
Applying Machine Learning to Predict the Exportome of Bovine and Canine Babesia Species That Cause Babesiosis


Abstract
1. Introduction
2. Results
2.1. Rule-Based Approach for Predicting an Exportome
2.2. Amino Acid Frequency
2.3. Machine Learning Approach for Predicting an Exportome
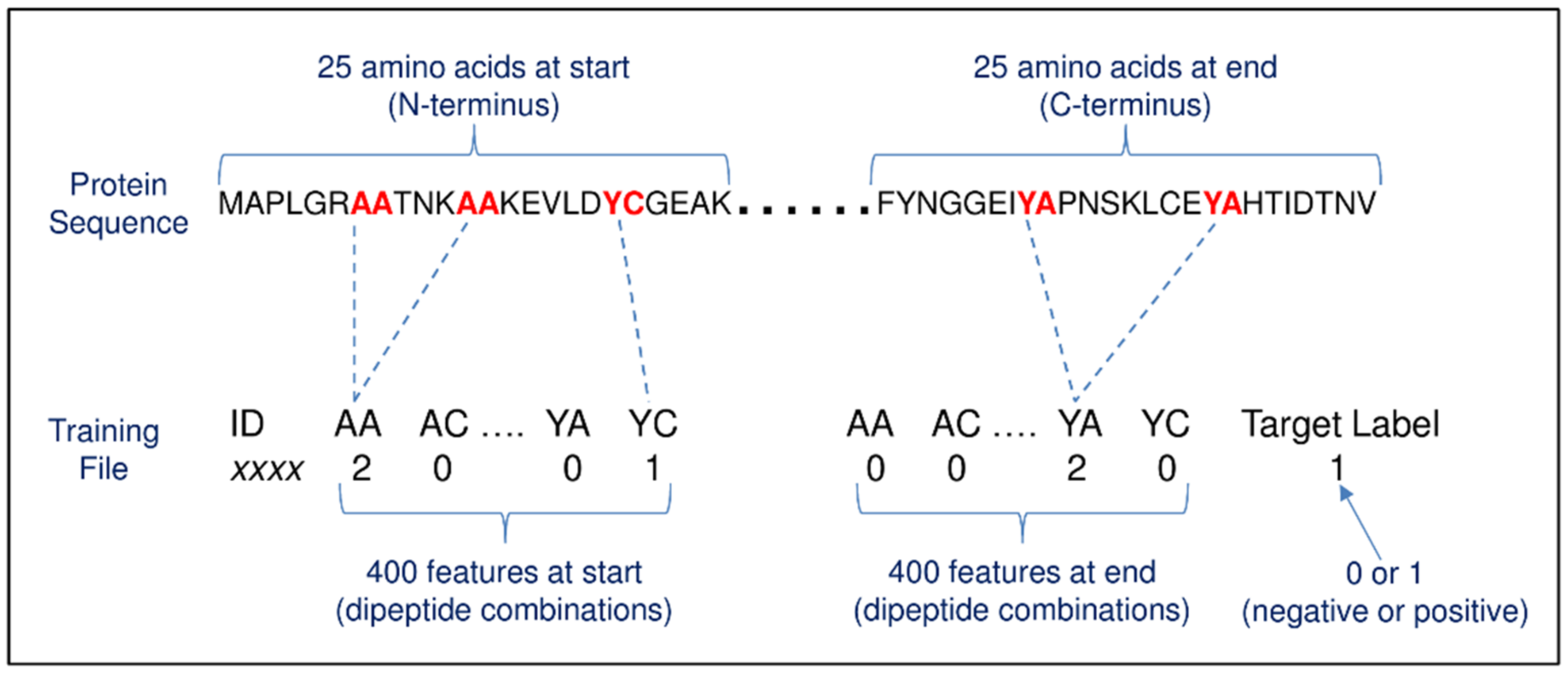
2.4. Method #1—Dipeptide Amino Acid Composition
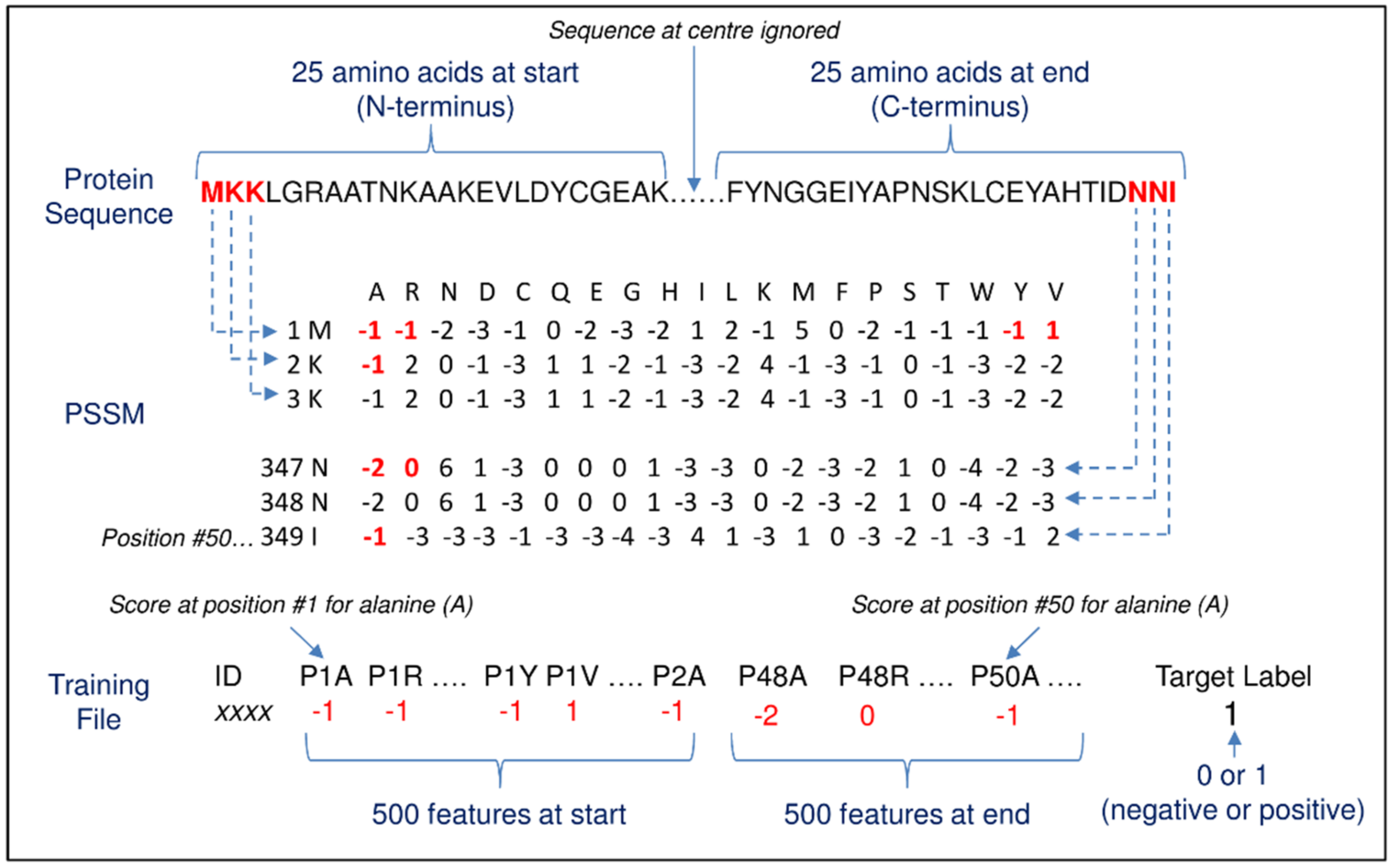
2.5. Method #2—Position-Specific Scoring Matrix (PSSM)
2.6. Method #3—Subcellular Location
2.7. Comparison of Classification Outcomes from the Three Methods
2.8. Full Babesia Bovis T2Bo Exportome Prediction
2.9. Full Babesia Bigemina BOND Exportome Prediction
2.10. Full Babesia Canis BcH-CHIPZ Exportome Prediction
2.11. Comparison between Babesia Exportome Predictions
2.12. Non-Classical Exported Proteins
2.13. Plasmodium Falciparum and Toxoplasma Gondii Exportome Prediction
3. Discussion
4. Materials and Methods
4.1. Data Source
4.2. Training Input Sequences for Machine Learning
4.3. Machine Learning Algorithms
4.4. Method #1—Dipeptide Amino Acid Composition
4.5. Method #2—Position-Specific Scoring Matrix (PSSM)
4.6. Method #3—Subcellular Location
4.7. Validation of Training Data
4.8. Predicting Non-Classical Exported Proteins
4.9. Plasmodium PEXEL Motifs
4.10. Method Implementation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Homer, M.J.; Aguilar-Delfin, I.; Telford, S.R.; Krause, P.J.; Persing, D.H. Babesiosis. Clin. Microbiol. Rev. 2000, 13, 451–469. [Google Scholar] [CrossRef] [PubMed]
- Eichenberger, R.M.; Ramakrishnan, C.; Russo, G.; Deplazes, P.; Hehl, A.B. Genome-wide analysis of gene expression and protein secretion of Babesia canis during virulent infection identifies potential pathogenicity factors. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Bock, R.; Jackson, L.; De Vos, A.; Jorgensen, W. Babesiosis of cattle. Parasitology 2004, 129, S247–S269. [Google Scholar] [CrossRef] [PubMed]
- Florin-Christensen, M.; Suarez, C.E.; Rodriguez, A.E.; Flores, D.A.; Schnittger, L. Vaccines against bovine babesiosis: Where we are now and possible roads ahead. Parasitology 2014, 141, 1563–1592. [Google Scholar] [CrossRef]
- Suarez, C.E.; Noh, S. Emerging perspectives in the research of bovine babesiosis and anaplasmosis. Vet. Parasitol. 2011, 180, 109–125. [Google Scholar] [CrossRef]
- Brayton, K.A.; Lau, A.O.T.; Herndon, D.R.; Hannick, L.; Kappmeyer, L.S.; Berens, S.J.; Bidwell, S.L.; Brown, W.C.; Crabtree, J.; Fadrosh, D.; et al. Genome sequence of babesia bovis and comparative analysis of apicomplexan hemoprotozoa. PLoS Pathog. 2007, 3, e148. [Google Scholar] [CrossRef]
- Irwin, P.J. Canine babesiosis: From molecular taxonomy to control. Parasites Vectors 2009, 2. [Google Scholar] [CrossRef]
