
β-lactam Resistance in Pseudomonas aeruginosa: Current Status, Future Prospects



Abstract
1. Introduction
2. Antibiotics Used against P. aeruginosa
3. The Problem of P. aeruginosa Antibiotic Resistance
4. Penicillin-Binding Proteins and Peptidoglycan Synthesis
5. β-lactam Antibiotics: Inhibitors of PBPs
6. Mechanisms of β-lactam Resistance
7. Target-Site Modification: Changes to PBP3
8. Reduced Uptake of β-lactams: The Role of Porins
9. P. aeruginosa Efflux Systems
10. Degradation of β-lactams by β-lactamases
11. β-lactamases Encoded by the Core Genome
12. β-lactamases Acquired by Horizontal Gene Transfer
13. Lifestyle and Metabolism: Other Contributors to Resistance
14. Conclusions and Prospects for the Future
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Crone, S.; Vives-Flórez, M.; Kvich, L.; Saunders, A.M.; Malone, M.; Nicolaisen, M.H.; Martínez-García, E.; Rojas-Acosta, C.; Catalina Gomez-Puerto, M.; Calum, H. The environmental occurrence of Pseudomonas aeruginosa. APMIS 2020, 128, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Bassetti, M.; Vena, A.; Croxatto, A.; Righi, E.; Guery, B. How to manage Pseudomonas aeruginosa infections. Drugs Context 2018, 7, 212527. [Google Scholar] [CrossRef] [PubMed]
- Bassetti, M.; Vena, A.; Russo, A.; Croxatto, A.; Calandra, T.; Guery, B. Rational approach in the management of Pseudomonas aeruginosa infections. Curr. Opin. Infect. Dis. 2018, 31, 578–586. [Google Scholar] [CrossRef] [PubMed]
- Sievert, D.M.; Ricks, P.; Edwards, J.R.; Schneider, A.; Patel, J.; Srinivasan, A.; Kallen, A.; Limbago, B.; Fridkin, S. Antimicrobial-resistant pathogens associated with healthcare-associated infections summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2009–2010. Infect. Control Hosp. Epidemiol. 2013, 34, 1–14. [Google Scholar] [CrossRef]
- Nathwani, D.; Raman, G.; Sulham, K.; Gavaghan, M.; Menon, V. Clinical and economic consequences of hospital-acquired resistant and multidrug-resistant Pseudomonas aeruginosa infections: A systematic review and meta-analysis. Antimicrob. Resist. Infect. Control 2014, 3, 32. [Google Scholar] [CrossRef]
- Luyt, C.-E.; Hékimian, G.; Koulenti, D.; Chastre, J. Microbial cause of ICU-acquired pneumonia: Hospital-acquired pneumonia versus ventilator-associated pneumonia. Curr. Opin. Crit. Care 2018, 24, 332–338. [Google Scholar] [CrossRef]
- Sarda, C.; Fazal, F.; Rello, J. Management of ventilator-associated pneumonia (VAP) caused by resistant gram-negative bacteria: Which is the best strategy to treat? Expert Rev. Respir. Med. 2019, 13, 787–798. [Google Scholar] [CrossRef]
- Raineri, E.; Porcella, L.; Acquarolo, A.; Crema, L.; Albertario, F.; Candiani, A. Ventilator-Associated Pneumonia Caused by Pseudomonas aeruginosa in Intensive Care Unit: Epidemiology and Risk Factors. J. Med. Microbiol. Diagn. 2014, 3, 1. [Google Scholar] [CrossRef]
- Hilliam, Y.; Kaye, S.; Winstanley, C. Pseudomonas aeruginosa and microbial keratitis. J. Med. Microbiol. 2020, 69, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Langan, K.M.; Kotsimbos, T.; Peleg, A.Y. Managing Pseudomonas aeruginosa respiratory infections in cystic fibrosis. Curr. Opin. Infect. Dis. 2015, 28, 547–556. [Google Scholar] [CrossRef]
- Garcia-Clemente, M.; de la Rosa, D.; Máiz, L.; Girón, R.; Blanco, M.; Olveira, C.; Canton, R.; Martinez-García, M.A. Impact of Pseudomonas aeruginosa Infection on Patients with Chronic Inflammatory Airway Diseases. J. Clin. Med. 2020, 9, 3800. [Google Scholar] [CrossRef]
- Gransden, W.R.; Leibovici, L.; Eykyn, S.J.; Pitlik, S.D.; Samra, Z.; Konisberger, H.; Drucker, M.; Phillips, I. Risk factors and a clinical index for diagnosis of Pseudomonas aeruginosa bacteremia. Clin. Microbiol. Infect. 1995, 1, 119–123. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tredget, E.E.; Shankowsky, H.A.; Rennie, R.; Burrell, R.E.; Logsetty, S. Pseudomonas infections in the thermally injured patient. Burns 2004, 30, 3–26. [Google Scholar] [CrossRef]
- Puchelle, E.; Bajolet, O.; Abély, M. Airway mucus in cystic fibrosis. Paediatr. Respir. Rev. 2002, 3, 115–119. [Google Scholar] [CrossRef]
- Mayer-Hamblett, N.; Kloster, M.; Rosenfeld, M.; Gibson, R.L.; Retsch-Bogart, G.Z.; Emerson, J.; Thompson, V.; Ramsey, B.W. Impact of sustained eradication of new Pseudomonas aeruginosa infection on long-term outcomes in cystic fibrosis. Clin. Infect. Dis. 2015, 61, 707–715. [Google Scholar] [CrossRef]
- Taccetti, G.; Campana, S.; Festini, F.; Mascherini, M.; Döring, G. Early eradication therapy against Pseudomonas aeruginosa in cystic fibrosis patients. Eur. Respir. J. 2005, 26, 458–461. [Google Scholar] [CrossRef]
- Zolin, A.; Bossi, A.; Cirilli, N.; Kashirskaya, N.; Padoan, R. Cystic Fibrosis Mortality in Childhood. Data from European Cystic Fibrosis Society Patient Registry. Int. J. Environ. Res. Public Health 2018, 15, 2020. [Google Scholar] [CrossRef]
- Williams, F.N.; Herndon, D.N.; Hawkins, H.K.; Lee, J.O.; Cox, R.A.; Kulp, G.A.; Finnerty, C.C.; Chinkes, D.L.; Jeschke, M.G. The leading causes of death after burn injury in a single pediatric burn center. Crit. Care 2009, 13, R183. [Google Scholar] [CrossRef] [PubMed]
- Norbury, W.; Herndon, D.N.; Tanksley, J.; Jeschke, M.G.; Finnerty, C.C. Infection in Burns. Surg. Infect. 2016, 17, 250–255. [Google Scholar] [CrossRef] [PubMed]
- Church, D.; Elsayed, S.; Reid, O.; Winston, B.; Lindsay, R. Burn wound infections. Clin. Microbiol. Rev. 2006, 19, 403–434. [Google Scholar] [CrossRef]
- Johnson, L.E.; D’Agata, E.M.C.; Paterson, D.L.; Clarke, L.; Qureshi, Z.A.; Potoski, B.A.; Peleg, A.Y. Pseudomonas aeruginosa bacteremia over a 10-year period: Multidrug resistance and outcomes in transplant recipients. Transpl. Infect. Dis. 2009, 11, 227–234. [Google Scholar] [CrossRef]
- Choi, Y.; Paik, J.H.; Kim, J.H.; Han, S.B.; Durey, A. Clinical predictors of Pseudomonas aeruginosa bacteremia in emergency department. Emerg. Med. Int. 2018, 2018, 7581036. [Google Scholar] [CrossRef]
- Kang, C.I.; Kim, S.H.; Kim, H.B.; Park, S.W.; Choe, Y.J.; Oh, M.D.; Kim, E.C.; Choe, K.W. Pseudomonas aeruginosa bacteremia: Risk factors for mortality and influence of delayed receipt of effective antimicrobial therapy on clinical outcome. Clin. Infect. Dis. 2003, 37, 745–751. [Google Scholar] [CrossRef] [PubMed]
- Joo, E.-J.; Kang, C.-I.; Ha, Y.E.; Kang, S.-J.; Park, S.Y.; Chung, D.R.; Peck, K.R.; Lee, N.Y.; Song, J.-H. Risk factors for mortality in patients with Pseudomonas aeruginosa bacteremia: Clinical impact of antimicrobial resistance on outcome. Microb. Drug Resist. 2011, 17, 305–312. [Google Scholar] [CrossRef]
- Lachiewicz, A.M.; Hauck, C.G.; Weber, D.J.; Cairns, B.A.; van Duin, D. Bacterial infections after burn injuries: Impact of multidrug resistance. Clin. Infect. Dis. 2017, 65, 2130–2136. [Google Scholar] [CrossRef] [PubMed]
- Bédard, E.; Prévost, M.; Déziel, E. Pseudomonas aeruginosa in premise plumbing of large buildings. MicrobiologyOpen 2016, 5, 937–956. [Google Scholar] [CrossRef] [PubMed]
- Trautmann, M.; Lepper, P.M.; Haller, M. Ecology of Pseudomonas aeruginosa in the intensive care unit and the evolving role of water outlets as a reservoir of the organism. Am. J. Infect. Control 2005, 33, S41–S49. [Google Scholar] [CrossRef]
- Parcell, B.J.; Oravcova, K.; Pinheiro, M.; Holden, M.T.G.; Phillips, G.; Turton, J.F.; Gillespie, S.H. Pseudomonas aeruginosa intensive care unit outbreak: Winnowing of transmissions with molecular and genomic typing. J. Hosp. Infect. 2018, 98, 282–288. [Google Scholar] [CrossRef]
- Quick, J.; Cumley, N.; Wearn, C.M.; Niebel, M.; Constantinidou, C.; Thomas, C.M.; Pallen, M.J.; Moiemen, N.S.; Bamford, A.; Oppenheim, B.; et al. Seeking the source of Pseudomonas aeruginosa infections in a recently opened hospital: An observational study using whole-genome sequencing. BMJ Open 2014, 4, e006278. [Google Scholar] [CrossRef]
- Lanini, S.; D’Arezzo, S.; Puro, V.; Martini, L.; Imperi, F.; Piselli, P.; Montanaro, M.; Paoletti, S.; Visca, P.; Ippolito, G. Molecular epidemiology of a Pseudomonas aeruginosa hospital outbreak driven by a contaminated disinfectant-soap dispenser. PLoS ONE 2011, 6, e17064. [Google Scholar] [CrossRef]
- Eckmanns, T.; Oppert, M.; Martin, M.; Amorosa, R.; Zuschneid, I.; Frei, U.; Ruden, H.; Weist, K. An outbreak of hospital-acquired Pseudomonas aeruginosa infection caused by contaminated bottled water in intensive care units. Clin. Microbiol. Infect. 2008, 14, 454–458. [Google Scholar] [CrossRef] [PubMed]
- Schelstraete, P.; Van daele, S.; De Boeck, K.; Proesmans, M.; Lebecque, P.; Leclercq-Foucart, J.; Malfroot, A.; Vaneechoutte, M.; De Baets, F. Pseudomonas aeruginosa in the home environment of newly infected cystic fibrosis patients. Eur. Respir. J. 2008, 31, 822–829. [Google Scholar] [CrossRef]
- Magiorakos, A.P.; Srinivasan, A.; Carey, R.B.; Carmeli, Y.; Falagas, M.E.; Giske, C.G.; Harbarth, S.; Hindler, J.F.; Kahlmeter, G.; Olsson-Liljequist, B.; et al. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: An international expert proposal for interim standard definitions for acquired resistance. Clin. Microbiol. Infect. 2012, 18, 268–281. [Google Scholar] [CrossRef] [PubMed]
- CDC. Antibiotic Resistance Threats in the United States, 2019; Centers for Disease Control and Prevention: Atlanta, GA, USA, 2019. [Google Scholar]
- Hirsch, E.B.; Tam, V.H. Impact of multidrug-resistant Pseudomonas aeruginosa infection on patient outcomes. Expert Rev. Pharm. Outcomes Res. 2010, 10, 441–451. [Google Scholar]
- Matos, E.C.O.d.; Andriolo, R.B.; Rodrigues, Y.C.; Lima, P.D.L.d.; Carneiro, I.C.d.R.S.; Lima, K.V.B. Mortality in patients with multidrug-resistant Pseudomonas aeruginosa infections: A meta-analysis. Rev. Soc. Bras. Med. Trop. 2018, 51, 415–420. [Google Scholar] [CrossRef]
- Bradley, P.; Gordon, N.C.; Walker, T.M.; Dunn, L.; Heys, S.; Huang, B.; Earle, S.; Pankhurst, L.J.; Anson, L.; De Cesare, M. Rapid antibiotic-resistance predictions from genome sequence data for Staphylococcus aureus and Mycobacterium tuberculosis. Nat. Commun. 2015, 6, 10063. [Google Scholar] [CrossRef]
- Pendleton, J.N.; Gorman, S.P.; Gilmore, B.F. Clinical relevance of the ESKAPE pathogens. Expert Rev. Anti-Infect. Ther. 2013, 11, 297–308. [Google Scholar] [CrossRef]
- De Oliveira, D.M.; Forde, B.M.; Kidd, T.J.; Harris, P.N.; Schembri, M.A.; Beatson, S.A.; Paterson, D.L.; Walker, M.J. Antimicrobial resistance in ESKAPE pathogens. Clin. Microbiol. Rev. 2020, 33, e00181-19. [Google Scholar] [CrossRef] [PubMed]
- Taconelli, E.; Magrini, N. Global Priority List of Antibiotic-Resistant Bacteria to Guide Research, Discovery and Development of New Antibiotics; World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- Elborn, J.S.; Vataire, A.-L.; Fukushima, A.; Aballea, S.; Khemiri, A.; Moore, C.; Medic, G.; Hemels, M.E.H. Comparison of inhaled antibiotics for the treatment of chronic Pseudomonas aeruginosa lung infection in patients with cystic fibrosis: Systematic literature review and network meta-analysis. Clin. Ther. 2016, 38, 2204–2226. [Google Scholar] [CrossRef]
- Horcajada, J.P.; Montero, M.; Oliver, A.; Sorlí, L.; Luque, S.; Gómez-Zorrilla, S.; Benito, N.; Grau, S. Epidemiology and treatment of multidrug-resistant and extensively drug-resistant Pseudomonas aeruginosa infections. Clin. Microbiol. Rev. 2019, 32, e00031-19. [Google Scholar] [CrossRef]
