Ecology Package. R Package Version 2.5-7. 2020. Available online: https://CRAN.R-project.org/package=vegan (accessed on 15 February 2021).
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-Learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Werner, J.J.; Koren, O.; Hugenholtz, P.; DeSantis, T.Z.; Walters, A.W.; Caporaso, J.G.; Angenent, L.T.; Knight, R.; Ley, E.R. Impact of training sets on classification of high-throughput bacterial 16s rRNA gene surveys. ISME J. 2012, 6, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Mandal, S.; Van Treuren, W.; White, R.A.; Eggesbø, M.; Knight, R.; Peddada, S.D. Analysis of Composition of Microbiomes: A Novel Method for Studying Microbial Composition. Microb. Ecol. Health Dis. 2015, 26, 27663. [Google Scholar] [CrossRef]
- Jiang, L.; Amir, A.; Morton, J.T.; Heller, R.; Arias-Castro, E.; Knight, R. Discrete False-Discovery Rate Improves Identification of Differentially Abundant Microbes. mSystems 2017, 2, e00092-17. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Housing | Birthplace | Sow-Contact | Perinatal Treatment | Reference |
|---|---|---|---|---|---|
| L3-NC | BSL3 | Farm (snatch farrowed) | No | Colistin and crystalline ceftiofur (N = 6) | This study |
| L3-LC | BSL3 | Farm | Less than 12 h | Crystalline ceftiofur (N = 7) Untreated (N = 6) | This study |
| L3-FC | BSL3 | BSL3 | Until weaning | Untreated (N = 11) | This study |
| FB-FR | Farm GM | Farm GM | Until weaning | Amoxicillin (N = 10) | [18] |
| Farm PT | Farm PT | Until weaning | Not available (N = 5) | ||
| Farm VL | Farm VL | Until weaning | Ceftiofur and tulathromycin (N = 5) |
| Taxonomy | Relative Abundance (%) | Statistics | |||
|---|---|---|---|---|---|
| Taxa * | L3-NC | L3-LC | L3-FC | DS-FDR (p) | ANCOM * (W) |
| Acidobacteria | 1.19 | 0.68 | 0.02 | 0.001 | |
| Actinobacteria | 1.72 | 0.71 | 6.21 | 0.001 | 30 |
| Actinomycetales | 0.97 | 0.38 | 5.02 | 0.001 | 122 |
| Corynebacteriaceae | 0.03 | 0.04 | 1.03 | 0.001 | 211 |
| Corynebacterium | 0.03 | 0.04 | 1.03 | 0.001 | |
| Micrococcaceae | 0.34 | 0.24 | 2.92 | 0.001 | |
| Rothia | 0.20 | 0.24 | 2.76 | 0.001 | |
| Coriobacteriales | 0.26 | 0.05 | 1.11 | 0.001 | 131 |
| Coriobacteriaceae | 0.26 | 0.05 | 1.11 | 0.001 | 206 |
| Bacteroidetes | 22.53 | 28.78 | 16.54 | 0.001 | 25 |
| Bacteroidales | 21.46 | 27.84 | 14.62 | 0.001 | |
| Bacteroidales;f__ | 1.85 | 4.00 | 2.36 | 0.001 | |
| Bacteroidales;f__;g__ | 1.85 | 4.00 | 2.36 | 0.001 | |
| Bacteroidaceae | 2.11 | 2.85 | 1.29 | 0.001 | |
| Bacteroides | 2.11 | 2.85 | 1.29 | 0.001 | |
| Porphyromonadaceae | 0.81 | 0.95 | 1.70 | 0.001 | |
| Porphyromonas | 0.06 | 0.01 | 1.09 | 0.001 | 448 |
| Prevotellaceae | 9.28 | 3.46 | 3.46 | 0.002 | |
| Prevotella | 9.28 | 3.46 | 3.46 | 0.002 | |
| Rikenellaceae | 0.56 | 1.61 | 0.13 | 0.001 | 216 |
| Rikenellaceae;g__ | 0.38 | 1.14 | 0.03 | 0.001 | 496 |
| S24-7 | 3.56 | 5.85 | 1.47 | 0.001 | |
| S24-7;g__ | 3.56 | 5.85 | 1.47 | 0.001 | |
| Paraprevotellaceae | 1.82 | 2.87 | 2.72 | 0.006 | |
| (Prevotella) | 1.69 | 2.34 | 1.46 | 0.001 | |
| p-2534-18B5 | 0.48 | 5.53 | 0.10 | 0.001 | 248 |
| p-2534-18B5;g__ | 0.48 | 5.53 | 0.10 | 0.001 | 505 |
| Flavobacteriales | 0.43 | 0.76 | 1.76 | 0.002 | |
| Flavobacteriaceae | 0.16 | 0.19 | 1.19 | 0.001 | |
| Cyanobacteria | 2.77 | 3.71 | 0.59 | 0.001 | 29 |
