Atomistic Simulations to Predict Favored Glass-Formation Composition and Ion-Beam-Mixing of Nano-Multiple-Metal-Layers to Produce Ternary Amorphous Films


Abstract
1. Introduction
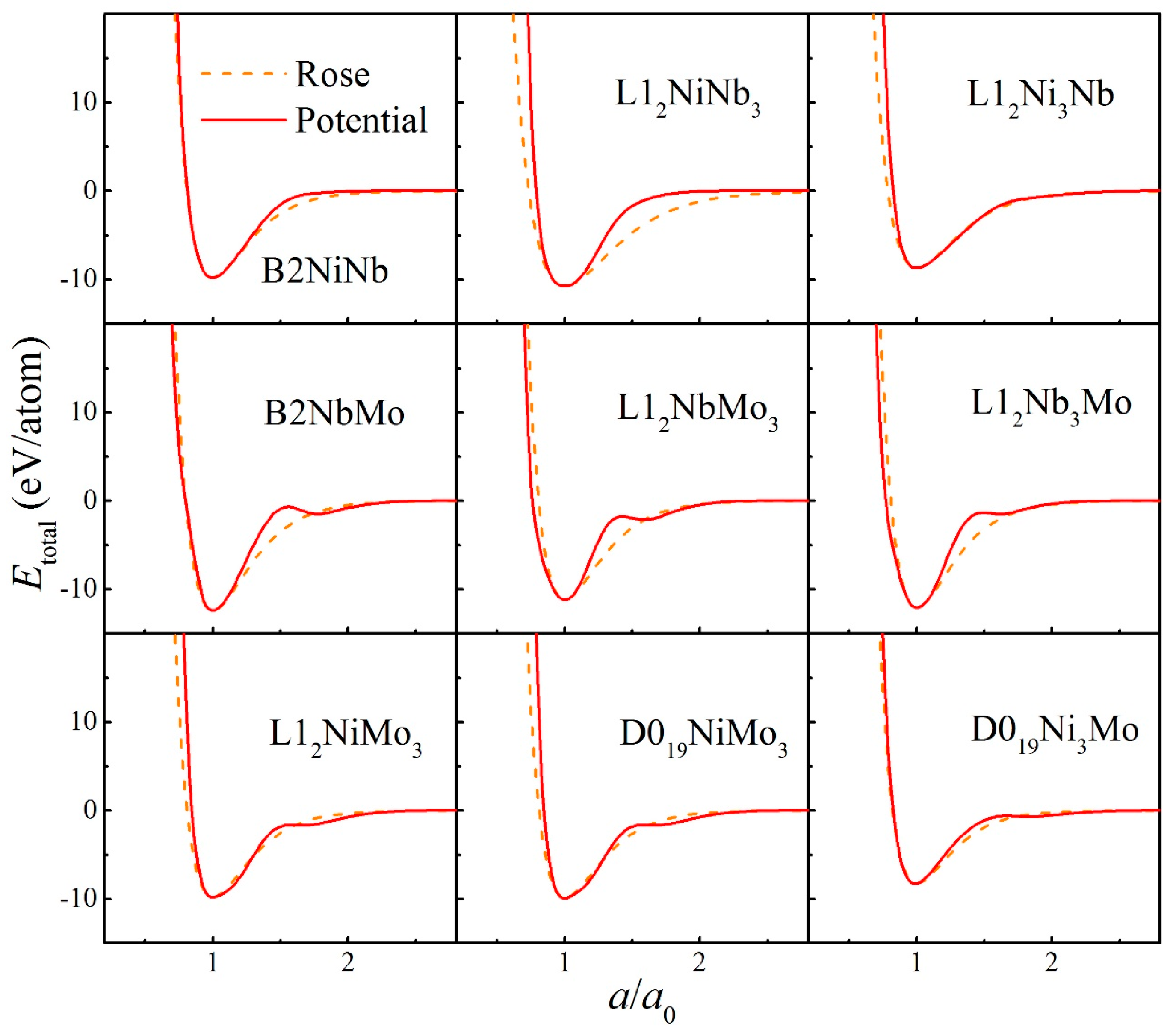
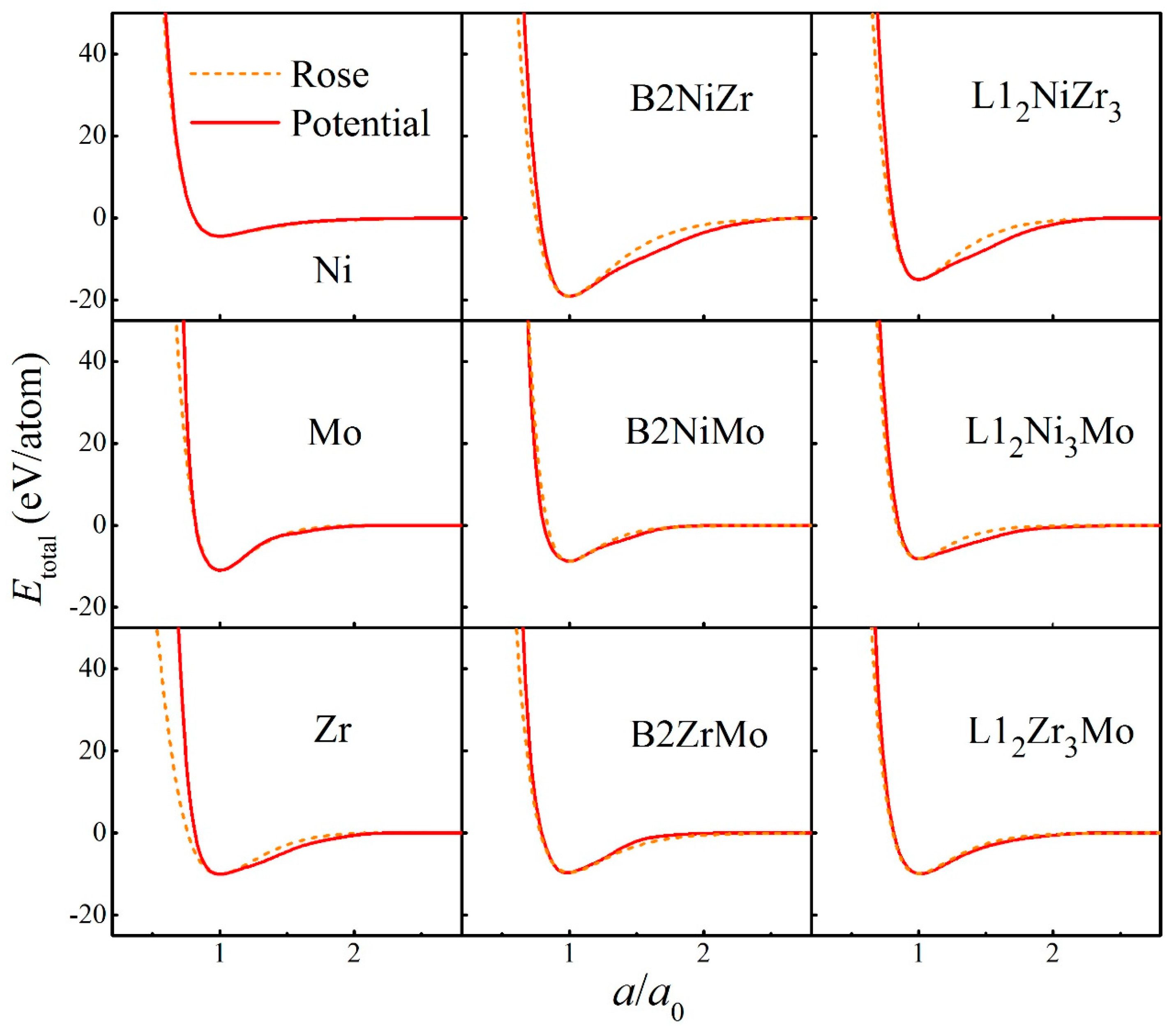
2. Construction of Ternary Interatomic Potential
3. Methods and Models
3.1. Atomistic Simulation
3.2. Ion Beam Mixing
4. Metallic Glass Formation Predicted by Atomistic Simulation
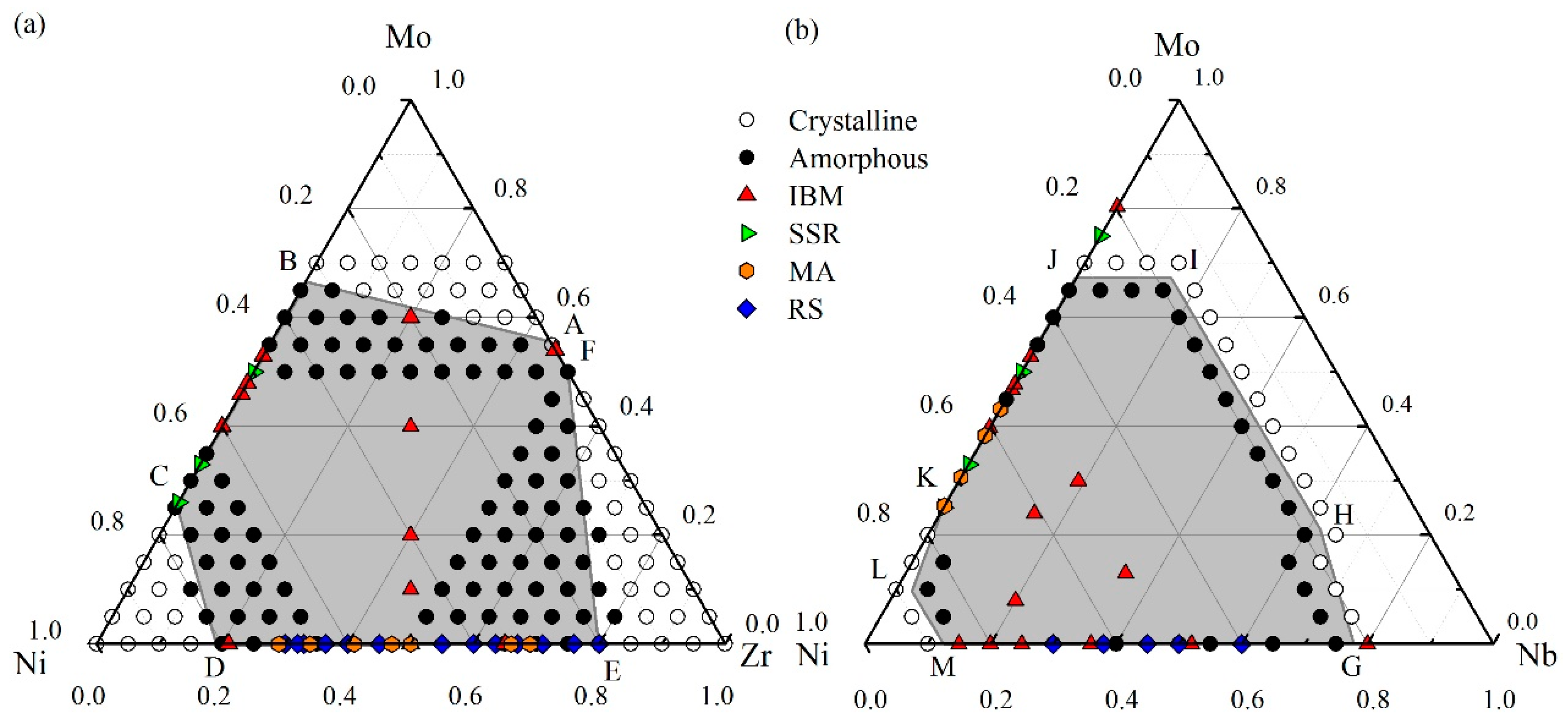
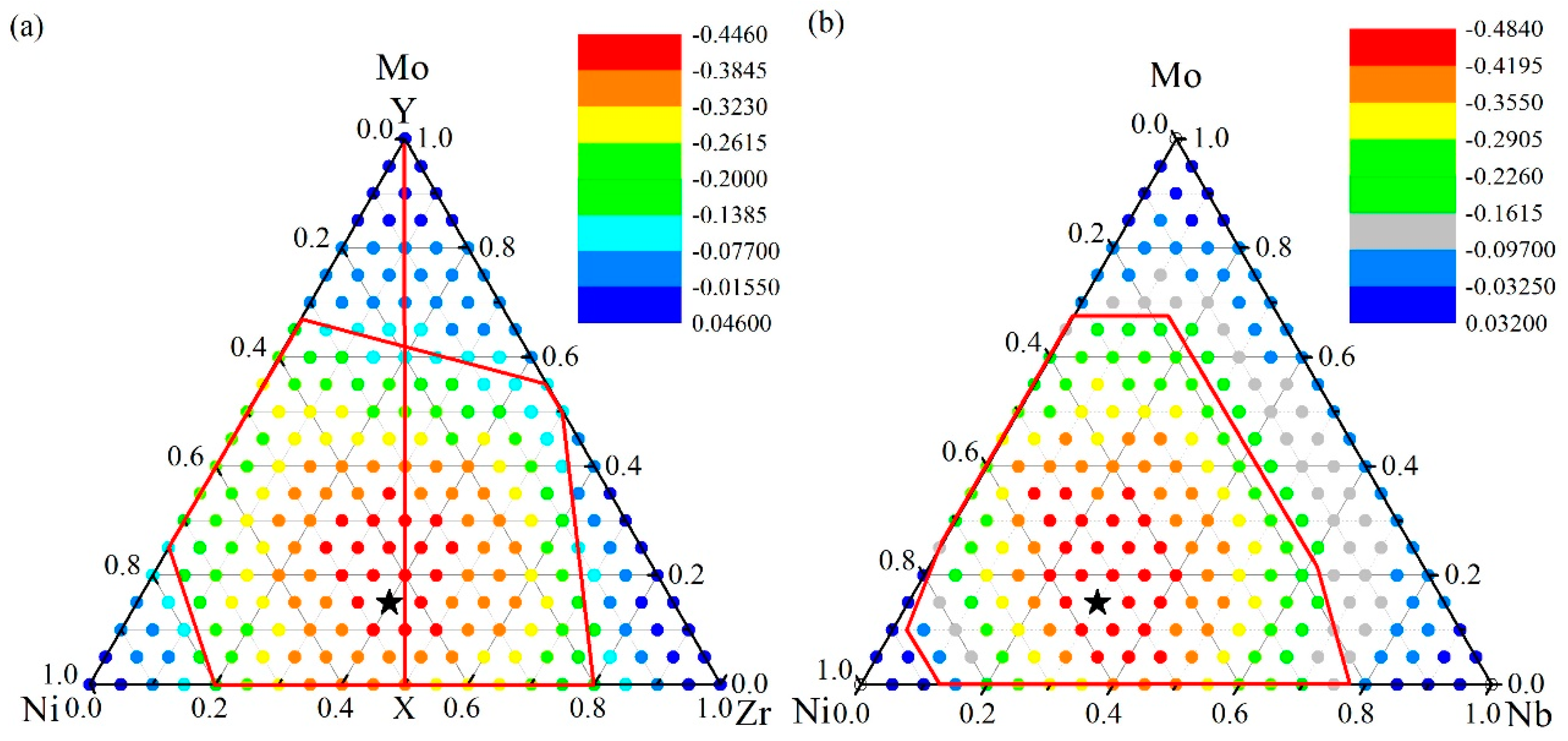
4.1. Glass Formation Region for the Ni-Zr-Mo System
4.2. Glass Formation Region for the Ni-Nb-Mo System
4.3. Glass Formation Ability of the Ternary Systems
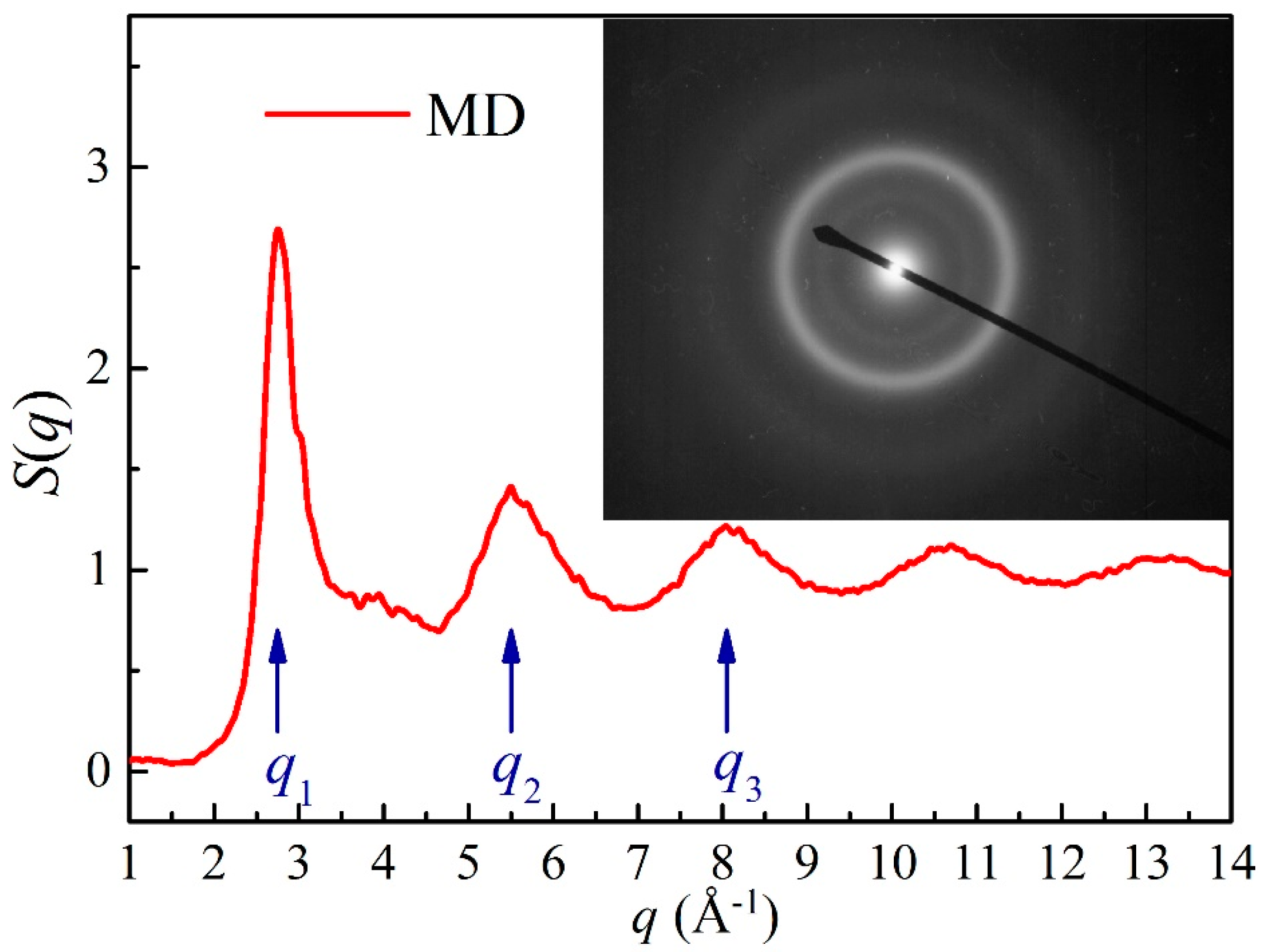
5. Experimental Verification of Atomic Simulation Prediction
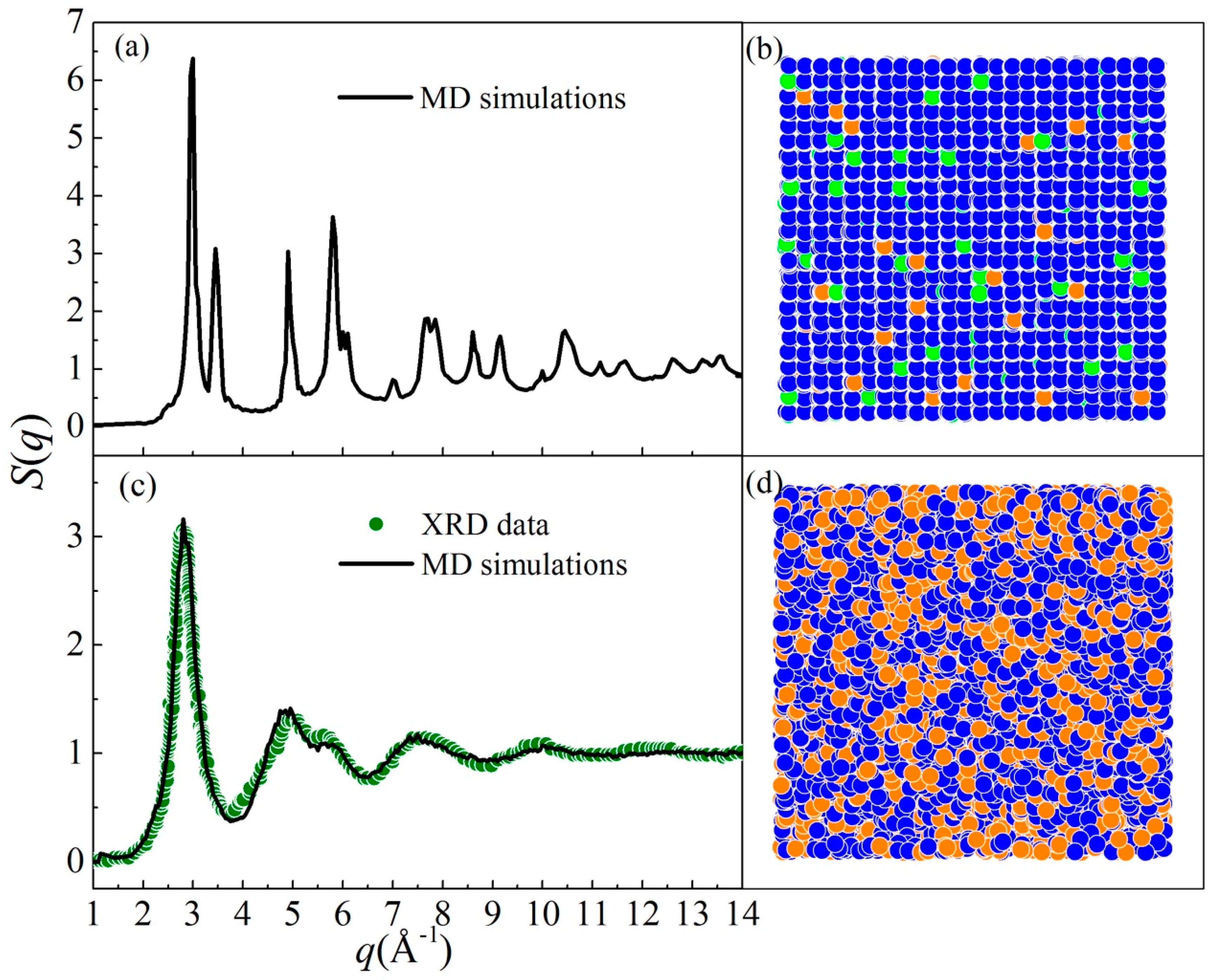
5.1. Metallic Glass Formation by Ion Beam Mixing
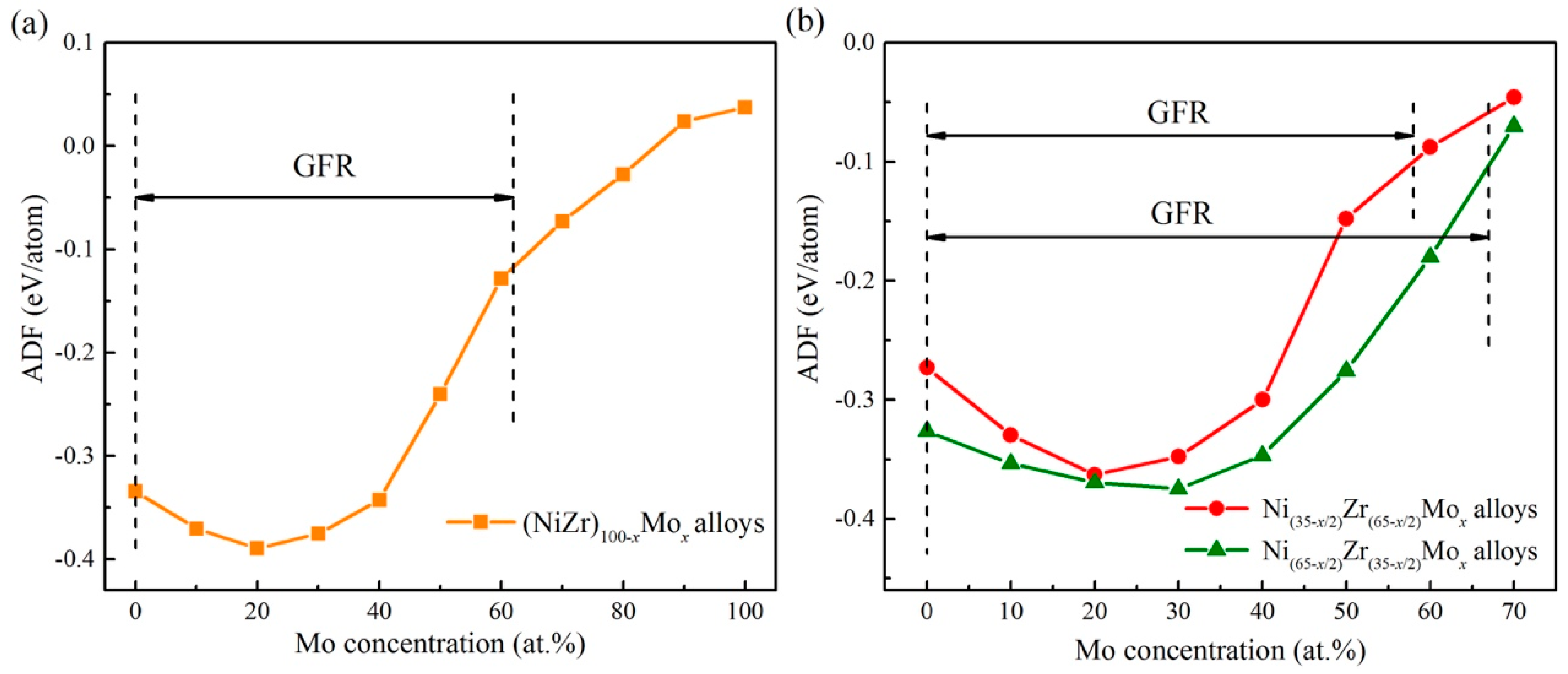
5.2. Connections between ADF and GFA
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sheng, H.W.; Luo, W.K.; Alamgir, F.M.; Bai, J.M.; Ma, E. Atomic packing and short-to-medium-range order in metallic glasses. Nature 2006, 439, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.Q.; Ma, E. Atomic-level structure and structure-property relationship in metallic glasses. Prog. Mater. Sci. 2011, 56, 379–473. [Google Scholar] [CrossRef]
- Schuh, C.A.; Hufnagel, T.C.; Ramamurty, U. Mechanical behavior of amorphous alloys. Acta Mater. 2007, 55, 4067–4109. [Google Scholar] [CrossRef]
- Yu, H.B.; Richert, R.; Samwer, K. Structural rearrangements governing Johari-Goldstein relaxations in metallic glasses. Sci. Adv. 2017, 3, e1701577. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhang, F.; Ye, Z.; Zhang, Y.; Fang, X.W.; Ding, Z.J.; Wang, C.Z.; Mendelev, M.I.; Ott, R.T.; Kramer, M.J.; et al. ‘Crystal Genes’ in Metallic Liquids and Glasses. Sci. Rep. 2016, 6, 23734. [Google Scholar] [CrossRef] [PubMed]
- Klement, W.K.; Willens, R.H.; Duwez, P. Non-crystalline Structure in Solidified Gold-Silicon Alloys. Nature 1960, 187, 869–870. [Google Scholar] [CrossRef]
- Liu, B.X.; Johnson, W.L.; Nicolet, M.-A.; Lau, S.S. Structural difference rule for amorphous alloy formation by ion mixing. Appl. Phys. Lett. 1983, 42, 45–47. [Google Scholar] [CrossRef]
- Liu, B.X.; Johnson, W.L.; Nicolet, M.-A.; Lau, S.S. Amorphous film formation by ion mixing in binary metal systems. Nucl. Instrum. Methods Phys. Res. 1983, 209, 229–234. [Google Scholar] [CrossRef]
