
Hepatitis C Virus Glycan-Dependent Interactions and the Potential for Novel Preventative Strategies


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
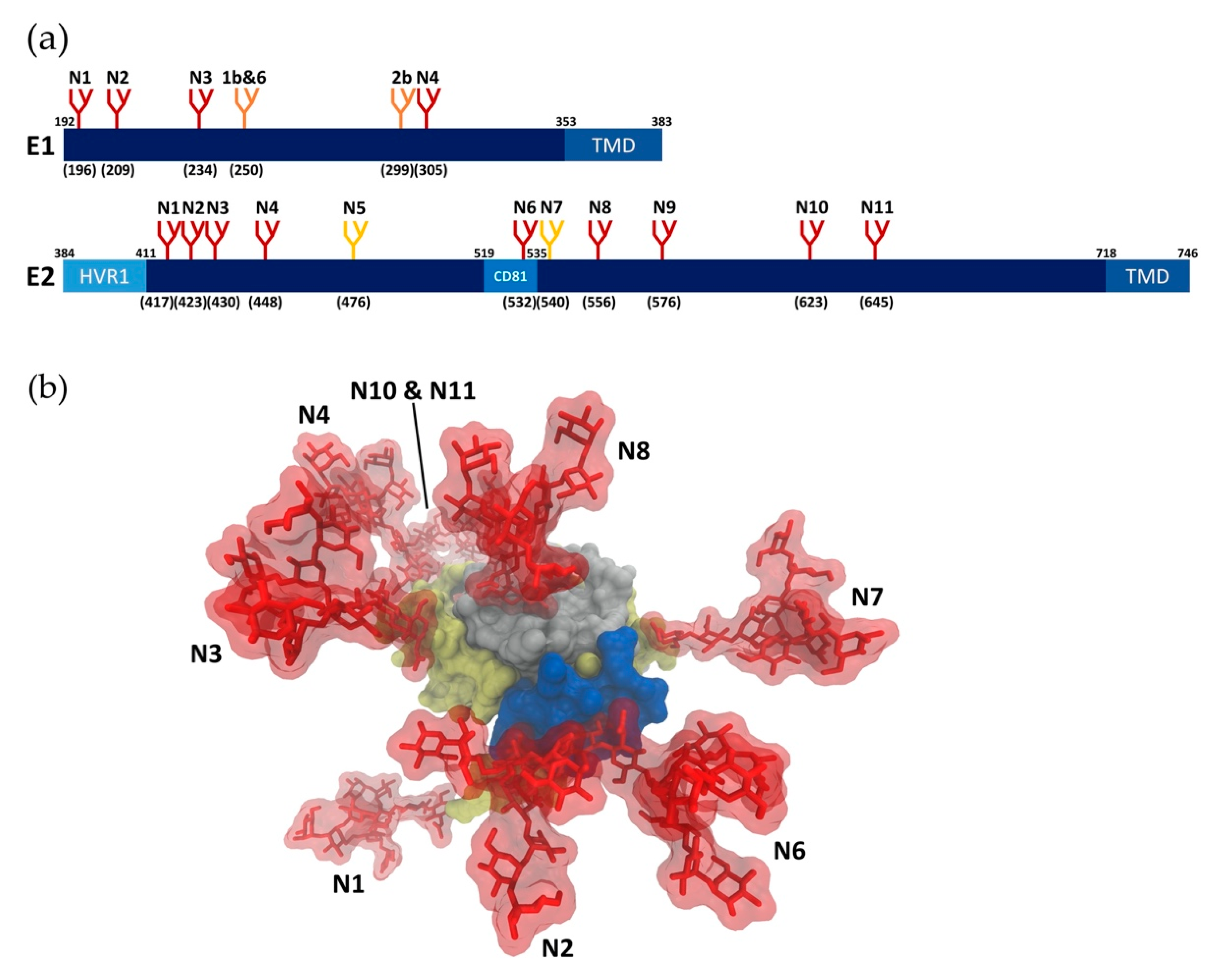
2. Viral Glycans
2.1. Glycan Profiling
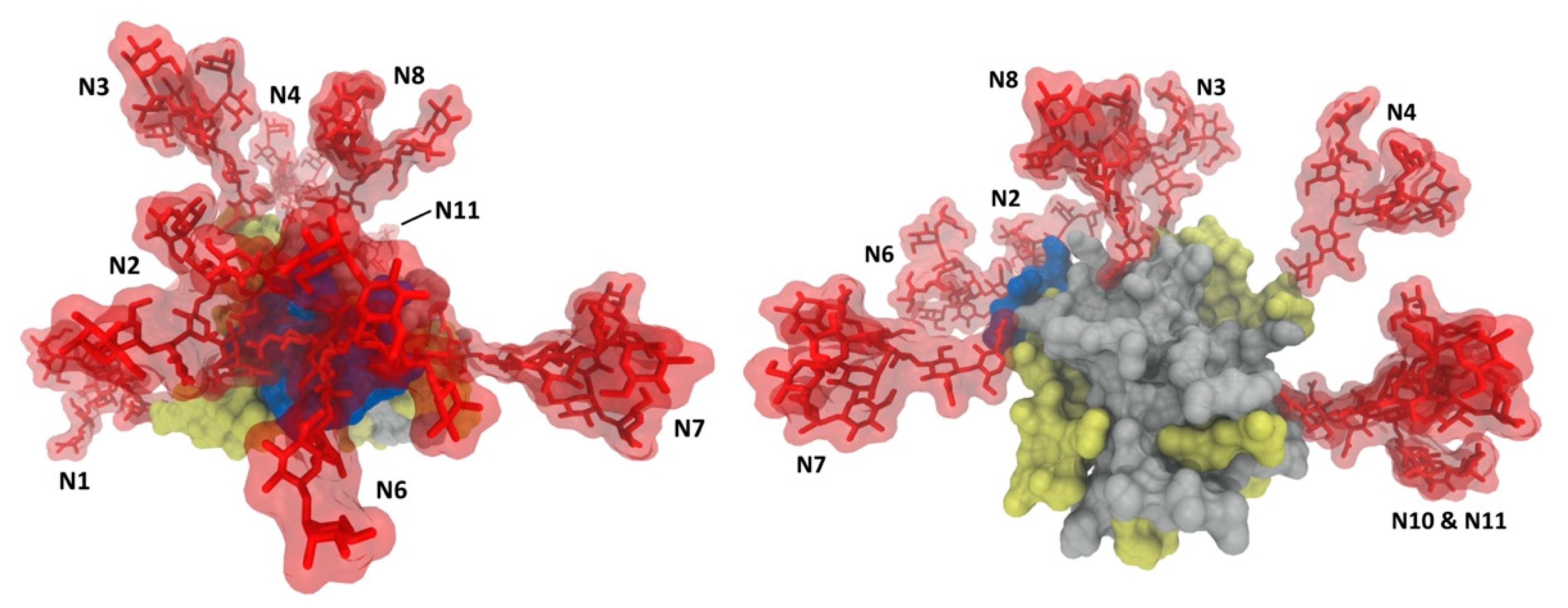
2.2. Glycan Functions
2.2.1. Glycans in Viral Entry & Infectivity
2.2.2. Glycans in Immune Evasion
2.2.3. Role of Glycans in the Envelope Breathing Model
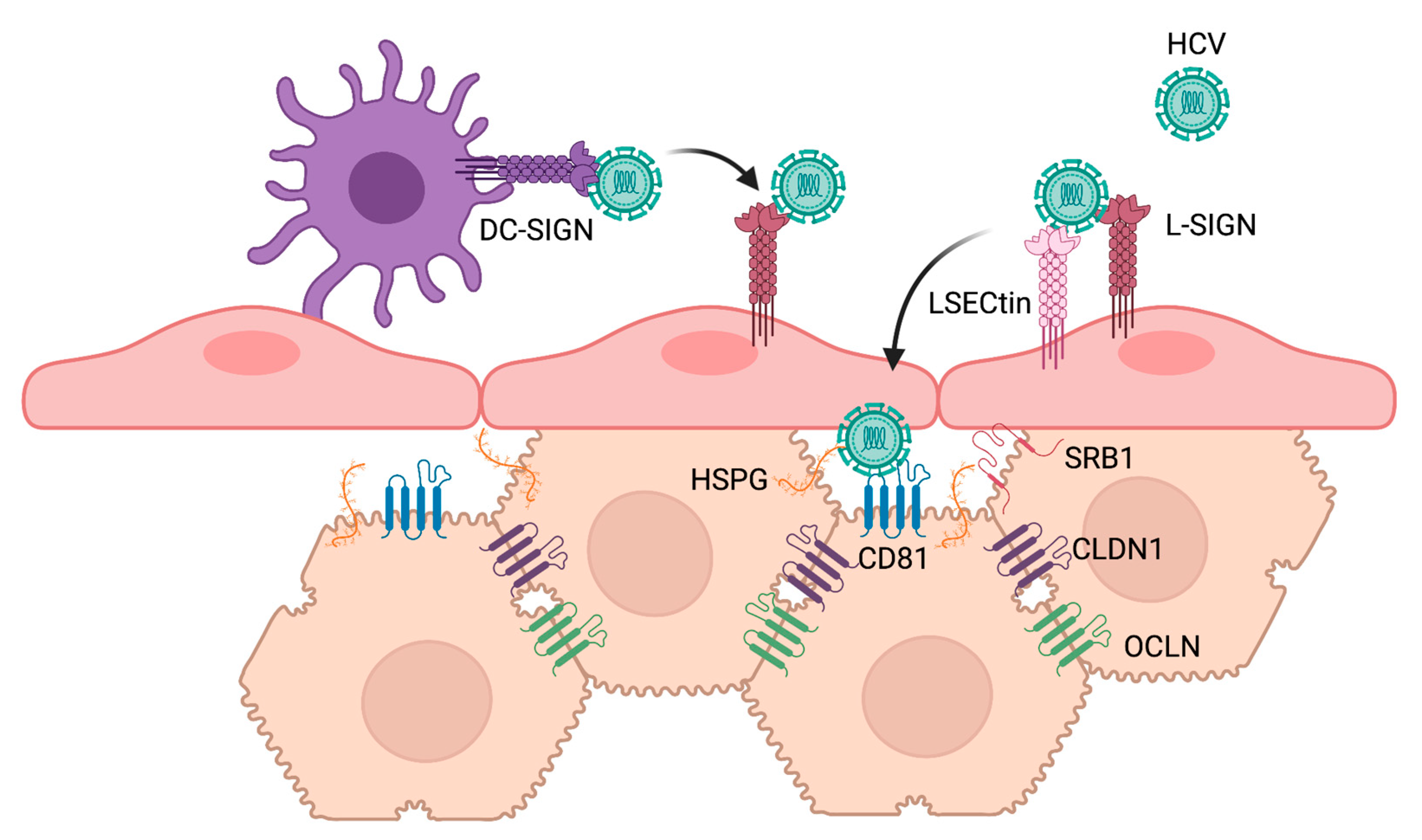
2.3. Viral Glycan Interactions with Cellular Lectin Receptors
3. Cellular Glycans
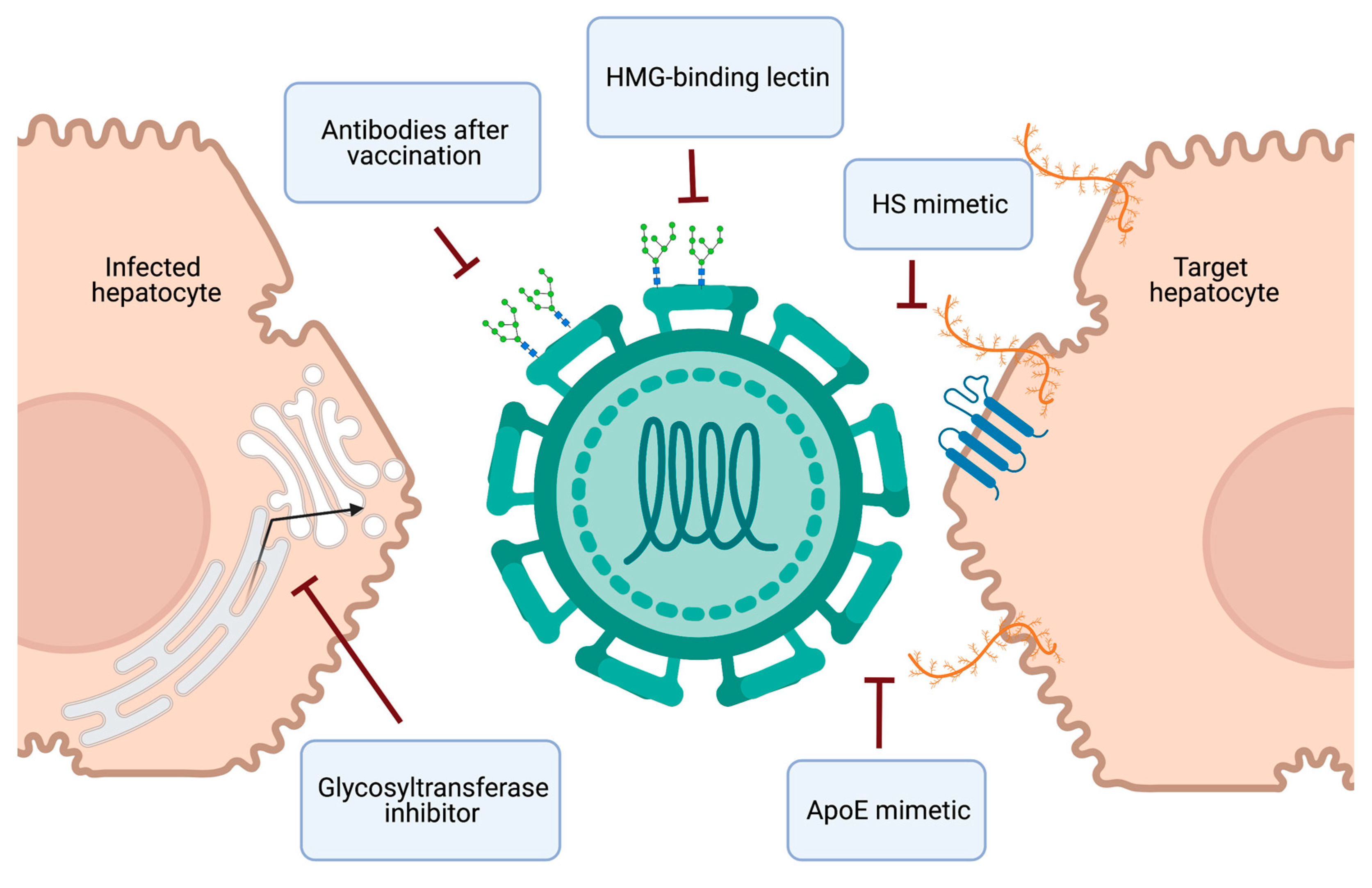
4. Therapeutics and Vaccine Development
4.1. Targeting the HCV-HSPG Interaction
4.2. Targeting Viral Glycans
4.3. Vaccine Development
5. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lauer, G.M.; Walker, B.D. Hepatitis C Virus Infection. N. Engl. J. Med. 2001, 345, 41–52. [Google Scholar] [CrossRef]
- Blach, S.; Zeuzem, S.; Manns, M.; Altraif, I.; Duberg, A.-S.; Muljono, D.H.; Waked, I.; Alavian, S.M.; Lee, M.-H.; Negro, F.; et al. Global prevalence and genotype distribution of hepatitis C virus infection in 2015: A modelling study. Lancet Gastroenterol. Hepatol. 2017, 2, 161–176. [Google Scholar] [CrossRef]
- Pileri, P.; Uematsu, Y.; Campagnoli, S.; Galli, G.; Falugi, F.; Petracca, R.; Weiner, A.J.; Houghton, M.; Rosa, D.; Grandi, G.; et al. Binding of Hepatitis C Virus to CD81. Science 1998, 282, 938–941. [Google Scholar] [CrossRef] [PubMed]
- Scarselli, E.; Ansuini, H.; Cerino, R.; Roccasecca, R.M.; Acali, S.; Filocamo, G.; Traboni, C.; Nicosia, A.; Cortese, R.; Vitelli, A. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J. 2002, 21, 5017–5025. [Google Scholar] [CrossRef]
- Ball, J.K.; Tarr, A.W.; McKeating, J.A. The past, present and future of neutralizing antibodies for hepatitis C virus. Antivir. Res. 2014, 105, 100–111. [Google Scholar] [CrossRef]
- Zając, M.; Muszalska, I.; Sobczak, A.; Dadej, A.; Tomczak, S.; Jelińska, A. Hepatitis C—New drugs and treatment prospects. Eur. J. Med. Chem. 2019, 165, 225–249. [Google Scholar] [CrossRef] [PubMed]
- De Monte, A.; Courjon, J.; Anty, R.; Cua, E.; Naqvi, A.; Mondain, V.; Cottalorda, J.; Ollier, L.; Giordanengo, V. Direct-acting antiviral treatment in adults infected with hepatitis C virus: Reactivation of hepatitis B virus coinfection as a further challenge. J. Clin. Virol. 2016, 78, 27–30. [Google Scholar] [CrossRef]
- Calvaruso, V.; Ferraro, D.; Licata, A.; Bavetta, M.G.; Petta, S.; Bronte, F.; Colomba, G.; Craxì, A.; Di Marco, V. HBV reactivation in patients with HCV/HBV cirrhosis on treatment with direct-acting antivirals. J. Viral Hepat. 2018, 25, 72–79. [Google Scholar] [CrossRef]
- Wang, C.; Ji, D.; Chen, J.; Shao, Q.; Li, B.; Liu, J.; Wu, V.; Wong, A.; Wang, Y.; Zhang, X.; et al. Hepatitis due to Reactivation of Hepatitis B Virus in Endemic Areas Among Patients With Hepatitis C Treated With Direct-acting Antiviral Agents. Clin. Gastroenterol. Hepatol. 2017, 15, 132–136. [Google Scholar] [CrossRef]
- Frazzoni, L.; Sikandar, U.; Metelli, F.; Sadalla, S.; Mazzella, G.; Bazzoli, F.; Fuccio, L.; Azzaroli, F. Hepatocellular Carcinoma Recurrence after Hepatitis C Virus Therapy with Direct-Acting Antivirals. A Systematic Review and Meta-Analysis. J. Clin. Med. 2021, 10, 1694. [Google Scholar] [CrossRef]
- Bartosch, B.; Dubuisson, J.; Cosset, F.-L. Infectious hepatitis C virus pseudo-particles containing functional E1-E2 envelope protein complexes. J. Exp. Med. 2003, 197, 633–642. [Google Scholar] [CrossRef]
- Lohmann, V.; Körner, F.; Koch, J.-O.; Herian, U.; Theilmann, L.; Bartenschlager, R. Replication of Subgenomic Hepatitis C Virus RNAs in a Hepatoma Cell Line. Science 1999, 285, 110–113. [Google Scholar] [CrossRef]
- Wakita, T.; Pietschmann, T.; Kato, T.; Date, T.; Miyamoto, M.; Zhao, Z.; Murthy, K.; Habermann, A.; Kräusslich, H.-G.; Mizokami, M.; et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 2005, 11, 791–796. [Google Scholar] [CrossRef]
- Crouchet, E.; Baumert, T.F.; Schuster, C. Hepatitis C virus-apolipoprotein interactions: Molecular mechanisms and clinical impact. Expert Rev. Proteom. 2017, 14, 593–606. [Google Scholar] [CrossRef] [PubMed]
- Colpitts, C.C.; Tsai, P.-L.; Zeisel, M.B. Hepatitis C Virus Entry: An Intriguingly Complex and Highly Regulated Process. Int. J. Mol. Sci. 2020, 21, 2091. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, M.B.; Felmlee, D.J.; Baumert, T.F. Hepatitis C virus entry. Curr. Top. Microbiol. Immunol. 2013, 369, 87–112. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Bowden, T.A.; Wilson, I.A.; Crispin, M. Exploitation of glycosylation in enveloped virus pathobiology. Biochim. Biophys. Acta Gen. Subj. 2019, 1480–1497. [Google Scholar] [CrossRef] [PubMed]
- Vigerust, D.J.; Shepherd, V.L. Virus glycosylation: Role in virulence and immune interactions. Trends Microbiol. 2007, 15, 211–218. [Google Scholar] [CrossRef]
