
The Role of Grain Boundary Precipitates during Intergranular Fracture in 6xxx Series Aluminium Alloys


Abstract
1. Introduction
2. Methods and Models
2.1. Experimental Details
2.2. Computational Details
2.3. Theoretical Background
3. Results and Discussion
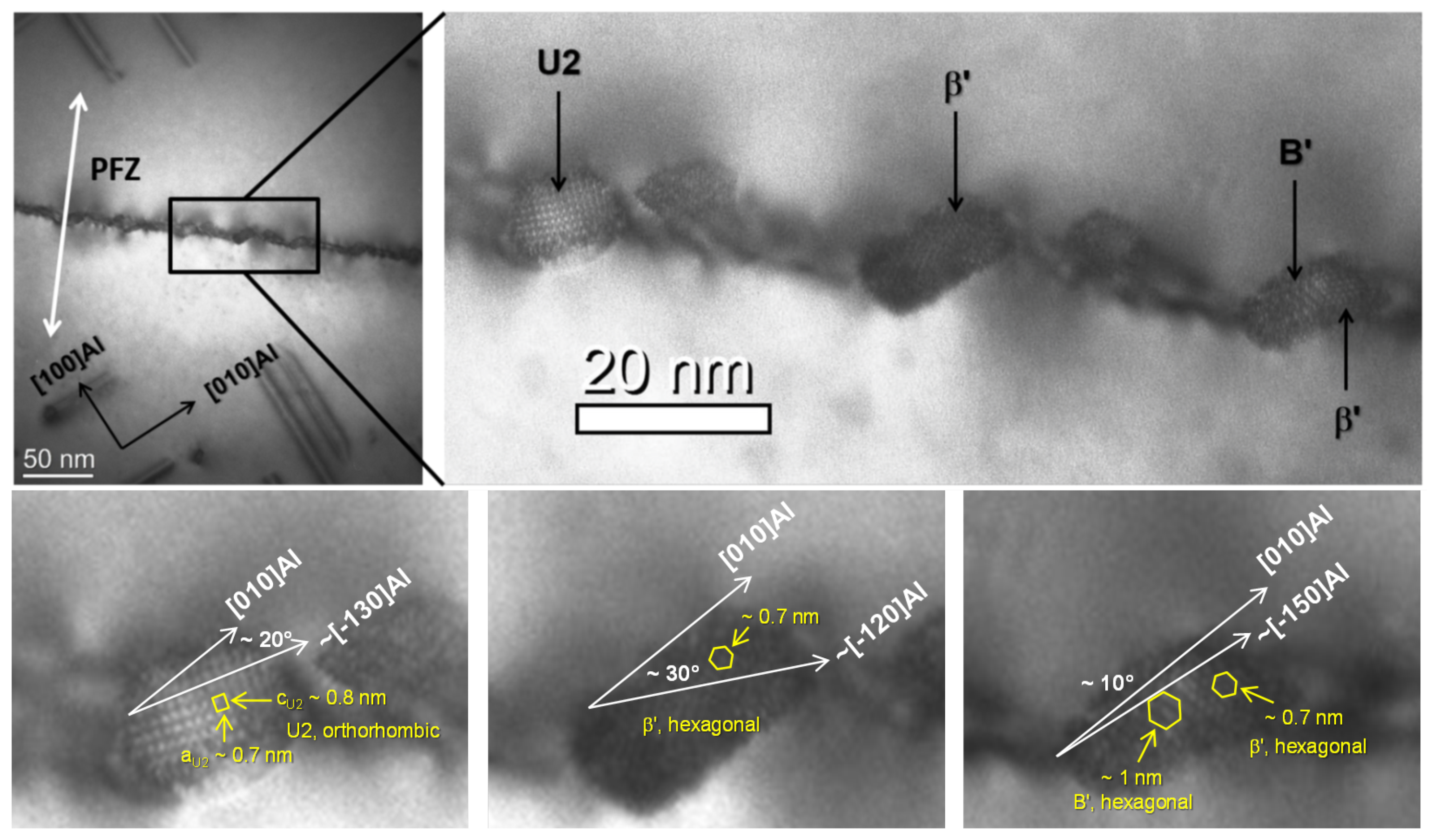
3.1. Experimental Observations of Grain Boundary Precipitates
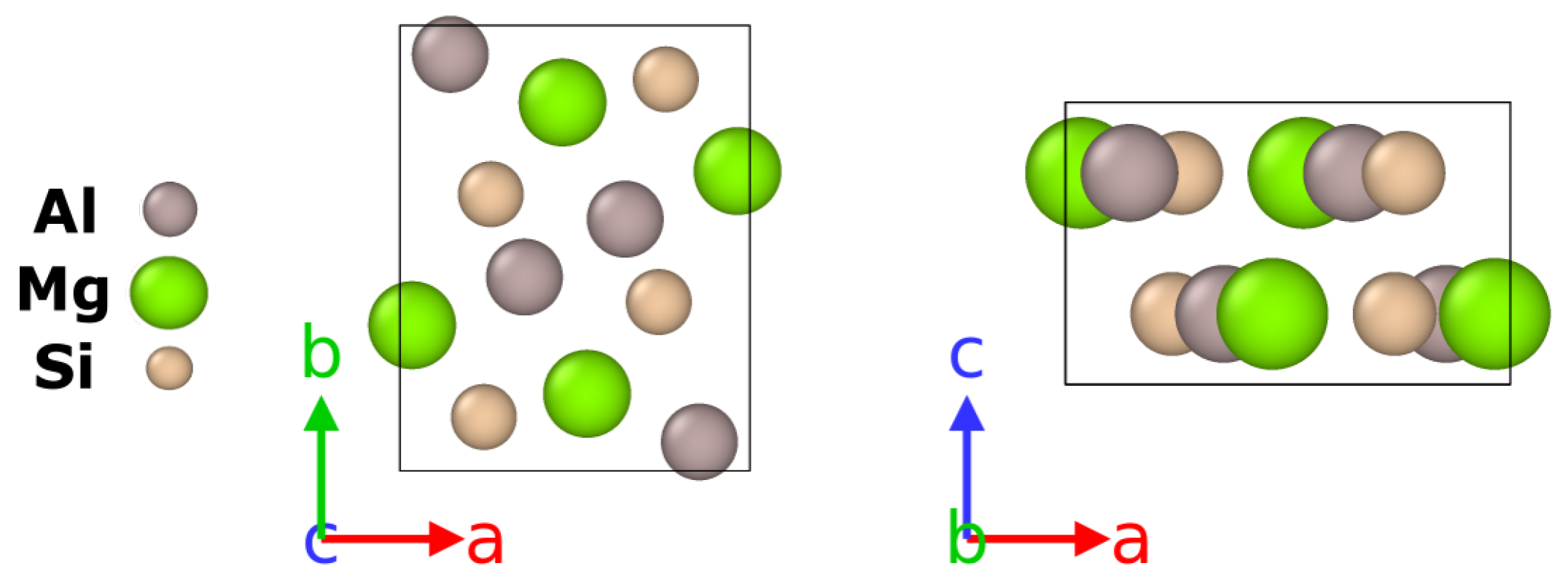
3.2. Bulk Properties of and U2
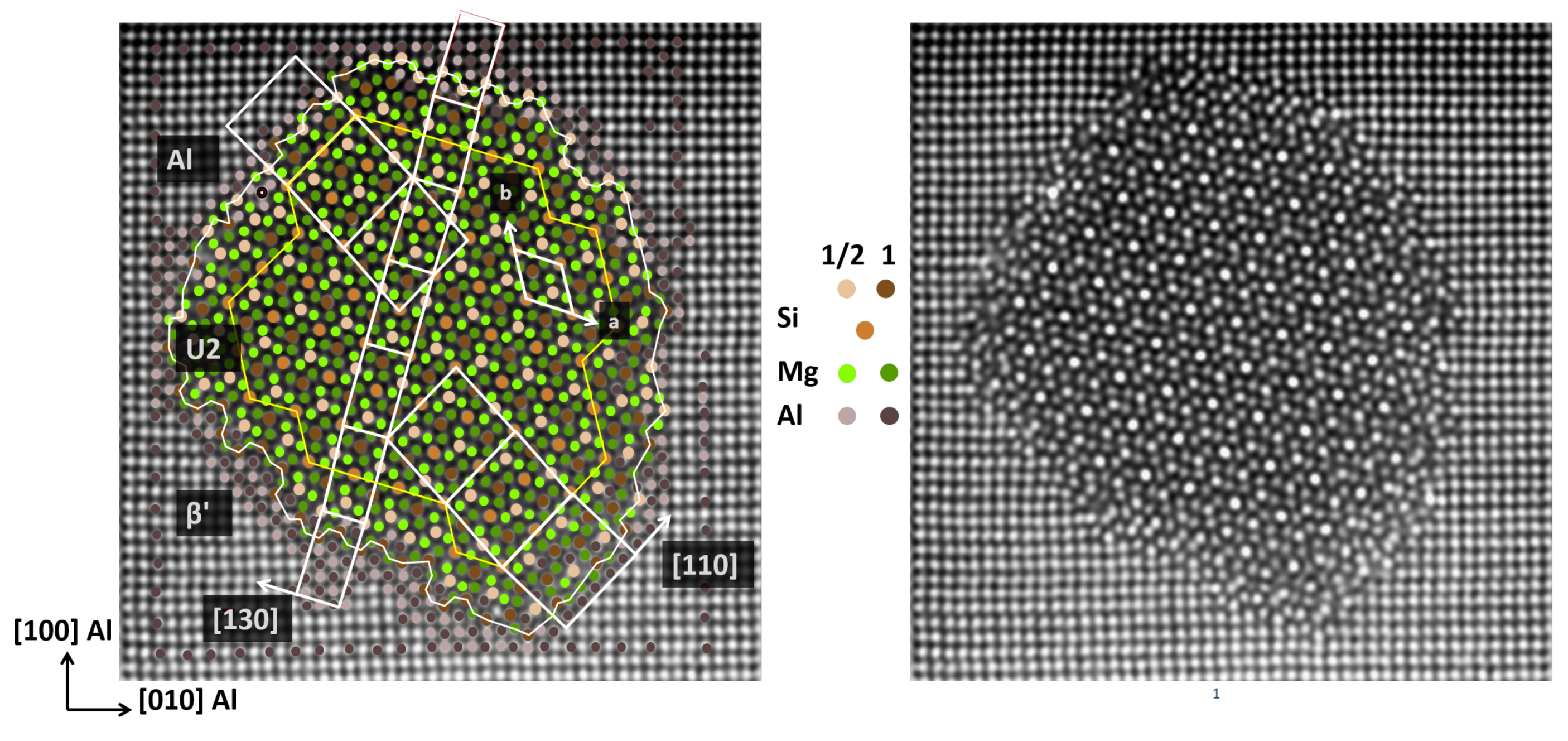
3.3. Construction of Realistic Interface Models
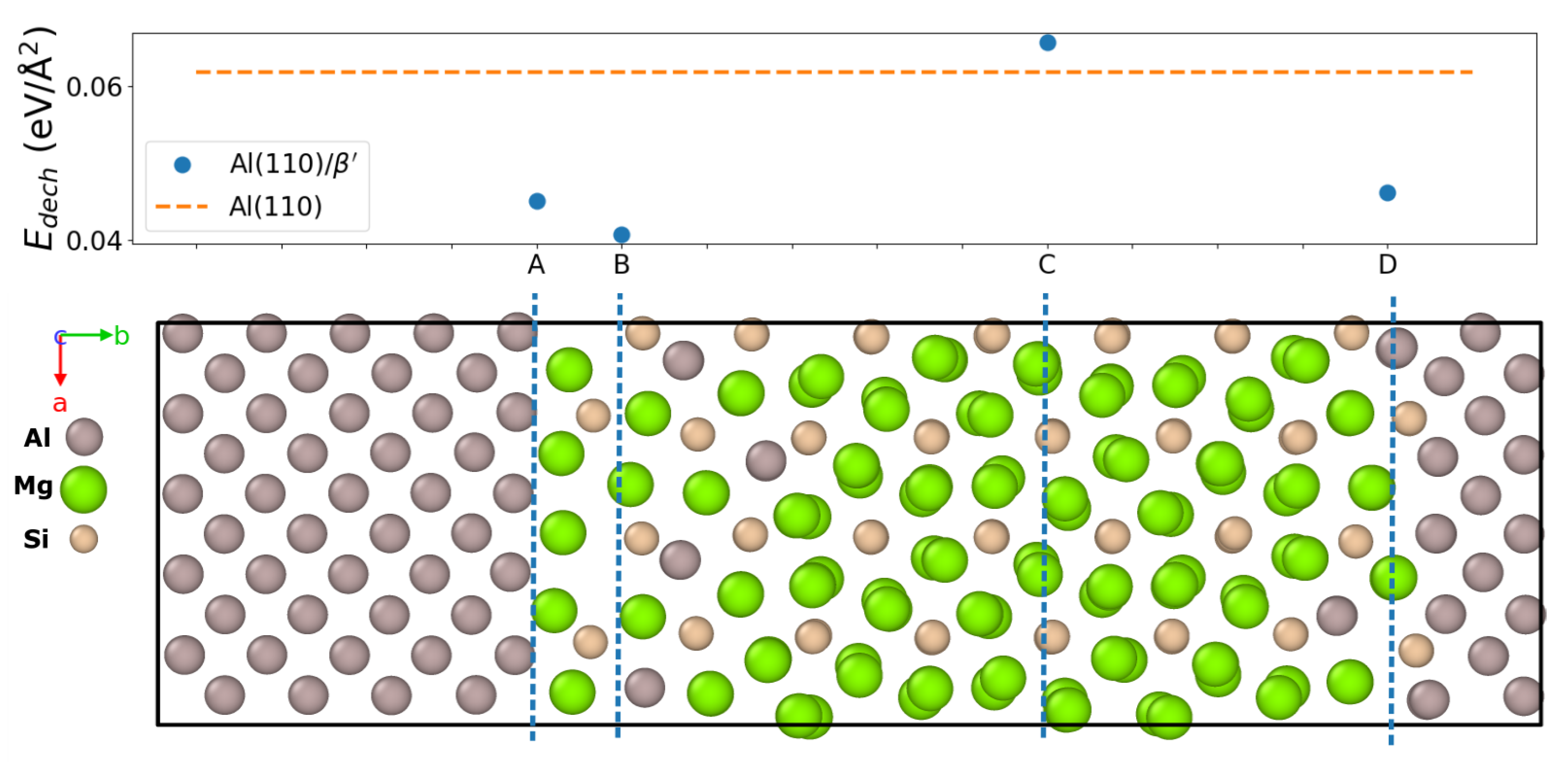
3.4. Decohesion Energies
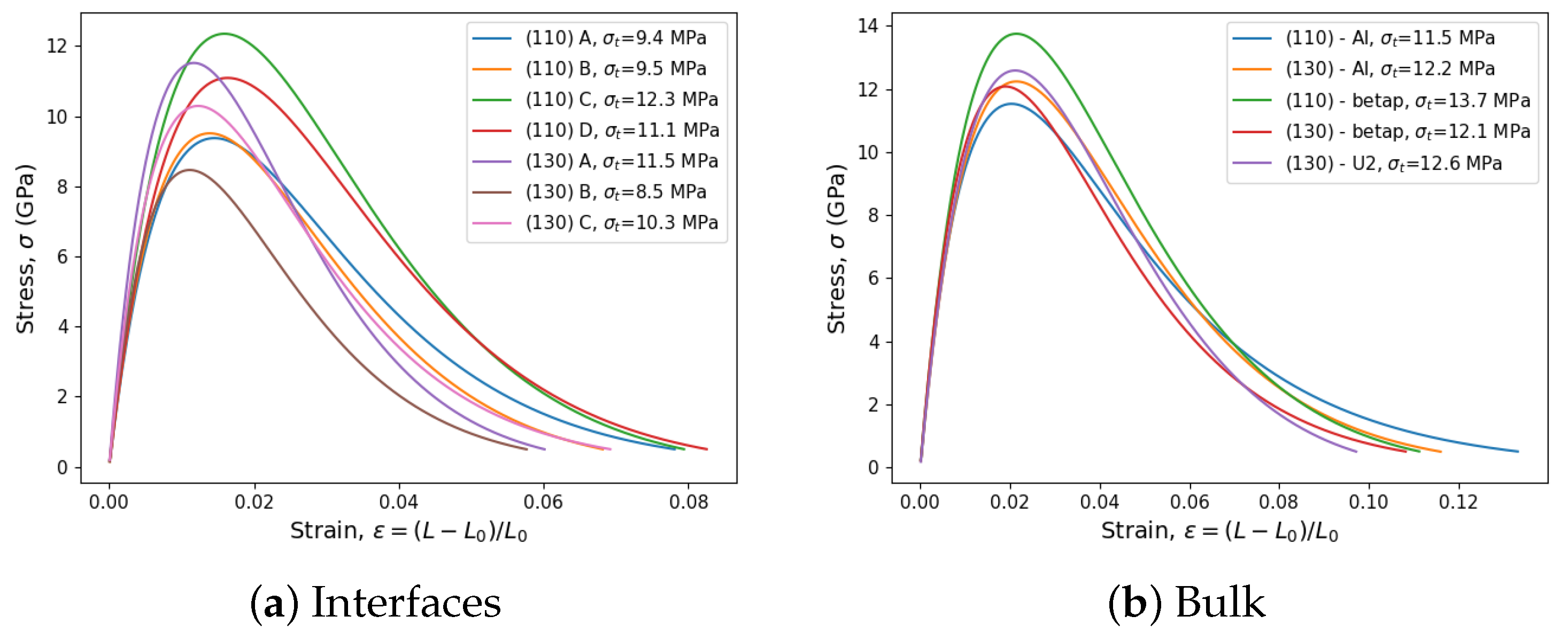
3.5. Tensile Tests and Tensile Strength
3.6. Interface Defects
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pedersen, K.O.; Westermann, I.; Furu, T.; Børvik, T.; Hopperstad, O.S. Influence of microstructure on work-hardening and ductile fracture of aluminium alloys. Mater. Des. 2015, 70, 31–44. [Google Scholar] [CrossRef]
