
Epidemiology, Genetic Characterization, and Evolution of Hunnivirus Carried by Rattus norvegicus and Rattus tanezumi: The First Epidemiological Evidence from Southern China


Abstract

1. Introduction
2. Results
2.1. Detection of Rat Hunnivirus
2.2. Phylogenetic Analysis of Partial 3CD Region Sequences
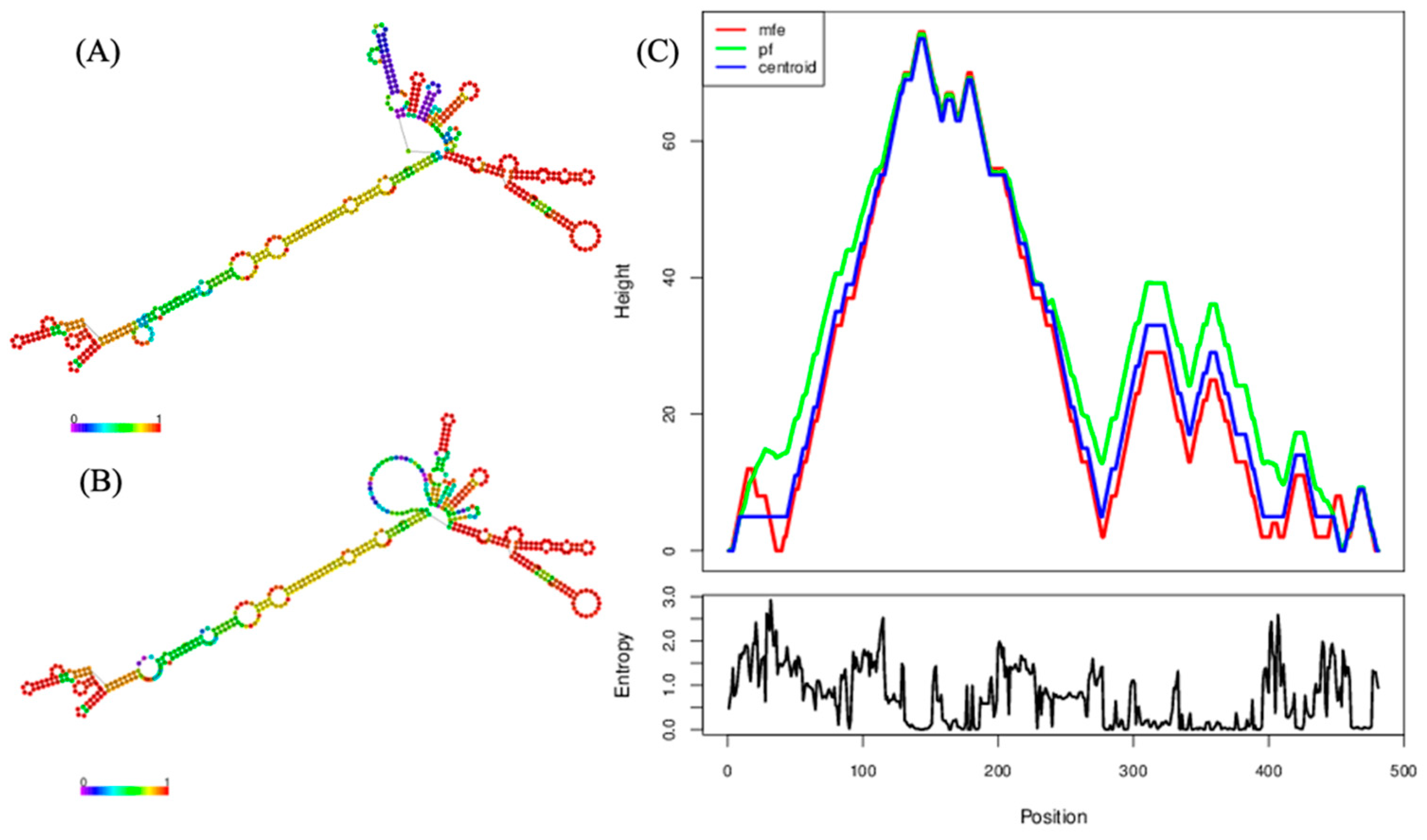
2.3. Genomic Analyses of the Near-Complete Sequence
2.4. Phylogenetic Analyses and Negative Selection during the Evolution of Hunnivirus
3. Discussion
4. Materials and Methods
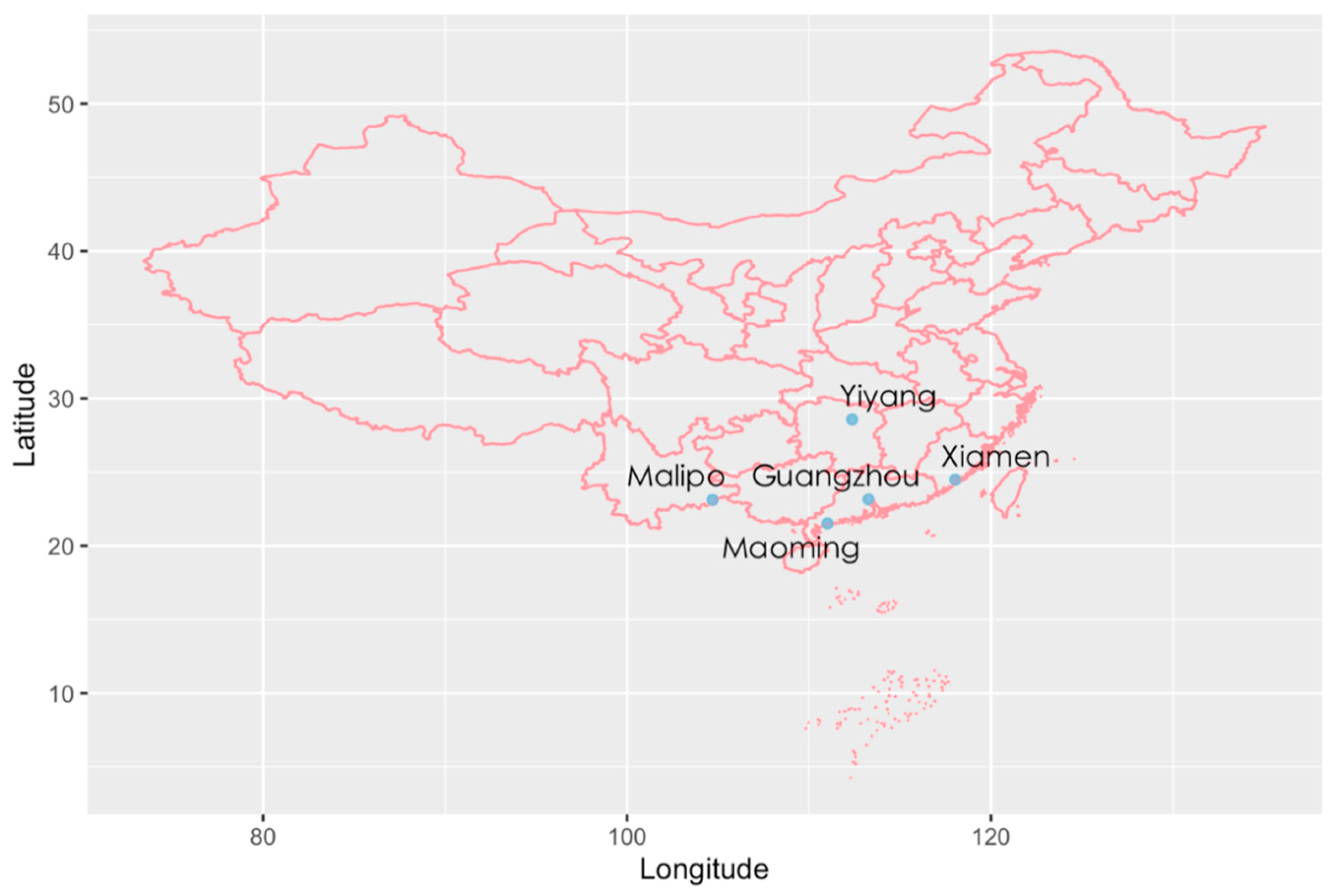
4.1. Sample Collection
4.2. Nucleic Acid Extraction and cDNA Synthesis
4.3. PCR Detection for Hunnivirus
4.4. Near-Complete Genome Amplification
4.5. Genetic and Phylogenetic Analyses
4.6. Selective Pressure Analyses
4.7. Data Summary
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Reuter, G.; Pankovics, P.; Knowles, N.J.; Boros, Á. Two closely related novel picornaviruses in cattle and sheep in Hungary from 2008 to 2009, proposed as members of a new genus in the family Picornaviridae. J. Virol. 2012, 86, 13295–13302. [Google Scholar] [CrossRef]
- Firth, C.; Bhat, M.; Firth, M.A.; Williams, S.H.; Frye, M.J.; Simmonds, P.; Conte, J.M.; Ng, J.; Garcia, J.; Bhuva, N.P.; et al. Detection of zoonotic pathogens and characterization of novel viruses carried by commensal Rattus norvegicus in New York City. MBio 2014, 5, e01933. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Lu, L.; Liu, F.; Su, H.; Dong, J.; Sun, L.; Zhu, Y.; Ren, X.; Yang, F.; Guo, F.; et al. Distribution and characteristics of rodent picornaviruses in China. Sci. Rep. 2016, 6, 34381. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Huang, M.; Chen, X.; Sun, Y.; Huang, J.; Hu, R.; Li, S. Identification and genome characterization of a novel feline picornavirus proposed in the Hunnivirus genus. Infect. Genet. Evol. 2019, 71, 47–50. [Google Scholar] [CrossRef]