- Schetters, T. Vaccination against canine babesiosis. Trends Parasitol. 2005, 21, 179–184. [Google Scholar] [CrossRef]
- Zhou, M.; Cao, S.; Luo, Y.; Liu, M.; Wang, G.; Moumouni, P.F.A.; Jirapattharasate, C.; Iguchi, A.; Vudriko, P.; Terkawi, M.A.; et al. Molecular identification and antigenic characterization of a merozoite surface antigen and a secreted antigen of Babesia canis (BcMSA1 and BcSA1). Parasites Vectors 2016, 9. [Google Scholar] [CrossRef]
- Hunfeld, K.P.; Hildebrandt, A.; Gray, J.S. Babesiosis: Recent insights into an ancient disease. Int. J. Parasitol. 2008, 38, 1219–1237. [Google Scholar] [CrossRef]
- Mosqueda, J.; Olvera-Ramirez, A.; Aguilar-Tipacamu, G.; Canto, G.J. Current Advances in Detection and Treatment of Babesiosis. Curr. Med. Chem. 2012, 19, 1504–1518. [Google Scholar] [CrossRef]
- Gohil, S.; Herrmann, S.; Gunther, S.; Cooke, B.M. Bovine babesiosis in the 21st century: Advances in biology and functional genomics. Int. J. Parasitol. 2013, 43, 125–132. [Google Scholar] [CrossRef]
- Kabanova, S.; Kleinbongard, P.; Volkmer, J.; Andree, B.; Kelm, M.; Jax, T.W. Gene expression analysis of human red blood cells. Int. J. Med. Sci. 2009, 6, 156–159. [Google Scholar] [CrossRef]
- Haase, S.; de Koning-Ward, T.F. New insights into protein export in malaria parasites. Cell. Microbiol. 2010, 12, 580–587. [Google Scholar] [CrossRef]
- Gohil, S.; Kats, L.M.; Sturm, A.; Cooke, B.M. Recent insights into alteration of red blood cells by Babesia bovis: Moovin’ forward. Trends Parasitol. 2010, 26, 591–599. [Google Scholar] [CrossRef]
- Schetters, T. Mechanisms Involved in the Persistence of Babesia canis Infection in Dogs. Pathogens 2019, 8, 94. [Google Scholar] [CrossRef]
- Seydel, K.B.; Milner, D.A., Jr.; Kamiza, S.B.; Molyneux, M.E.; Taylor, T.E. The distribution and intensity of parasite sequestration in comatose malawian children. J. Infect. Dis. 2006, 194, 208–215. [Google Scholar] [CrossRef]
- Aikawa, M.; Pongponratn, E.; Tegoshi, T.; Nakamura, K.I.; Nagatake, T.; Cochrane, A.; Ozaki, L.S. A study on the pathogenesis of human cerebral malaria and cerebral babesiosis. Mem. Do Inst. Oswaldo Cruz 1992, 87, 297–301. [Google Scholar] [CrossRef]
- Hutchings, C.L.; Li, A.; Fernandez, K.M.; Fletcher, T.; Jackson, L.A.; Molloy, J.B.; Jorgensen, W.K.; Lim, C.T.; Cooke, B.M. New insights into the altered adhesive and mechanical properties of red blood cells parasitized by Babesia bovis. Mol. Microbiol. 2007, 65, 1092–1105. [Google Scholar] [CrossRef]
- Suarez, C.E.; Alzan, H.F.; Silva, M.G.; Rathinasamy, V.; Poole, W.A.; Cooke, B.M. Unravelling the cellular and molecular pathogenesis of bovine babesiosis: Is the sky the limit? Int. J. Parasitol. 2019, 49, 183–197. [Google Scholar] [CrossRef]