- Chen, Y.; Pedersen, K.; Clausen, A.; Hopperstad, O. An experimental study on the dynamic fracture of extruded AA6xxx and AA7xxx aluminium alloys. Mater. Sci. Eng. A 2009, 523, 253–262. [Google Scholar] [CrossRef]
- Westermann, I.; Pedersen, K.; Børvik, T.; Hopperstad, O. Work-hardening and ductility of artificially aged AA6060 aluminium alloy. Mech. Mater. 2016, 97, 100–117. [Google Scholar] [CrossRef]
- Teichmann, K.; Marioara, C.D.; Andersen, S.J.; Marthinsen, K. TEM study of β′ precipitate interaction mechanisms with dislocations and β′ interfaces with the aluminium matrix in Al-Mg-Si alloys. Mater. Charact. 2013, 75, 1–7. [Google Scholar] [CrossRef]
- Anderson, T.L. Fracture Mechanics: Fundamentals and Applications; CRC Press: Boca Raton, FL, USA, 2005. [Google Scholar]
- Benzerga, A.A.; Leblond, J.B. Ductile fracture by void growth to coalescence. Adv. Appl. Mech. 2010, 44, 169–305. [Google Scholar]
- Besson, J. Continuum models of ductile fracture: A review. Int. J. Damage Mech. 2010, 19, 3–52. [Google Scholar] [CrossRef]