- Goffard, A.; Dubuisson, J. Glycosylation of hepatitis C virus envelope proteins. Biochimie 2003, 85, 295–301. [Google Scholar] [CrossRef]
- Helle, F.; Goffard, A.; Morel, V.; Duverlie, G.; McKeating, J.; Keck, Z.-Y.; Foung, S.; Penin, F.; Dubuisson, J.; Voisset, C. The neutralizing activity of antihepatitis C virus antibodies is modulated by specific glycans on the E2 envelope protein. J. Virol. 2007, 81, 8101–8111. [Google Scholar] [CrossRef]
- Meunier, J.C.; Fournillier, A.; Choukhi, A.; Cahour, A.; Cocquerel, L.; Dubuisson, J.; Wychowski, C. Analysis of the glycosylation sites of hepatitis C virus (HCV) glycoprotein E1 and the influence of E1 glycans on the formation of the HCV glycoprotein complex. J. Gen. Virol. 1999, 80, 887–896. [Google Scholar] [CrossRef]
- Goffard, A.; Callens, N.; Bartosch, B.; Wychowski, C.; Cosset, F.-L.; Montpellier, C.; Dubuisson, J. Role of N-linked glycans in the functions of hepatitis C virus envelope glycoproteins. J. Virol. 2005, 79, 8400–8409. [Google Scholar] [CrossRef] [PubMed]
- Helle, F.; Vieyres, G.; Elkrief, L.; Popescu, C.-I.; Wychowski, C.; Descamps, V.; Castelain, S.; Roingeard, P.; Duverlie, G.; Dubuisson, J. Role of N-linked glycans in the functions of HCV envelope proteins incorporated into infectious virions. J. Virol. 2010, 84, 11905–11915. [Google Scholar] [CrossRef] [PubMed]
- Anjum, S.; Wahid, A.; Afzal, M.S.; Albecka, A.; Alsaleh, K.; Ahmad, T.; Baumert, T.F.; Wychowski, C.; Qadri, I.; Penin, F.; et al. Additional glycosylation within a specific hypervariable region of subtype 3a of hepatitis C virus protects against virus neutralization. J. Infect. Dis. 2013, 208, 1888–1897. [Google Scholar] [CrossRef] [PubMed]
- Fénéant, L.; Potel, J.; François, C.; Sané, F.; Douam, F.; Belouzard, S.; Calland, N.; Vausselin, T.; Rouillé, Y.; Descamps, V.; et al. New insights into the understanding of hepatitis C virus entry and cell to cell transmission by using the ionophore monensin A. J. Virol. 2015, 89, 8346–8364. [Google Scholar] [CrossRef] [PubMed]
- Steinmann, E.; Pietschmann, T. Cell culture systems for hepatitis C virus. In Hepatitis C Virus: From Molecular Virology to Antiviral Therapy. Current Topics in Microbiology and Immunology; Ralf, B., Ed.; Springer: Berlin/Heidelberg, Germany, 2013; Volume 369, pp. 17–48. [Google Scholar] [CrossRef]
- De Beeck, A.O.; Voisset, C.; Bartosch, B.; Ciczora, Y.; Cocquerel, L.; Keck, Z.; Foung, S.; Cosset, F.-L.; Dubuisson, J. Characterization of functional hepatitis C virus envelope glycoproteins. J. Virol. 2004, 78, 2994–3002. [Google Scholar] [CrossRef] [PubMed]
- Beyene, A.; Basu, A.; Meyer, K.; Ray, R. Influence of N-linked glycans on intracellular transport of hepatitis C virus E1 chimeric glycoprotein and its role in pseudotype virus infectivity. Virology 2004, 324, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Flint, M.; Logvinoff, C.; Rice, C.M.; McKeating, J.A. Characterization of infectious retroviral pseudotype particles bearing hepatitis C virus glycoproteins. J. Virol. 2004, 78, 6875–6882. [Google Scholar] [CrossRef]
- Iacob, R.E.; Perdivara, I.; Przybylski, M.; Tomer, K.B. Mass spectrometric characterization of glycosylation of hepatitis C virus E2 envelope glycoprotein reveals extended microheterogeneity of N-glycans. J. Am. Soc. Mass Spectrom. 2007, 19, 428–444. [Google Scholar] [CrossRef]
- Bräutigam, J.; Scheidig, A.J.; Egge-Jacobsen, W. Mass spectrometric analysis of hepatitis C viral envelope protein E2 reveals extended microheterogeneity of mucin-type O-linked glycosylation. Glycobiology 2013, 23, 453–474. [Google Scholar] [CrossRef]
- Miyanari, Y.; Atsuzawa, K.; Usuda, N.; Watashi, K.; Hishiki, T.; Zayas, M.; Bartenschlager, R.; Wakita, T.; Hijikata, M.; Shimotohno, K. The lipid droplet is an important organelle for hepatitis C virus production. Nat. Cell Biol. 2007, 9, 1089–1097. [Google Scholar] [CrossRef] [PubMed]
- Sandrin, V.; Boulanger, P.; Penin, F.; Granier, C.; Cosset, F.-L.; Bartosch, B. Assembly of functional hepatitis C virus glycoproteins on infectious pseudoparticles occurs intracellularly and requires concomitant incorporation of E1 and E2 glycoproteins. J. Gen. Virol. 2005, 86, 3189–3199. [Google Scholar] [CrossRef] [PubMed]
- Popescu, C.-I.; Riva, L.; Vlaicu, O.; Farhat, R.; Rouillé, Y.; Dubuisson, J. Hepatitis C virus life cycle and lipid metabolism. Biology 2014, 3, 892–921. [Google Scholar] [CrossRef]
- Guo, Y.; Yu, H.; Zhong, Y.; He, Y.; Qin, X.; Qin, Y.; Zhou, Y.; Zhang, P.; Zhang, Y.; Li, Z.; et al. Lectin microarray and mass spectrometric analysis of hepatitis C proteins reveals N-linked glycosylation. Medicine 2018, 97, e0208. [Google Scholar] [CrossRef] [PubMed]
- Whidby, J.; Mateu, G.; Scarborough, H.; Demeler, B.; Grakoui, A.; Marcotrigiano, J. Blocking hepatitis C virus infection with recombinant form of envelope protein 2 ectodomain. J. Virol. 2009, 83, 11078–11089. [Google Scholar] [CrossRef] [PubMed]