- Gohil, S.; Kats, L.M.; Seemann, T.; Fernandez, K.M.; Siddiqui, G.; Cooke, B.M. Bioinformatic prediction of the exportome of Babesia bovis and identification of novel proteins in parasite-infected red blood cells. Int. J. Parasitol. 2013, 43, 409–416. [Google Scholar] [CrossRef]
- Rathinasamy, V.; Poole, W.A.; Bastos, R.G.; Suarez, C.E.; Cooke, B.M. Babesiosis Vaccines: Lessons Learned, Challenges Ahead, and Future Glimpses. Trends Parasitol. 2019, 35, 622–635. [Google Scholar] [CrossRef]
- Moxon, C.A.; Grau, G.E.; Craig, A.G. Malaria: Modification of the red blood cell and consequences in the human host. Br. J. Haematol. 2011, 154, 670–679. [Google Scholar] [CrossRef]
- Verma, R.; Tiwari, A.; Kaur, S.; Varshney, G.C.; Raghava, G.P.S. Identification of proteins secreted by malaria parasite into erythrocyte using SVM and PSSM profiles. BMC Bioinform. 2008, 9. [Google Scholar] [CrossRef]
- Sargeant, T.J.; Marti, M.; Caler, E.; Carlton, J.M.; Simpson, K.; Speed, T.P.; Cowman, A.F. Lineage-specific expansion of proteins exported to erythrocytes in malaria parasites. Genome Biol. 2006, 7. [Google Scholar] [CrossRef]
- Marti, M.; Good, R.T.; Rug, M.; Knuepfer, E.; Cowman, A.F. Targeting malaria virulence and remodeling proteins to the host erythrocyte. Science 2004, 306, 1930–1933. [Google Scholar] [CrossRef]
- Hiller, N.L.; Bhattacharjee, S.; van Ooij, C.; Liolios, K.; Harrison, T.; Lopez-Estrano, C.; Haldar, K. A host-targeting signal in virulence proteins reveals a secretome in malarial infection. Science 2004, 306, 1934–1937. [Google Scholar] [CrossRef] [PubMed]
- Jonsdottir, T.K.; Gabriela, M.; Crabb, B.S.; de Koning-Ward, T.; Gilson, P.R. Defining the Essential Exportome of the Malaria Parasite. Trends Parasitol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Maier, A.G.; Rug, M.; O’Neill, M.T.; Brown, M.; Chakravorty, S.; Szestak, T.; Chesson, J.; Wu, Y.; Hughes, K.; Coppel, R.L.; et al. Exported proteins required for virulence and rigidity of Plasmodium falciparum-infected human erythrocytes. Cell 2008, 134, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Cooke, B.M.; Buckingham, D.W.; Glenister, F.K.; Fernandez, K.M.; Bannister, L.H.; Marti, M.; Mohandas, N.; Coppel, R.L. A Maurer’s cleft-associated protein is essential for expression of the major malaria virulence antigen on the surface of infected red blood cells. J. Cell Biol. 2006, 172, 899–908. [Google Scholar] [CrossRef] [PubMed]
- Hines, S.A.; Palmer, G.H.; Brown, W.C.; McElwain, T.F.; Suarez, C.E.; Vidotto, O.; Riceficht, A.C. Genetic and antigenic characterization of Babesia bovis merozoite spherical body protein Bb-1. Mol. Biochem. Parasitol. 1995, 69, 149–159. [Google Scholar] [CrossRef]