- Vasudevan, A.K.; Doherty, R. Grain boundary ductile fracture in precipitation hardened aluminum alloys. Acta Metall. 1987, 35, 1193–1219. [Google Scholar] [CrossRef]
- Pardoen, T.; Dumont, D.; Deschamps, A.; Brechet, Y. Grain boundary versus transgranular ductile failure. J. Mech. Phys. Solids 2003, 51, 637–665. [Google Scholar] [CrossRef]
- Christiansen, E.; Marioara, C.D.; Marthinsen, K.; Hopperstad, O.S.; Holmestad, R. Lattice rotations in precipitate free zones in an Al-Mg-Si alloy. Mater. Charact. 2018, 144, 522–531. [Google Scholar] [CrossRef]
- Frodal, B.H.; Christiansen, E.; Myhr, O.R.; Hopperstad, O.S. The role of quench rate on the plastic flow and fracture of three aluminium alloys with different grain structure and texture. Int. J. Eng. Sci. 2020, 150, 103257. [Google Scholar] [CrossRef]
- Jacobs, M.H. The structure of the metastable precipitates formed during ageing of an Al-Mg-Si alloy. Phil. Mag. A 1972, 26, 1–13. [Google Scholar] [CrossRef]
- Ravi, C.; Wolverton, C. First-principles study of crystal structure and stability of Al-Mg-Si-(Cu) precipitates. Acta Mater. 2004, 52, 4213–4227. [Google Scholar] [CrossRef]
- Vissers, R.; van Huis, M.A.; Jansen, J.; Zandbergen, H.W.; Marioara, C.D.; Andersen, S.J. The crystal structure of the β′ phase in Al-Mg-Si alloys. Acta Mater. 2007, 55, 3815–3823. [Google Scholar] [CrossRef]
- Matsuda, K.; Sakaguchi, Y.; Miyata, Y.; Uetani, Y.; Sato, T.; Kamio, A.; Ikeno, S. Precipitation sequence of various kinds of metastable phases in Al-1.0mass% Mg2Si-0.4mass% Si alloy. J. Mater. Sci. 2000, 35, 179–189. [Google Scholar] [CrossRef]
- Frøseth, A.G.; Høier, R.; Derlet, P.M.; Andersen, S.J.; Marioara, C.D. Bonding in MgSi and Al-Mg-Si compounds relevant to Al-Mg-Si alloys. Phys. Rev. B 2003, 67, 224106. [Google Scholar] [CrossRef]
- Andersen, S.J.; Marioara, C.D.; Frøseth, A.; Vissers, R.; Zandbergen, H.W. Crystal structure of the orthorhombic U2-Al4Mg4Si4 precipitate in the AlMgSi alloy system and its relation to the β′ and β″ phases. Mater. Sci. Eng. A 2005, 390, 127–138. [Google Scholar] [CrossRef]