- Pond, S.L.; Frost, S.D. Datamonkey: Rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics 2005, 21, 2531–2533. [Google Scholar] [CrossRef]
- Reuter, G.; Boldizsár, A.; Pankovics, P. Complete nucleotide and amino acid sequences and genetic organization of porcine kobuvirus, a member of a new species in the genus Kobuvirus, family Picornaviridae. Arch. Virol. 2009, 154, 101–108. [Google Scholar] [CrossRef]
- Rivadulla, E.; Romalde, J.L. A comprehensive review on human Aichi virus. Virol. Sin. 2020, 35, 501–516. [Google Scholar] [CrossRef] [PubMed]
- Reuter, G.; Boros, Á.; Pankovics, P.; Egyed, L. Kobuvirus in domestic sheep, Hungary. Emerg. Infect. Dis. 2010, 16, 869–870. [Google Scholar] [CrossRef]
- Khamrin, P.; Maneekarn, N.; Okitsu, S.; Ushijima, H. Epidemiology of human and animal kobuviruses. Virusdisease 2014, 25, 195–200. [Google Scholar] [CrossRef]
- Verma, H.; Mor, S.K.; Abdel-Glil, M.Y.; Goyal, S.M. Identification and molecular characterization of porcine kobuvirus in US swine. Virus Genes 2013, 46, 551–553. [Google Scholar] [CrossRef]
- Chung, J.Y.; Kim, S.H.; Kim, Y.H.; Lee, M.H.; Lee, K.K.; Oem, J.K. Detection and genetic characterization of feline kobuviruses. Virus Genes 2013, 47, 559–562. [Google Scholar] [CrossRef]
- Choi, J.W.; Lee, M.H.; Lee, K.K.; Oem, J.K. Genetic characteristics of the complete feline kobuvirus genome. Virus Genes. 2015, 50, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Di Martino, B.; Di Felice, E.; Ceci, C.; Di Profio, F.; Marsilio, F. Canine kobuviruses in diarrhoeic dogs in Italy. Vet. Microbiol. 2013, 166, 246–249. [Google Scholar] [CrossRef] [PubMed]
- Di Martino, B.; Di Profio, F.; Melegari, I.; Di Felice, E.; Robetto, S.; Guidetti, C.; Orusa, R.; Martella, V.; Marsilio, F. Molecular detection of kobuviruses in European roe deer (Capreolus capreolus) in Italy. Arch. Virol. 2015, 160, 2083–2086. [Google Scholar] [CrossRef] [PubMed]