- Allred, D.R.; Carlton, J.M.R.; Satcher, R.L.; Long, J.A.; Brown, W.C.; Patterson, P.E.; O’Connor, R.M.; Stroup, S.E. The ves multigene family of B. bovis encodes components of rapid antigenic variation at the infected erythrocyte surface. Mol. Cell 2000, 5, 153–162. [Google Scholar] [CrossRef]
- Ruef, B.J.; Dowling, S.C.; Conley, P.G.; Perryman, L.E.; Brown, W.C.; Jasmer, D.P.; Rice-Ficht, A.C. A unique Babesia bovis spherical body protein is conserved among geographic isolates and localizes to the infected erythrocyte membrane. Mol. Biochem. Parasitol. 2000, 105, 1–12. [Google Scholar] [CrossRef]
- Montoya, J.G.; Liesenfeld, O. Toxoplasmosis. Lancet 2004, 363, 1965–1976. [Google Scholar] [CrossRef]
- Kim, K.; Weiss, L.M. Toxoplasma gondii: The model apicomplexan. Int. J. Parasitol. 2004, 34, 423–432. [Google Scholar] [CrossRef]
- Sibley, L.D. Toxoplasma gondii: Perfecting an intracellular life style. Traffic 2003, 4, 581–586. [Google Scholar] [CrossRef]
- Mundwiler-Pachlatko, E.; Beck, H.-P. Maurer’s clefts, the enigma of Plasmodium falciparum. Proc. Natl. Acad. Sci. USA 2013, 110, 19987–19994. [Google Scholar] [CrossRef]
- Romisch, K. Diversion at the ER: How Plasmodium falciparum exports proteins into host erythrocytes. F1000Research 2012, 1, 12. [Google Scholar] [CrossRef]
- Radisky, D.C.; Stallings-Mann, M.; Hirai, Y.; Bissell, M.J. Single proteins might have dual but related functions in intracellular and extracellular microenvironments. Nat. Rev. Mol. Cell Biol. 2009, 10, 228–234. [Google Scholar] [CrossRef]
- Nickel, W. The mystery of nonclassical protein secretion—A current view on cargo proteins and potential export routes. Eur. J. Biochem. 2003, 270, 2109–2119. [Google Scholar] [CrossRef]
- Horton, P.; Park, K.J.; Obayashi, T.; Fujita, N.; Harada, H.; Adams-Collier, C.J.; Nakai, K. WoLF PSORT: Protein localization predictor. Nucleic Acids Res. 2007, 35, W585–W587. [Google Scholar] [CrossRef] [PubMed]
- Vonheijne, G. The Signal Peptide. J. Membr. Biol. 1990, 115, 195–201. [Google Scholar] [CrossRef]
- Emanuelsson, O.; Brunak, S.; von Heijne, G.; Nielsen, H. Locating proteins in the cell using TargetP, SignalP and related tools. Nat. Protoc. 2007, 2, 953–971. [Google Scholar] [CrossRef]
- Yokoyama, N.; Okamura, M.; Igarashi, I. Erythrocyte invasion by Babesia parasites: Current advances in the elucidation of the molecular interactions between the protozoan ligands and host receptors in the invasion stage. Vet. Parasitol. 2006, 138, 22–32. [Google Scholar] [CrossRef]
- Elisa Rodriguez, A.; Florin-Christensen, M.; Agustina Flores, D.; Echaide, I.; Suarez, C.E.; Schnittger, L. The glycosylphosphatidylinositol-anchored protein repertoire of Babesia bovis and its significance for erythrocyte invasion. Ticks Tick-Borne Dis. 2014, 5, 343–348. [Google Scholar] [CrossRef]