- Talwalkar, J.S.; Murray, T.S. The approach to Pseudomonas aeruginosa in cystic fibrosis. Clin. Chest Med. 2016, 37, 69–81. [Google Scholar] [CrossRef]
- Castle, S.S. Ticarcillin. In xPharm: The Comprehensive Pharmacology Reference; Enna, S.J., Bylund, D.B., Eds.; Elsevier: New York, NY, USA, 2007; pp. 1–5. [Google Scholar] [CrossRef]
- Castle, S.S. Piperacillin. In xPharm: The Comprehensive Pharmacology Reference; Enna, S.J., Bylund, D.B., Eds.; Elsevier: New York, NY, USA, 2007; pp. 1–5. [Google Scholar] [CrossRef]
- Scholar, E. Aztreonam. In xPharm: The Comprehensive Pharmacology Reference; Enna, S.J., Bylund, D.B., Eds.; Elsevier: New York, NY, USA, 2007; pp. 1–5. [Google Scholar] [CrossRef]
- Hansen, C.; Skov, M. Evidence for the efficacy of aztreonam for inhalation solution in the management of Pseudomonas aeruginosa in patients with cystic fibrosis. Ther. Adv. Respir. Dis. 2015, 9, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Taccetti, G.; Francalanci, M.; Pizzamiglio, G.; Messore, B.; Carnovale, V.; Cimino, G.; Cipolli, M. Cystic Fibrosis: Recent Insights into Inhaled Antibiotic Treatment and Future Perspectives. Antibiotics 2021, 10, 338. [Google Scholar] [CrossRef]
- Blanco-Aparicio, M.; Saleta Canosa, J.L.; Valiño López, P.; Martín Egaña, M.T.; Vidal Garcia, I.; Montero Martínez, C. Eradication of Pseudomonas aeruginosa with inhaled colistin in adults with non-cystic fibrosis bronchiectasis. Chronic Respir. Dis. 2019, 16, 1–9. [Google Scholar] [CrossRef]
- King, D.E.; Malone, R.; Lilley, S.H. New classification and update on the quinolone antibiotics. Am. Fam. Physician 2000, 61, 2741–2748. [Google Scholar]
- Smith, S.; Rowbotham, N.J.; Charbek, E. Inhaled antibiotics for pulmonary exacerbations in cystic fibrosis. Cochrane Database Syst. Rev. 2018, 10, CD008319. [Google Scholar] [CrossRef] [PubMed]
- Pandey, N.; Cascella, M. Beta Lactam Antibiotics. In StatPearls; StatPearls: Treasure Island, FL, USA, 2021. [Google Scholar]
- Campanella, T.A.; Gallagher, J.C. A clinical review and critical evaluation of Imipenem-Relebactam: Evidence to date. Infect. Drug Resist. 2020, 13, 4297. [Google Scholar] [CrossRef]
- Kong, K.-F.; Schneper, L.; Mathee, K. β-lactam antibiotics: From antibiosis to resistance and bacteriology. APMIS 2010, 118, 1–36. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Ortega, C.; Wiegand, I.; Olivares, J.; Hancock, R.E.; Martinez, J.L. Genetic determinants involved in the susceptibility of Pseudomonas aeruginosa to β-lactam antibiotics. Antimicrob. Agents Chemother. 2010, 54, 4159–4167. [Google Scholar] [CrossRef]
- Breidenstein, E.B.M.; de la Fuente-Núñez, C.; Hancock, R.E.W. Pseudomonas aeruginosa: All roads lead to resistance. Trends Microbiol. 2011, 19, 419–426. [Google Scholar] [CrossRef]
- Ciofu, O.; Tolker-Nielsen, T. Tolerance and Resistance of Pseudomonas aeruginosa Biofilms to Antimicrobial Agents-How P. aeruginosa Can Escape Antibiotics. Front. Microbiol. 2019, 10, 913. [Google Scholar] [CrossRef]
- Povolotsky, T.L.; Keren-Paz, A.; Kolodkin-Gal, I. Metabolic Microenvironments Drive Microbial Differentiation and Antibiotic Resistance. Trends Genet. 2021, 37, 4–8. [Google Scholar] [CrossRef]
- Soares, A.; Alexandre, K.; Etienne, M. Tolerance and Persistence of Pseudomonas aeruginosa in Biofilms Exposed to Antibiotics: Molecular Mechanisms, Antibiotic Strategies and Therapeutic Perspectives. Front. Microbiol. 2020, 11, 2057. [Google Scholar] [CrossRef] [PubMed]
- Hengzhuang, W.; Ciofu, O.; Yang, L.; Wu, H.; Song, Z.; Oliver, A.; Hoiby, N. High β-lactamase levels change the pharmacodynamics of β-lactam antibiotics in Pseudomonas aeruginosa biofilms. Antimicrob. Agents Chemother. 2013, 57, 196–204. [Google Scholar] [CrossRef]
- Ramsay, K.A.; Wardell, S.J.T.; Patrick, W.M.; Brockway, B.; Reid, D.W.; Winstanley, C.; Bell, S.C.; Lamont, I.L. Genomic and phenotypic comparison of environmental and patient-derived isolates of Pseudomonas aeruginosa suggest that antimicrobial resistance is rare within the environment. J. Med. Microbiol. 2019, 68, 1591–1595. [Google Scholar] [CrossRef]
- Nordmann, P.; Poirel, L. Epidemiology and Diagnostics of Carbapenem Resistance in Gram-negative Bacteria. Clin. Infect. Dis. 2019, 69, S521–S528. [Google Scholar] [CrossRef]
- Shortridge, D.; Gales, A.C.; Streit, J.M.; Huband, M.D.; Tsakris, A.; Jones, R.N. Geographic and Temporal Patterns of Antimicrobial Resistance in Pseudomonas aeruginosa Over 20 Years From the SENTRY Antimicrobial Surveillance Program, 1997–2016. Open Forum Infect. Dis. 2019, 6, S63–S68. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimpour, M.; Nikokar, I.; Ghasemi, Y.; Sedigh Ebrahim-Saraie, H.; Araghian, A.; Farahbakhsh, M.; Ghassabi, F. Antibiotic resistance and frequency of class 1 integrons among Pseudomonas aeruginosa isolates obtained from wastewaters of a burn center in Northern Iran. Ann. Ig. 2018, 30, 112–119. [Google Scholar] [CrossRef]
- Feretzakis, G.; Loupelis, E.; Sakagianni, A.; Skarmoutsou, N.; Michelidou, S.; Velentza, A.; Martsoukou, M.; Valakis, K.; Petropoulou, S.; Koutalas, E. A 2-year single-centre audit on antibiotic resistance of Pseudomonas aeruginosa, Acinetobacter baumannii and Klebsiella pneumoniae strains from an intensive care unit and other wards in a general public hospital in Greece. Antibiotics 2019, 8, 62. [Google Scholar] [CrossRef] [PubMed]
- Lake, J.G.; Weiner, L.M.; Milstone, A.M.; Saiman, L.; Magill, S.S.; See, I. Pathogen distribution and antimicrobial resistance among pediatric healthcare-associated infections reported to the National Healthcare Safety Network, 2011–2014. Infect. Control Hosp. Epidemiol. 2018, 39, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, X.-L.; Huang, A.-W.; Liu, S.-L.; Liu, W.-J.; Zhang, N.; Lu, X.-Z. Mortality attributable to carbapenem-resistant Pseudomonas aeruginosa bacteremia: A meta-analysis of cohort studies. Emerg. Microbes Infect. 2016, 5, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Li, X.; Li, W.; Du, X.; He, J.-Q.; Tao, C.; Feng, Y. Influence of carbapenem resistance on mortality of patients with Pseudomonas aeruginosa infection: A meta-analysis. Sci. Rep. 2015, 5, 11715. [Google Scholar] [CrossRef] [PubMed]
- Balkhair, A.; Al-Muharrmi, Z.; Al’Adawi, B.; Al Busaidi, I.; Taher, H.B.; Al-Siyabi, T.; Al Amin, M.; Hassan, K.S. Prevalence and 30-day all-cause mortality of carbapenem-and colistin-resistant bacteraemia caused by Acinetobacter baumannii, Pseudomonas aeruginosa, and Klebsiella pneumoniae: Description of a decade-long trend. Int. J. Infect. Dis. 2019, 85, 10–15. [Google Scholar] [CrossRef]
- Lin, K.Y.; Lauderdale, T.L.; Wang, J.T.; Chang, S.C. Carbapenem-resistant Pseudomonas aeruginosa in Taiwan: Prevalence, risk factors, and impact on outcome of infections. J. Microbiol. Immunol. Infect. 2016, 49, 52–59. [Google Scholar] [CrossRef]
- Cassini, A.; Högberg, L.D.; Plachouras, D.; Quattrocchi, A.; Hoxha, A.; Simonsen, G.S.; Colomb-Cotinat, M.; Kretzschmar, M.E.; Devleesschauwer, B.; Cecchini, M.; et al. Attributable deaths and disability-adjusted life-years caused by infections with antibiotic-resistant bacteria in the EU and the European Economic Area in 2015: A population-level modelling analysis. Lancet Infect. Dis. 2019, 19, 56–66. [Google Scholar] [CrossRef]
- Kaier, K.; Heister, T.; Gotting, T.; Wolkewitz, M.; Mutters, N.T. Measuring the in-hospital costs of Pseudomonas aeruginosa pneumonia: Methodology and results from a German teaching hospital. BMC Infect. Dis. 2019, 19, 1028. [Google Scholar] [CrossRef] [PubMed]
- Lister Philip, D.; Wolter Daniel, J.; Hanson Nancy, D. Antibacterial-Resistant Pseudomonas aeruginosa: Clinical Impact and Complex Regulation of Chromosomally Encoded Resistance Mechanisms. Clin. Microbiol. Rev. 2009, 22, 582–610. [Google Scholar] [CrossRef]
- López-Causapé, C.; Cabot, G.; del Barrio-Tofiño, E.; Oliver, A. The Versatile Mutational Resistome of Pseudomonas aeruginosa. Front. Microbiol. 2018, 9, 685. [Google Scholar] [CrossRef]
- López-Causapé, C.; Rojo-Molinero, E.; Macià, M.D.; Oliver, A. The problems of antibiotic resistance in cystic fibrosis and solutions. Expert Rev. Respir. Med. 2015, 9, 73–88. [Google Scholar] [CrossRef]
- Llanes, C.; Pourcel, C.; Richardot, C.; Plesiat, P.; Fichant, G.; Cavallo, J.D.; Merens, A.; Group, G.S. Diversity of β-lactam resistance mechanisms in cystic fibrosis isolates of Pseudomonas aeruginosa: A French multicentre study. J. Antimicrob. Chemother. 2013, 68, 1763–1771. [Google Scholar] [CrossRef]
- Pitten, F.A.; Panzig, B.; Schröder, G.; Tietze, K.; Kramer, A. Transmission of a multiresistant Pseudomonas aeruginosa strain at a German University Hospital. J. Hosp. Infect. 2001, 47, 125–130. [Google Scholar] [CrossRef]
- Aguilera-Sáez, J.; Andreu-Solà, V.; Larrosa Escartín, N.; Rodríguez Garrido, V.; Armadans Gil, L.; Sánchez García, J.M.; Campins, M.; Baena Caparrós, J.; Barret, J.P. Extensively drug-resistant Pseudomonas aeruginosa outbreak in a burn unit: Management and solutions. Ann. Burn. Fire Disasters 2019, 32, 47–55. [Google Scholar]
- Douglas, M.W.; Mulholland, K.; Denyer, V.; Gottlieb, T. Multi-drug resistant Pseudomonas aeruginosa outbreak in a burns unit—An infection control study. Burns 2001, 27, 131–135. [Google Scholar] [CrossRef]
- Vollmer, W.; Holtje, J.V. The architecture of the murein (peptidoglycan) in gram-negative bacteria: Vertical scaffold or horizontal layer(s)? J. Bacteriol. 2004, 186, 5978–5987. [Google Scholar] [CrossRef]
- Torrens, G.; Escobar-Salom, M.; Pol-Pol, E.; Camps-Munar, C.; Cabot, G.; López-Causapé, C.; Rojo-Molinero, E.; Oliver, A.; Juan, C. Comparative analysis of peptidoglycans from Pseudomonas aeruginosa isolates recovered from chronic and acute infections. Front. Microbiol. 2019, 10, 1868. [Google Scholar] [CrossRef]
- Vollmer, W.; Blanot, D.; De Pedro, M.A. Peptidoglycan structure and architecture. FEMS Microbiol. Rev. 2008, 32, 149–167. [Google Scholar] [CrossRef]
- Akira Yanai, K.K. Teruhiko Beppu and Kei Arima. Peptidoglycan of Pseudomonas aeruginosa. Agric. Biol. Chem. 1975, 40, 1505–1508. [Google Scholar]
- Sauvage, E.; Kerff, F.; Terrak, M.; Ayala, J.A.; Charlier, P. The penicillin-binding proteins: Structure and role in peptidoglycan biosynthesis. FEMS Microbiol. Rev. 2008, 32, 234–258. [Google Scholar] [CrossRef]
- Heywood, A.; Lamont, I.L. Cell envelope proteases and peptidases of Pseudomonas aeruginosa: Multiple roles, multiple mechanisms. FEMS Microbiol. Rev. 2020, 44, 857–873. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zhang, Y.-M.; Davies, C. Penicillin-binding protein 3 is essential for growth of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2017, 61, e01651-16. [Google Scholar] [CrossRef] [PubMed]
- Goffin, C.; Ghuysen, J.M. Biochemistry and comparative genomics of SxxK superfamily acyltransferases offer a clue to the mycobacterial paradox: Presence of penicillin-susceptible target proteins versus lack of efficiency of penicillin as therapeutic agent. Microbiol. Mol. Biol. Rev. 2002, 66, 702–738. [Google Scholar] [CrossRef] [PubMed]
- Van Heijenoort, J.; Gutmann, L. Correlation between the structure of the bacterial peptidoglycan monomer unit, the specificity of transpeptidation, and susceptibility to β-lactams. Proc. Natl. Acad. Sci. USA 2000, 97, 5028–5030. [Google Scholar] [CrossRef] [PubMed]