- Koch, C.C.; Cavin, O.B.; McKamey, C.G.; Scarbrough, J.O. Preparation of “amorphous” Ni60Nb40 by mechanical alloying. Appl. Phys. Lett. 1983, 43, 1017–1019. [Google Scholar] [CrossRef]
- Suryanarayana, C. Mechanical alloying and milling. Prog. Mater. Sci. 2001, 46, 1–184. [Google Scholar] [CrossRef]
- Bordeenithikasem, P.; Liu, J.B.; Kube, S.A.; Li, Y.L.; Ma, T.X.; Scanley, B.E.; Broadbridge, C.C.; Vlassak, J.J.; Singer, J.P.; Schroers, J. Determination of critical cooling rates in metallic glass forming alloy libraries through laser spike annealing. Sci. Rep. 2017, 7, 7155. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.K.; Cheng, K.C.; Huang, J.C. Effects of laser annealing parameters on optical and electrical properties of ITO/metallic glass alloy Bi-layer films. Nanoscale Res. Lett. 2015, 10, 274. [Google Scholar] [CrossRef] [PubMed]
- Ruffino, F.; Torrisi, V.; Grillo, R.; Cacciato, G.; Zimbone, M.; Piccitto, G.; Grimaldi, M.G. Nanoporous Au structures by dealloying Au/Ag thermal- or laser-dewetted bilayers on surfaces. Superlattice Microst. 2017, 103, 28–47. [Google Scholar] [CrossRef]
- Martin, G.; Bellon, P. Driven Alloys. Solid State Phys. 1996, 50, 189–331. [Google Scholar]
- Liu, B.X.; Lai, W.S.; Zhang, Z.J. Solid-state crystal-to-amorphous transition in metal-metal multilayers and its thermodynamic and atomistic modelling. Adv. Phys. 2001, 50, 367–429. [Google Scholar] [CrossRef]
- Suryanarayana, C. Non-Equilibrium Processing of Materials; Pergamon: Oxford, UK, 1999. [Google Scholar]
- Haasen, P.; Jaffee, R.I. Amorphous Metals and Semiconductors: Proceedings of an International Workshop; Pergamon Press: Coronado, CA, USA, 1985. [Google Scholar]
- Lv, Z.P.; Bei, H.; Liu, C.T. Recent progress in quantifying glass-forming ability of bulk metallic glasses. Intermetallics 2007, 15, 618–624. [Google Scholar]
- Wen, T.Q.; Tang, L.; Sun, Y.; Ho, K.M.; Wang, C.Z.; Wang, N. Crystal genes in a marginal glass-forming system of Ni50Zr50. Phys. Chem. Chem. Phys. 2017, 19, 30429–30438. [Google Scholar] [CrossRef] [PubMed]
- Turnbull, D. Amorphous Solid Formation and Interstitial Solution Behavior in Metallic Alloy Systems. J. Phys. 1974, 35, C4.1–C4.10. [Google Scholar]
- Liu, B.X. Further studies of ion mixing in binary metal system. Nucl. Instrum. Methods Phys. Res. 1985, 7–8, 547–551. [Google Scholar]
- Liu, B.X. Ion mixing and metallic alloy phase formation. Phys. Status Solidi A 1986, 94, 11–34. [Google Scholar] [CrossRef]