- Falkowska, E.; Kajumo, F.; Garcia, E.; Reinus, J.; Dragic, T. Hepatitis C virus envelope glycoprotein E2 glycans modulate entry, CD81 binding, and neutralization. J. Virol. 2007, 81, 8072–8079. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Giang, E.; Nieusma, T.; Kadam, R.U.; Cogburn, K.E.; Hua, Y.; Dai, X.; Stanfield, R.L.; Burton, D.R.; Ward, A.B.; et al. Hepatitis C virus E2 envelope glycoprotein core structure. Science 2013, 342, 1090–1094. [Google Scholar] [CrossRef]
- Delgrange, D.; Pillez, A.; Castelain, S.; Cocquerel, L.; Rouillé, Y.; Dubuisson, J.; Wakita, T.; Duverlie, G.; Wychowski, C. Robust production of infectious viral particles in Huh7 cells by introducing mutations in hepatitis C virus structural proteins. J. Gen. Virol. 2007, 88, 2495–2503. [Google Scholar] [CrossRef]
- Bungyoku, Y.; Shoji, I.; Makine, T.; Adachi, T.; Hayashida, K.; Nagano-Fujii, M.; Ide, Y.-H.; Deng, L.; Hotta, H. Efficient production of infectious hepatitis C virus with adaptive mutations in cultured hepatoma cells. J. Gen. Virol. 2009, 90, 1681–1691. [Google Scholar] [CrossRef]
- Prentoe, J.; Velázquez-Moctezuma, R.; Augestad, E.H.; Galli, A.; Wang, R.; Law, M.; Alter, H.; Bukh, J. Hypervariable region 1 and N-linked glycans of HCV regulate viron neutralization by modulating envelope conformations. Proc. Natl. Acad. Sci. USA 2019, 116, 10039–10047. [Google Scholar] [CrossRef]
- Huang, H.; Sun, F.; Owen, D.M.; Li, W.; Chen, Y.; Michael Gale, J.; Ye, J. Hepatitis C virus production by human hepatocytes dependent on assembly and secretion of very low density lipoproteins. Proc. Natl. Acad. Sci. USA 2007, 104, 5848–5853. [Google Scholar] [CrossRef]
- Gastaminza, P.; Cheng, G.; Wieland, S.; Zhong, J.; Liao, W.; Chisari, F.V. Cellular determinants of hepatitis C virus assembly, maturation, degradation, and secretion. J. Virol. 2008, 82, 2120–2129. [Google Scholar] [CrossRef]
- Wasilewski, L.N.; Ray, S.C.; Bailey, J.R. Hepatitis C virus resistance to broadly neutralizing antibodies measured using replication competent virus and pseudo particles. J. Gen. Virol. 2016, 97, 2883–2893. [Google Scholar] [CrossRef]
- Li, Y.; Pierce, B.G.; Wang, Q.; Keck, Z.-Y.; Fuerst, T.R.; Foung, S.K.H.; Mariuzza, R.A. Structural basis for penetration of the glycan shield of hepatitis C virus E2 glycoprotein by a broadly neutralizing human antibody. J. Biol. Chem. 2015, 290, P10117–P10125. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Gaschen, B.; Blay, W.; Foley, B.; Haigwood, N.; Kuiken, C.; Korber, B. Tracking global patterns of N-linked glycosylation site variation in highly variable viral glycoproteins: HIV, SIV, and HCV envelopes and influenza hemagglutinin. Glycobiology 2004, 14, 1229–1246. [Google Scholar] [CrossRef] [PubMed]
- Lavie, M.; Hanoulle, X.; Dubuisson, J. Glycan shielding and modulation of hepatitis C virus neutralizing antibodies. Front. Immunol. 2018, 9, 910. [Google Scholar] [CrossRef] [PubMed]
- Dustin, L.B.; Rice, C.M. Flying under the radar: The immunobiology of hepatitis C. Annu. Rev. Immunol. 2007, 25, 71–99. [Google Scholar] [CrossRef] [PubMed]
- Pierce, B.G.; Keck, Z.-Y.; Lau, P.; Fauvelle, C.; Gowthaman, R.; Baumert, T.F.; Fuerst, T.R.; Mariuzza, R.A.; Foung, S.K.H. Global mapping of antibody recognition of the hepatitis C virus E2 glycoprotein: Implications for vaccine design. Proc. Natl. Acad. Sci. USA 2016, 113, E6946–E6954. [Google Scholar] [CrossRef]
- Gopal, R.; Jackson, K.; Tzarum, N.; Kong, L.; Ettenger, A.; Guest, J.; Pfaff, J.M.; Barnes, T.; Honda, A.; Giang, E.; et al. Probing the antigenicity of hepatitis C virus envelope glycoprotein complex by high-throughput mutagenesis. PLoS Pathog. 2017, 13, e1006735. [Google Scholar] [CrossRef]
- Helle, F.; Duverlie, G.; Dubuisson, J. The hepatitis C virus glycan shield and evasion of the humoral immune response. Viruses 2011, 3, 1909–1932. [Google Scholar] [CrossRef]
- Pantua, H.; Diao, J.; Ultsch, M.; Hazen, M.; Mathieu, M.; McCutcheon, K.; Takeda, K.; Date, S.; Cheung, T.K.; Phung, Q.; et al. Glycan shifting on hepatitis C virus (HCV) E2 glycoprotein is a mechanism for escape from broadly neutralizing antibodies. J. Mol. Biol. 2013, 425, 1899–1914. [Google Scholar] [CrossRef] [PubMed]
- Flyak, A.I.; Ruiz, S.; Colbert, M.; Luong, T.; Crowe, J.E., Jr.; Bailey, J.R.; Bjorkman, P.J. Broadly neutralizing antibodies use a CDRH3 disulfide motif to recognize an E2 glycoprotein site that can be targeted for vaccine design. Cell Host Microbe 2018, 24, 703–716.e3. [Google Scholar] [CrossRef] [PubMed]
- Tzarum, N.; Giang, E.; Kong, L.; He, L.; Prentoe, J.; Augestad, E.; Hua, Y.; Castillo, S.; Lauer, G.M.; Bukh, J.; et al. Genetic and structural insights into broad neutralization of hepatitis C virus by human VH1-69 antibodies. Sci. Adv. 2019, 5, eaav1882. [Google Scholar] [CrossRef] [PubMed]
- Keck, Z.-Y.; Sung, V.M.H.; Perkins, S.; Rowe, J.; Paul, S.; Liang, T.J.; Lai, M.M.C.; Foung, S.K.H. Human monoclonal antibody to hepatitis C virus E1 glycoprotein that blocks virus attachment and viral infectivity. J. Virol. 2004, 78, 7257–7263. [Google Scholar] [CrossRef] [PubMed]