- Gaffar, F.R.; Yatsuda, A.P.; Franssen, F.F.J.; de Vries, E. A Babesia bovis merozoite protein with a domain architecture highly similar to the thrombospondin-related anonymous protein (TRAP) present in Plasmodium sporozoites. Mol. Biochem. Parasitol. 2004, 136, 25–34. [Google Scholar] [CrossRef]
- Brown, W.C.; McElwain, T.F.; Palmer, G.H.; Chantler, S.E.; Estes, D.M. Bovine CD4(+) T-lymphocyte clones specific for rhoptry-associated protein 1 of Babesia bigemina stimulate enhanced immunoglobulin G1 (IgG1) and IgG2 synthesis. Infect. Immun. 1999, 67, 155–164. [Google Scholar] [CrossRef]
- Norimine, J.; Mosqueda, J.; Suarez, C.; Palmer, G.H.; McElwain, T.F.; Mbassa, G.; Brown, W.C. Stimulation of T-helper cell gamma interferon and immunoglobulin G responses specific for Babesia bovis rhoptry-associated protein 1 (RAP-1) or a RAP-1 protein lacking the carboxy-terminal repeat region is insufficient to provide protective immunity against virulent B. bovis challenge. Infect. Immun. 2003, 71, 5021–5032. [Google Scholar] [CrossRef]
- Nakashima, H.; Nishikawa, K. Discrimination of intracellular and extracellular proteins using amino acid composition and residue-pair frequencies. J. Mol. Biol. 1994, 238, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Bendtsen, J.D.; Jensen, L.J.; Blom, N.; von Heijne, G.; Brunak, S. Feature-based prediction of non-classical and leaderless protein secretion. Protein Eng. Des. Sel. 2004, 17, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Poschmann, G.; Waldera-Lupa, D.; Rafiee, N.; Kollmann, M.; Stuehler, K. OutCyte: A novel tool for predicting unconventional protein secretion. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef]
- Bendtsen, J.D.; Nielsen, H.; von Heijne, G.; Brunak, S. Improved prediction of signal peptides: SignalP 3.0. J. Mol. Biol. 2004, 340, 783–795. [Google Scholar] [CrossRef]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L.L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef]
- Pierleoni, A.; Martelli, P.L.; Casadio, R. PredGPI: A GPI-anchor predictor. BMC Bioinform. 2008, 9, 392. [Google Scholar] [CrossRef]
- Jones, D.T. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 1999, 292, 195–202. [Google Scholar] [CrossRef]
- Armenteros, J.J.A.; Tsirigos, K.D.; Sonderby, C.K.; Petersen, T.N.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019, 37, 420–423. [Google Scholar] [CrossRef]
- Kall, L.; Krogh, A.; Sonnhammer, E.L.L. A combined transmembrane topology and signal peptide prediction method. J. Mol. Biol. 2004, 338, 1027–1036. [Google Scholar] [CrossRef]
