Abstract
The water gas shift (WGS) is an equilibrium exothermic reaction, whose corresponding industrial process is normally carried out in two adiabatic stages, to overcome the thermodynamic and kinetic limitations. The high temperature stage makes use of iron/chromium-based catalysts, while the low temperature stage employs copper/zinc-based catalysts. Nevertheless, both these systems have several problems, mainly dealing with safety issues and process efficiency. Accordingly, in the last decade abundant researches have been focused on the study of alternative catalytic systems. The best performances have been obtained with noble metal-based catalysts, among which, platinum-based formulations showed a good compromise between performance and ease of preparation. These catalytic systems are extremely attractive, as they have numerous advantages, including the feasibility of intermediate temperature (250–400 °C) applications, the absence of pyrophoricity, and the high activity even at low loadings. The particle size plays a crucial role in determining their catalytic activity, enhancing the performance of the nanometric catalytic systems: the best activity and stability was reported for particle sizes < 1.7 nm. Moreover the optimal Pt loading seems to be located near 1 wt%, as well as the optimal Pt coverage was identified in 0.25 ML. Kinetics and mechanisms studies highlighted the low energy activation of Pt/Mo2C-based catalytic systems (Ea of 38 kJ·mol−1), the associative mechanism is the most encountered on the investigated studies. This review focuses on a selection of recent published articles, related to the preparation and use of unstructured platinum-based catalysts in water gas shift reaction, and is organized in five main sections: comparative studies, kinetics, reaction mechanisms, sour WGS and electrochemical promotion. Each section is divided in paragraphs, at the end of the section a summary and a summary table are provided.
1. Introduction
Water-gas shift (WGS) is a mildly exothermic equilibrium reaction (ΔH0298K = −41.2 kJ·mol−1), which can be conducted both heterogeneously and homogeneously, although the homogeneous process is not of commercial interest [1]. The WGS is involved in the production of important chemicals and in numerous chemical transformation processes, such as production of ammonia [2,3] (Figure 1) and methanol [4] as well as hydrogen, the likely energy carrier of the future [5]. The WGS reaction, besides taking part in the syngas production processes, is normally used to reduce the CO concentration, increasing the hydrogen content or to tune the H2/CO ratio in the gas stream [6]. The major issue of the WGS process is the exothermicity of the reaction: as the reaction proceeds the temperature increases due to the heat of the reaction, but, at the same time, the equilibrium conversion decreases and therefore a thermodynamic limitation occurs. On the other hand, too low temperatures slow down the reaction kinetics and, in any case, the threshold temperatures of the catalysts play a fundamental role in the process parameters choice. The exothermic reactions are normally carried out in multistage processes [7], to achieve a compromise between acceptable reaction rates and good conversion values. In the case of WGS, two shift processes are enough to reach a CO concentration, in the gas stream, less than 0.2% [8]: the high temperature shift (HTS) operates in the temperature range 300–450 °C to take advantage of the high reaction rate, while the low temperature shift (LTS) operates in the temperature range 200–250 °C to reach higher conversions [9]; moreover, an intermediate cooling stage is also operated. The conventionally used catalysts in HTS processes are iron–chromium oxide based; until the 1980s the original unsupported formulation remained almost unvaried [10], with the most efforts focused on improving the synthesis.
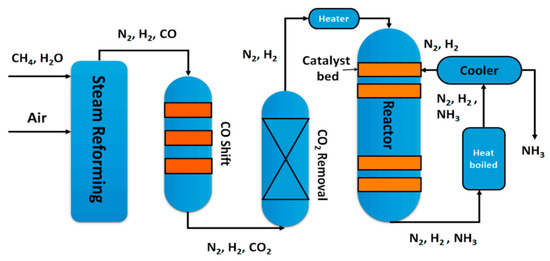
Figure 1.
A flow scheme of the Haber–Bosch process, reproduced from [3], with permission from MDPI, 2018.
Only later, the effect of promoters was investigated to increase the activity, and thermally stabilize the catalysts under WGS reaction conditions [11]. The conventionally used LTS catalysts are copper–zinc oxide based [12], usually Cu supported on alumina stabilized with zinc oxide promoter [13]. The use of these catalysts, despite the good performance in WGS reaction, presents a series of critical issues. In particular, HTS catalysts may suffer from overreduction [11], they are not active at low temperature and present toxicity problems due to the presence of Cr(VI) in the waste. On the other hand, LTS catalysts are pyrophoric and sensitive to the temperature, deactivating over 350 °C [14]; moreover, they are sulphur, halogen and unsaturated hydrocarbon intolerant [15]. In the last decades a growing interest has been focused on the use of noble metals-based catalysts, in particular to platinum supported on highly reducible oxides [16]. These catalysts are attractive for strategic reasons: the high activity in a wide range of temperature allows their use both in HTS and LTS processes; the non-pyrophoricity makes them suitable for oxidative start up and shut down cyclic operations; the high compatibility with conductive structures make them the ideal candidates for the design of structured reactors for a single-stage WGS process [17].