- Janisch, R.; Ahmed, N.; Hartmaier, A. Ab initio tensile tests of Al bulk crystals and grain boundaries: Universality of mechanical behaviour. Phys. Rev. B 2010, 81, 184108. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comp. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Perdew, J.P.; Wang, Y. Pair-distribution function and its coupling-constant average for the spin-polarized electron gas. Phys. Rev. B 1992, 46, 12947. [Google Scholar] [CrossRef] [PubMed]
- Tahir, A.; Janisch, R.; Hartmaier, A. Hydrogen embrittlement of a carbon segregated Σ5 (310)[001] symmetrical tilt grain boundary in α-Fe. Mater. Sci. Eng. A 2014, 612, 462–467. [Google Scholar] [CrossRef]
- Yamaguchi, M. First-principles study on the grain boundary embrittlement of metals by solute segregation: Part I. iron (Fe)-solute (B, C, P, and S) systems. Metall. Mater. Trans. A 2011, 42, 319–329. [Google Scholar] [CrossRef]
- Rose, J.H.; Smith, J.R.; Ferrante, J. Universal features of bonding in metals. Phys. Rev. B 1983, 28, 1835–1845. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Unit Cell | Occ | a [Å] | b [Å] | c [Å] | [eV/Atom] |
|---|---|---|---|---|---|
| fixed | 2/3 | 7.13 | 7.13 | 12.10 | −0.337 |
| relaxed | 2/3 | 7.17 | 7.17 | 12.28 | −0.340 |
| fixed | 1 | 7.13 | 7.13 | 12.10 | −0.039 |
| relaxed | 1 | 7.18 | 7.18 | 13.70 | −0.206 |
| U2 relaxed | - | 6.57 | 7.99 | 4.09 | −0.212 |
| U2 fixed | - | 6.37 | 8.20 | 4.03 | −0.207 |
| Relation | a [Å] | a [Å] | a [Å] |
|---|---|---|---|
| 6.37 | 6.56 | 7.17 | |
| 14.25 | - | 14.34 |
| Decohesion Plane | [GPa] | ||
|---|---|---|---|
| [eV/Å] | |||
| Al (110) | 0.061 | 0.060 | 11.5 |
| Al (130) | 0.063 | 0.064 | 12.2 |
| U2 (130) | 0.046 | 0.048 | 12.6 |
| (110) | 0.070 | 0.072 | 13.7 |
| (130) | 0.062 | 0.072 | 12.1 |
| (110) A—Al/U2 | 0.045 | 0.043 | 9.4 |
| (110) B—U2 | 0.040 | 0.041 | 9.5 |
| (110) C— | 0.060 | 0.067 | 12.3 |
| (110) D—U2/Al | 0.046 | 0.046 | 11.1 |
| (130) A—Al/U2 | 0.058 | 0.055 | 11.5 |
| (130) B—U2 | 0.044 | 0.048 | 8.5 |
| (130) C— | 0.055 | 0.069 | 10.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ringdalen, I.G.; Jensen, I.J.T.; Marioara, C.D.; Friis, J. The Role of Grain Boundary Precipitates during Intergranular Fracture in 6xxx Series Aluminium Alloys. Metals 2021, 11, 894. https://doi.org/10.3390/met11060894
Ringdalen IG, Jensen IJT, Marioara CD, Friis J. The Role of Grain Boundary Precipitates during Intergranular Fracture in 6xxx Series Aluminium Alloys. Metals. 2021; 11(6):894. https://doi.org/10.3390/met11060894
Chicago/Turabian StyleRingdalen, Inga G., Ingvild J. T. Jensen, Calin D. Marioara, and Jesper Friis. 2021. "The Role of Grain Boundary Precipitates during Intergranular Fracture in 6xxx Series Aluminium Alloys" Metals 11, no. 6: 894. https://doi.org/10.3390/met11060894
APA StyleRingdalen, I. G., Jensen, I. J. T., Marioara, C. D., & Friis, J. (2021). The Role of Grain Boundary Precipitates during Intergranular Fracture in 6xxx Series Aluminium Alloys. Metals, 11(6), 894. https://doi.org/10.3390/met11060894