- Meunier, J.-C.; Russell, R.S.; Goossens, V.; Priem, S.; Walter, H.; Depla, E.; Union, A.; Faulk, K.N.; Bukh, J.; Emerson, S.U.; et al. Isolation and characterization of broadly neutralizing human monoclonal antibodies to the e1 glycoprotein of hepatitis C virus. J. Virol. 2008, 82, 966–973. [Google Scholar] [CrossRef] [PubMed]
- Prentoe, J.; Jensen, T.B.; Meuleman, P.; Serre, S.B.N.; Scheel, T.K.H.; Leroux-Roels, G.; Gottwein, J.M.; Bukh, J. Hypervariable region 1 differentially impacts viability of hepatitis C virus strains of genotypes 1 to 6 and impairs virus neutralization. J. Virol. 2011, 85, 2224–2234. [Google Scholar] [CrossRef]
- Prentoe, J.; Verhoye, L.; Moctezuma, R.V.; Buysschaert, C.; Farhoudi, A.; Wang, R.; Alter, H.; Meuleman, P.; Bukh, J. HVR1-mediated antibody evasion of highly infectious in vivo adapted HCV in humanised mice. Gut 2016, 65, 1988–1997. [Google Scholar] [CrossRef]
- Prentoe, J.; Velázquez-Moctezuma, R.; Foung, S.K.H.; Law, M.; Bukh, J. Hypervariable region 1 shielding of hepatitis C virus is a main contributor to genotypic differences in neutralization sensitivity. Hepatology 2016, 64, 1881–1892. [Google Scholar] [CrossRef]
- Prentoe, J.; Bukh, J. Hypervariable region 1 in envelope protein 2 of hepatitis C virus: A linchpin in neutralizing antibody evasion and viral entry. Front. Immunol. 2018, 9, 2146. [Google Scholar] [CrossRef]
- Khera, T.; Behrendt, P.; Bankwitz, D.; Brown, R.J.P.; Todt, D.; Doepke, M.; Khan, A.G.; Schulze, K.; Law, J.; Logan, M.; et al. Functional and immunogenic characterization of diverse HCV glycoprotein E2 variants. J. Hepatol. 2019, 70, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Dowd, K.A.; Jost, C.A.; Durbin, A.P.; Whitehead, S.S.; Pierson, T.C. A dynamic landscape for antibody binding modulates antibody-mediated neutralization of West Nile virus. PLoS Pathog. 2011, 7, e1002111. [Google Scholar] [CrossRef] [PubMed]
- Dowd, K.A.; Mukherjee, S.; Kuhn, R.J.; Pierson, T.C. Combined effects of the structural heterogeneity and dynamics of flaviviruses on antibody recognition. J. Virol. 2014, 88, 11726–11737. [Google Scholar] [CrossRef] [PubMed]
- Sabo, M.C.; Luca, V.C.; Ray, S.C.; Bukh, J.; Fremont, D.H.; Diamond, M.S. Hepatitis C virus epitope exposure and neutralization by antibodies is affected by time and temperature. Virology 2012, 422, 174–184. [Google Scholar] [CrossRef]
- Vieyres, G.; Descamps, V.; Duverlie, G.; Patel, A.H.; Dubuisson, J.; Thomas, X. Characterization of the envelope glycoproteins associated with infectious hepatitis C virus. J. Virol. 2010, 84, 10159–10168. [Google Scholar] [CrossRef]
- Tscherne, D.M.; Jones, C.T.; Evans, M.J.; Lindenbach, B.D.; McKeating, J.A.; Rice, C.M. Time- and temperature-dependent activation of hepatitis C virus for low-pH-triggered entry. J. Virol. 2006, 80, 1734–1741. [Google Scholar] [CrossRef]
- Khan, A.G.; Whidby, J.; Miller, M.T.; Scarborough, H.; Zatorski, A.V.; Cygan, A.; Price, A.A.; Yost, S.A.; Bohannon, C.D.; Jacob, J.; et al. Structure of the core ectodomain of the hepatitis C virus envelope glycoprotein 2. Nature 2014, 509, 381–384. [Google Scholar] [CrossRef]
- Gastaminza, P.; Dryden, K.A.; Boyd, B.; Wood, M.R.; Law, M.; Yeager, M.; Chisari, F.V. Ultrastructural and biophysical characterization of hepatitis C virus particles produced in cell culture. J. Virol. 2010, 84, 10999–11009. [Google Scholar] [CrossRef]
- Catanese, M.T.; Uryu, K.; Kopp, M.; Edwards, T.J.; Andrus, L.; Rice, W.J.; Silvestry, M.; Kuhn, R.J.; Rice, C.M. Ultrastructural analysis of hepatitis C virus particles. Proc. Natl. Acad. Sci. USA 2013, 110, 9505–9510. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, R.J.; Dowd, K.A.; Post, C.B.; Pierson, T.C. Shake, rattle, and roll: Impact of the dynamics of flavivirus particles on their interactions with the host. Virology 2015, 479–480, 508–517. [Google Scholar] [CrossRef]
- Pöhlmann, S.; Zhang, J.; Baribaud, F.; Chen, Z.; Leslie, G.J.; Lin, G.; Granelli-Piperno, A.; Doms, R.W.; Rice, C.M.; McKeating, J.A. Hepatitis C Virus Glycoproteins Interact with DC-SIGN and DC-SIGNR. J. Virol. 2003, 77, 4070–4080. [Google Scholar] [CrossRef]
- Gardner, J.P.; Durso, R.J.; Arrigale, R.R.; Donovan, G.P.; Maddon, P.J.; Dragic, T.; Olson, W.C. L-SIGN (CD 209L) is a liver-specific capture receptor for hepatitis C virus. Proc. Natl. Acad. Sci. USA 2003, 100, 4498–4503. [Google Scholar] [CrossRef]
- Lozach, P.-Y.; Lortat-Jacob, H.; De Lacroix De Lavalette, A.; Staropoli, I.; Foung, S.; Amara, A.; Houlès, C.; Fieschi, F.; Schwartz, O.; Virelizier, J.-L.; et al. DC-SIGN and L-SIGN Are High Affinity Binding Receptors for Hepatitis C Virus Glycoprotein E2. J. Biol. Chem. 2003, 278, 20358–20366. [Google Scholar] [CrossRef]