Several papers have been published on the use and the role of platinum as catalyst in the WGS reaction, focusing on the structural characteristics and morphology of the catalysts, on the chemical nature of the species and intermediates in the reaction mechanisms and on the effect of promoters in reaction conditions [18]. These works have involved both theoretical and experimental studies and have obtained a level of understanding that constitutes the basis for the design of highly selective catalysts and innovative reactor configurations. The studies on this kind of catalysts have highlighted the strong correlation between the reaction performances and the active metal particle size; the possibility of using increasingly powerful and faster computers, the recent advances in the sophisticated synthetic methodology and in atomic resolution microscopy, allowing the synthesis and study of nanoparticle and sub-nanoparticle catalytic based systems up to the single atom level [19]. Understanding the reaction mechanisms at the atomic level for sure provides fundamental information; however, most of the catalytic systems, in addition to the active metal, have a non-inert support; therefore the catalytic activity is strongly related to the metal/support interactions. For this reason, the design of innovative reactor configurations [20], as well as new catalytic formulations [21], cannot ignore the understanding of phenomena at the molecular level, as well as the roles that regulate the transition from microscopic to macroscopic phenomena.
It is also worthy to highlight the particular issue of the catalyst stability, which is a determinant factor in the WGS process. Together with an eventual poisoning effect due to the presence of sour gasses, such as H2S, in the process stream, most of the deactivation problems are mainly related to the cycling operation which the process requires. For this reason, an interesting topic in the research of new catalytic formulations is the catalyst stability under different reaction conditions and operating cycles. This aspect is further detailed in Section 5.
In this short review, the main results published in recent selected articles (from 2011 up to now) on the Pt-based heterogeneous unstructured catalysts for WGS reaction will be summarized; Pt-based structured catalysts have recently been the objective of a comprehensive review on structured catalysts for WGS reaction [22]. The review article has been divided in five main sections, while the articles have been distributed, considering the closest correspondence to the topic: comparative studies (monometallic, polymetallic and promoted catalysts), kinetics studies, reaction mechanism studies, deactivation studies and electrochemical promotion.
2. Comparative Studies
2.1. Monometallic Catalysts: The Supports
This section is devoted to the Pt-based monometallic catalysts literature survey, focusing on the effect of the support preparation on the WGS reaction. The section is divided into four paragraphs, the first (Section 2.1.1) is devoted to the effect of ceria support, the second (Section 2.1.2) to the effect of ceria-based solid solutions and mixed oxide support, the third (Section 2.1.3) to the effect of supports different from ceria, finally the forth (Section 2.1.4) to commercial supports; moreover a brief conclusion is also provided. At the end of the section Table 1 reports the performance of one selected catalyst per article.
2.1.1. Effect of the Preparation Method of the Ceria Support
Pastor-Perez et al. [23] compared the catalytic activity of three platinum-based catalysts obtained by impregnation of ceria support with three different techniques: urea homogeneous precipitation (Pt/CeO2-U), microwave-assisted hydrothermal synthesis (Pt/CeO2-MW) and polymer-assisted hydrothermal synthesis (Pt/CeO2-T). The catalytic tests were carried out by using two different feeding conditions: an ideal gas stream and a real gas stream. The Pt/CeO2-T catalyst showed the worst catalytic activity, due to the poorest reducibility and lowest Pt dispersion. Under ideal stream conditions, Pt/CeO2-U and Pt/CeO2-MW catalysts showed similar trend, while under realistic stream conditions the Pt/CeO2-U catalyst showed the best catalytic activity. TPR (temperature programmed reduction) and XPS (X-ray photoelectron spectroscopy) analyses showed that the ceria surface reduction, on Pt/CeO2-U catalyst, took place at a lower temperature; the enhanced redox properties were related to the electronics perturbations resulting from the strong metal-support interaction, which could explain the excellent catalytic activity. In addition, the smallest band gap was observed for the CeO2-U support, due to a higher concentration of Ce3+ ions and oxygen vacancies in the sample. Such structural defects play an important role in the catalytic activity, acting as adsorption sites for water activation and assisting the noble metal to complete the catalytic cycle. Furthermore, Pt/CeO2-U had the higher Pt/Ce ratio, corresponding to the best metallic dispersion, which positively affected the catalytic activity. In further studies, the effect of a cold Ar radio frequency (RF) plasma treatment on the catalytic activity of a Pt/CeO2 catalyst was evaluated [24]. The results of the activity tests showed that the plasma treatment alone did not improve the CO conversion, compared to the conventional reduction treatment, while higher catalytic activity was observed when both treatments were combined, particularly in the calcined sample. This result was attributed to the high electron density of Pt particles on the support and to the strong interaction between the Pt particles and the support, which took place at a lower temperature than the conventional Pt/CeO2 catalyst, as well to the increased metal-support interaction obtained.