- Goffin, C.; Ghuysen, J.M. Multimodular penicillin-binding proteins: An enigmatic family of orthologs and paralogs. Microbiol. Mol. Biol. Rev. 1998, 62, 1079–1093. [Google Scholar] [CrossRef]
- Tomberg, J.; Temple, B.; Fedarovich, A.; Davies, C.; Nicholas, R.A. A highly conserved interaction involving the middle residue of the SXN active-site motif is crucial for function of class B penicillin-binding proteins: Mutational and computational analysis of PBP 2 from N. gonorrhoeae. Biochemistry 2012, 51, 2775–2784. [Google Scholar] [CrossRef]
- Macheboeuf, P.; Contreras-Martel, C.; Job, V.; Dideberg, O.; Dessen, A. Penicillin Binding Proteins: Key players in bacterial cell cycle and drug resistance processes. FEMS Microbiol. Rev. 2006, 30, 673–691. [Google Scholar] [CrossRef] [PubMed]
- Sauvage, E.; Terrak, M. Glycosyltransferases and transpeptidases/penicillin-binding proteins: Valuable targets for new antibacterials. Antibiotics 2016, 5, 12. [Google Scholar] [CrossRef]
- Ropy, A.; Cabot, G.; Sánchez-Diener, I.; Aguilera, C.; Moya, B.; Ayala, J.A.; Oliver, A. Role of Pseudomonas aeruginosa low-molecular-mass penicillin-binding proteins in AmpC expression, β-lactam resistance, and peptidoglycan structure. Antimicrob. Agents Chemother. 2015, 59, 3925–3934. [Google Scholar] [CrossRef]
- Ghosh, A.S.; Chowdhury, C.; Nelson, D.E. Physiological functions of D-alanine carboxypeptidases in Escherichia coli. Trends Microbiol. 2008, 16, 309–317. [Google Scholar] [CrossRef]
- Torrens, G.; Hernandez, S.B.; Ayala, J.A.; Moya, B.; Juan, C.; Cava, F.; Oliver, A. Regulation of AmpC-driven β-lactam resistance in Pseudomonas aeruginosa: Different pathways, different signaling. mSystems 2019, 4, e00524-19. [Google Scholar] [CrossRef]
- Vollmer, W.; Joris, B.; Charlier, P.; Foster, S. Bacterial peptidoglycan (murein) hydrolases. FEMS Microbiol. Rev. 2008, 32, 259–286. [Google Scholar] [CrossRef]
- Aguilera Rossi, C.G.; Gómez-Puertas, P.; Ayala Serrano, J.A. In vivo functional and molecular characterization of the Penicillin-Binding Protein 4 (DacB) of Pseudomonas aeruginosa. BMC Microbiol. 2016, 16, 234. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Hesek, D.; Blázquez, B.; Lastochkin, E.; Boggess, B.; Fisher, J.F.; Mobashery, S. Catalytic spectrum of the penicillin-binding protein 4 of Pseudomonas aeruginosa, a nexus for the induction of β-lactam antibiotic resistance. J. Am. Chem. Soc. 2015, 137, 190–200. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, J.; Oesch, G.; Kuster, S.P. Bacteriostatic versus bactericidal antibiotics for patients with serious bacterial infections: Systematic review and meta-analysis. J. Antimicrob. Chemother. 2014, 70, 382–395. [Google Scholar] [CrossRef]
- Dumancas, G.G.; Hikkaduwa Koralege, R.S.; Mojica, E.R.E.; Murdianti, B.S.; Pham, P.J. Penicillins. In Encyclopedia of Toxicology, 3rd ed.; Wexler, P., Ed.; Academic Press: Oxford, UK, 2014; pp. 768–772. [Google Scholar] [CrossRef]
- Zeng, X.; Lin, J. β-lactamase induction and cell wall metabolism in gram-negative bacteria. Front. Microbiol. 2013, 4, 128. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Zaniewski, R.P.; Marr, E.S.; Lacey, B.M.; Tomaras, A.P.; Evdokimov, A.; Miller, J.R.; Shanmugasundaram, V. Structural basis for effectiveness of siderophore-conjugated monocarbams against clinically relevant strains of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 2010, 107, 22002–22007. [Google Scholar] [CrossRef]
- Beadle, B.M.; Nicholas, R.A.; Shoichet, B.K. Interaction energies between β-lactam antibiotics and E. coli penicillin-binding protein 5 by reversible thermal denaturation. Protein Sci. 2001, 10, 1254–1259. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Uehara, T.; Bernhardt, T.G. β-lactam antibiotics induce a lethal malfunctioning of the bacterial cell wall synthesis machinery. Cell 2014, 159, 1300–1311. [Google Scholar] [CrossRef]
- Yao, Z.; Kahne, D.; Kishony, R. Distinct single-cell morphological dynamics under β-lactam antibiotics. Mol. Cell 2012, 48, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Horii, T.; Ichiyama, S.; Ohta, M.; Kobayashi, M. Relationship between morphological changes and endotoxin release induced by carbapenems in Pseudomonas aeruginosa. J. Med. Microbiol. 1999, 48, 309–315. [Google Scholar] [CrossRef][Green Version]
- Lee, D.; Das, S.; Dawson, N.L.; Dobrijevic, D.; Ward, J.; Orengo, C. Novel computational protocols for functionally classifying and characterising serine β-lactamases. PLoS Comput. Biol. 2016, 12, e1004926. [Google Scholar] [CrossRef]
- Liras, P.; Martín, J.F. β-lactam Antibiotics. In Encyclopedia of Microbiology, 3rd ed.; Schaechter, M., Ed.; Academic Press: Oxford, UK, 2009; pp. 274–289. [Google Scholar] [CrossRef]
- Okamoto, K.; Gotoh, N.; Nishino, T. Pseudomonas aeruginosa reveals high intrinsic resistance to penem antibiotics: Penem resistance mechanisms and their interplay. Antimicrob. Agents Chemother. 2001, 45, 1964–1971. [Google Scholar] [CrossRef] [PubMed]
- Davies, T.A.; Shang, W.; Bush, K.; Flamm, R.K. Affinity of doripenem and comparators to penicillin-binding proteins in Escherichia coli and Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2008, 52, 1510–1512. [Google Scholar] [CrossRef]
- Davies, T.A.; Page, M.G.; Shang, W.; Andrew, T.; Kania, M.; Bush, K. Binding of ceftobiprole and comparators to the penicillin-binding proteins of Escherichia coli, Pseudomonas aeruginosa, Staphylococcus aureus, and Streptococcus pneumoniae. Antimicrob. Agents Chemother. 2007, 51, 2621–2624. [Google Scholar] [CrossRef]
- Fontana, R.; Cornaglia, G.; Ligozzi, M.; Mazzariol, A. The final goal: Penicillin-binding proteins and the target of cephalosporins. Clin. Microbiol. Infect. 2000, 6 (Suppl. S3), 34–40. [Google Scholar] [CrossRef] [PubMed]
- Dik, D.A.; Madukoma, C.S.; Tomoshige, S.; Kim, C.; Lastochkin, E.; Boggess, W.C.; Fisher, J.F.; Shrout, J.D.; Mobashery, S. Slt, MltD, and MltG of Pseudomonas aeruginosa as targets of Bulgecin A in potentiation of β-lactam antibiotics. ACS Chem. Biol. 2019, 14, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Lamers, R.P.; Nguyen, U.T.; Nguyen, Y.; Buensuceso, R.N.C.; Burrows, L.L. Loss of membrane-bound lytic transglycosylases increases outer membrane permeability and β-lactam sensitivity in Pseudomonas aeruginosa. MicrobiologyOpen 2015, 4, 879–895. [Google Scholar] [CrossRef]
- Lee, M.; Batuecas, M.T.; Tomoshige, S.; Domínguez-Gil, T.; Mahasenan, K.V.; Dik, D.A.; Hesek, D.; Millán, C.; Usón, I.; Lastochkin, E. Exolytic and endolytic turnover of peptidoglycan by lytic transglycosylase Slt of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 2018, 115, 4393–4398. [Google Scholar] [CrossRef]
- Domenig, C.; Traunmüller, F.; Kozek, S.; Wisser, W.; Klepetko, W.; Steininger, R.; Spiss, C.; Thalhammer, F. Continuous β-lactam antibiotic therapy in a double-lung transplanted patient with a multidrug-resistant Pseudomonas aeruginosa infection. Transplantation 2001, 71, 744–745. [Google Scholar] [CrossRef]
- Moriyama, B.; Henning, S.A.; Childs, R.; Holland, S.M.; Anderson, V.L.; Morris, J.C.; Wilson, W.H.; Drusano, G.L.; Walsh, T.J. High-dose continuous infusion β-lactam antibiotics for the treatment of resistant Pseudomonas aeruginosa infections in immunocompromised patients. Ann. Pharmacother. 2010, 44, 929–935. [Google Scholar] [CrossRef]
- Yu, Z.; Pang, X.; Wu, X.; Shan, C.; Jiang, S. Clinical outcomes of prolonged infusion (extended infusion or continuous infusion) versus intermittent bolus of meropenem in severe infection: A meta-analysis. PLoS ONE 2018, 13, e0201667. [Google Scholar] [CrossRef]
- Siriyong, T.; Murray, R.M.; Bidgood, L.E.; Young, S.A.; Wright, F.; Parcell, B.J.; Voravuthikunchai, S.P.; Coote, P.J. Dual β-lactam combination therapy for multi-drug resistant Pseudomonas aeruginosa infection: Enhanced efficacy in vivo and comparison with monotherapies of penicillin-binding protein inhibition. Sci. Rep. 2019, 9, 9098. [Google Scholar] [CrossRef] [PubMed]
- Mikhail, S.; Singh, N.B.; Kebriaei, R.; Rice, S.A.; Stamper, K.C.; Castanheira, M.; Rybak, M.J. Evaluation of the synergy of ceftazidime-avibactam in combination with meropenem, amikacin, aztreonam, colistin, or fosfomycin against well-characterized multidrug-resistant Klebsiella pneumoniae and Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2019, 63, e00779-19. [Google Scholar] [CrossRef] [PubMed]
- Reygaert, W.C. An overview of the antimicrobial resistance mechanisms of bacteria. AIMS Microbiol. 2018, 4, 482–501. [Google Scholar] [CrossRef]
- Munita, J.M.; Arias, C.A. Mechanisms of antibiotic resistance. Microbiol. Spectr. 2016, 4, 4.2.15. [Google Scholar] [CrossRef]
- Karballaei Mirzahosseini, H.; Hadadi-Fishani, M.; Morshedi, K.; Khaledi, A. Meta-analysis of biofilm formation, antibiotic resistance pattern, and biofilm-related genes in Pseudomonas aeruginosa isolated from clinical samples. Microb. Drug Resist. 2020, 26, 815–824. [Google Scholar] [CrossRef] [PubMed]
- Wardell, S.J.T.; Rehman, A.; Martin, L.W.; Winstanley, C.; Patrick, W.M.; Lamont, I.L. A large-scale whole-genome comparison shows that experimental evolution in response to antibiotics predicts changes in naturally evolved clinical Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2019, 63, e01619-19. [Google Scholar] [CrossRef]
- Yen, P.; Papin, J.A. History of antibiotic adaptation influences microbial evolutionary dynamics during subsequent treatment. PLoS Biol. 2017, 15, e2001586. [Google Scholar] [CrossRef]
- Jorth, P.; McLean, K.; Ratjen, A.; Secor, P.R.; Bautista, G.E.; Ravishankar, S.; Rezayat, A.; Garudathri, J.; Harrison, J.J.; Harwood, R.A. Evolved aztreonam resistance is multifactorial and can produce hypervirulence in Pseudomonas aeruginosa. mBio 2017, 8, e00517-17. [Google Scholar] [CrossRef] [PubMed]
- Sanz-García, F.; Hernando-Amado, S.; Martínez, J.L. Mutation-Driven Evolution of Pseudomonas aeruginosa in the Presence of either Ceftazidime or Ceftazidime-Avibactam. Antimicrob. Agents Chemother. 2018, 62, e01379-18. [Google Scholar] [CrossRef]
- Marvig, R.L.; Sommer, L.M.; Molin, S.; Johansen, H.K. Convergent evolution and adaptation of Pseudomonas aeruginosa within patients with cystic fibrosis. Nat. Genet. 2015, 47, 57–64. [Google Scholar] [CrossRef]
- Sumita, Y.; Fakasawa, M. Potent activity of meropenem against Escherichia coli arising from its simultaneous binding to penicillin-binding proteins 2 and 3. J. Antimicrob. Chemother. 1995, 36, 53–64. [Google Scholar] [CrossRef]
- Kocaoglu, O.; Carlson, E.E. Profiling of β-Lactam selectivity for penicillin-binding proteins in Escherichia coli strain DC2. Antimicrob. Agents Chemother. 2015, 59, 2785–2790. [Google Scholar] [CrossRef] [PubMed]
- Diawara, I.; Nayme, K.; Katfy, K.; Barguigua, A.; Kettani-Halabi, M.; Belabbes, H.; Timinouni, M.; Zerouali, K.; Elmdaghri, N. Analysis of amino acid motif of penicillin-binding proteins 1a, 2b, and 2x in invasive Streptococcus pneumoniae nonsusceptible to penicillin isolated from pediatric patients in Casablanca, Morocco. BMC Res. Notes 2018, 11, 632. [Google Scholar] [CrossRef][Green Version]
- Cabot, G.; Zamorano, L.; Moya, B.; Juan, C.; Navas, A.; Blazquez, J.; Oliver, A. Evolution of Pseudomonas aeruginosa antimicrobial resistance and fitness under low and high mutation rates. Antimicrob. Agents Chemother. 2016, 60, 1767–1778. [Google Scholar] [CrossRef] [PubMed]