- Inoue, A. Stabilization of metallic supercooled liquid and bulk amorphous alloys. Acta Mater. 2000, 48, 279–306. [Google Scholar] [CrossRef]
- Johnson, W.L. Bulk glass-forming metallic alloys: Science and technology. MRS Bull. 1999, 24, 42–56. [Google Scholar] [CrossRef]
- Li, J.H.; Dai, X.D.; Liang, S.H.; Tai, K.P.; Kong, Y.; Liu, B.X. Interatomic potentials of the binary transition metal systems and some applications in materials physics. Phys. Rep. 2008, 455, 1–134. [Google Scholar] [CrossRef]
- Li, J.H.; Dai, Y.; Cui, Y.Y.; Liu, B.X. Atomistic theory for predicting the binary metallic glass formation. Mater. Sci. Eng. R 2011, 72, 1–28. [Google Scholar] [CrossRef]
- Luo, S.Y.; Li, J.H.; Liu, J.B.; Liu, B.X. Atomic modeling to design favored compositions for the ternary Ni-Nb-Zr metallic glass formation. Acta Mater. 2014, 76, 482–492. [Google Scholar] [CrossRef]
- Sun, Y.; Zhang, Y.; Zhang, F.; Ye, Z.; Ding, Z.J.; Wang, C.Z.; Ho, K.M. Cooling rate dependence of structural order in Al90Sm10 metallic glass. J. Appl. Phys. 2016, 120, 015901. [Google Scholar] [CrossRef]
- Thompson, M.W. Defects and Radiation Damage in Metals; Cambridge University Press: Cambidge, UK, 1969. [Google Scholar]
- Russell, K.C. Phase-stability under irradiation. Prog. Mater. Sci. 1984, 28, 229–434. [Google Scholar] [CrossRef]
- Bransden, B.H. Atomic Collision Theory; Benjamin-Cummings Pub.: London, UK, 1983. [Google Scholar]
- Sigmund, P. Particle Penetration and Radiation Effects; Springer: Berlin, Germany, 2006. [Google Scholar]
- Greer, J.R.; Hosson, J.T.M.D. Plasticity in small-sized metallic systems: Intrinsic versus extrinsic size effect. Prog. Mater. Sci. 2011, 56, 654–724. [Google Scholar] [CrossRef]
- Jang, D.C.; Greer, J.R. Transition from a strong-yet-brittle to a stronger-and-ductile state by size reduction of metallic glasses. Nat. Mater. 2010, 9, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Ghidelli, M.; Gravier, S.; Blandin, J.-J.; Djemia, P.; Mompiou, F.; Abadias, G.; Raskin, J.-P.; Pardoen, T. Extrinsic mechanical size effects in thin ZrNi metallic glass films. Acta Mater. 2015, 90, 232–241. [Google Scholar] [CrossRef]
- Ghidelli, M.; Idrissi, H.; Gravier, S.; Blandin, J.-J.; Raskin, J.-P.; Schryvers, D.; Pardoen, T. Homogeneous flow and size dependent mechanical behavior in highly ductile Zr65Ni35 metallic glass films. Acta Mater. 2017, 131, 246–259. [Google Scholar] [CrossRef]
- Ghidelli, M.; Volland, A.; Bandin, J.-J.; Pardoen, T.; Raskin, J.-P.; Mompiou, F.; Djemia, P.; Gravier, S. Exploring the mechanical size effects in Zr65Ni35 thin film metallic glasses. J. Alloys Compd. 2014, 615, S90–S92. [Google Scholar] [CrossRef]