- Melegari, I.; Sarchese, V.; Di Profio, F.; Robetto, S.; Carella, E.; Sanchez, S.B.; Orusa, R.; Martella, V.; Marsilio, F.; Di Martino, B. First molecular identification of kobuviruses in wolves (Canis lupus) in Italy. Arch. Virol. 2018, 163, 509–513. [Google Scholar] [CrossRef] [PubMed]
- You, F.F.; Zhang, M.Y.; He, H.; He, W.Q.; Li, Y.Z.; Chen, Q. Kobuviruses carried by Rattus norvegicus in Guangdong, China. BMC Microbiol. 2020, 20, 94. [Google Scholar] [CrossRef]
- Jackova, A.; Sliz, I.; Mandelik, R.; Salamunova, S.; Novotny, J.; Kolesarova, M.; Vlasakova, M.; Vilcek, S. Porcine kobuvirus 1 in healthy and diarrheic pigs: Genetic detection and characterization of virus and co-infection with rotavirus A. Infect. Genet. Evol. 2017, 49, 73–77. [Google Scholar] [CrossRef]
- Rossmann, M.G.; Arnold, E.; Erickson, J.W.; Frankenberger, E.A.; Griffith, J.P.; Hecht, H.J.; Johnson, J.E.; Kamer, G.; Luo, M.; Mosser, A.G.; et al. Structure of a human common cold virus and functional relationship to other picornaviruses. Nature 1985, 317, 145–153. [Google Scholar] [CrossRef]
- Miller, M.E.; Reboussin, D.M.; Antony, A.C. Methods for the prediction of cleavage sites of the polypeptide in certain proteins. J. Agric. Biol. Environ. Stat. 2000, 151–167. [Google Scholar] [CrossRef]
- Hirose, T.; Mishima, Y.; Tomari, Y. Elements and machinery of non-coding RNAs: Toward their taxonomy. EMBO Rep. 2014, 15, 489–507. [Google Scholar] [CrossRef]
- Sato, K.; Akiyama, M.; Sakakibara, Y. RNA secondary structure prediction using deep learning with thermodynamic integration. Nat. Commun. 2021, 12, 941. [Google Scholar] [CrossRef] [PubMed]