- Castanheira, M.; Doyle, T.B.; Smith, C.J.; Mendes, R.E.; Sader, H.S. Combination of MexAB-OprM overexpression and mutations in efflux regulators, PBPs and chaperone proteins is responsible for ceftazidime/avibactam resistance in Pseudomonas aeruginosa clinical isolates from US hospitals. J. Antimicrob. Chemother. 2019, 74, 2588–2595. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.T.; Sinha, U.; Zhang, Y.; Wang, P.W.; Donaldson, S.L.; Coburn, B.; Waters, V.J.; Yau, Y.C.W.; Tullis, D.E.; Guttman, D.S.; et al. Penicillin-binding protein 3 is a common adaptive target among Pseudomonas aeruginosa isolates from adult cystic fibrosis patients treated with β-lactams. Int. J. Antimicrob. Agents 2019, 53, 620–628. [Google Scholar] [CrossRef]
- López-Causapé, C.; Sommer, L.M.; Cabot, G.; Rubio, R.; Ocampo-Sosa, A.A.; Johansen, H.K.; Figuerola, J.; Cantón, R.; Kidd, T.J.; Molin, S. Evolution of the Pseudomonas aeruginosa mutational resistome in an international cystic fibrosis clone. Sci. Rep. 2017, 7, 5555. [Google Scholar] [CrossRef]
- Ren, J.; Nettleship, J.E.; Males, A.; Stuart, D.I.; Owens, R.J. Crystal structures of penicillin-binding protein 3 in complexes with azlocillin and cefoperazone in both acylated and deacylated forms. FEBS Lett. 2016, 590, 288–297. [Google Scholar] [CrossRef]
- Sainsbury, S.; Bird, L.; Rao, V.; Shepherd, S.M.; Stuart, D.I.; Hunter, W.N.; Owens, R.J.; Ren, J. Crystal structures of penicillin-binding protein 3 from Pseudomonas aeruginosa: Comparison of native and antibiotic-bound forms. J. Mol. Biol. 2011, 405, 173–184. [Google Scholar] [CrossRef] [PubMed]
- del Barrio-Tofiño, E.; López-Causapé, C.; Cabot, G.; Rivera, A.; Benito, N.; Segura, C.; Montero, M.M.; Sorlí, L.; Tubau, F.; Gómez-Zorrilla, S. Genomics and susceptibility profiles of extensively drug-resistant Pseudomonas aeruginosa isolates from Spain. Antimicrob. Agents Chemother. 2017, 61, e01589-17. [Google Scholar] [CrossRef] [PubMed]
- Cabot, G.; Lopez-Causape, C.; Ocampo-Sosa, A.A.; Sommer, L.M.; Dominguez, M.A.; Zamorano, L.; Juan, C.; Tubau, F.; Rodriguez, C.; Moya, B.; et al. Deciphering the resistome of the widespread Pseudomonas aeruginosa sequence type 175 international high-risk clone through whole-genome sequencing. Antimicrob. Agents Chemother. 2016, 60, 7415–7423. [Google Scholar] [CrossRef]
- Diaz Caballero, J.; Clark, S.T.; Coburn, B.; Zhang, Y.; Wang, P.W.; Donaldson, S.L.; Tullis, D.E.; Yau, Y.C.; Waters, V.J.; Hwang, D.M.; et al. Selective sweeps and parallel pathoadaptation drive Pseudomonas aeruginosa evolution in the cystic fibrosis lung. mBio 2015, 6, e00981-15. [Google Scholar] [CrossRef]
- Jorth, P.; Staudinger, B.J.; Wu, X.; Hisert, K.B.; Hayden, H.; Garudathri, J.; Harding, C.L.; Radey, M.C.; Rezayat, A.; Bautista, G.; et al. Regional isolation drives bacterial diversification within cystic fibrosis lungs. Cell Host Microbe 2015, 18, 307–319. [Google Scholar] [CrossRef]
- Markussen, T.; Marvig, R.L.; Gómez-Lozano, M.; Aanæs, K.; Burleigh, A.E.; Høiby, N.; Johansen, H.K.; Molin, S.; Jelsbak, L. Environmental heterogeneity drives within-host diversification and evolution of Pseudomonas aeruginosa. mBio 2014, 5, e01592-14. [Google Scholar] [CrossRef]
- Marvig, R.L.; Johansen, H.K.; Molin, S.; Jelsbak, L. Genome analysis of a transmissible lineage of Pseudomonas aeruginosa reveals pathoadaptive mutations and distinct evolutionary paths of hypermutators. PLoS Genet. 2013, 9, e1003741. [Google Scholar] [CrossRef]
- Yang, L.; Jelsbak, L.; Marvig, R.L.; Damkiaer, S.; Workman, C.T.; Rau, M.H.; Hansen, S.K.; Folkesson, A.; Johansen, H.K.; Ciofu, O.; et al. Evolutionary dynamics of bacteria in a human host environment. Proc. Natl. Acad. Sci. USA 2011, 108, 7481–7486. [Google Scholar] [CrossRef]
- Chotirmall, S.H.; Sherrard, L.J.; Tai, A.S.; Wee, B.A.; Ramsay, K.A.; Kidd, T.J.; Ben Zakour, N.L.; Whiley, D.M.; Beatson, S.A.; Bell, S.C. Within-host whole genome analysis of an antibiotic resistant Pseudomonas aeruginosa strain sub-type in cystic fibrosis. PLoS ONE 2017, 12, e0172179. [Google Scholar] [CrossRef]
- Colque, C.A.; Albarracin Orio, A.G.; Feliziani, S.; Marvig, R.L.; Tobares, A.R.; Johansen, H.K.; Molin, S.; Smania, A.M. Hypermutator Pseudomonas aeruginosa exploits multiple genetic pathways to develop multidrug resistance during long-term infections in the airways of cystic fibrosis patients. Antimicrob. Agents Chemother. 2020, 64, e02142-19. [Google Scholar] [CrossRef]
- McLean, K.; Lee, D.; Holmes, E.A.; Penewit, K.; Waalkes, A.; Ren, M.; Lee, S.A.; Gasper, J.; Manoil, C.; Salipante, S.J. Genomic analysis identifies novel Pseudomonas aeruginosa resistance genes under selection during inhaled aztreonam therapy in vivo. Antimicrob. Agents Chemother. 2019, 63, e00866-19. [Google Scholar] [CrossRef]
- Feng, Y.; Jonker, M.J.; Moustakas, I.; Brul, S.; Ter Kuile, B.H. Dynamics of Mutations during Development of Resistance by Pseudomonas aeruginosa against Five Antibiotics. Antimicrob. Agents Chemother. 2016, 60, 4229–4236. [Google Scholar] [CrossRef]
- Thegerström, J.; Matuschek, E.; Su, Y.-C.; Riesbeck, K.; Resman, F. A novel PBP3 substitution in Haemophilus influenzae confers reduced aminopenicillin susceptibility. BMC Microbiol. 2018, 18, 1–7. [Google Scholar] [CrossRef]
- Behmard, E.; Ahmadi, A.; Najafi, A. Influence of the T to S mutation at the STMK motif on antibiotic resistance of penicillin binding protein 1A: A comprehensive computational study. J. Mol. Graph. Model. 2019, 87, 185–191. [Google Scholar] [CrossRef]
- Nagai, K.; Davies, T.A.; Jacobs, M.R.; Appelbaum, P.C. Effects of amino acid alterations in penicillin-binding proteins (PBPs) 1a, 2b, and 2x on PBP affinities of penicillin, ampicillin, amoxicillin, cefditoren, cefuroxime, cefprozil, and cefaclor in 18 clinical isolates of penicillin-susceptible, -intermediate, and -resistant pneumococci. Antimicrob. Agents Chemother. 2002, 46, 1273–1280. [Google Scholar] [CrossRef]
- Giske, C.G.; Buaro, L.; Sundsfjord, A.; Wretlind, B. Alterations of porin, pumps, and penicillin-binding proteins in carbapenem resistant clinical isolates of Pseudomonas aeruginosa. Microb. Drug Resist. 2008, 14, 23–30. [Google Scholar] [CrossRef]
- Li, H.; Luo, Y.-F.; Williams, B.J.; Blackwell, T.S.; Xie, C.-M. Structure and function of OprD protein in Pseudomonas aeruginosa: From antibiotic resistance to novel therapies. Int. J. Med. Microbiol. 2012, 302, 63–68. [Google Scholar] [CrossRef]
- Choudhury, D.; Talukdar, A.D.; Choudhury, M.D.; Maurya, A.P.; Chanda, D.D.; Chakravorty, A.; Bhattacharjee, A. Carbapenem nonsusceptibility with modified OprD in clinical isolates of Pseudomonas aeruginosa from India. Indian J. Med. Microbiol. 2017, 35, 137–139. [Google Scholar] [CrossRef]
- Shu, J.C.; Kuo, A.J.; Su, L.H.; Liu, T.P.; Lee, M.H.; Su, I.N.; Wu, T.L. Development of carbapenem resistance in Pseudomonas aeruginosa is associated with OprD polymorphisms, particularly the amino acid substitution at codon 170. J. Antimicrob. Chemother. 2017, 72, 2489–2495. [Google Scholar] [CrossRef]
- Lee, J.-Y.; Ko, K.S. OprD mutations and inactivation, expression of efflux pumps and AmpC, and metallo-β-lactamases in carbapenem-resistant Pseudomonas aeruginosa isolates from South Korea. Int. J. Antimicrob. Agents 2012, 40, 168–172. [Google Scholar] [CrossRef]
- Kos, V.N.; McLaughlin, R.E.; Gardner, H.A. Identification of unique in-frame deletions in OprD among clinical isolates of Pseudomonas aeruginosa. Pathog. Dis. 2016, 74, ftw031. [Google Scholar] [CrossRef][Green Version]
- Ochs, M.M.; McCusker, M.P.; Bains, M.; Hancock, R.E.W. Negative regulation of the Pseudomonas aeruginosa outer membrane porin OprD selective for imipenem and basic amino acids. Antimicrob. Agents Chemother. 1999, 43, 1085–1090. [Google Scholar] [CrossRef]
- Iyer, R.; Sylvester, M.A.; Velez-Vega, C.; Tommasi, R.; Durand-Reville, T.F.; Miller, A.A. Whole-cell-based assay to evaluate structure permeation relationships for carbapenem passage through the Pseudomonas aeruginosa porin OprD. ACS Infect. Dis. 2017, 3, 310–319. [Google Scholar] [CrossRef]
- Muramatsu, H.; Horii, T.; Morita, M.; Hashimoto, H.; Kanno, T.; Maekawa, M. Effect of basic amino acids on susceptibility to carbapenems in clinical Pseudomonas aeruginosa isolates. Int. J. Med. Microbiol. 2003, 293, 191–197. [Google Scholar] [CrossRef]
- Skurnik, D.; Roux, D.; Cattoir, V.; Danilchanka, O.; Lu, X.; Yoder-Himes, D.R.; Han, K.; Guillard, T.; Jiang, D.; Gaultier, C.; et al. Enhanced in vivo fitness of carbapenem-resistant oprD mutants of Pseudomonas aeruginosa revealed through high-throughput sequencing. Proc. Natl. Acad. Sci. USA 2013, 110, 20747–20752. [Google Scholar] [CrossRef]
- Ude, J.; Tripathi, V.; Buyck, J.M.; Söderholm, S.; Cunrath, O.; Fanous, J.; Claudi, B.; Egli, A.; Schleberger, C.; Hiller, S.; et al. Outer membrane permeability: Antimicrobials and diverse nutrients bypass porins in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 2021, 118, e2107644118. [Google Scholar] [CrossRef]
- Isabella, V.M.; Campbell, A.J.; Manchester, J.; Sylvester, M.; Nayar, A.S.; Ferguson, K.E.; Tommasi, R.; Miller, A.A. Toward the rational design of carbapenem uptake in Pseudomonas aeruginosa. Chem. Biol. 2015, 22, 535–547. [Google Scholar] [CrossRef]
- Tamber, S.; Maier, E.; Benz, R.; Hancock Robert, E.W. Characterization of OpdH, a Pseudomonas aeruginosa Porin Involved in the Uptake of Tricarboxylates. J. Bacteriol. 2007, 189, 929–939. [Google Scholar] [CrossRef]
- Samanta, S.; Bodrenko, I.; Acosta-Gutiérrez, S.; D’Agostino, T.; Pathania, M.; Ghai, I.; Schleberger, C.; Bumann, D.; Wagner, R.; Winterhalter, M.; et al. Getting drugs through small pores: Exploiting the porins pathway in Pseudomonas aeruginosa. ACS Infect. Dis. 2018, 4, 1519–1528. [Google Scholar] [CrossRef]
- Chalhoub, H.; Pletzer, D.; Weingart, H.; Braun, Y.; Tunney, M.M.; Elborn, J.S.; Rodriguez-Villalobos, H.; Plésiat, P.; Kahl, B.C.; Denis, O. Mechanisms of intrinsic resistance and acquired susceptibility of Pseudomonas aeruginosa isolated from cystic fibrosis patients to temocillin, a revived antibiotic. Sci. Rep. 2017, 7, 40208. [Google Scholar] [CrossRef]
- Buyck, J.M.; Guénard, S.; Plésiat, P.; Tulkens, P.M.; Van Bambeke, F. Role of MexAB-OprM in intrinsic resistance of Pseudomonas aeruginosa to temocillin and impact on the susceptibility of strains isolated from patients suffering from cystic fibrosis. J. Antimicrob. Chemother. 2012, 67, 771–775. [Google Scholar] [CrossRef][Green Version]
- Ferrer-Espada, R.; Shahrour, H.; Pitts, B.; Stewart, P.S.; Sánchez-Gómez, S.; Martínez-de-Tejada, G. A permeability-increasing drug synergizes with bacterial efflux pump inhibitors and restores susceptibility to antibiotics in multi-drug resistant Pseudomonas aeruginosa strains. Sci. Rep. 2019, 9, 3452. [Google Scholar] [CrossRef]
- Fernando, D.M.; Kumar, A. Resistance-nodulation-division multidrug efflux pumps in gram-negative bacteria: Role in virulence. Antibiotics 2013, 2, 163–181. [Google Scholar] [CrossRef]
- Köhler, T.; Michea-Hamzehpour, M.; Plesiat, P.; Kahr, A.L.; Pechere, J.C. Differential selection of multidrug efflux systems by quinolones in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 1997, 41, 2540–2543. [Google Scholar] [CrossRef]