- Buschow, K.H.J.; Beekmans, N.M. Thermal stability and electronic properties of Zr-Co and Zr-Ni alloys. Phys. Rev. B 1979, 19, 3843. [Google Scholar] [CrossRef]
- Ghidelli, M.; Gravier, S.; Blandin, J.-J.; Pardoen, T.; Raskin, J.-P.; Mompiou, F. Compositional-induced structural change in ZrxNi100-x thin film metallic glasses. J. Alloys Compd. 2014, 615, S348–S351. [Google Scholar] [CrossRef]
- Guo, G.-Q.; Yang, L.; Huang, C.-L.; Chen, D. Structural origin of the different glass-forming abilities in ZrCu and ZrNi metallic glasses. J. Mater. Res. 2011, 26, 2098. [Google Scholar]
- Mattern, N.; Bednarčik, J.; Pauly, S.; Wang, G.; Das, J.; Eckert, J. Structural evolution of Cu-Zr glasses under tension. Acta Mater. 2009, 57, 4133–4139. [Google Scholar] [CrossRef]
- Zhang, Z.J.; Bai, H.Y.; Qiu, Q.L.; Yang, T.; Tao, K.; Liu, B.X. Phase evolution upon ion mixing and solid-state reaction and thermo-dynamic interpretation in the Ni-Nb system. J. Appl. Phys. 1993, 73, 1702–1710. [Google Scholar] [CrossRef]
- Kimura, H.; Inoue, A.; Yamaura, S.; Sasamori, K.; Nishida, M.; Shinpo, Y.; Okouchi, H. Thermal stability and mechanical properties of glassy and amorphous Ni-Nb-Zr alloys produced by rapid solidification. Mater. Trans. 2003, 44, 1167–1171. [Google Scholar] [CrossRef]
- Oreshkin, A.I.; Mantsevich, V.N.; Savinov, S.V.; Oreshkin, S.I.; Panov, V.I.; Yavari, A.R.; Miracle, D.B.; Louzguine-Luzgin, D.V. In situ visualization of Ni-Nb bulk metallic glasses phase transition. Acta Mater. 2013, 61, 5216–5222. [Google Scholar] [CrossRef]
- Daw, M.S.; Baskes, M.I. Semiempirical, quantum mechanical calculation of hydrogen embrittlement in metals. Phys. Rev. Lett. 1983, 50, 1285. [Google Scholar] [CrossRef]
- Daw, M.S.; Baskes, M.I. Embedded-atom method-Derivation and application to impurities, surface, and other defects in metals. Phys. Rev. B 1984, 29, 6443–6453. [Google Scholar] [CrossRef]
- Finnis, M.W.; Sinclair, J.E. A simple empirical n-body potential for transition-metals. Philos. Mag. A 1984, 50, 45–55. [Google Scholar] [CrossRef]
- Tomanek, D.; Aligia, A.A.; Balseiro, C.A. Calculation of elastic strain and electronic effects on surface segregation. Phys. Rev. B 1985, 32, 5051–5056. [Google Scholar] [CrossRef]
- Baskes, M.I.; Johnson, R.A. Modified embedded-atom potentials for hcp metals. Model. Simul. Mater. Sci. Eng. 1994, 2, 147–163. [Google Scholar] [CrossRef]