- Drexler, J.F.; Baumgarte, S.; de Souza Luna, L.K.; Eschbach-Bludau, M.; Lukashev, A.N.; Drosten, C. Aichi virus shedding in high concentrations in patients with acute diarrhea. Emerg. Infect. Dis. 2011, 17, 1544–1548. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.Z.W.; Shen, Q.; Yang, Z.; Zhu, J.; Cui, L.; Hua, X. Aichi virus strains in children with gastroenteritis, China. Emerg. Infect. Dis. 2009, 15, 1702–1703. [Google Scholar] [CrossRef]
- Kapoor, A.; Victoria, J.; Simmonds, P.; Slikas, E.; Chieochansin, T.; Naeem, A.; Shaukat, S.; Sharif, S.; Alam, M.M.; Angez, M.; et al. A highly prevalent and genetically diversified Picornaviridae genus in South. Asian children. Proc. Natl. Acad. Sci. USA 2008, 105, 20482–20487. [Google Scholar] [CrossRef]
- Nielsen, A.C.; Gyhrs, M.L.; Nielsen, L.P.; Pedersen, C.; Böttiger, B. Gastroenteritis and the novel picornaviruses aichi virus, cosavirus, saffold virus, and salivirus in young children. J. Clin. Virol. 2013, 57, 239–242. [Google Scholar] [CrossRef]
- Greninger, A.L.; Runckel, C.; Chiu, C.Y.; Haggerty, T.; Parsonnet, J.; Ganem, D.; DeRisi, J.L. The complete genome of klassevirus—A novel picornavirus in pediatric stool. Virol. J. 2009, 6, 82. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Victoria, J.; Kapoor, A.; Blinkova, O.; Wang, C.; Babrzadeh, F.; Mason, C.J.; Pandey, P.; Triki, H.; Bahri, O.; et al. A novel picornavirus associated with gastroenteritis. J. Virol. 2009, 83, 12002–12006. [Google Scholar] [CrossRef]
- Park, S.J.; Kim, H.K.; Moon, H.J.; Song, D.S.; Rho, S.M.; Han, J.Y.; Nguyen, V.G.; Park, B.K. Molecular detection of porcine kobuviruses in pigs in Korea and their association with diarrhea. Arch. Virol. 2010, 155, 1803–1811. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Fredrickson, R.; Duncan, M.; Samuelson, J.; Hsiao, S.H. Bovine Kobuvirus in calves with diarrhea, United States. Emerg. Infect. Dis. 2020, 26, 176–178. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Region | Rattus Norvegicus | Rattus Tanezumi | Total | ||
|---|---|---|---|---|---|
| Number of Samples | Prevalence | Number of Samples | Prevalence | ||