- Guénard, S.; Muller, C.; Monlezun, L.; Benas, P.; Broutin, I.; Jeannot, K.; Plésiat, P. Multiple mutations lead to MexXY-OprM-dependent aminoglycoside resistance in clinical strains of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2014, 58, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Llanes, C.; Köhler, T.; Patry, I.; Dehecq, B.; van Delden, C.; Plésiat, P. Role of the MexEF-OprN efflux system in low-level resistance of Pseudomonas aeruginosa to ciprofloxacin. Antimicrob. Agents Chemother. 2011, 55, 5676–5684. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-Z.; Zhang, L.; Poole, K. Interplay between the MexA-MexB-OprM multidrug efflux system and the outer membrane barrier in the multiple antibiotic resistance of Pseudomonas aeruginosa. J. Antimicrob. Chemother. 2000, 45, 433–436. [Google Scholar] [CrossRef]
- Masuda, N.; Sakagawa, E.; Ohya, S.; Gotoh, N.; Tsujimoto, H.; Nishino, T. Substrate specificities of MexAB-OprM, MexCD-OprJ, and MexXY-oprM efflux pumps in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2000, 44, 3322–3327. [Google Scholar] [CrossRef] [PubMed]
- Ayoub Moubareck, C.; Hammoudi Halat, D.; Akkawi, C.; Nabi, A.; AlSharhan, M.A.; AlDeesi, Z.O.; Peters, C.C.; Celiloglu, H.; Karam Sarkis, D. Role of outer membrane permeability, efflux mechanism, and carbapenemases in carbapenem-nonsusceptible Pseudomonas aeruginosa from Dubai hospitals: Results of the first cross-sectional survey. Int. J. Infect. Dis. 2019, 84, 143–150. [Google Scholar] [CrossRef]
- Choudhury, D.; Das Talukdar, A.; Dutta Choudhury, M.; Maurya, A.P.; Paul, D.; Dhar Chanda, D.; Chakravorty, A.; Bhattacharjee, A. Transcriptional analysis of MexAB-OprM efflux pumps system of Pseudomonas aeruginosa and its role in carbapenem resistance in a tertiary referral hospital in India. PLoS ONE 2015, 10, e0133842. [Google Scholar] [CrossRef] [PubMed]
- Köhler, T.; Michéa-Hamzehpour, M.; Henze, U.; Gotoh, N.; Kocjancic Curty, L.; Pechère, J.-C. Characterization of MexE–MexF–OprN, a positively regulated multidrug efflux system of Pseudomonas aeruginosa. Mol. Microbiol. 1997, 23, 345–354. [Google Scholar] [CrossRef]
- Moyá, B.; Beceiro, A.; Cabot, G.; Juan, C.; Zamorano, L.; Alberti, S.; Oliver, A. Pan-β-lactam resistance development in Pseudomonas aeruginosa clinical strains: Molecular mechanisms, penicillin-binding protein profiles, and binding affinities. Antimicrob. Agents Chemother. 2012, 56, 4771–4778. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Xu, Y.; Wang, Z.; Fang, Y.; Shen, J. Overexpression of MexAB-OprM efflux pump in carbapenem-resistant Pseudomonas aeruginosa. Arch. Microbiol. 2016, 198, 565–571. [Google Scholar] [CrossRef]
- Aires, J.R.; Köhler, T.; Nikaido, H.; Plésiat, P. Involvement of an active efflux system in the natural resistance of Pseudomonas aeruginosa to aminoglycosides. Antimicrob. Agents Chemother. 1999, 43, 2624–2628. [Google Scholar] [CrossRef] [PubMed]
- Morita, Y.; Tomida, J.; Kawamura, Y. MexXY multidrug efflux system of Pseudomonas aeruginosa. Front. Microbiol. 2012, 3, 408. [Google Scholar] [CrossRef]
- Köhler, T.; Michea-Hamzehpour, M.; Epp, S.F.; Pechere, J.C. Carbapenem activities against Pseudomonas aeruginosa: Respective contributions of OprD and efflux systems. Antimicrob. Agents Chemother. 1999, 43, 424–427. [Google Scholar] [CrossRef] [PubMed]
- Sobel Mara, L.; Neshat, S.; Poole, K. Mutations in PA2491 (mexS) promote MexT-dependent mexEF-oprN expression and multidrug resistance in a clinical Strain of Pseudomonas aeruginosa. J. Bacteriol. 2005, 187, 1246–1253. [Google Scholar] [CrossRef]
- Köhler, T.; Epp, S.F.; Curty, L.K.; Pechère, J.C. Characterization of MexT, the regulator of the MexE-MexF-OprN multidrug efflux system of Pseudomonas aeruginosa. J. Bacteriol. 1999, 181, 6300–6305. [Google Scholar] [CrossRef]
- Saito, K.; Yoneyama, H.; Nakae, T. nalB-type mutations causing the overexpression of the MexAB-OprM efflux pump are located in the mexR gene of the Pseudomonas aeruginosa chromosome. FEMS Microbiol. Lett. 1999, 179, 67–72. [Google Scholar] [CrossRef]
- Sobel, M.L.; Hocquet, D.; Cao, L.; Plesiat, P.; Poole, K. Mutations in PA3574 (nalD) lead to increased MexAB-OprM expression and multidrug resistance in laboratory and clinical isolates of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2005, 49, 1782–1786. [Google Scholar] [CrossRef]
- Morita, Y.; Cao, L.; Gould, V.C.; Avison, M.B.; Poole, K. nalD encodes a second repressor of the mexAB-oprM multidrug efflux operon of Pseudomonas aeruginosa. J. Bacteriol. 2006, 188, 8649–8654. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Yi, C.; Zhang, J.; Zhang, W.; Ge, Z.; Yang, C.G.; He, C. Structural insight into the oxidation-sensing mechanism of the antibiotic resistance of regulator MexR. EMBO Rep. 2010, 11, 685–690. [Google Scholar] [CrossRef] [PubMed]
- Evans, K.; Adewoye, L.; Poole, K. MexR repressor of the mexAB-oprM multidrug efflux operon of Pseudomonas aeruginosa identification of MexR binding sites in the mexA-mexR intergenic region. J. Bacteriol. 2001, 183, 807–812. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Akama, H.; Yoshihara, E.; Nakae, T. Mutations affecting DNA-binding activity of the MexR repressor of mexR-mexA-mexB-oprM operon expression. J. Bacteriol. 2003, 185, 6195–6198. [Google Scholar] [CrossRef]
- Cao, L.; Srikumar, R.; Poole, K. MexAB-OprM hyperexpression in NalC-type multidrug-resistant Pseudomonas aeruginosa: Identification and characterization of the nalC gene encoding a repressor of PA3720-PA3719. Mol. Microbiol. 2004, 53, 1423–1436. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, Y.; Eda, S.; Gotoh, N.; Yoshihara, E.; Nakae, T. MexZ-mediated regulation of mexXY multidrug efflux pump expression in Pseudomonas aeruginosa by binding on the mexZ-mexX intergenic DNA. FEMS Microbiol. Lett. 2004, 238, 23–28. [Google Scholar] [CrossRef]
- Masuda, N.; Sakagawa, E.; Ohya, S.; Gotoh, N.; Tsujimoto, H.; Nishino, T. Contribution of the MexX-MexY-oprM efflux system to intrinsic resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2000, 44, 2242–2246. [Google Scholar] [CrossRef]
- Frimodt-Møller, J.; Rossi, E.; Haagensen, J.A.J.; Falcone, M.; Molin, S.; Johansen, H.K. Mutations causing low level antibiotic resistance ensure bacterial survival in antibiotic-treated hosts. Sci. Rep. 2018, 8, 12512. [Google Scholar] [CrossRef]
- Seupt, A.; Schniederjans, M.; Tomasch, J.; Haussler, S. Expression of the MexXY aminoglycoside efflux pump and presence of an aminoglycoside-modifying enzyme in clinical Pseudomonas aeruginosa isolates are highly correlated. Antimicrob. Agents Chemother. 2020, 65, e01166-20. [Google Scholar] [CrossRef]
- Gotoh, N.; Tsujimoto, H.; Tsuda, M.; Okamoto, K.; Nomura, A.; Wada, T.; Nakahashi, M.; Nishino, T. Characterization of the MexC-MexD-OprJ multidrug efflux system in ΔmexA-mexB-oprM mutants of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 1998, 42, 1938–1943. [Google Scholar] [CrossRef]
- Fraud, S.; Campigotto, A.J.; Chen, Z.; Poole, K. MexCD-OprJ multidrug efflux system of Pseudomonas aeruginosa: Involvement in chlorhexidine resistance and induction by membrane-damaging agents dependent upon the AlgU stress response sigma factor. Antimicrob. Agents Chemother. 2008, 52, 4478–4482. [Google Scholar] [CrossRef] [PubMed]
- Purssell, A.; Poole, K. Functional characterization of the NfxB repressor of the mexCD–oprJ multidrug efflux operon of Pseudomonas aeruginosa. Microbiology 2013, 159, 2058–2073. [Google Scholar] [CrossRef]
- Higgins, P.G.; Fluit, A.C.; Milatovic, D.; Verhoef, J.; Schmitz, F.J. Mutations in GyrA, ParC, MexR and NfxB in clinical isolates of Pseudomonas aeruginosa. Int. J. Antimicrob. Agents 2003, 21, 409–413. [Google Scholar] [CrossRef]
- Gomis-Font, M.A.; Pitart, C.; Del Barrio-Tofiño, E.; Zboromyrska, Y.; Cortes-Lara, S.; Mulet, X.; Marco, F.; Vila, J.; López-Causapé, C.; Oliver, A. Emergence of resistance to novel cephalosporin-β-lactamase inhibitor combinations through the modification of the Pseudomonas aeruginosa MexCD-OprJ efflux pump. Antimicrob. Agents Chemother. 2021, 65, e0008921. [Google Scholar] [CrossRef]
- Köhler, T.; van Delden, C.; Curty, L.K.; Hamzehpour, M.M.; Pechere, J.C. Overexpression of the MexEF-OprN multidrug efflux system affects cell-to-cell signaling in Pseudomonas aeruginosa. J. Bacteriol. 2001, 183, 5213–5222. [Google Scholar] [CrossRef]
- Maseda, H.; Yoneyama, H.; Nakae, T. Assignment of the substrate-selective subunits of the MexEF-OprN multidrug efflux pump of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2000, 44, 658–664. [Google Scholar] [CrossRef]
- Maseda, H.; Uwate, M.; Nakae, T. Transcriptional regulation of the mexEF-oprN multidrug efflux pump operon by MexT and an unidentified repressor in nfxC-type mutant of Pseudomonas aeruginosa. FEMS Microbiol. Lett. 2010, 311, 36–43. [Google Scholar] [CrossRef]
- Jeannot, K.; Elsen, S.; Köhler, T.; Attree, I.; Delden, C.v.; Plésiat, P. Resistance and virulence of Pseudomonas aeruginosa clinical strains overproducing the MexCD-OprJ efflux pump. Antimicrob. Agents Chemother. 2008, 52, 2455–2462. [Google Scholar] [CrossRef]
- Tooke, C.L.; Hinchliffe, P.; Bragginton, E.C.; Colenso, C.K.; Hirvonen, V.H.A.; Takebayashi, Y.; Spencer, J. β-lactamases and β-lactamase Inhibitors in the 21st Century. J. Mol. Biol. 2019, 431, 3472–3500. [Google Scholar] [CrossRef] [PubMed]
- Bush, K.; Jacoby, G.A. Updated functional classification of β-lactamases. Antimicrob. Agents Chemother. 2010, 54, 969–976. [Google Scholar] [CrossRef]
- Boyd, S.E.; Livermore, D.M.; Hooper, D.C.; Hope, W.W. Metallo-β-lactamases: Structure, function, epidemiology, treatment options, and the development pipeline. Antimicrob. Agents Chemother. 2020, 64, e00397-20. [Google Scholar] [CrossRef] [PubMed]
- Palzkill, T. Metallo-β-lactamase structure and function. Ann. N. Y. Acad. Sci. 2013, 1277, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Queenan, A.M.; Bush, K. Carbapenemases: The versatile β-lactamases. Clin. Microbiol. Rev. 2007, 20, 440–458. [Google Scholar] [CrossRef]
- Adam, M.A.; Elhag, W.I. Prevalence of metallo-β-lactamase acquired genes among carbapenems susceptible and resistant Gram-negative clinical isolates using multiplex PCR, Khartoum hospitals, Khartoum Sudan. BMC Infect. Dis. 2018, 18, 668. [Google Scholar] [CrossRef]
- Meletis, G. Carbapenem resistance: Overview of the problem and future perspectives. Ther. Adv. Infect. Dis. 2016, 3, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Pintos-Pascual, I.; Cantero-Caballero, M.; Munez Rubio, E.; Sanchez-Romero, I.; Asensio-Vegas, A.; Ramos-Martinez, A. Epidemiology and clinical of infections and colonizations caused by Enterobacterales producing carbapenemases in a tertiary hospital. Rev. Española Quimioter. 2020, 33, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Betancur, J.C.; Appel, T.M.; Esparza, G.; Gales, A.C.; Levy-Hara, G.; Cornistein, W.; Vega, S.; Nunez, D.; Cuellar, L.; Bavestrello, L.; et al. Update on the epidemiology of carbapenemases in Latin America and the Caribbean. Expert Rev. Anti-Infect. Ther. 2021, 19, 197–213. [Google Scholar] [CrossRef] [PubMed]
- Fuster, B.; Tormo, N.; Salvador, C.; Gimeno, C. Detection of two simultaneous outbreaks of Klebsiella pneumoniae coproducing OXA-48 and NDM-1 carbapenemases in a tertiary-care hospital in Valencia, Spain. New Microbes New Infect. 2020, 34, 100660. [Google Scholar] [CrossRef] [PubMed]