- Hu, W.; Zhang, B.; Huang, B.; Gao, F.; Bacon, D.J. Analytic modified embedded atom potentials for hcp metals. J. Phys. Condens. Matter 2001, 13, 1193–1213. [Google Scholar] [CrossRef]
- Johnson, R.A.; Oh, D.J. Analytic modified embedded atom potentials for bcc metals. J. Mater. Res. 1989, 4, 1195–1201. [Google Scholar] [CrossRef]
- Dai, X.D.; Li, J.H.; Kong, Y. Long-range empirical potential for the bcc structured transition metals. Phys. Rev. B 2007, 75, 052102. [Google Scholar] [CrossRef]
- Dai, X.D.; Kong, Y.; Li, J.H. Long-range empirical potential model: Application to fcc transition metals and alloys. Phys. Rev. B 2007, 75, 104101. [Google Scholar] [CrossRef]
- Dai, Y.; Li, J.H.; Liu, B.X. Long-range empirical potential model: Extension to hexagonal close-packed metals. J. Phys. Condens. Matter 2009, 21, 385402. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Luo, S.Y.; Li, J.H.; Liu, J.B.; Liu, B.X. Interatomic potential to predict the glass-forming ability of Ni-Nb-Mo ternary alloys. J. Mater. Sci. 2014, 49, 7263–7272. [Google Scholar] [CrossRef]
- Yang, M.H.; Li, S.N.; Li, Y.; Li, J.H.; Liu, B.X. Atomistic modeling to optimize composition and characterize structure of Ni-Zr-Mo metallic glasses. Phys. Chem. Chem. Phys. 2015, 17, 13355. [Google Scholar] [CrossRef] [PubMed]
- Lide, D.R.; Bruno, T.L. CRC Handbook of Chemistry and Physics; CRC Press: New York, NY, USA, 2012. [Google Scholar]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.I.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Z. Kristallogr.-Cryst. Mater. 2005, 220, 567–570. [Google Scholar] [CrossRef]
- Segall, M.D.; Lindan, P.J.D.; Probert, M.J.; Pickard, C.J.; Hasnip, P.J.; Clark, S.J. First-principles simulation: Ideas, illustrations and the CASTEP code. J. Phys. Condens. Matter 2002, 14, 2717–2744. [Google Scholar] [CrossRef]
- Blochl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Perdew, J.P.; Wang, Y. Pair-distribution function and its coupling-constant average for the spin-polarized electron-gas. Phys. Rev. B 1992, 46, 12947–12954. [Google Scholar] [CrossRef]
- Ikehata, H.; Nagasako, N.; Furuta, T.; Fukumoto, A.; Miwa, K.; Saito, T. First-principles calculations for development of low elastic modulus Ti alloys. Phys. Rev. B 2004, 70, 174113. [Google Scholar] [CrossRef]
- Brandes, E.A.; Brook, G.B. Smithells Metals Reference Book, 8th ed.; Elsevier Butterworth-Heinemann Ltd.: Oxford, UK, 2004. [Google Scholar]