- Saunier, B.; Triyatni, M.; Ulianich, L.; Maruvada, P.; Yen, P.; Kohn, L.D. Role of the asialoglycoprotein receptor in binding and entry of hepatitis C virus structural proteins in cultured human hepatocytes. J. Virol. 2003, 77, 546–559. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Ali, M.A.M.; Shi, Y.; Zhao, Y.; Luo, F.; Yu, J.; Xiang, T.; Tang, J.; Li, D.; Hu, Q.; et al. Specifically binding of L-ficolin to N-glycans of HCV envelope glycoproteins E1 and E2 leads to complement activation. Cell. Mol. Immunol. 2009, 6, 235–244. [Google Scholar] [CrossRef]
- Hamed, M.R.; Brown, R.J.P.; Zothner, C.; Urbanowicz, R.A.; Mason, C.P.; Krarup, A.; McClure, C.P.; Irving, W.L.; Ball, J.K.; Harris, M.; et al. Recombinant human L-ficolin directly neutralizes hepatitis C virus entry. J. Innate Immun. 2014, 6, 676–684. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hao, B.; Kuai, X.; Xing, G.; Yang, J.; Chen, J.; Tang, L.; Zhang, L.; He, F. C-type lectin LSECtin interacts with DC-SIGNR and is involved in hepatitis C virus binding. Mol. Cell. Biochem. 2009, 327, 183–190. [Google Scholar] [CrossRef]
- Brown, K.S.; Keogh, M.J.; Owsianka, A.M.; Adair, R.; Patel, A.H.; Arnold, J.N.; Ball, J.K.; Sim, R.B.; Tarr, A.W.; Hickling, T.P. Specific interaction of hepatitis C virus glycoproteins with mannan binding lectin inhibits virus entry. Protein Cell 2010, 1, 664–674. [Google Scholar] [CrossRef] [PubMed]
- Gramberg, T.; Hofmann, H.; Möller, P.; Lalor, P.F.; Marzi, A.; Geier, M.; Krumbiegel, M.; Winkler, T.; Kirchhoff, F.; Adams, D.H.; et al. LSECtin interacts with filovirus glycoproteins and the spike protein of SARS coronavirus. Virology 2005, 340, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.-C.; Chuang, P.-K.; Chen, C.-H.; Chan, Y.-T.; Chen, J.-R.; Lin, S.-W.; Ma, C.; Hsu, T.-L.; Wong, C.-H. Role of N-Linked Glycans in the Interactions of Recombinant HCV Envelope Glycoproteins with Cellular Receptors. ACS Chem. Biol. 2014, 9, 1437–1443. [Google Scholar] [CrossRef] [PubMed]
- Lozach, P.-Y.; Amara, A.; Bartosch, B.; Virelizier, J.-L.; Arenzana-Seisdedos, F.; Cosset, F.-L.; Altmeyer, R. C-type Lectins L-SIGN and DC-SIGN Capture and Transmit Infectious Hepatitis C Virus Pseudotype Particles. J. Biol. Chem. 2004, 279, 32035–32045. [Google Scholar] [CrossRef] [PubMed]
- Cormier, E.G.; Durso, R.J.; Tsamis, F.; Boussemart, L.; Manix, C.; Olson, W.C.; Gardner, J.P.; Dragic, T. L-SIGN (CD209L) and DC-SIGN (CD209) mediate transinfection of liver cells by hepatitis C virus. Proc. Natl. Acad. Sci. USA 2004, 101, 14067–14072. [Google Scholar] [CrossRef]
- Ishibashi, M.; Morita, N.; Nomura-Kawaguchi, C.; Shimizu, Y.; Wakita, T.; Esumi, M. CLEC4M-positive and CD81-negative Huh7 cells are not susceptible to JFH-1 HCVcc infection but mediate transinfection. Arch. Virol. 2014, 159, 2949–2955. [Google Scholar] [CrossRef] [PubMed]
- Barth, H.; Schäfer, C.; Adah, M.I.; Zhang, F.; Linhardt, R.J.; Toyoda, H.; Kinoshita-Toyoda, A.; Toida, T.; van Kuppevelt, T.H.; Depla, E.; et al. Cellular Binding of Hepatitis C Virus Envelope Glycoprotein E2 Requires Cell Surface Heparan Sulfate. J. Biol. Chem. 2003, 278, 41003–41012. [Google Scholar] [CrossRef]
- Barth, H.; Schnober, E.K.; Zhang, F.; Linhardt, R.J.; Depla, E.; Boson, B.; Cosset, F.-L.; Patel, A.H.; Blum, H.E.; Baumert, T.F. Viral and Cellular Determinants of the Hepatitis C Virus Envelope-Heparan Sulfate Interaction. J. Virol. 2006, 80, 10579–10590. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Kanda, T.; Beyene, A.; Saito, K.; Meyer, K.; Ray, R. Sulfated Homologues of Heparin Inhibit Hepatitis C Virus Entry into Mammalian Cells. J. Virol. 2007, 81, 3933–3941. [Google Scholar] [CrossRef]
- Jiang, J.; Cun, W.; Wu, X.; Shi, Q.; Tang, H.; Luo, G. Hepatitis C Virus Attachment Mediated by Apolipoprotein E Binding to Cell Surface Heparan Sulfate. J. Virol. 2012, 86, 7256–7267. [Google Scholar] [CrossRef]
- Lefèvre, M.; Felmlee, D.J.; Parnot, M.; Baumert, T.F.; Schuster, C. Syndecan 4 Is Involved in Mediating HCV Entry through Interaction with Lipoviral Particle-Associated Apolipoprotein E. PLoS ONE 2014, 9, e95550. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Martinez, P.; Séron, K.; Luo, G.; Allain, F.; Dubuisson, J.; Belouzard, S. Characterization of Hepatitis C Virus Interaction with Heparan Sulfate Proteoglycans. J. Virol. 2015, 89, 3846–3858. [Google Scholar] [CrossRef] [PubMed]
- Lyon, M.; Deakin, J.A.; Gallagher, J.T. Liver heparan sulfate structure. A novel molecular design. J. Biol. Chem. 1994, 269, 11208–11215. [Google Scholar] [CrossRef]
- Toida, T.; Yoshida, H.; Toyoda, H.; Koshiishi, I.; Imanari, T.; Hileman, R.E.; Fromm, J.R.; Linhardt, R.J. Structural differences and the presence of unsubstituted amino groups in heparan sulphates from different tissues and species. Biochem. J. 1997, 322, 499–506. [Google Scholar] [CrossRef]
- Shi, Q.; Jiang, J.; Luo, G. Syndecan-1 Serves as the Major Receptor for Attachment of Hepatitis C Virus to the Surfaces of Hepatocytes. J. Virol. 2013, 87, 6866–6875. [Google Scholar] [CrossRef]
- Fan, H.; Qiao, L.; Kang, K.-D.; Fan, J.; Wei, W.; Luo, G. Attachment and Postattachment Receptors Important for Hepatitis C Virus Infection and Cell-to-Cell Transmission. J. Virol. 2017, 91, e00280-17. [Google Scholar] [CrossRef]