- Kollenda, H.; Frickmann, H.; Ben Helal, R.; Wiemer, D.F.; Naija, H.; El Asli, M.S.; Egold, M.; Bugert, J.J.; Handrick, S.; Wolfel, R.; et al. Screening for Carbapenemases in Ertapenem-Resistant Enterobacteriaceae Collected at a Tunisian Hospital Between 2014 and 2018. Eur. J. Microbiol. Immunol. 2019, 9, 9–13. [Google Scholar] [CrossRef]
- Marathe, N.P.; Berglund, F.; Razavi, M.; Pal, C.; Droge, J.; Samant, S.; Kristiansson, E.; Larsson, D.G.J. Sewage effluent from an Indian hospital harbors novel carbapenemases and integron-borne antibiotic resistance genes. Microbiome 2019, 7, 97. [Google Scholar] [CrossRef]
- Hammoudi Halat, D.; Ayoub Moubareck, C. The current burden of carbapenemases: Review of significant properties and dissemination among gram-negative bacteria. Antibiotics 2020, 9, 186. [Google Scholar] [CrossRef]
- Lahiri, S.D.; Johnstone, M.R.; Ross, P.L.; McLaughlin, R.E.; Olivier, N.B.; Alm, R.A. Avibactam and class C β-lactamases: Mechanism of inhibition, conservation of the binding pocket, and implications for resistance. Antimicrob. Agents Chemother. 2014, 58, 5704–5713. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.P.; Saraiva, L.; Pinto, M.; Sousa, M.E. Boronic acids and their derivatives in medicinal chemistry: Synthesis and biological applications. Molecules 2020, 25, 4323. [Google Scholar] [CrossRef]
- Rodríguez, D.; Maneiro, M.; Vázquez-Ucha, J.C.; Beceiro, A.; González-Bello, C. 6-arylmethylidene penicillin-based sulfone inhibitors for repurposing antibiotic efficiency in priority pathogens. J. Med. Chem. 2020, 63, 3737–3755. [Google Scholar] [CrossRef]
- Drawz, S.M.; Bethel, C.R.; Doppalapudi, V.R.; Sheri, A.; Pagadala, S.R.; Hujer, A.M.; Skalweit, M.J.; Anderson, V.E.; Chen, S.G.; Buynak, J.D.; et al. Penicillin sulfone inhibitors of class D β-lactamases. Antimicrob. Agents Chemother. 2010, 54, 1414–1424. [Google Scholar] [CrossRef] [PubMed]
- Ju, L.C.; Cheng, Z.; Fast, W.; Bonomo, R.A.; Crowder, M.W. The continuing challenge of metallo-β-lactamase inhibition: Mechanism matters. Trends Pharmacol. Sci. 2018, 39, 635–647. [Google Scholar] [CrossRef]
- Poole, K. Pseudomonas aeruginosa: Resistance to the max. Front. Microbiol. 2011, 2, 65. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Gallego, M.; Torrens, G.; Castillo-Vera, J.; Moya, B.; Zamorano, L.; Cabot, G.; Hultenby, K.; Albertí, S.; Mellroth, P.; Henriques-Normark, B. Impact of AmpC derepression on fitness and virulence: The mechanism or the pathway? mBio 2016, 7, e01783-16. [Google Scholar] [CrossRef]
- Rafiee, R.; Eftekhar, F.; Tabatabaei, S.A.; Minaee Tehrani, D. Prevalence of extended-spectrum and metallo β-lactamase production in AmpC β-lactamase producing Pseudomonas aeruginosa isolates from burns. Jundishapur J. Microbiol. 2014, 7, e16436. [Google Scholar] [CrossRef] [PubMed]
- Fisher, J.F.; Mobashery, S. Constructing and deconstructing the bacterial cell wall. Protein Sci. 2020, 29, 629–646. [Google Scholar] [CrossRef] [PubMed]
- Vadlamani, G.; Thomas, M.D.; Patel, T.R.; Donald, L.J.; Reeve, T.M.; Stetefeld, J.; Standing, K.G.; Vocadlo, D.J.; Mark, B.L. The β-lactamase gene regulator AmpR is a tetramer that recognizes and binds the d-Ala-d-Ala motif of its repressor UDP-N-acetylmuramic ccid (MurNAc)-pentapeptide. J. Biol. Chem. 2015, 290, 2630–2643. [Google Scholar] [CrossRef]
- Dik, D.A.; Domínguez-Gil, T.; Lee, M.; Hesek, D.; Byun, B.; Fishovitz, J.; Boggess, B.; Hellman, L.M.; Fisher, J.F.; Hermoso, J.A.; et al. Muropeptide Binding and the X-ray Structure of the Effector Domain of the Transcriptional Regulator AmpR of Pseudomonas aeruginosa. J. Am. Chem. Soc. 2017, 139, 1448–1451. [Google Scholar] [CrossRef]
- Juan, C.; Macia, M.D.; Gutierrez, O.; Vidal, C.; Perez, J.L.; Oliver, A. Molecular mechanisms of β-lactam resistance mediated by AmpC hyperproduction in Pseudomonas aeruginosa clinical strains. Antimicrob. Agents Chemother. 2005, 49, 4733–4738. [Google Scholar] [CrossRef] [PubMed]
- Schmidtke, A.J.; Hanson, N.D. Role of ampD homologs in overproduction of AmpC in clinical isolates of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2008, 52, 3922–3927. [Google Scholar] [CrossRef] [PubMed]
- Kong, K.F.; Jayawardena, S.R.; Indulkar, S.D.; Del Puerto, A.; Koh, C.L.; Hoiby, N.; Mathee, K. Pseudomonas aeruginosa AmpR is a global transcriptional factor that regulates expression of AmpC and PoxB β-lactamases, proteases, quorum sensing, and other virulence factors. Antimicrob. Agents Chemother. 2005, 49, 4567–4575. [Google Scholar] [CrossRef]
- Caille, O.; Zincke, D.; Merighi, M.; Balasubramanian, D.; Kumari, H.; Kong, K.-F.; Silva-Herzog, E.; Narasimhan, G.; Schneper, L.; Lory, S.; et al. Structural and functional characterization of Pseudomonas aeruginosa global regulator AmpR. J. Bacteriol. 2014, 196, 3890–3902. [Google Scholar] [CrossRef] [PubMed]
- Domitrovic, T.N.; Hujer, A.M.; Perez, F.; Marshall, S.H.; Hujer, K.M.; Woc-Colburn, L.E.; Parta, M.; Bonomo, R.A. Multidrug resistant Pseudomonas aeruginosa causing prosthetic valve endocarditis: A genetic-based chronicle of evolving antibiotic resistance. Open Forum Infect Dis. 2016, 3, ofw188. [Google Scholar] [CrossRef]
- Bagge, N.; Ciofu, O.; Hentzer, M.; Campbell, J.I.; Givskov, M.; Hoiby, N. Constitutive high expression of chromosomal β-lactamase in Pseudomonas aeruginosa caused by a new insertion sequence (IS1669) located in ampD. Antimicrob. Agents Chemother. 2002, 46, 3406–3411. [Google Scholar] [CrossRef]
- Moya, B.; Dötsch, A.; Juan, C.; Blázquez, J.; Zamorano, L.; Haussler, S.; Oliver, A. β-lactam resistance response triggered by inactivation of a nonessential penicillin-binding protein. PLoS Pathog. 2009, 5, e1000353. [Google Scholar] [CrossRef]
- Zamorano, L.; Moya, B.; Juan, C.; Mulet, X.; Blazquez, J.; Oliver, A. The Pseudomonas aeruginosa CreBC two-component system plays a major role in the response to β-lactams, fitness, biofilm growth, and global regulation. Antimicrob. Agents Chemother. 2014, 58, 5084–5095. [Google Scholar] [CrossRef] [PubMed]
- Simner, P.J.; Beisken, S.; Bergman, Y.; Posch, A.E.; Cosgrove, S.E.; Tamma, P.D. Cefiderocol activity against clinical Pseudomonas aeruginosa isolates exhibiting ceftolozane-tazobactam resistance. Open Forum Infect Disease. 2021, 8, ofab311. [Google Scholar] [CrossRef] [PubMed]
- Tam, V.H.; Schilling, A.N.; LaRocco, M.T.; Gentry, L.O.; Lolans, K.; Quinn, J.P.; Garey, K.W. Prevalence of AmpC over-expression in bloodstream isolates of Pseudomonas aeruginosa. Clin. Microbiol. Infect. 2007, 13, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Slater, C.L.; Winogrodzki, J.; Fraile-Ribot, P.A.; Oliver, A.; Khajehpour, M.; Mark, B.L. Adding insult to injury: Mechanistic basis for how AmpC mutations allow Pseudomonas aeruginosa to accelerate cephalosporin hydrolysis and evade avibactam. Antimicrob. Agents Chemother. 2020, 64, e00894-20. [Google Scholar] [CrossRef]
- Berrazeg, M.; Jeannot, K.; Ntsogo Enguene, V.Y.; Broutin, I.; Loeffert, S.; Fournier, D.; Plesiat, P. Mutations in β-lactamase AmpC increase resistance of Pseudomonas aeruginosa isolates to antipseudomonal cephalosporins. Antimicrob. Agents Chemother. 2015, 59, 6248–6255. [Google Scholar] [CrossRef]
- Compain, F.; Debray, A.; Adjadj, P.; Dorchene, D.; Arthur, M. Ceftazidime-avibactam resistance mediated by the N346Y substitution in various AmpC β-lactamases. Antimicrob. Agents Chemother. 2020, 64, e02311-19. [Google Scholar] [CrossRef]
- Fernandez-Esgueva, M.; Lopez-Calleja, A.I.; Mulet, X.; Fraile-Ribot, P.A.; Cabot, G.; Huarte, R.; Rezusta, A.; Oliver, A. Characterization of AmpC β-lactamase mutations of extensively drug-resistant Pseudomonas aeruginosa isolates that develop resistance to ceftolozane/tazobactam during therapy. Enferm. Infecc. Microbiol. Clínica 2020, 38, 474–478. [Google Scholar] [CrossRef]
- Fajardo, A.; Hernando-Amado, S.; Oliver, A.; Ball, G.; Filloux, A.; Martinez, J.L. Characterization of a novel Zn(2)(+)-dependent intrinsic imipenemase from Pseudomonas aeruginosa. J. Antimicrob. Chemother. 2014, 69, 2972–2978. [Google Scholar] [CrossRef] [PubMed]
- Girlich, D.; Naas, T.; Nordmann, P. Biochemical characterization of the naturally occurring oxacillinase OXA-50 of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2004, 48, 2043–2048. [Google Scholar] [CrossRef] [PubMed]
- Zincke, D.; Balasubramanian, D.; Silver, L.L.; Mathee, K. Characterization of a Carbapenem-Hydrolyzing Enzyme, PoxB, in Pseudomonas aeruginosa PAO1. Antimicrob. Agents Chemother. 2016, 60, 936–945. [Google Scholar] [CrossRef] [PubMed]
- Lahiri, S.D.; Mangani, S.; Durand-Reville, T.; Benvenuti, M.; Luca, F.D.; Sanyal, G.; Docquier, J.-D. Structural Insight into Potent Broad-Spectrum Inhibition with Reversible Recyclization Mechanism: Avibactam in Complex with CTX-M-15 and Pseudomonas aeruginosa AmpC β-Lactamases. Antimicrob. Agents Chemother. 2013, 57, 2496–2505. [Google Scholar] [CrossRef]
- Watanabe, M.; Iyobe, S.; Inoue, M.; Mitsuhashi, S. Transferable imipenem resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 1991, 35, 147–151. [Google Scholar] [CrossRef] [PubMed]
- Dortet, L.; Flonta, M.; Boudehen, Y.M.; Creton, E.; Bernabeu, S.; Vogel, A.; Naas, T. Dissemination of carbapenemase-producing Enterobacteriaceae and Pseudomonas aeruginosa in Romania. Antimicrob. Agents Chemother. 2015, 59, 7100–7103. [Google Scholar] [CrossRef]
- Evans, B.A.; Amyes, S.G. OXA β-lactamases. Clin. Microbiol. Rev. 2014, 27, 241–263. [Google Scholar] [CrossRef] [PubMed]
- Dehbashi, S.; Tahmasebi, H.; Alikhani, M.Y.; Keramat, F.; Arabestani, M.R. Distribution of Class B and Class A β-Lactamases in Clinical Strains of Pseudomonas aeruginosa: Comparison of Phenotypic Methods and High-Resolution Melting Analysis (HRMA) Assay. Infect. Drug Resist. 2020, 13, 2037–2052. [Google Scholar] [CrossRef]
- De Champs, C.; Chanal, C.; Sirot, D.; Baraduc, R.; Romaszko, J.P.; Bonnet, R.; Plaidy, A.; Boyer, M.; Carroy, E.; Gbadamassi, M.C.; et al. Frequency and diversity of Class A extended-spectrum β-lactamases in hospitals of the Auvergne, France: A 2 year prospective study. J. Antimicrob. Chemother. 2004, 54, 634–639. [Google Scholar] [CrossRef]
- Schauer, J.; Gatermann, S.G.; Hoffmann, D.; Hupfeld, L.; Pfennigwerth, N. GPC-1, a novel class A carbapenemase detected in a clinical Pseudomonas aeruginosa isolate. J. Antimicrob. Chemother. 2020, 75, 911–916. [Google Scholar] [CrossRef]
- Zhao, W.-H.; Hu, Z.-Q. β-Lactamases identified in clinical isolates of Pseudomonas aeruginosa. Crit. Rev. Microbiol. 2010, 36, 245–258. [Google Scholar] [CrossRef] [PubMed]
- Bush, K.; Bradford, P.A. Epidemiology of β-Lactamase-Producing Pathogens. Clin. Microbiol. Rev. 2020, 33, e00047-19. [Google Scholar] [CrossRef]
- Wang, W.; Wang, X. Prevalence of metallo-β-lactamase genes among Pseudomonas aeruginosa isolated from various clinical samples in China. J. Lab. Med. 2020, 44, 197–203. [Google Scholar] [CrossRef]
- Philippon, A.; Arlet, G.; Jacoby, G.A. Plasmid-determined AmpC-type β-lactamases. Antimicrob. Agents Chemother. 2002, 46, 1–11. [Google Scholar] [CrossRef]