- Zhang, Q.; Lai, W.S.; Liu, B.X. Molecular dynamics study of solid state interfacial reaction in the Ni-Mo system. J. Comput.-Aided Mater. Des. 1999, 6, 103–116. [Google Scholar] [CrossRef]
- Rose, J.H.; Smith, J.R.; Guinea, F.; Ferrante, J. Universal features of the equation of state of metals. Phys. Rev. B 1984, 29, 2963–2969. [Google Scholar] [CrossRef]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Yang, M.H.; Li, J.H.; Liu, B.X. Proposed correlation of structure network inherited from producing techniques and deformation behavior for Ni-Ti-Mo metallic glasses via atomistic simulations. Sci. Rep. 2016, 6, 29722. [Google Scholar] [CrossRef] [PubMed]
- Suryanarayana, C.; Norton, M.G. X-ray Diffraction: A Practical Approach; Plenum Press: New York, NY, USA, 1998. [Google Scholar]
- Liu, B.X.; Lai, W.S.; Zhang, Q. Irradiation induced amorphization in metallic multilayers and calculation of glass-forming ability from atomistic potential in the binary metal systems. Mater. Sci. Eng. Rep. 2000, 29, 1–48. [Google Scholar] [CrossRef]
- Ziegler, J.F.; Biersack, J.P.; Littmark, U. The Stopping and Range of Ions in Solids; Pergamon Press: New York, NY, USA, 1985. [Google Scholar]
- Li, Y.; Wang, T.L.; Ding, N.; Liu, J.B.; Liu, B.X. Metallic glass formation in the ternary Ni-Nb-Mo system by ion beam mixing. Sci. China Technol. Sci. 2012, 55, 2206–2211. [Google Scholar] [CrossRef]
- Yang, M.H.; Li, N.; Li, J.H.; Liu, B.X. Effects of Mo addition on the metallic glass formation of (ZrNi)100−xMox alloys. J. Alloys Compd. 2014, 606, 7–10. [Google Scholar] [CrossRef]
- Kaban, I.; Jόvári, P.; Kokotin, V.; Shuleshova, O.; Beuneu, B.; Saksl, K.; Mattern, N.; Eckert, J.; Greer, A.L. Local atomic arrangements and their topology in Ni-Zr and Cu-Zr glassy and crystalline alloys. Acta Mater. 2013, 61, 2509–2520. [Google Scholar] [CrossRef]
- Zhang, Z.J.; Liu, B.X. Solid-state reaction to synthesize Ni-Mo metastable alloys. J. Appl. Phys. 1994, 76, 3351–3356. [Google Scholar] [CrossRef]
- Li, J.H.; Zhao, S.Z.; Dai, Y.; Cui, Y.Y.; Liu, B.X. Formation and structure of Al-Zr metallic glasses studied by Monte Carlo simulations. J. Appl. Phys. 2011, 109, 113538. [Google Scholar] [CrossRef]
- Weeber, A.W.; Bakker, H. Extension of the glass-forming range of Ni-Zr by mechanical alloying. J. Phys. F Met. Phys. 1998, 18, 1359–1369. [Google Scholar] [CrossRef]
- Yang, M.H.; Li, J.H.; Liu, B.X. Proposed correlation of glass formation ability with critical dosage for the amorphous alloys formed by ion beam mixing. RSC Adv. 2015, 5, 16400. [Google Scholar] [CrossRef]