| Xiamen | 18 | 5.6% (1/18) | 13 | - | 3.2% (1/31) |
| Malipo | 47 | 21.3% (10/47) | 1 | 100% (1/1) | 22.9% (11/48) |
| Yiyang | 68 | 27.9% (19/68) | 15 | 33.3% (5/15) | 28.9% (24/83) |
| Guangzhou | 139 | 17.3% (24/139) | 7 | 14.3% (1/7) | 17.1% (25/146) |
| Maoming | 87 | 11.5% (10/87) | 9 | - | 10.4% (10/96) |
| Total | 359 | 17.8% (64/359) | 45 | 15.6% (7/45) | 17.6% (71/404) |
| Gene Region | Rat | Feline | Bovine | Ovine | |||
|---|---|---|---|---|---|---|---|
| Rat/NYc-E21/USA/2012 | 83GR-70-RAT106/Vietnam/2012 | 05VZ-75-RAT099/Vietnam/2013 | Rat/Wencheng-Nc42–1/China/2012 | FeHuV-1/GZ/2017 | BHUV1/HUN/2008 | OHUV1/HUN/2009 | |
| L | 97.2/92.9 | 94.5/88.2 | 95.3/88.2 | 79.9/61.2 | 97.2/91.8 | 63.8/45.9 | 74.0/56.5 |
| P1 | 85.2/93.1 | 61.8/63.5 | 61.3/63.5 | 65.3/70.3 | 85.2/84.6 | 65.2/69.9 | 63.4/68.0 |
| P2 | 90.7/97.4 | 80.4/87.6 | 80.0/89.3 | 67.5/70.4 | 90.7/86.7 | 63.0/66.8 | 64.3/67.5 |
| P3 | 94.9/96.9 | 91.0/94.1 | 91.3/95.1 | 76.6/81.2 | 94.9/97.0 | 73.8/80.5 | 73.9/80.6 |
| Polyprotein | 91.2/92.7 | 79.5/78.3 | 78.7/79.1 | 70.3/71.5 | 85.1/86.5 | 67.6/70.1 | 67.7/69.9 |
| Reaction Number | Primer Name | Primer Sequences (5′-3′) | Size of PCR Products (bp) |
|---|---|---|---|
| 1 | UNIV-kobu-F | TGGAYTACAAG(/R)TGTTTTGATGC | 216 |
| UNIV-kobu-R | ATGTTGTTRATGATGGTGTTGA | ||
| 2 | RHuV-F | TGGTGACCGGACTGATGGACCC | 254 |
| RHuV-R | TCAGTTCAGCATGCAGCACCGG | ||
| 3 | RHuV-3CD-F | GGATATTTYCCCCGCGGCAARG | 1610 |
| RHuV-3CD-R | ATAGTCTTGC TCCCCGCGGTGT | ||
| 4 | RHuV-S-F1 | AGTGACCCCATGCGAAGTGCTG | 1044 |
| RHuV-S-F2 | CCCTTGTGTGTCTGAGCGCCAC | 899 | |
| RHuV-837-R | GACGAGTCGCCACCTCCAGCAC | ||
| 5 | RHuV-335-F | ATCTGGGGCCCTGTCTGGAGTG | 1191 |
| RHuV-1508-R | GAGTCCAAGGGGCACTCTGGGT | ||
| 6 | RHuV-1204-F1 | TAAGAACACACCAGCTGCCGCG | 1701 |
| RHuV-2171-F2 | GCAAATCCCAGCAGTGGTGGCT | 734 | |
| RHuV-2883-R | GCCCCCATGGTGTTGAGTTGGG | ||