- Doi, Y.; Paterson, D.L. Detection of plasmid-mediated class C β-lactamases. Int. J. Infect. Dis. 2007, 11, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Fraile-Ribot, P.A.; Del Rosario-Quintana, C.; López-Causapé, C.; Gomis-Font, M.A.; Ojeda-Vargas, M.; Oliver, A. Emergence of resistance to novel β-lactam-β-lactamase inhibitor combinations due to horizontally acquired AmpC (FOX-4) in Pseudomonas aeruginosa sequence type 308. Antimicrob. Agents Chemother. 2019, 64, e02112-19. [Google Scholar] [CrossRef]
- Upadhyay, S.; Mishra, S.; Sen, M.R.; Banerjee, T.; Bhattacharjee, A. Co-existence of Pseudomonas-derived cephalosporinase among plasmid encoded CMY-2 harbouring isolates of Pseudomonas aeruginosa in north India. Indian J. Med Microbiol. 2013, 31, 257–260. [Google Scholar] [CrossRef]
- Antunes, N.T.; Fisher, J.F. Acquired Class D β-lactamases. Antibiotics 2014, 3, 398–434. [Google Scholar] [CrossRef]
- Nasser, M.; Gayen, S.; Kharat, A.S. Prevalence of β-lactamase and antibiotic-resistant Pseudomonas aeruginosa in the Arab region. J. Glob. Antimicrob. Resist. 2020, 22, 152–160. [Google Scholar] [CrossRef]
- Amirkamali, S.; Naserpour-Farivar, T.; Azarhoosh, K.; Peymani, A. Distribution of the bla OXA, bla VEB-1, and bla GES-1 genes and resistance patterns of ESBL-producing Pseudomonas aeruginosa isolated from hospitals in Tehran and Qazvin, Iran. Rev. Soc. Bras. Med. Trop. 2017, 50, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Esenkaya Taşbent, F.; Özdemir, M. The presence of OXA type carbapenemases in Pseudomonas strains: First report from Turkey. Mikrobiyoloji Bul. 2015, 49, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Yoon, E.-J.; Jeong, S.H. Mobile carbapenemase genes in Pseudomonas aeruginosa. Front. Microbiol. 2021, 12, 30. [Google Scholar] [CrossRef]
- Perez-Vazquez, M.; Sola-Campoy, P.J.; Zurita, Á.M.; Avila, A.; Gomez-Bertomeu, F.; Solis, S.; Lopez-Urrutia, L.; GÓnzalez-BarberÁ, E.M.; Cercenado, E.; Bautista, V. Carbapenemase-producing Pseudomonas aeruginosa in Spain: Interregional dissemination of the high-risk clones ST175 and ST244 carrying blaVIM-2, blaVIM-1, blaIMP-8, blaVIM-20 and blaKPC-2. Int. J. Antimicrob. Agents 2020, 56, 106026. [Google Scholar] [CrossRef]
- Bonnin, R.A.; Bogaerts, P.; Girlich, D.; Huang, T.-D.; Dortet, L.; Glupczynski, Y.; Naas, T. Molecular characterization of OXA-198 carbapenemase-producing Pseudomonas aeruginosa clinical isolates. Antimicrob. Agents Chemother. 2018, 62, e02496-17. [Google Scholar] [CrossRef]
- Botelho, J.; Grosso, F.; Peixe, L. Unravelling the genome of a Pseudomonas aeruginosa isolate belonging to the high-risk clone ST235 reveals an integrative conjugative element housing a blaGES-6 carbapenemase. J. Antimicrob. Chemother. 2018, 73, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Botelho, J.; Schulenburg, H. The role of integrative and conjugative elements in antibiotic resistance evolution. Trends Microbiol. 2021, 29, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Nordmann, P.; Gniadkowski, M.; Giske, C.G.; Poirel, L.; Woodford, N.; Miriagou, V.; European Network on, C. Identification and screening of carbapenemase-producing Enterobacteriaceae. Clin. Microbiol. Infect. 2012, 18, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Yang, H.; Li, J.; Chen, F.; Hu, L.; Jing, Y.; Luo, X.; Yin, Z.; Zou, M.; Zhou, D. Novel chromosome-borne accessory genetic elements carrying multiple antibiotic resistance genes in Pseudomonas aeruginosa. Front. Cell. Infect. Microbiol. 2021, 11, 139. [Google Scholar] [CrossRef] [PubMed]
- Botelho, J.; Roberts, A.P.; León-Sampedro, R.; Grosso, F.; Peixe, L. Carbapenemases on the move: It’s good to be on ICEs. Mob. DNA 2018, 9, 1–11. [Google Scholar] [CrossRef]
- Domingues, S.; da Silva, G.J.; Nielsen, K.M. Integrons: Vehicles and pathways for horizontal dissemination in bacteria. Mob. Genet. Elem. 2012, 2, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Ma, J.; Jia, W.; Li, W. Antimicrobial resistance and molecular characterization of gene cassettes from class 1 integrons in Pseudomonas aeruginosa strains. Microb. Drug Resist. 2020, 26, 670–676. [Google Scholar] [CrossRef] [PubMed]
- Ahmadian, L.; Haghshenas, M.R.; Mirzaei, B.; Norouzi Bazgir, Z.; Goli, H.R. Distribution and molecular characterization of resistance gene cassettes containing class 1 integrons in multi-drug resistant (MDR) clinical isolates of Pseudomonas aeruginosa. Infect. Drug Resist. 2020, 13, 2773–2781. [Google Scholar] [CrossRef] [PubMed]
- Jamal, A.J.; Mataseje, L.F.; Brown, K.A.; Katz, K.; Johnstone, J.; Muller, M.P.; Allen, V.G.; Borgia, S.; Boyd, D.A.; Ciccotelli, W.; et al. Carbapenemase-producing Enterobacterales in hospital drains in Southern Ontario, Canada. J. Hosp. Infect. 2020, 106, 820–827. [Google Scholar] [CrossRef]
- Park, S.C.; Parikh, H.; Vegesana, K.; Stoesser, N.; Barry, K.E.; Kotay, S.M.; Dudley, S.; Peto, T.E.A.; Crook, D.W.; Walker, A.S.; et al. Risk factors associated with carbapenemase-producing enterobacterales (CPE) positivity in the hospital wastewater environment. Appl. Environ. Microbiol. 2020, 86, e01715-20. [Google Scholar] [CrossRef] [PubMed]
- Snitkin, E.S. Contamination of Hospital Plumbing: A Source or a Sink for Antibiotic-Resistant Organisms? JAMA Netw. Open 2019, 2, e187660. [Google Scholar] [CrossRef]
- Cahill, N.; O’Connor, L.; Mahon, B.; Varley, A.; McGrath, E.; Ryan, P.; Cormican, M.; Brehony, C.; Jolley, K.A.; Maiden, M.C.; et al. Hospital effluent: A reservoir for carbapenemase-producing Enterobacterales? Sci. Total Environ. 2019, 672, 618–624. [Google Scholar] [CrossRef]
- Cherak, Z.; Loucif, L.; Moussi, A.; Rolain, J.-M. Carbapenemase-producing Gram-negative bacteria in aquatic environments: A review. J. Glob. Antimicrob. Resist. 2021, 25, 287–309. [Google Scholar] [CrossRef] [PubMed]
- Bleichenbacher, S.; Stevens, M.J.A.; Zurfluh, K.; Perreten, V.; Endimiani, A.; Stephan, R.; Nüesch-Inderbinen, M. Environmental dissemination of carbapenemase-producing Enterobacteriaceae in rivers in Switzerland. Environ. Pollut. 2020, 265, 115081. [Google Scholar] [CrossRef]
- Colosi, I.A.; Baciu, A.M.; Opris, R.V.; Peca, L.; Gudat, T.; Simon, L.M.; Colosi, H.A.; Costache, C. Prevalence of ESBL, AmpC and carbapenemase-producing enterobacterales isolated from raw vegetables retailed in Romania. Foods 2020, 9, 1726. [Google Scholar] [CrossRef]
- Borlee, B.R.; Goldman, A.D.; Murakami, K.; Samudrala, R.; Wozniak, D.J.; Parsek, M.R. Pseudomonas aeruginosa uses a cyclic-di-GMP-regulated adhesin to reinforce the biofilm extracellular matrix. Mol. Microbiol. 2010, 75, 827–842. [Google Scholar] [CrossRef]
- Gloag, E.S.; Marshall, C.W.; Snyder, D.; Lewin, G.R.; Harris, J.S.; Santos-Lopez, A.; Chaney, S.B.; Whiteley, M.; Cooper, V.S.; Wozniak, D.J. Pseudomonas aeruginosa Interstrain Dynamics and Selection of Hyperbiofilm Mutants during a Chronic Infection. mBio 2019, 10, e01698-19. [Google Scholar] [CrossRef]
- Kovach, K.; Davis-Fields, M.; Irie, Y.; Jain, K.; Doorwar, S.; Vuong, K.; Dhamani, N.; Mohanty, K.; Touhami, A.; Gordon, V.D. Evolutionary adaptations of biofilms infecting cystic fibrosis lungs promote mechanical toughness by adjusting polysaccharide production. npj Biofilms Microbiomes 2017, 3, 1–9. [Google Scholar] [CrossRef]
- Starkey, M.; Hickman, J.H.; Ma, L.; Zhang, N.; De Long, S.; Hinz, A.; Palacios, S.; Manoil, C.; Kirisits, M.J.; Starner, T.D.; et al. Pseudomonas aeruginosa rugose small-colony variants have adaptations that likely promote persistence in the cystic fibrosis lung. J. Bacteriol. 2009, 191, 3492–3503. [Google Scholar] [CrossRef]
- Hickman, J.W.; Tifrea, D.F.; Harwood, C.S. A chemosensory system that regulates biofilm formation through modulation of cyclic diguanylate levels. Proc. Natl. Acad. Sci. USA 2005, 102, 14422–14427. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.; Pan, Y.; Cai, Z.; Liu, Y.; Zhang, Y.; Liu, M.; Liu, Y.; Wang, K.; Zhang, L.; Yang, L. rpoS-mutation variants are selected in Pseudomonas aeruginosa biofilms under imipenem pressure. Cell Biosci. 2021, 11, 138. [Google Scholar] [CrossRef]
- Cochran, W.L.; Suh, S.J.; McFeters, G.A.; Stewart, P.S. Role of RpoS and AlgT in Pseudomonas aeruginosa biofilm resistance to hydrogen peroxide and monochloramine. J. Appl. Microbiol. 2000, 88, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Fernández, L.; Álvarez-Ortega, C.; Wiegand, I.; Olivares, J.; Kocíncová, D.; Lam, J.S.; Martínez, J.L.; Hancock, R.E.W. Characterization of the polymyxin B resistome of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2013, 57, 110–119. [Google Scholar] [CrossRef] [PubMed]
- De Groote, V.N.; Verstraeten, N.; Fauvart, M.; Kint, C.I.; Verbeeck, A.M.; Beullens, S.; Cornelis, P.; Michiels, J. Novel persistence genes in Pseudomonas aeruginosa identified by high-throughput screening. FEMS Microbiol. Lett. 2009, 297, 73–79. [Google Scholar] [CrossRef]
- Wright, R.C.T.; Friman, V.-P.; Smith, M.C.M.; Brockhurst, M.A. Resistance evolution against phage combinations depends on the timing and order of exposure. mBio 2019, 10, e01652-19. [Google Scholar] [CrossRef] [PubMed]
- La Rosa, R.; Rossi, E.; Feist, A.M.; Johansen, H.K.; Molin, S. Compensatory evolution of Pseudomonas aeruginosa’s slow growth phenotype suggests mechanisms of adaptation in cystic fibrosis. Nat. Commun. 2021, 12, 3186. [Google Scholar] [CrossRef]
- Lopatkin Allison, J.; Bening Sarah, C.; Manson Abigail, L.; Stokes Jonathan, M.; Kohanski Michael, A.; Badran Ahmed, H.; Earl Ashlee, M.; Cheney Nicole, J.; Yang Jason, H.; Collins James, J. Clinically relevant mutations in core metabolic genes confer antibiotic resistance. Science 2021, 371, eaba0862. [Google Scholar] [CrossRef]
- Malone, J.G. Role of small colony variants in persistence of Pseudomonas aeruginosa infections in cystic fibrosis lungs. Infect. Drug Resist. 2015, 8, 237–247. [Google Scholar] [CrossRef]
- Al Ahmar, R.; Kirby, B.D.; Yu, H.D. Pyrimidine biosynthesis regulates the small-colony variant and mucoidy in Pseudomonas aeruginosa through sigma factor competition. J. Bacteriol. 2019, 201, e00575-18. [Google Scholar] [CrossRef]
- Jeukens, J.; Freschi, L.; Kukavica-Ibrulj, I.; Emond-Rheault, J.-G.; Tucker, N.P.; Levesque, R.C. Genomics of antibiotic-resistance prediction in Pseudomonas aeruginosa. Ann. N. Y. Acad. Sci. 2019, 1435, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Khaledi, A.; Weimann, A.; Schniederjans, M.; Asgari, E.; Kuo, T.-H.; Oliver, A.; Cabot, G.; Kola, A.; Gastmeier, P.; Hogardt, M.; et al. Predicting antimicrobial resistance in Pseudomonas aeruginosa with machine learning-enabled molecular diagnostics. EMBO Mol. Med. 2020, 12, e10264. [Google Scholar] [CrossRef] [PubMed]
- Geha, D.J.; Uhl, J.R.; Gustaferro, C.A.; Persing, D.H. Multiplex PCR for identification of methicillin-resistant staphylococci in the clinical laboratory. J. Clin. Microbiol. 1994, 32, 1768–1772. [Google Scholar] [CrossRef]
- Pournajaf, A.; Ardebili, A.; Goudarzi, L.; Khodabandeh, M.; Narimani, T.; Abbaszadeh, H. PCR-based identification of methicillin-resistant Staphylococcus aureus strains and their antibiotic resistance profiles. Asian Pac. J. Trop. Biomed. 2014, 4, S293–S297. [Google Scholar] [CrossRef]
- Nguyen, T.N.A.; Anton-Le Berre, V.; Bañuls, A.-L.; Nguyen, T.V.A. Molecular Diagnosis of Drug-Resistant Tuberculosis; A Literature Review. Front. Microbiol. 2019, 10, 794. [Google Scholar] [CrossRef] [PubMed]
- Cole, K.A.; Rivard, K.R.; Dumkow, L.E. Antimicrobial stewardship interventions to combat antibiotic resistance: An update on targeted strategies. Curr. Infect. Dis. Rep. 2019, 21, 33. [Google Scholar] [CrossRef]