- Xi, X.K.; Li, L.L.; Zhang, B.; Wang, W.H.; Wu, Y. Correlation of atomic cluster symmetry and glass-forming ability of metallic glass. Phys. Rev. Lett. 2007, 99, 095501. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.H.; Duan, G.; Johnson, W.L. Unusual Glass-Forming Ability of Bulk Amorphous Alloys Based on Ordinary Metal Copper. Phys. Rev. Lett. 2004, 92, 245504. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Fang, S.S.; Wang, Q.; Dong, Y.D. Thermodynamic modeling of glass formation in metallic glasses. Appl. Rev. Lett. 2006, 88, 171905. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Ni-Ni | Nb-Nb | Mo-Mo | Ni-Nb | Ni-Mo | Nb-Mo |
|---|---|---|---|---|---|---|
| m | 4 | 4 | 4 | 4 | 4 | 4 |
| n | 5 | 8 | 6 | 8 | 6 | 6 |
| rc1 (Å) | 5.769 | 4.803 | 4.758 | 5.188 | 5.245 | 5.259 |
| rc2 (Å) | 7.245 | 6.819 | 7.242 | 5.150 | 5.017 | 7.550 |
| c0 (eV) | 313.309 | 5561.735 | 10,271.293 | 2246.718 | 2297.975 | 2837.964 |
| c1 (eV/Å) | −381.137 | −7613.754 | −14,753.967 | −2878.560 | −3065.951 | −4301.152 |
| c2 (eV/Å2) | 172.524 | 3927.589 | 7943.743 | 1410.824 | 1548.758 | 2449.419 |
| c3 (eV/Å3) | −34.507 | −896.592 | −1896.380 | −308.742 | −351.481 | −618.445 |
| c4 (eV/Å4) | 2.597 | 76.071 | 169.334 | 25.062 | 30.267 | 58.207 |
| α (eV) | 4.091 | 29.385 | 11.828 | 58.520 | 19.195 | 8.480 |
| Parameters | Ni-Ni | Zr-Zr | Mo-Mo | Ni-Zr | Ni-Mo | Zr-Mo |
|---|---|---|---|---|---|---|
| m | 4 | 4 | 4 | 4 | 4 | 4 |
| n | 6 | 4 | 6 | 5 | 6 | 5 |
| rc1 (Å) | 5.750 | 6.438 | 4.618 | 4.783 | 5.268 | 4.944 |
| rc2 (Å) | 7.172 | 7.223 | 6.414 | 7.385 | 6.217 | 5.312 |
| c0 (eV) | 322.065 | 1353.855 | 11,867.251 | 331.755 | 1444.553 | 1582.121 |
| c1 (eV/Å) | −391.987 | −1453.094 | −17,112.524 | −430.706 | −1776.802 | −2021.254 |
| c2 (eV/Å2) | 178.332 | 586.519 | 9255.419 | 254.058 | 810.354 | 977.406 |
| c3 (eV/Å3) | −36.094 | −104.936 | −2218.683 | −74.642 | −161.714 | −207.188 |
| c4 (eV/Å4) | 2.770 | 7.009 | 198.426 | 8.798 | 11.686 | 15.759 |
| α (eV) | 5.390 | 7.830 | −11.879 | 8.343 | −3.227 | −11.513 |
| Compounds | NbNi | NbMo | Nb3Ni | NbNi3 | Nb3Mo | NbMo3 | Mo3Ni | MoNi3 |
|---|---|---|---|---|---|---|---|---|
| Structure | B2 | B2 | L12 | L12 | L12 | L12 | L12 | L12 |
| a (Å) | 3.111 | 3.198 | 3.929 | 3.679 | 4.183 | 4.004 | 3.761 | 3.644 |
| 3.095 | 3.226 | 4.100 | 3.700 | 4.159 | 4.052 | 3.871 | 3.647 | |
| Ec (eV) | 6.121 | 7.522 | 6.714 | 6.121 | 7.522 | 6.803 | 6.113 | 5.057 |
| 6.047 | 7.652 | 6.707 | 6.047 | 7.652 | 6.800 | 6.150 | 4.994 | |