- Armenteros, J.J.A.; Sonderby, C.K.; Sonderby, S.K.; Nielsen, H.; Winther, O. DeepLoc: Prediction of protein subcellular localization using deep learning. Bioinformatics 2017, 33, 3387–3395. [Google Scholar] [CrossRef]
- Ferreri, L.M.; Brayton, K.A.; Sondgeroth, K.S.; Lau, A.O.T.; Suarez, C.E.; McElwain, T.F. Expression and strain variation of the novel “small open reading frame” (smorf) multigene family in Babesia bovis. Int. J. Parasitol. 2012, 42, 131–138. [Google Scholar] [CrossRef]
- Hakimi, H.; Templeton, T.J.; Sakaguchi, M.; Yamagishi, J.; Miyazaki, S.; Yahata, K.; Uchihashi, T.; Kawazu, S.-I.; Kaneko, O.; Asada, M. Novel Babesia bovis exported proteins that modify properties of infected red blood cells. PLoS Pathog. 2020, 16, e1008917. [Google Scholar] [CrossRef]
- Maier, A.G.; Cooke, B.M.; Cowman, A.F.; Tilley, L. Malaria parasite proteins that remodel the host erythrocyte. Nat. Rev. Microbiol. 2009, 7, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Paoletta, M.S.; Laughery, J.M.; Arias, L.S.L.; Ortiz, J.M.J.; Montenegro, V.N.; Petrigh, R.; Ueti, M.W.; Suarez, C.E.; Farber, M.D.; Wilkowsky, S.E. The key to egress? Babesia bovis perforin-like protein 1 (PLP1) with hemolytic capacity is required for blood stage replication and is involved in the exit of the parasite from the host cell. Int. J. Parasitol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Pei, X.; Guo, X.; Coppel, R.; Bhattacharjee, S.; Haldar, K.; Gratzer, W.; Mohandas, N.; An, X. The ring-infected erythrocyte surface antigen (RESA) of Plasmodium falciparum stabilizes spectrin tetramers and suppresses further invasion. Blood 2007, 110, 1036–1042. [Google Scholar] [CrossRef] [PubMed]
- Dorin-Semblat, D.; Demarta-Gatsi, C.; Hamelin, R.; Armand, F.; Carvalho, T.G.; Moniatte, M.; Doerig, C. Malaria Parasite-Infected Erythrocytes Secrete PfCK1, the Plasmodium Homologue of the Pleiotropic Protein Kinase Casein Kinase 1. PLoS ONE 2015, 10, e0139591. [Google Scholar] [CrossRef]
- Khattab, A.; Klinkert, M.-Q. Maurer’s clefts-restricted localization, orientation and export of a Plasmodium falciparum RIFIN. Traffic 2006, 7, 1654–1665. [Google Scholar] [CrossRef]
- Hakimi, M.-A.; Olias, P.; Sibley, L.D. Toxoplasma Effectors Targeting Host Signaling and Transcription. Clin. Microbiol. Rev. 2017, 30, 615–645. [Google Scholar] [CrossRef]
- De Koning-Ward, T.F.; Dixon, M.W.A.; Tilley, L.; Gilson, P.R. Plasmodium species: Master renovators of their host cells. Nat. Rev. Microbiol. 2016, 14, 494–507. [Google Scholar] [CrossRef]
- Hsiao, C.-H.C.; Hiller, N.L.; Haldar, K.; Knoll, L.J. A HT/PEXEL Motif in Toxoplasma Dense Granule Proteins is a Signal for Protein Cleavage but not Export into the Host Cell. Traffic 2013, 14, 519–531. [Google Scholar] [CrossRef]