- Wang, M.; Wei, H.; Zhao, Y.; Shang, L.; Di, L.; Lyu, C.; Liu, J. Analysis of multidrug-resistant bacteria in 3223 patients with hospital-acquired infections (HAI) from a tertiary general hospital in China. Bosn. J. Basic Med. Sci. 2019, 19, 86–93. [Google Scholar] [CrossRef]
- Stultz, J.S.; Arnold, S.R.; Shelton, C.M.; Bagga, B.; Lee, K.R. Antimicrobial stewardship impact on Pseudomonas aeruginosa susceptibility to meropenem at a tertiary pediatric institution. Am. J. Infect. Control 2019, 47, 1513–1515. [Google Scholar] [CrossRef] [PubMed]
- Raymond, B. Five rules for resistance management in the antibiotic apocalypse, a road map for integrated microbial management. Evol. Appl. 2019, 12, 1079–1091. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Shen, W.; Zhong, Q.; Chen, Q.; He, X.; Baker, J.L.; Xiong, K.; Jin, X.; Wang, J.; Hu, F.; et al. Development of a Bacteriophage Cocktail to Constrain the Emergence of Phage-Resistant Pseudomonas aeruginosa. Front. Microbiol. 2020, 11, 327. [Google Scholar] [CrossRef]
- El Haddad, L.; Harb, C.P.; Gebara, M.A.; Stibich, M.A.; Chemaly, R.F. A systematic and critical review of bacteriophage therapy against multidrug-resistant ESKAPE organisms in humans. Clin. Infect. Dis. 2019, 69, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; He, Y.; Wang, Z.; Wei, J.; Hu, T.; Si, J.; Tao, G.; Zhang, L.; Xie, L.; Abdalla, A.E. A combination therapy of phages and antibiotics: Two is better than one. Int. J. Biol. Sci. 2021, 17, 3573. [Google Scholar] [CrossRef]
- Oechslin, F.; Piccardi, P.; Mancini, S.; Gabard, J.; Moreillon, P.; Entenza, J.M.; Resch, G.; Que, Y.-A. Synergistic interaction between phage therapy and antibiotics clears Pseudomonas aeruginosa infection in endocarditis and reduces virulence. J. Infect. Dis. 2017, 215, 703–712. [Google Scholar] [CrossRef]
- Gu Liu, C.; Green, S.I.; Min, L.; Clark, J.R.; Salazar, K.C.; Terwilliger, A.L.; Kaplan, H.B.; Trautner, B.W.; Ramig, R.F.; Maresso, A.W. Phage-antibiotic synergy is driven by a unique combination of antibacterial mechanism of action and stoichiometry. mBio 2020, 11, e01462-20. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Chang, R.Y.K.; Lin, Y.; Morales, S.; Kutter, E.; Chan, H.-K. Phage cocktail powder for Pseudomonas aeruginosa respiratory infections. Int. J. Pharm. 2021, 596, 120200. [Google Scholar] [CrossRef] [PubMed]
- Pletzer, D.; Coleman, S.R.; Hancock, R.E. Anti-biofilm peptides as a new weapon in antimicrobial warfare. Curr. Opin. Microbiol. 2016, 33, 35–40. [Google Scholar] [CrossRef]
- Butler, M.S.; Paterson, D.L. Antibiotics in the clinical pipeline in October 2019. J. Antibiot. 2020, 73, 329–364. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.-Y.; Lv, Y.; Zhu, Y.; Wei, M.-J.; Liu, M.-Y.; Ji, X.-W.; Kang, Z.-S.; Xia, Y.-H.; Tian, J.-H.; Ma, Y. A first-in-human safety, tolerability, and pharmacokinetics study of benapenem in healthy Chinese volunteers. Antimicrob. Agents Chemother. 2019, 63, e02188-18. [Google Scholar] [CrossRef]
- Reck, F.; Bermingham, A.; Blais, J.; Capka, V.; Cariaga, T.; Casarez, A.; Colvin, R.; Dean, C.R.; Fekete, A.; Gong, W. Optimization of novel monobactams with activity against carbapenem-resistant Enterobacteriaceae–identification of LYS228. Bioorganic Med. Chem. Lett. 2018, 28, 748–755. [Google Scholar] [CrossRef] [PubMed]
- Tümmler, B. Emerging therapies against infections with Pseudomonas aeruginosa. F1000Research 2019, 8, 1371. [Google Scholar] [CrossRef]
- Vázquez-Ucha, J.C.; Arca-Suárez, J.; Bou, G.; Beceiro, A. New Carbapenemase Inhibitors: Clearing the Way for the β-lactams. Int. J. Mol. Sci. 2020, 21, 9308. [Google Scholar] [CrossRef] [PubMed]
- Everett, M.; Sprynski, N.; Coelho, A.; Castandet, J.; Bayet, M.; Bougnon, J.; Lozano, C.; Davies, D.T.; Leiris, S.; Zalacain, M.; et al. Discovery of a novel metallo-β-lactamase inhibitor that potentiates meropenem activity against carbapenem-resistant enterobacteriaceae. Antimicrob. Agents Chemother. 2018, 62, e00074-18. [Google Scholar] [CrossRef]
- Davies, D.T.; Leiris, S.; Sprynski, N.; Castandet, J.; Lozano, C.; Bousquet, J.; Zalacain, M.; Vasa, S.; Dasari, P.K.; Pattipati, R.; et al. ANT2681: SAR studies leading to the identification of a metallo-β-lactamase inhibitor with potential for clinical use in combination with meropenem for the treatment of infections caused by NDM-producing enterobacteriaceae. ACS Infect. Dis. 2020, 6, 2419–2430. [Google Scholar] [CrossRef]
- Bush, K.; Page, M.G.P. What we may expect from novel antibacterial agents in the pipeline with respect to resistance and pharmacodynamic principles. J. Pharmacokinet. Pharmacodyn. 2017, 44, 113–132. [Google Scholar] [CrossRef]
- Durand-Reville, T.F.; Miller, A.A.; O’Donnell, J.P.; Wu, X.; Sylvester, M.A.; Guler, S.; Iyer, R.; Shapiro, A.B.; Carter, N.M.; Velez-Vega, C.; et al. Rational design of a new antibiotic class for drug-resistant infections. Nature 2021, 597, 698–702. [Google Scholar] [CrossRef]
- Möllmann, U.; Heinisch, L.; Bauernfeind, A.; Köhler, T.; Ankel-Fuchs, D. Siderophores as drug delivery agents: Application of the “Trojan Horse” strategy. BioMetals 2009, 22, 615–624. [Google Scholar] [CrossRef]
- Miller, M.J.; Zhu, H.; Xu, Y.; Wu, C.; Walz, A.J.; Vergne, A.; Roosenberg, J.M.; Moraski, G.; Minnick, A.A.; McKee-Dolence, J.; et al. Utilization of microbial iron assimilation processes for the development of new antibiotics and inspiration for the design of new anticancer agents. BioMetals 2009, 22, 61–75. [Google Scholar] [CrossRef] [PubMed]
- Zhanel, G.G.; Golden, A.R.; Zelenitsky, S.; Wiebe, K.; Lawrence, C.K.; Adam, H.J.; Idowu, T.; Domalaon, R.; Schweizer, F.; Zhanel, M.A. Cefiderocol: A siderophore cephalosporin with activity against carbapenem-resistant and multidrug-resistant Gram-negative bacilli. Drugs 2019, 79, 271–289. [Google Scholar] [CrossRef] [PubMed]
- Hori, T.; Owusu, Y.B.; Sun, D. US FDA-Approved Antibiotics During the 21st Century. In Reference Module in Biomedical Sciences; Elsevier: Amsterdam, The Netherlands, 2021. [Google Scholar] [CrossRef]
- Kresken, M.; Korte-Berwanger, M.; Gatermann, S.G.; Pfeifer, Y.; Pfennigwerth, N.; Seifert, H.; Werner, G. In vitro activity of cefiderocol against aerobic Gram-negative bacterial pathogens from Germany. Int. J. Antimicrob. Agents 2020, 56, 106128. [Google Scholar] [CrossRef] [PubMed]
- Luscher, A.; Moynié, L.; Auguste, P.S.; Bumann, D.; Mazza, L.; Pletzer, D.; Naismith, J.H.; Köhler, T. TonB-dependent receptor repertoire of Pseudomonas aeruginosa for uptake of siderophore-drug conjugates. Antimicrob. Agents Chemother. 2018, 62, e00097-18. [Google Scholar] [CrossRef] [PubMed]
- Streling, A.P.; Al Obaidi, M.M.; Lainhart, W.D.; Zangeneh, T.; Khan, A.; Dinh, A.Q.; Hanson, B.; Arias, C.A.; Miller, W.R. Evolution of Cefiderocol Non-Susceptibility in Pseudomonas aeruginosa in a Patient Without Previous Exposure to the Antibiotic. Clin. Infect. Dis. 2021, 73, e4472–e4474. [Google Scholar] [CrossRef]
- Gonzalez Gomez, A.; Hosseinidoust, Z. Liposomes for antibiotic encapsulation and delivery. ACS Infect. Dis. 2020, 6, 896–908. [Google Scholar] [CrossRef]
- Wang, Y.; Yuan, Q.; Feng, W.; Pu, W.; Ding, J.; Zhang, H.; Li, X.; Yang, B.; Dai, Q.; Cheng, L. Targeted delivery of antibiotics to the infected pulmonary tissues using ROS-responsive nanoparticles. J. Nanobiotechnology 2019, 17, 103. [Google Scholar] [CrossRef]
- Lamers, R.P.; Cavallari, J.F.; Burrows, L.L. The efflux inhibitor phenylalanine-arginine β-naphthylamide (PAβN) permeabilizes the outer membrane of gram-negative bacteria. PLoS ONE 2013, 8, e60666. [Google Scholar] [CrossRef]
- Rampioni, G.; Pillai, C.R.; Longo, F.; Bondì, R.; Baldelli, V.; Messina, M.; Imperi, F.; Visca, P.; Leoni, L. Effect of efflux pump inhibition on Pseudomonas aeruginosa transcriptome and virulence. Sci. Rep. 2017, 7, 11392. [Google Scholar] [CrossRef] [PubMed]







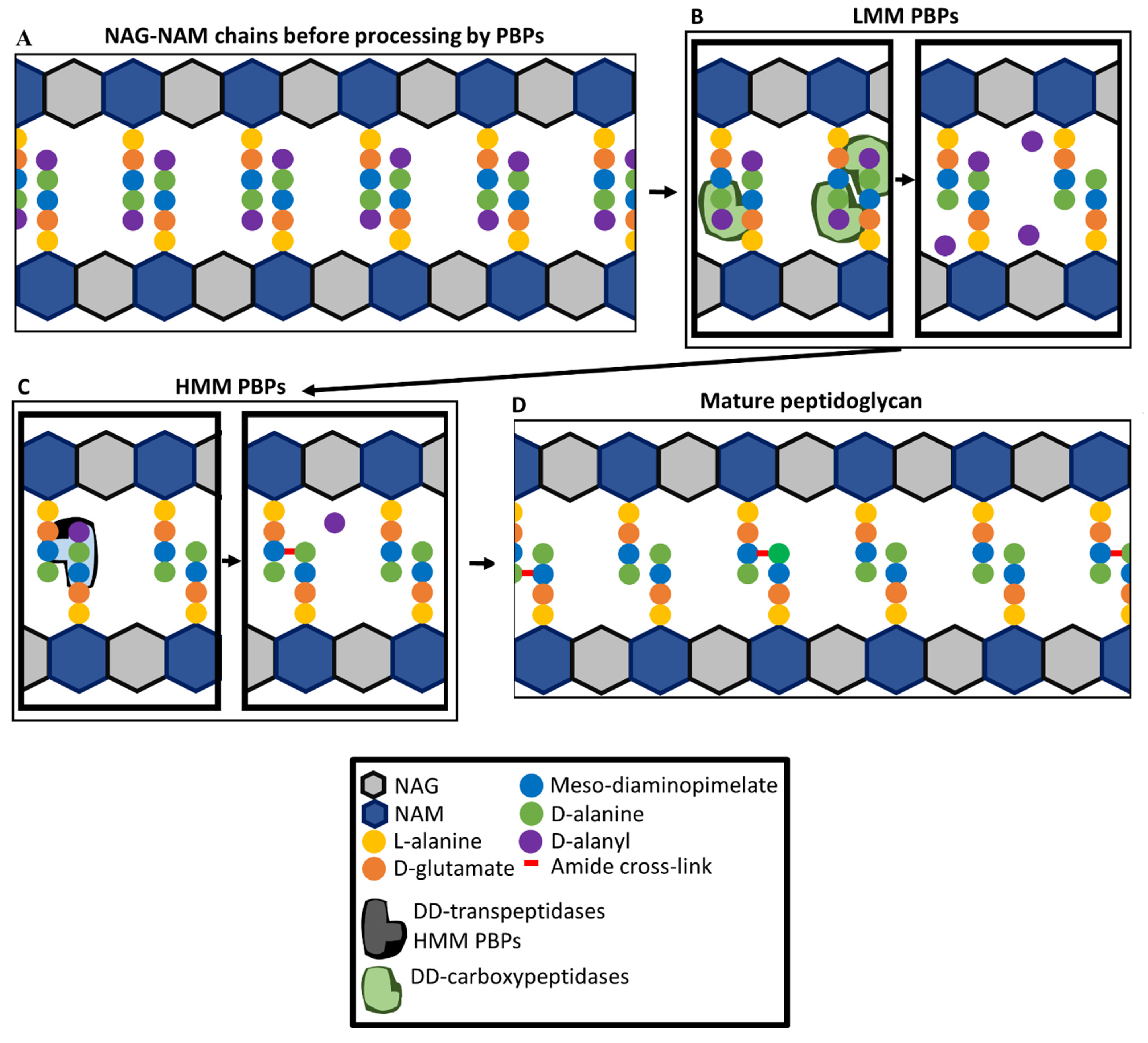{kind=link}
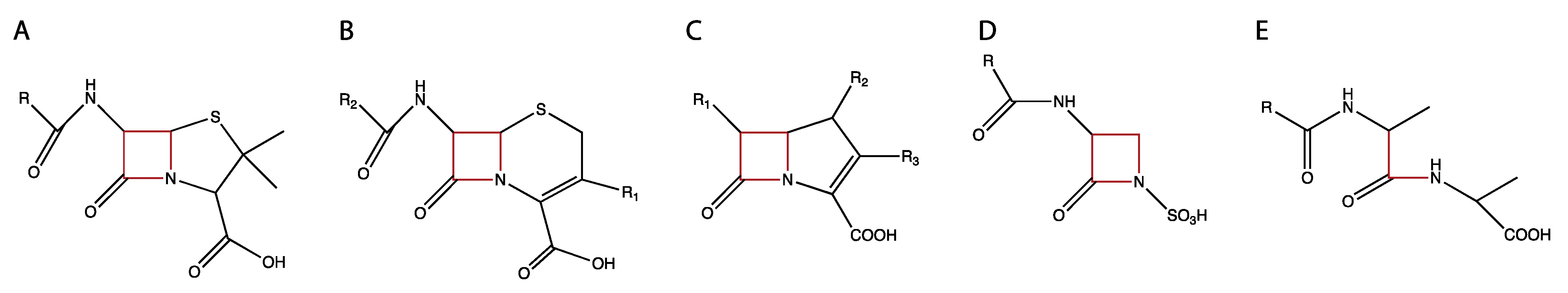{kind=link}
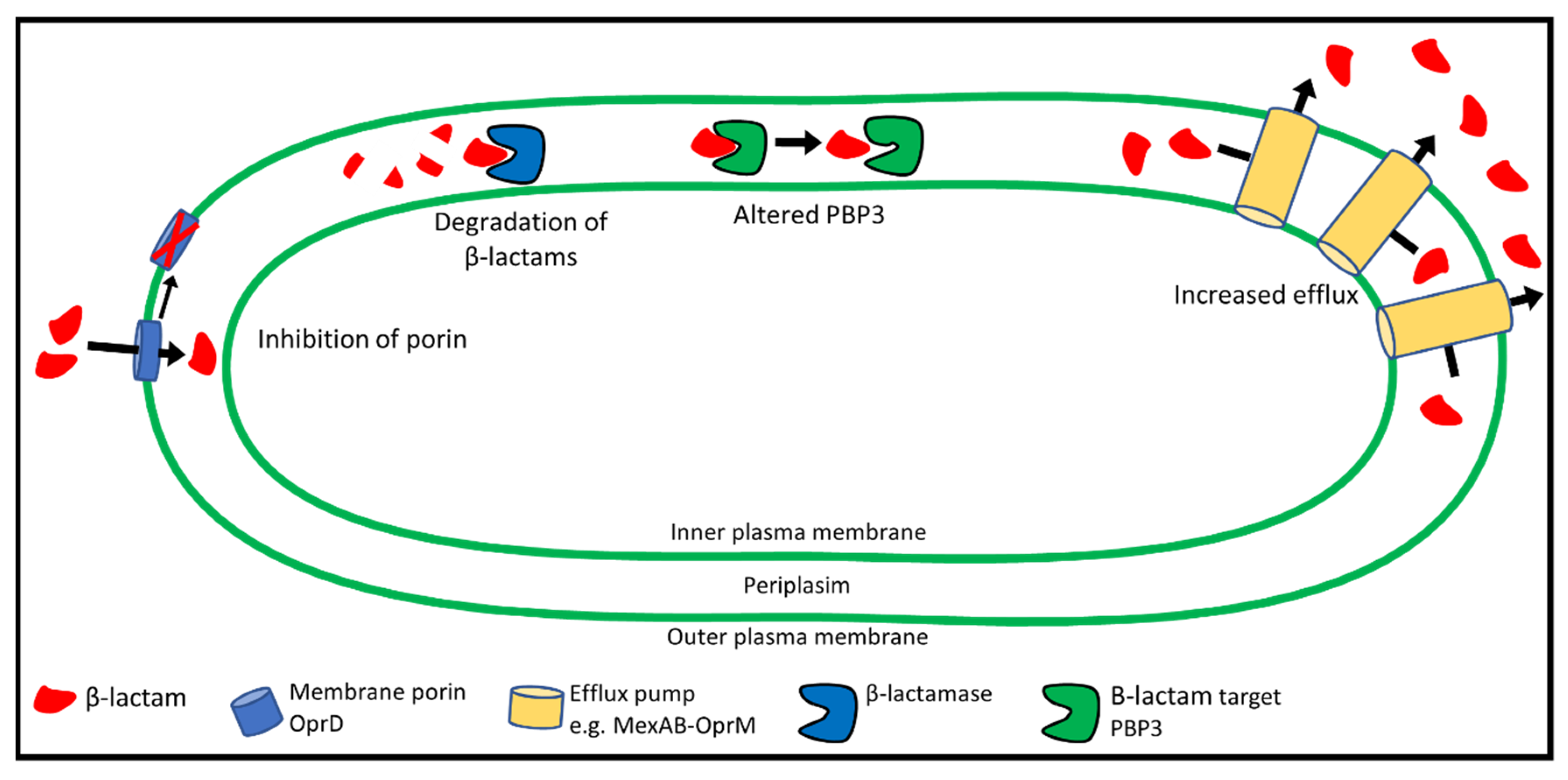{kind=link}
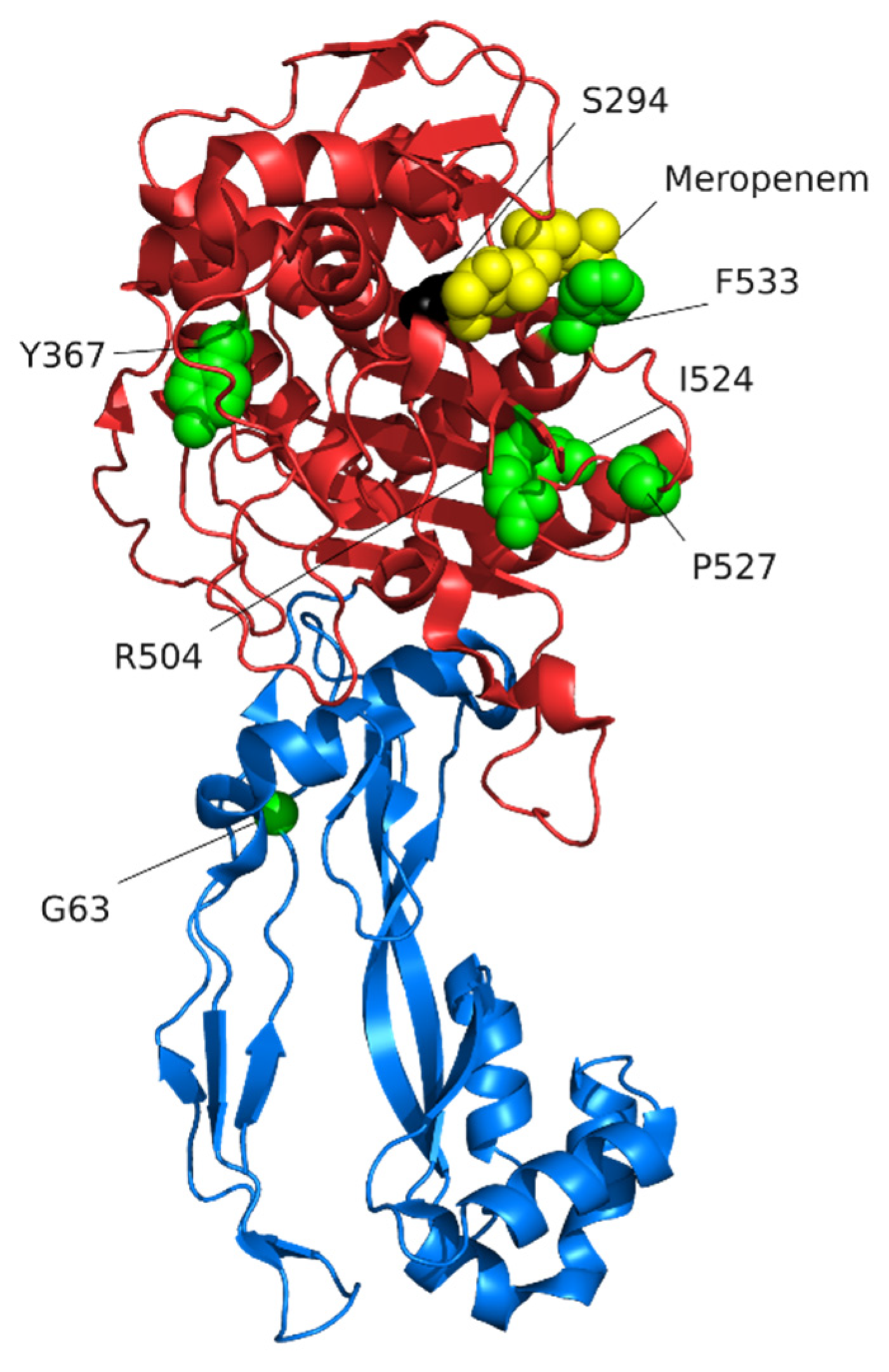{kind=link}
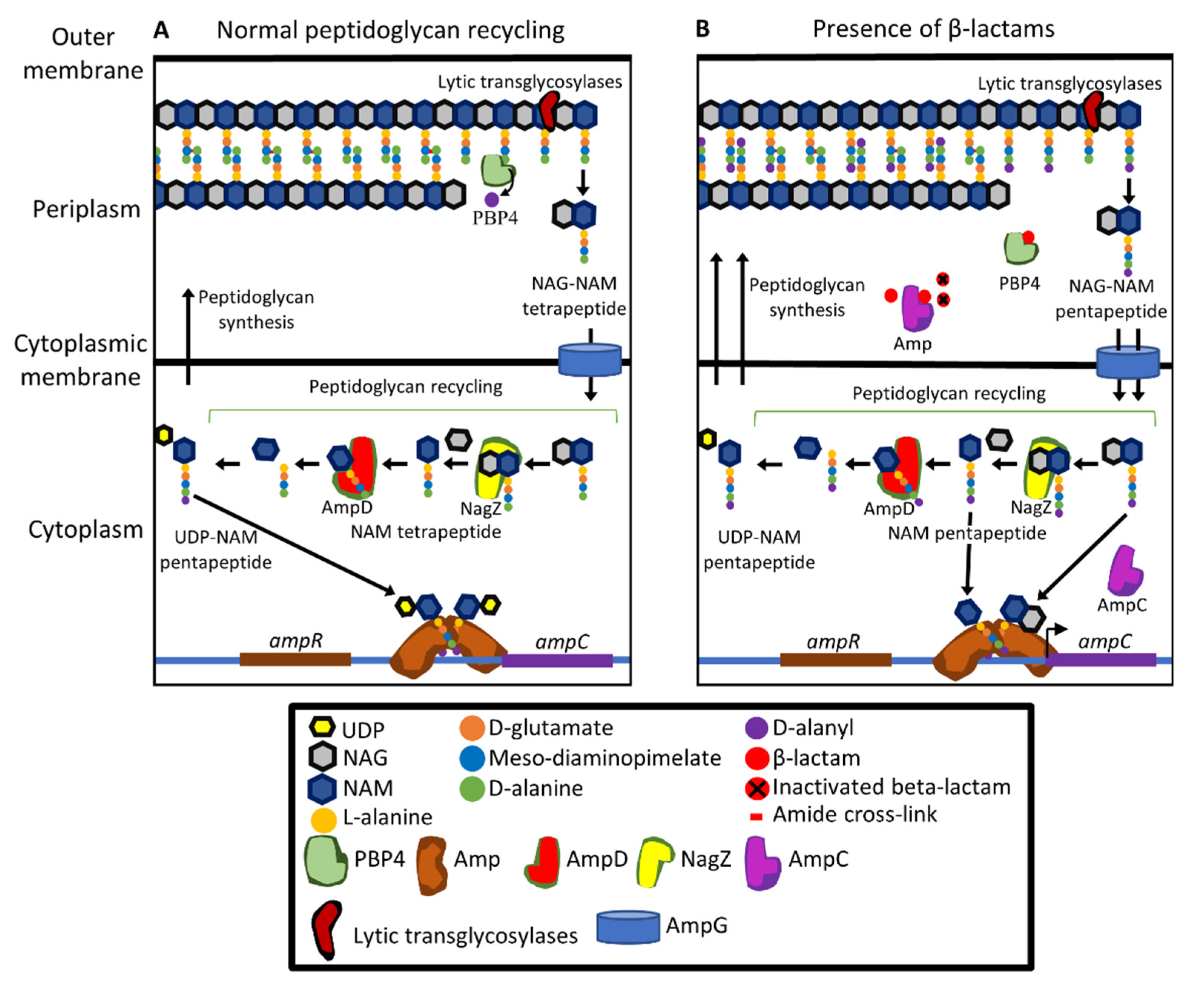{kind=link}
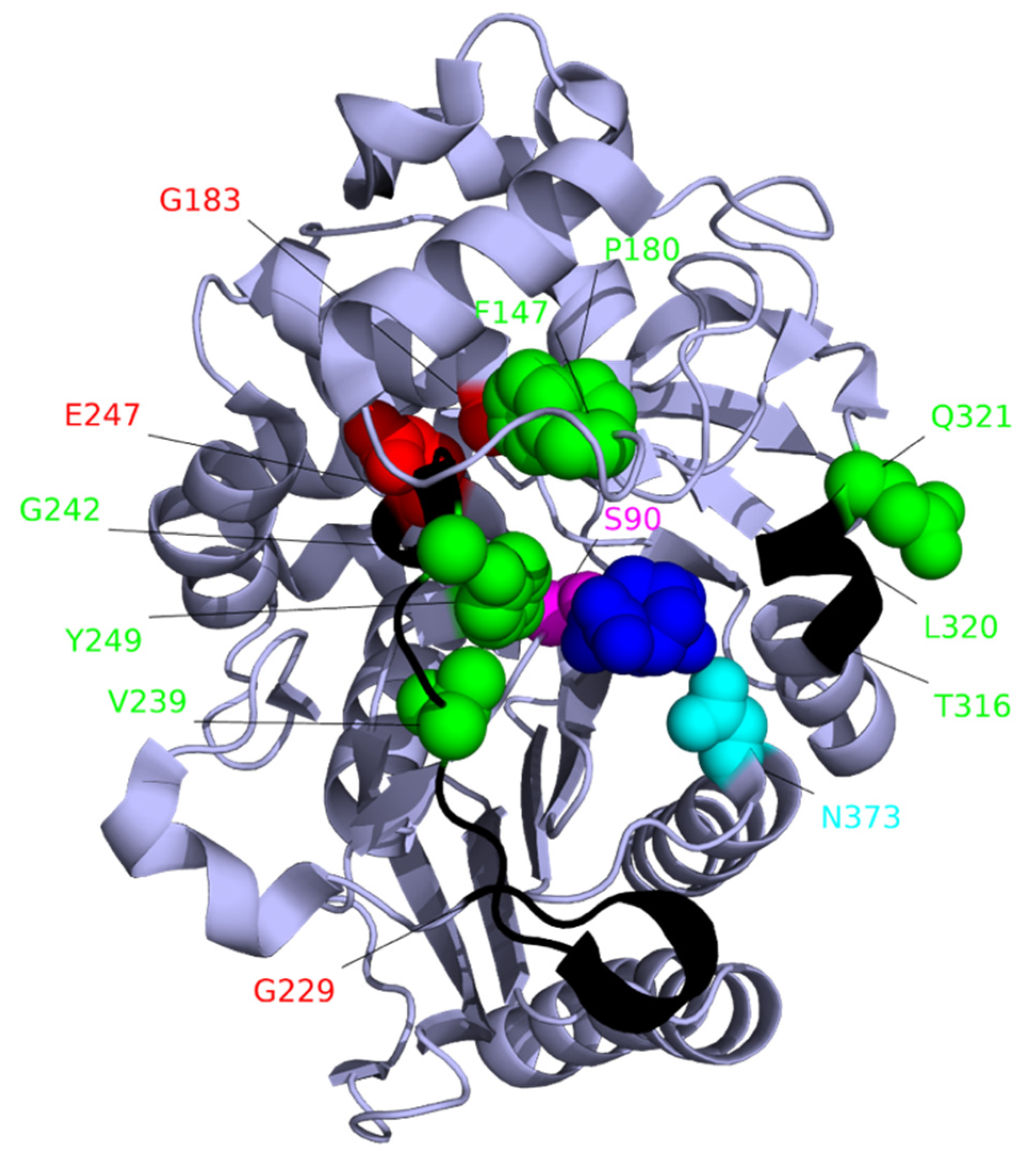{kind=link}
| Antibiotic Class | Antibiotic Subclass | Antibiotic | Antibiotic Use |
|---|---|---|---|
| β-lactams a | Penicillins | Ticarcillin Piperacillin | Parenteral or intravenous for treatment of infections |
| Monobactams | Aztreonam | Inhalation for long-term treatment of chronic lung infections and intramuscular injection for the treatment of acute infections | |
| Carbapenems | Imipenem b meropenem | Intravenous for treatment of acute or chronic infections | |
| Cephalosporins | Ceftazidime Cefepime Ceftolozone | Inhalation or intravenous for treatment of acute or chronic infections | |
| Aminoglycosides | 4,6-di-substituted deoxystreptamine ring | Tobramycin Gentamicin Amikacin | Inhalation or intravenous for treatment of acute or chronic infections |
| Quinolones | Fluoroquinolones | Ciprofloxacin Levofloxacin | Oral or intravenous intake for treatment of acute infections |
| Lipopeptides | Polymyxins | Colistin | Inhalation for treatment of chronic lung infections |
| Antibiotic a | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DOR | IMI | MER | CEF | AZT | CEN | CEP | FAR | |||||
| PBP | Binding Affinity IC50 (μg/mL) b | |||||||||||
| 1a | 0.5 | 0.1 | 0.5 | 0.5 | 0.2 | 0.2 | 0.8 | 2 | 2 | 19 | 35 | 0.23 |
| 1b | 0.6 | 0.2 | 0.5 | 0.5 | 3 | 5 | 6 | 2 | 2 | 2 | 0.7 | 0.15 |
| 2 | 0.06 | 0.1 | 0.1 | 0.05 | >32 | >32 | 25 | 16 | 16 | >250 | ND | 0.19 |
| 3 | 0.07 | 0.09 | 0.1 | 0.08 | 0.1 | 0.1 | 0.1 | 0.03 | 0.03 | 0.3 | 41 | 0.20 |
| 4 | 0.008 | 0.008 | 0.01 | 0.008 | 2 | 2 | ND | 16 | 16 | ND | ND | 0.13 |
| 5 | 8 | 2 | 2 | 16 | >32 | >32 | ND | >16 | >16 | ND | ND | >1 |
| MIC (μg/mL) for P. aeruginosa c | ||||||||||||
| 0.25 | 1 | 1 | 0.5 | 1 | 1 | 0.5 | 4 | 4 | 0.03 | 16 | 512 | |
| Reference | [110] | [110] | [111] | [110] | [110] | [111] | [112] | [110] | [111] | [112] | [112] | [109] |
| Efflux System | Antibiotics Affected by Increased Expression |
|---|---|
| MexAB-OprM | Aztreonam, other β-lactams a, quinolones, tetracyclines, macrolides, novobiocin and chloramphenicol [172,173] |
| MexXY-OprM | Aminoglycosides, tetracyclines, β-lactams b and macrolides [171,172,174,180,181] |
| MexCD-OprJ | β-lactams c and fluoroquinolones [170,174] |
| MexEF-OprN | Imipenem d and fluoroquinolones [172,177] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Glen, K.A.; Lamont, I.L. β-lactam Resistance in Pseudomonas aeruginosa: Current Status, Future Prospects. Pathogens 2021, 10, 1638. https://doi.org/10.3390/pathogens10121638
Glen KA, Lamont IL. β-lactam Resistance in Pseudomonas aeruginosa: Current Status, Future Prospects. Pathogens. 2021; 10(12):1638. https://doi.org/10.3390/pathogens10121638
Chicago/Turabian StyleGlen, Karl A., and Iain L. Lamont. 2021. "β-lactam Resistance in Pseudomonas aeruginosa: Current Status, Future Prospects" Pathogens 10, no. 12: 1638. https://doi.org/10.3390/pathogens10121638
APA StyleGlen, K. A., & Lamont, I. L. (2021). β-lactam Resistance in Pseudomonas aeruginosa: Current Status, Future Prospects. Pathogens, 10(12), 1638. https://doi.org/10.3390/pathogens10121638