| B0 (Mbar) | 2.237 | 1.846 | 1.444 | 2.237 | 1.846 | 2.298 | 2.389 | 2.287 |
| 2.166 | 1.810 | 1.765 | 2.166 | 1.810 | 2.144 | 2.412 | 2.230 |
| Compounds | NiZr | NiMo | ZrMo | Ni3Zr | NiZr3 | Ni3Mo | NiMo3 | Zr3Mo |
|---|---|---|---|---|---|---|---|---|
| Structure | B2 | B2 | B2 | L12 | L12 | L12 | L12 | L12 |
| a (Å) | 3.162 | 3.044 | 3.284 | 3.770 | 4.297 | 3.584 | 3.678 | 4.428 |
| 3.212 | 2.998 | 3.349 | 3.759 | 4.279 | 3.647 | 3.871 | 4.379 | |
| Ec (eV) | 5.717 | 5.447 | 6.032 | 5.344 | 5.767 | 5.060 | 6.001 | 6.172 |
| 5.720 | 5.390 | 6.054 | 5.342 | 5.803 | 4.994 | 6.150 | 6.175 | |
| B0 (Mbar) | 1.463 | 2.625 | 1.524 | 1.687 | 1.267 | 2.227 | 1.316 | 1.211 |
| 1.422 | 2.328 | 1.491 | 1.697 | 1.071 | 2.230 | 2.412 | 1.172 |
| Dosage | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| (Xe+/cm2) | Ni52Nb35Mo13 | Ni61Nb15Mo24 | Ni72Nb20Mo8 | Ni51Nb19Mo30 |
| 8 × 1014 | A | A + Bcc | A + Fcc | A + Bcc |
| 1 × 1015 | A | A | A + Fcc | A + Bcc |
| 2 × 1015 | A | A | A + Fcc | A + Bcc |
| 3 × 1015 | A | A | A | A + Bcc |
| 5 × 1015 | A | A | A | A |
| Dosage | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| (Xe+/cm2) | (NiZr)90Mo10 | (NiZr)80Mo20 | (NiZr)60Mo40 | (NiZr)40Mo60 |
| 1 × 1015 | A + Bcc | A + Bcc | A + Bcc | A + Bcc |
| 3 × 1015 | A | A + Bcc | A + Bcc | A + Bcc |
| 5 × 1015 | A | A | A + Bcc | A + Bcc |
| 7 × 1015 | A | A | A | A + Bcc |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, M.H.; Li, J.H.; Liu, B.X.; Liu, J.B. Atomistic Simulations to Predict Favored Glass-Formation Composition and Ion-Beam-Mixing of Nano-Multiple-Metal-Layers to Produce Ternary Amorphous Films. Metals 2018, 8, 129. https://doi.org/10.3390/met8020129
Yang MH, Li JH, Liu BX, Liu JB. Atomistic Simulations to Predict Favored Glass-Formation Composition and Ion-Beam-Mixing of Nano-Multiple-Metal-Layers to Produce Ternary Amorphous Films. Metals. 2018; 8(2):129. https://doi.org/10.3390/met8020129
Chicago/Turabian StyleYang, M. H., J. H. Li, B. X. Liu, and J. B. Liu. 2018. "Atomistic Simulations to Predict Favored Glass-Formation Composition and Ion-Beam-Mixing of Nano-Multiple-Metal-Layers to Produce Ternary Amorphous Films" Metals 8, no. 2: 129. https://doi.org/10.3390/met8020129
APA StyleYang, M. H., Li, J. H., Liu, B. X., & Liu, J. B. (2018). Atomistic Simulations to Predict Favored Glass-Formation Composition and Ion-Beam-Mixing of Nano-Multiple-Metal-Layers to Produce Ternary Amorphous Films. Metals, 8(2), 129. https://doi.org/10.3390/met8020129