- Asada, M.; Goto, Y.; Yahata, K.; Yokoyama, N.; Kawai, S.; Inoue, N.; Kaneko, O.; Kawazu, S.-I. Gliding Motility of Babesia bovis Merozoites Visualized by Time-Lapse Video Microscopy. PLoS ONE 2012, 7, e35227. [Google Scholar] [CrossRef]
- Bateman, A.; Martin, M.J.; O’Donovan, C.; Magrane, M.; Apweiler, R.; Alpi, E.; Antunes, R.; Arganiska, J.; Bely, B.; Bingley, M.; et al. UniProt: A hub for protein information. Nucleic Acids Res. 2015, 43, D204–D212. [Google Scholar] [CrossRef]
- Banumathy, G.; Singh, V.; Tatu, U. Host chaperones are recruited in membrane-bound complexes by Plasmodium falciparum. J. Biol. Chem. 2002, 277, 3902–3912. [Google Scholar] [CrossRef]
- Kuelzer, S.; Charnaud, S.; Dagan, T.; Riedel, J.; Mandal, P.; Pesce, E.R.; Blatch, G.L.; Crabb, B.S.; Gilson, P.R.; Przyborski, J.M. Plasmodium falciparum-encoded exported hsp70/hsp40 chaperone/co-chaperone complexes within the host erythrocyte. Cell. Microbiol. 2012, 14, 1784–1795. [Google Scholar] [CrossRef]
- Aurrecoechea, C.; Brestelli, J.; Brunk, B.P.; Fischer, S.; Gajria, B.; Gao, X.; Gingle, A.; Grant, G.; Harb, O.S.; Heiges, M.; et al. EuPathDB: A portal to eukaryotic pathogen databases. Nucleic Acids Res. 2010, 38, D415–D419. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef]
- Freund, Y.; Schapire, R.E. A decision-theoretic generalization of on-line learning and an application to boosting. J. Comput. Syst. Sci. 1997, 55, 119–139. [Google Scholar] [CrossRef]
- Friedman, J.; Hastie, T.; Tibshirani, R. Additive logistic regression: A statistical view of boosting. Ann. Stat. 2000, 28, 337–374. [Google Scholar] [CrossRef]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Horton, P.; Nakai, K. Better prediction of protein cellular localization sites with the k nearest neighbors classifier. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1997, 5, 142–152. [Google Scholar]
- Ng, A.Y.; Jordan, M.I. On Discriminative vs. Generative classifiers: A comparison of logistic regression and naive Bayes. In Proceedings of the 14th International Conference on Neural Information Processing Systems: Natural and Synthetic, Vancouver, BC, Canada, 3–8 December 2001; pp. 841–848. [Google Scholar]
- Hinton, G.E.; Salakhutdinov, R.R. Reducing the dimensionality of data with neural networks. Science 2006, 313, 504–507. [Google Scholar] [CrossRef]
- Burges, C.J.C. A tutorial on Support Vector Machines for pattern recognition. Data Min. Knowl. Discov. 1998, 2, 121–167. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.H.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Matthews, K.M.; Pitman, E.L.; de Koning-Ward, T.F. Illuminating how malaria parasites export proteins into host erythrocytes. Cell. Microbiol. 2019, 21, e13009. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Algorithm | Accuracy (%) | Error Rate (%) | Sensitivity (%) | False Positive Rate (%) | Specificity (%) | Positive Predictive Value (%) | Negative Predictive Value (%) |