- Grigorov, B.; Reungoat, E.; Gentil dit Maurin, A.; Varbanov, M.; Blaising, J.; Michelet, M.; Manuel, R.; Parent, R.; Bartosch, B.; Zoulim, F.; et al. Hepatitis C virus infection propagates through interactions between Syndecan-1 and CD81 and impacts the hepatocyte glycocalyx. Cell. Microbiol. 2017, 19, e12711. [Google Scholar] [CrossRef]
- Timpe, J.M.; Stamataki, Z.; Jennings, A.; Hu, K.; Farquhar, M.J.; Harris, H.J.; Schwarz, A.; Desombere, I.; Roels, G.L.; Balfe, P.; et al. Hepatitis C virus cell-cell transmission in hepatoma cells in the presence of neutralizing antibodies. Hepatology 2008, 47, 17–24. [Google Scholar] [CrossRef]
- Vervaeke, P.; Alen, M.; Noppen, S.; Schols, D.; Oreste, P.; Liekens, S. Sulfated Escherichia coli K5 polysaccharide derivatives inhibit dengue virus infection of human microvascular endothelial cells by interacting with the viral envelope protein E domain III. PLoS ONE 2013, 8, e74035. [Google Scholar] [CrossRef] [PubMed]
- Hidari, K.I.P.J.; Ikeda, K.; Watanabe, I.; Abe, T.; Sando, A.; Itoh, Y.; Tokiwa, H.; Morita, K.; Suzuki, T. 3-O-sulfated glucuronide derivative as a potential anti-dengue virus agent. Biochem. Biophys. Res. Commun. 2012, 424, 573–578. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Li, B.; Linhardt, R.J. Pathogenesis and Inhibition of Flaviviruses from a Carbohydrate Perspective. Pharmaceuticals 2017, 10, 44. [Google Scholar] [CrossRef] [PubMed]
- Colpitts, C.C.; Schang, L.M. A Small Molecule Inhibits Virion Attachment to Heparan Sulfate- or Sialic Acid-Containing Glycans. J. Virol. 2014, 88, 7806–7817. [Google Scholar] [CrossRef] [PubMed]
- Ciesek, S.; von Hahn, T.; Colpitts, C.C.; Schang, L.M.; Friesland, M.; Steinmann, J.; Manns, M.P.; Ott, M.; Wedemeyer, H.; Meuleman, P.; et al. The green tea polyphenol, epigallocatechin-3-gallate, inhibits hepatitis C virus entry. Hepatology 2011, 54, 1947–1955. [Google Scholar] [CrossRef]
- Calland, N.; Albecka, A.; Belouzard, S.; Wychowski, C.; Duverlie, G.; Descamps, V.; Hober, D.; Dubuisson, J.; Rouillé, Y.; Séron, K. (−)-Epigallocatechin-3-gallate is a new inhibitor of hepatitis C virus entry. Hepatology 2012, 55, 720–729. [Google Scholar] [CrossRef]
- Bhat, R.; Adam, A.T.; Lee, J.J.; Deloison, G.; Rouillé, Y.; Séron, K.; Rotella, D.P. Structure–activity studies of (−)-epigallocatechin gallate derivatives as HCV entry inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 4162–4165. [Google Scholar] [CrossRef]
- O’Shea, D.; Law, J.; Egli, A.; Douglas, D.; Lund, G.; Forester, S.; Lambert, J.; Law, M.; Burton, D.R.; Tyrrell, D.L.J.; et al. Prevention of hepatitis C virus infection using a broad cross-neutralizing monoclonal antibody (AR4A) and epigallocatechin gallate. Liver Transpl. 2016, 22, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Shiha, G.; Soliman, R.; Elbasiony, M.; Darwish, N.H.E.; Mousa, S.A. Addition of Epigallocatechin Gallate 400 mg to Sofosbuvir 400 mg + Daclatisvir 60 mg With or Without Ribavirin in Treatment of Patients with Chronic Hepatitis C Improves the Safety Profile: A Pilot Study. Sci. Rep. 2019, 9, 13593. [Google Scholar] [CrossRef]
- Liu, S.; McCormick, K.D.; Zhao, W.; Zhao, T.; Fan, D.; Wang, T. Human apolipoprotein E peptides inhibit hepatitis C virus entry by blocking virus binding. Hepatology 2012, 56, 484–491. [Google Scholar] [CrossRef]
- Nikoulin, I.R.; Curtiss, L.K. An apolipoprotein E synthetic peptide targets to lipoproteins in plasma and mediates both cellular lipoprotein interactions in vitro and acute clearance of cholesterol-rich lipoproteins in vivo. J. Clin. Investig. 1998, 101, 223–234. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lynch, J.R.; Tang, W.; Wang, H.; Vitek, M.P.; Bennett, E.R.; Sullivan, P.M.; Warner, D.S.; Laskowitz, D.T. ApoE Genotype and an ApoE-mimetic Peptide Modify the Systemic and Central Nervous System Inflammatory Response. J. Biol. Chem. 2003, 278, 48529–48533. [Google Scholar] [CrossRef]
- Krol, E.; Wandzik, I.; Szeja, W.; Grynkiewicz, G.; Szewczyk, B. In vitro antiviral activity of some uridine derivatives of 2-deoxy sugars against classical swine fever virus. Antiviral Res. 2010, 86, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Krol, E.; Wandzik, I.; Krejmer-Rabalska, M.; Szewczyk, B. Biological Evaluation of Uridine Derivatives of 2-Deoxy Sugars as Potential Antiviral Compounds against Influenza a Virus. Int. J. Mol. Sci. 2017, 18, 1700. [Google Scholar] [CrossRef]
- Krol, E.; Wandzik, I.; Pastuch-Gawolek, G.; Szewczyk, B. Anti-Hepatitis C Virus Activity of Uridine Derivatives of 2-Deoxy Sugars. Molecules 2018, 23, 1547. [Google Scholar] [CrossRef]
- Swanson, M.D.; Boudreaux, D.M.; Salmon, L.; Chugh, J.; Winter, H.C.; Meagher, J.L.; André, S.; Murphy, P.V.; Oscarson, S.; Roy, R.; et al. Engineering a Therapeutic Lectin by Uncoupling Mitogenicity from Antiviral Activity. Cell 2015, 163, 746–758. [Google Scholar] [CrossRef]