| 7 | RHuV-2659-F | CTACCCACCAGGTTCACACGTA | 1970 |
| RHuV-4607-R | CTGTCTTGTAGGCGGCACCAGC | ||
| 8 | RHuV-3647-F | GACTTGAGCAACTTTGGACCAA | 1584 |
| RHuV-5207-R | CTCTCTTGCCGGCRGARTTRCC | ||
| 9 | RHuV-4608-F | CTGGTGCCGCCTACAAGACAGC | 1148 |
| RHuV-5734-R | CCCAGGTGCTGTTTGGGCTCTG | ||
| 10 | RHuV-5192-F | CAGGGTGCCTATGGCGGTAACT | 1138 |
| RHuV-6308-R | TCAGTTCAGCATGCAGCACCGG | ||
| 11 | RHuV-6194-F | GGGACTGACAACCTGGACCCGA | 984 |
| RHuV-7156-R | ATAGTCTTGCTCCCCGCGGTGT | ||
| 12 | RHuV-6526-F | ATCCGCCGTTGGGACAAATCCG | 866 |
| RHuV-E-F | TGCTCTGGGGAAAAATAACCCT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, M.; Li, Q.; Wu, F.; Ou, Z.; Li, Y.; You, F.; Chen, Q. Epidemiology, Genetic Characterization, and Evolution of Hunnivirus Carried by Rattus norvegicus and Rattus tanezumi: The First Epidemiological Evidence from Southern China. Pathogens 2021, 10, 661. https://doi.org/10.3390/pathogens10060661
Zhang M, Li Q, Wu F, Ou Z, Li Y, You F, Chen Q. Epidemiology, Genetic Characterization, and Evolution of Hunnivirus Carried by Rattus norvegicus and Rattus tanezumi: The First Epidemiological Evidence from Southern China. Pathogens. 2021; 10(6):661. https://doi.org/10.3390/pathogens10060661
Chicago/Turabian StyleZhang, Minyi, Qiushuang Li, Fei Wu, Zejin Ou, Yongzhi Li, Fangfei You, and Qing Chen. 2021. "Epidemiology, Genetic Characterization, and Evolution of Hunnivirus Carried by Rattus norvegicus and Rattus tanezumi: The First Epidemiological Evidence from Southern China" Pathogens 10, no. 6: 661. https://doi.org/10.3390/pathogens10060661
APA StyleZhang, M., Li, Q., Wu, F., Ou, Z., Li, Y., You, F., & Chen, Q. (2021). Epidemiology, Genetic Characterization, and Evolution of Hunnivirus Carried by Rattus norvegicus and Rattus tanezumi: The First Epidemiological Evidence from Southern China. Pathogens, 10(6), 661. https://doi.org/10.3390/pathogens10060661