|---|---|---|---|---|---|---|---|
| Ensemble | 90.89 | 9.11 | 91.12 | 9.35 | 90.65 | 90.7 | 91.08 |
| SVM | 90.19 | 9.81 | 90.65 | 10.28 | 89.72 | 89.81 | 90.57 |
| adaBoost | 89.49 | 10.51 | 88.79 | 9.81 | 90.19 | 90.05 | 88.94 |
| RF | 89.25 | 10.75 | 91.59 | 13.08 | 86.92 | 87.5 | 91.18 |
| ANN | 87.15 | 12.85 | 87.38 | 13.08 | 86.92 | 86.98 | 87.32 |
| Algorithm | Accuracy (%) | Error Rate (%) | Sensitivity (%) | False Positive Rate (%) | Specificity (%) | Positive Predictive Value (%) | Negative Predictive Value (%) |
|---|---|---|---|---|---|---|---|
| RF | 89.95 | 10.05 | 88.79 | 8.88 | 91.12 | 90.91 | 89.04 |
| adaBoost | 89.25 | 10.75 | 88.79 | 10.28 | 89.72 | 89.62 | 88.89 |
| Ensemble | 88.32 | 11.68 | 87.85 | 11.21 | 88.79 | 88.68 | 87.96 |
| SVM | 88.08 | 11.92 | 87.85 | 11.68 | 88.32 | 88.26 | 87.91 |
| ANN | 84.58 | 15.42 | 85.05 | 15.89 | 84.11 | 84.26 | 84.91 |
| Algorithm | Accuracy (%) | Error Rate (%) | Sensitivity (%) | False Positive Rate (%) | Specificity (%) | Positive Predictive Value (%) | Negative Predictive Value (%) |
|---|---|---|---|---|---|---|---|
| Ensemble | 96.26 | 3.74 | 98.13 | 5.61 | 94.39 | 94.59 | 98.06 |
| RF | 95.56 | 4.44 | 97.66 | 6.54 | 93.46 | 93.72 | 97.56 |
| SVM | 95.09 | 4.91 | 96.73 | 6.54 | 93.46 | 93.67 | 96.62 |
| adaBoost | 94.86 | 5.14 | 96.26 | 6.54 | 93.46 | 93.64 | 96.15 |
| NB | 93.46 | 6.54 | 96.26 | 9.35 | 90.65 | 91.15 | 96.04 |
| ANN | 93.22 | 6.78 | 92.99 | 6.54 | 93.46 | 93.43 | 93.02 |
| kNN | 89.72 | 10.28 | 90.19 | 10.75 | 89.25 | 89.35 | 90.09 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goodswen, S.J.; Kennedy, P.J.; Ellis, J.T. Applying Machine Learning to Predict the Exportome of Bovine and Canine Babesia Species That Cause Babesiosis. Pathogens 2021, 10, 660. https://doi.org/10.3390/pathogens10060660
Goodswen SJ, Kennedy PJ, Ellis JT. Applying Machine Learning to Predict the Exportome of Bovine and Canine Babesia Species That Cause Babesiosis. Pathogens. 2021; 10(6):660. https://doi.org/10.3390/pathogens10060660
Chicago/Turabian StyleGoodswen, Stephen J., Paul J. Kennedy, and John T. Ellis. 2021. "Applying Machine Learning to Predict the Exportome of Bovine and Canine Babesia Species That Cause Babesiosis" Pathogens 10, no. 6: 660. https://doi.org/10.3390/pathogens10060660
APA StyleGoodswen, S. J., Kennedy, P. J., & Ellis, J. T. (2021). Applying Machine Learning to Predict the Exportome of Bovine and Canine Babesia Species That Cause Babesiosis. Pathogens, 10(6), 660. https://doi.org/10.3390/pathogens10060660