- Hamorsky, K.T.; Kouokam, J.C.; Dent, M.W.; Grooms, T.N.; Husk, A.S.; Hume, S.D.; Rogers, K.A.; Villinger, F.; Morris, M.K.; Hanson, C.V.; et al. Engineering of a Lectibody Targeting High-Mannose-Type Glycans of the HIV Envelope. Mol. Ther. 2019, 27, 2038–2052. [Google Scholar] [CrossRef]
- Dent, M.; Hamorsky, K.; Vausselin, T.; Dubuisson, J.; Miyata, Y.; Morikawa, Y.; Matoba, N. Safety and Efficacy of Avaren-Fc Lectibody Targeting HCV High-Mannose Glycans in a Human Liver Chimeric Mouse Model. Cell. Mol. Gastroenterol. Hepatol. 2021, 11, 185–198. [Google Scholar] [CrossRef]
- Fauvelle, C.; Colpitts, C.C.; Keck, Z.-Y.; Pierce, B.G.; Foung, S.K.H.; Baumert, T.F. Hepatitis C virus vaccine candidates inducing protective neutralizing antibodies. Expert Rev. Vaccines 2016, 15, 1535–1544. [Google Scholar] [CrossRef]
- Zingaretti, C.; De Francesco, R.; Abrignan, S. Why is it so difficult to develop a hepatitis C virus preventive vaccine? Clin. Microbiol. Infect. 2014, 20, 103–109. [Google Scholar] [CrossRef]
- Baumert, T.F.; Fauvelle, C.; Chen, D.Y.; Lauer, G.M. A prophylactic hepatitis C virus vaccine: A distant peak still worth climbing. J. Hepatol. 2014, 61, S34–S44. [Google Scholar] [CrossRef]
- Ghasemi, F.; Rostami, S.; Meshkat, Z. Progress in the development of vaccines for hepatitis C virus infection. World J. Gastroenterol. 2015, 21, 11984–12002. [Google Scholar] [CrossRef]
- Ogholikhan, S.; Schwarz, K.B. Hepatitis Vaccines. Vaccines 2016, 4, 6. [Google Scholar] [CrossRef] [PubMed]
- Vieyres, G.; Dubuisson, J.; Patel, A.H. Characterization of antibody-mediated neutralization directed against the hypervariable region 1 of hepatitis C virus E2 glycoprotein. J. Gen. Virol. 2011, 92, 494–506. [Google Scholar] [CrossRef] [PubMed]
- Rappuoli, R. Glycoconjugate vaccines: Principles and mechanisms. Sci. Transl. Med. 2018, 10, eaat4615. [Google Scholar] [CrossRef] [PubMed]
- Calarese, D.A.; Lee, H.-K.; Huang, C.-Y.; Best, M.D.; Astronomo, R.D.; Stanfield, R.L.; Katinger, H.; Burton, D.R.; Wong, C.-H.; Wilson, I.A. Dissection of the carbohydrate specificity of the broadly neutralizing anti-HIV-1 antibody 2G12. Proc. Natl. Acad. Sci. USA 2005, 102, 13372–13377. [Google Scholar] [CrossRef] [PubMed]
- Stanfield, R.L.; De Castro, C.; Marzaioli, A.M.; Wilson, I.A.; Pantophlet, R. Crystal structure of the HIV neutralizing antibody 2G12 in complex with a bacterial oligosaccharide analog of mammalian oligomannose. Glycobiology 2015, 25, 412–419. [Google Scholar] [CrossRef]
- Binley, J.M.; Wrin, T.; Korber, B.; Zwick, M.B.; Wang, M.; Chappey, C.; Stiegler, G.; Kunert, R.; Zolla-Pazner, S.; Katinger, H.; et al. Comprehensive cross-clade neutralization analysis of a panel of anti-human immunodeficiency virus type 1 monoclonal antibodies. J. Virol. 2004, 78, 13232–13252. [Google Scholar] [CrossRef] [PubMed]
- Bastida, I.; Fernández-Tejada, A. Synthetic carbohydrate-based HIV-1 vaccines. Drug Discov. Today Technol. 2020, 35–36, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Fournillier, A.; Wychowski, C.; Boucreux, D.; Baumert, T.F.; Meunier, J.-C.; Jacobs, D.; Muguet, S.; Depla, E.; Inchauspé, G. Induction of hepatitis C virus E1 envelope protein-specific immune response can be enhanced by mutation of N-glycosylation sites. J. Virol. 2001, 75, 12088–12097. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Chen, H.; Luo, F.; Li, P.; Pan, Q.; Xia, B.; Qi, Z.; Ho, W.-Z.; Zhang, X.-L. Deletion of N-glycosylation sites of hepatitis C virus envelope protein E1 enhances specific cellular and humoral immune responses. Vaccine 2007, 25, 6572–6580. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Wan, Q.; Feng, Y.; Liu, M.; Wu, J.; Chen, X.; Zhang, X.-L. Engineering of N-glycosylation of hepatitis C virus envelope protein E2 enhances T cell responses for DNA immunization. Vaccine 2007, 25, 1544–1551. [Google Scholar] [CrossRef]
- Ren, Y.; Min, Y.-Q.; Liu, M.; Chi, L.; Zhao, P.; Zhang, X.-L. N-glycosylation mutated HCV envelope glycoprotein complex enhances antigen presenting activity and cellular and neutralizing antibody responses. Biochim. Biophys. Acta 2016, 1860, 1764–1775. [Google Scholar] [CrossRef]
- Liu, J.C.; Paruch, L.; Dobrica, M.-O.; Caras, I.; Tucureanu, C.; Onu, A.; Ciulean, S.; Stavaru, C.; Eerde, A.; Wang, Y.; et al. Lettuce produced hepatitis C virus E1E2 heterodimer triggers immune responses in mice and antibody production after oral vaccination. Plant Biotechnol. J. 2017, 15, 1611–1621. [Google Scholar] [CrossRef]
- Pierce, B.G.; Keck, Z.-Y.; Wang, R.; Lau, P.; Garagusi, K.; Elkholy, K.; Toth, E.A.; Urbanowicz, R.A.; Guest, J.D.; Agnihotri, P.; et al. Structure-Based Design of Hepatitis C Virus E2 Glycoprotein Improves Serum Binding and Cross-Neutralization. J. Virol. 2020, 94, e00704-20. [Google Scholar] [CrossRef]
- Ströh, L.J.; Nagarathinam, K.; Krey, T. Conformational Flexibility in the CD81-Binding Site of the Hepatitis C virus Glycoprotein E2. Front. Immunol. 2018, 9, 1396. [Google Scholar] [CrossRef]