| Firmicutes | 58.43 | 50.69 | 37.69 | 0.027 | |
| Bacillales | 0.36 | 0.56 | 1.53 | 0.001 | |
| Staphylococcaceae | 0.26 | 0.55 | 1.12 | 0.001 | |
| Lactobacillales | 1.23 | 1.68 | 5.00 | 0.001 | |
| Aerococcaceae | 0.06 | 0.01 | 1.31 | 0.001 | 224 |
| Lactobacillaceae | 0.20 | 0.76 | 1.01 | 0.001 | |
| Lactobacillus | 0.20 | 0.76 | 1.00 | 0.001 | |
| Streptococcaceae | 0.73 | 0.49 | 1.62 | 0.001 | |
| Streptococcus | 0.59 | 0.42 | 1.52 | 0.001 | |
| Clostridiales | 54.65 | 45.83 | 28.82 | 0.001 | |
| Clostridiales;f__ | 7.58 | 8.77 | 2.46 | 0.001 | |
| Clostridiales;f__;g__ | 7.58 | 8.77 | 2.46 | 0.001 | |
| Christensenellaceae | 1.51 | 0.26 | 0.92 | 0.001 | |
| Christensenellaceae;g__ | 1.51 | 0.26 | 0.92 | 0.001 | |
| Clostridiaceae | 1.14 | 0.67 | 1.53 | 0.001 | |
| Lachnospiraceae | 17.25 | 11.67 | 5.94 | 0.001 | |
| Lachnospiraceae;__ | 3.77 | 2.79 | 1.23 | 0.001 | |
| Lachnospiraceae;g__ | 5.76 | 4.68 | 2.07 | 0.001 | |
| Blautia | 1.23 | 0.41 | 0.50 | 0.001 | |
| Coprococcus | 2.93 | 1.48 | 0.91 | 0.001 | |
| Roseburia | 1.22 | 0.17 | 0.23 | 0.001 | |
| Ruminococcaceae | 21.77 | 19.88 | 10.86 | 0.001 | |
| Ruminococcaceae;__ | 1.61 | 1.71 | 0.75 | 0.001 | |
| Oscillospira | 3.87 | 6.03 | 1.24 | 0.001 | |
| Ruminococcus | 4.41 | 3.52 | 1.27 | 0.001 | |
| (Mogibacteriaceae) | 0.89 | 0.52 | 1.20 | 0.001 | |
| (Mogibacteriaceae);g__ | 0.87 | 0.52 | 1.19 | 0.001 | |
| (Tissierellaceae) | 0.14 | 0.05 | 1.15 | 0.007 | |
| Proteobacteria | 8.21 | 9.46 | 31.06 | 0.001 | 27 |
| Rhizobiales | 0.95 | 1.18 | 0.16 | 0.001 | 123 |
| Neisseriales | 0.05 | 0.37 | 1.03 | 0.001 | 143 |
| Neisseriaceae | 0.05 | 0.37 | 1.03 | 0.005 | 241 |
| Pseudomonadales | 1.38 | 2.19 | 26.17 | 0.001 | 135 |
| Moraxellaceae | 1.15 | 2.01 | 26.01 | 0.002 | 216 |
| Moraxellaceae;__ | 0.12 | 0.37 | 1.41 | 0.001 | |
| Enhydrobacter | 0.34 | 0.23 | 18.32 | 0.001 | 450 |
| Moraxella | 0.44 | 0.77 | 5.98 | 0.001 | |
| Spirochaetes | 0.38 | 0.77 | 2.56 | 0.001 | 29 |
| Spirochaetales | 0.09 | 0.64 | 2.01 | 0.001 | 145 |
| Spirochaetaceae | 0.09 | 0.64 | 2.01 | 0.001 | 249 |
| Treponema | 0.09 | 0.64 | 2.01 | 0.001 | 493 |
| Tenericutes | 1.38 | 1.66 | 0.66 | 0.001 | 27 |
| Verrucomicrobia | 0.68 | 0.59 | 1.55 | 0.001 | |
| WCHB1-41 | 0.00 | 0.16 | 1.18 | 0.001 | 144 |
| RFP12 | 0.00 | 0.15 | 1.08 | 0.001 | 249 |
| RFP12;g__ | 0.00 | 0.15 | 1.08 | 0.001 | 522 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Obregon-Gutierrez, P.; Aragon, V.; Correa-Fiz, F. Sow Contact Is a Major Driver in the Development of the Nasal Microbiota of Piglets. Pathogens 2021, 10, 697. https://doi.org/10.3390/pathogens10060697
Obregon-Gutierrez P, Aragon V, Correa-Fiz F. Sow Contact Is a Major Driver in the Development of the Nasal Microbiota of Piglets. Pathogens. 2021; 10(6):697. https://doi.org/10.3390/pathogens10060697
Chicago/Turabian StyleObregon-Gutierrez, Pau, Virginia Aragon, and Florencia Correa-Fiz. 2021. "Sow Contact Is a Major Driver in the Development of the Nasal Microbiota of Piglets" Pathogens 10, no. 6: 697. https://doi.org/10.3390/pathogens10060697
APA StyleObregon-Gutierrez, P., Aragon, V., & Correa-Fiz, F. (2021). Sow Contact Is a Major Driver in the Development of the Nasal Microbiota of Piglets. Pathogens, 10(6), 697. https://doi.org/10.3390/pathogens10060697