- Meola, A.; Tarr, A.W.; England, P.; Meredith, L.W.; McClure, C.P.; Foung, S.K.H.; McKeating, J.A.; Ball, J.K.; Rey, F.A.; Krey, T. Structural flexibility of a conserved broadly neutralizing epitope in hepatitis C virus glycoprotein E2. J. Virol. 2015, 89, 2170–2181. [Google Scholar] [CrossRef]
- Chen, L.; Do Kwon, Y.; Zhou, T.; Wu, X.; O’Dell, S.; Cavacini, L.; Hessell, A.J.; Pancera, M.; Tang, M.; Xu, L.; et al. Structural Basis of Immune Evasion at the Site of CD4 Attachment on HIV-1 gp120. Science 2009, 326, 1123–1127. [Google Scholar] [CrossRef]
- Li, D.; von Schaewen, M.; Wang, X.; Tao, W.; Zhang, Y.; Li, L.; Heller, B.; Hrebikova, G.; Deng, Q.; Ploss, A.; et al. Altered glycosylation patterns increase immunogenicity of a subunit hepatitis C virus vaccine, inducing neutralizing antibodies which confer protection in mice. J. Virol. 2016, 90, 10486–10498. [Google Scholar] [CrossRef] [PubMed]
- Urbanowicz, R.A.; Wang, R.; Schiel, J.E.; Keck, Z.; Kerzic, M.C.; Lau, P.; Rangarajan, S.; Garagusi, K.J.; Tan, L.; Guest, J.D.; et al. Antigenicity and immunogenicity of differentially glycosylated HCV E2 envelop proteins expressed in mammalian and insect cells. J. Virol. 2019, 93, e01403-18. [Google Scholar] [CrossRef] [PubMed]
- Pantophlet, R.; Wilson, I.A.; Burton, D.R. Hyperglycosylated mutants of human immunodeficiency virus (HIV) type 1 monomeric gp120 as novel antigens for HIV vaccine design. J. Virol. 2003, 77, 5889–5901. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ingale, J.; Tran, K.; Kong, L.; Dey, B.; McKee, K.; Schief, W.; Kwong, P.D.; Mascola, J.R.; Wyatt, R.T. Hyperglycosylated stable core immunogens designed to present the CD4 binding site are preferentially recognized by broadly neutralizing antibodies. J. Virol. 2014, 88, 14002–14016. [Google Scholar] [CrossRef]
- Ringe, R.P.; Ozorowski, G.; Rantalainen, K.; Struwe, W.B.; Matthews, K.; Torres, J.L.; Yasmeen, A.; Cottrell, C.A.; Ketas, T.J.; LaBranche, C.C.; et al. Reducing V3 antigenicity and immunogenicity on soluble, native-like HIV-1 Env SOSIP trimers. J. Virol. 2017, 91, e00677-17. [Google Scholar] [CrossRef] [PubMed]
- Kachko, A.; Frey, S.E.; Sirota, L.; Ray, R.; Wells, F.; Zubkova, I.; Zhang, P.; Major, M.E. Antibodies to an interfering epitope in hepatitis C virus E2 can mask vaccine-induced neutralizing activity. Hepatology 2015, 62, 1670–1682. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Zhong, L.; Struble, E.B.; Watanabe, H.; Kachko, A.; Mihalik, K.; Virata-Theimer, M.L.; Alter, H.J.; Feinstone, S.; Major, M. Depletion of interfering antibodies in chronic hepatitis C patients and vaccinated chimpanzees reveals broad cross-genotype neutralizing activity. Proc. Natl. Acad. Sci. USA 2009, 106, 7537–7541. [Google Scholar] [CrossRef]
- Hajarizadeh, B.; Grebely, J.; Byrne, M.; Marks, P.; Amin, J.; McManus, H.; Butler, T.; Cunningham, E.B.; Vickerman, P.; Martin, N.K.; et al. Evaluation of hepatitis C treatment-as-prevention within Australian prisons (SToP-C): A prospective cohort study. Lancet Gastroenterol. Hepatol. 2021. [Google Scholar] [CrossRef]
- Hu, T.-H.; Su, W.-W.; Yang, C.-C.; Yang, C.-C.; Kuo, W.-H.; Chen, Y.-Y.; Yeh, Y.-H.; Chen, S.-S.; Tsao, Y.-Y.; Chen, K.-M.; et al. Elimination of Hepatitis C Virus in a Dialysis Population: A Collaborative Care Model in Taiwan. Am. J. Kidney Dis. 2021. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Ren, Y.; Zhang, X.; Zhao, P.; Tao, W.; Zhong, J.; Li, Q.; Zhang, X.-L. Ficolin-2 Inhibits Hepatitis C Virus Infection, whereas Apolipoprotein E3 Mediates Viral Immune Escape. J. Immunol. 2014, 193, 783–796. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo, L.; Oliveira, C.; Fournier, C.; Descamps, V.; Morel, V.; Dubuisson, J.; Brochot, E.; Francois, C.; Castelain, S.; Duverlie, G.; et al. Hepatitis C Virus Resistance to Carbohydrate-Binding Agents. PLoS ONE 2016, 11, e0149064. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
LeBlanc, E.V.; Kim, Y.; Capicciotti, C.J.; Colpitts, C.C. Hepatitis C Virus Glycan-Dependent Interactions and the Potential for Novel Preventative Strategies. Pathogens 2021, 10, 685. https://doi.org/10.3390/pathogens10060685
LeBlanc EV, Kim Y, Capicciotti CJ, Colpitts CC. Hepatitis C Virus Glycan-Dependent Interactions and the Potential for Novel Preventative Strategies. Pathogens. 2021; 10(6):685. https://doi.org/10.3390/pathogens10060685
Chicago/Turabian StyleLeBlanc, Emmanuelle V., Youjin Kim, Chantelle J. Capicciotti, and Che C. Colpitts. 2021. "Hepatitis C Virus Glycan-Dependent Interactions and the Potential for Novel Preventative Strategies" Pathogens 10, no. 6: 685. https://doi.org/10.3390/pathogens10060685
APA StyleLeBlanc, E. V., Kim, Y., Capicciotti, C. J., & Colpitts, C. C. (2021). Hepatitis C Virus Glycan-Dependent Interactions and the Potential for Novel Preventative Strategies. Pathogens, 10(6), 685. https://doi.org/10.3390/pathogens10060685