Palma et al. [25] investigated the WGS activity of a platinum-based catalyst supported on nanocrystalline ceria (PtCeSAS), prepared through the CO2 supercritical antisolvent process (SAS) using cerium acetylacetonate (Ce(acac)3) as precursor. The PtCeSAS catalyst showed higher CO conversion than the PtCeComm catalyst, prepared by impregnation of commercial ceria, in all the investigated temperature range. The better performance of PtCeSAS was ascribed to the higher specific surface area and to the smaller crystallite size, confirming the beneficial role of the SAS process. In further studies, the superior activity of platinum-based catalysts supported on nano-ceria prepared by the SAS process (Pt/CeO2_S), with respect the platinum-based catalysts supported on commercial nanoceria (Pt/CeO2_A), was attributed to the higher reducibility of the Pt/CeO2_S catalyst compared to Pt/CeO2_A [26]. The Raman spectra and TEM (transmission electron microscopy) images (Figure 2) demonstrated a much higher presence of the oxygen vacancies in the CeO2_S support, suggesting high distortions of the lattice and consequently, higher oxygen storage capacity, directly involved in the reaction mechanism.

Figure 2.
TEM images of Pt/CeO2_S and Pt/CeO2_A samples, reproduced from [26], with permission from Elsevier, 2018.
Potdar et al. [27] developed a nano-sized Pt/CeO2 catalyst, in which the support was obtained by a precipitation/digestion technique, using different digestion times (2, 4, 8 h). The 1wt%Pt/CeO2 catalyst, obtained by depositing Pt on the 4 h digested-ceria, approached CO equilibrium conversion at the reaction temperature of 320 °C and showed in general higher conversion values than the catalyst obtained with the ceria without the digestion phase in the whole temperature range. This result highlighted the beneficial effect of the digestion phase, controlling both the BET (Brunauer–Emmett–Teller) surface area of the pre-calcined CeO2 support and the activity of the same Pt/CeO2 catalyst. Roh et al. [28] studied, in time-on stream tests, the catalyst obtained with 4 h digested-ceria, showing a slightly decrease of the CO conversion during the reaction time. The deactivation was ascribed to the Pt sintering and to the carbonate species formation, which blocked the active sites. The high activity and stability of this catalyst was attributed to the weakening of the superficial Ce-O bond by the nano-sized Pt species, thus releasing reactive and available oxygen for the oxidation of CO to CO2, with a simultaneous creation of oxygen vacancies on CeO2 supports re-filled by H2O.
Im et al. [29] compared nanorod and particle shape CeO2 as a support of platinum-based catalysts. The supports were synthesized through hydrothermal process (for nanorod support) and through precipitation method (for particle shape support). Three different reaction times (12, 48, and 96 h) for hydrothermal preparation ceria nanorods were applied, obtaining some differences in physical characteristics, such as aspect ratio, BET surface area, pore diameter and pore volume. The Pt/CeO2 catalyst, obtained by loading the ceria, synthesized with 12 h of hydrothermal process time, showed the highest catalytic activity, due to the high concentration of Ce3+ and to the strong interaction between Pt and Ce. Furthermore, the reduced pores size hindered to internal diffusion, with a consequent negative effect on the catalytic activity.
Mei et al. [30] investigated nanometric ceria with different shapes (rod, cube, polyhedron), to evaluate the effect of preparation method and a sodium cyanide (NaCN) leaching on the catalytic activity. Platinum loading was performed in three different ways: deposition/precipitation method with (NH4)2CO3, deposition/precipitation method with NH4OH and impregnation method. The results demonstrated that the catalyst obtained by loading the ceria nanorod had the highest CO conversion (Figure 3) when the preparation method was deposition/precipitation with NH4OH or impregnation, while in the case of the catalyst obtained by loading the polyhedron ceria, the highest CO conversion was obtained when the preparation method was deposition/precipitation with (NH4)2CO3. The characterization results suggested a higher Pt dispersion and stabilization on ceria nanorods than on the other kind of support, probably due to a better anchoring of the fine Pt clusters. The leaching with NaCN of the catalysts prepared by impregnation method resulted in the increase of the CO conversions at low temperature, due to the strengthening of the Pt-ceria interaction, with the rod- ceria based catalyst showing the highest catalytic activity.
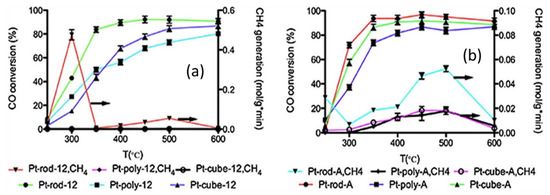
Figure 3.
CO conversion of the catalysts prepared by (a) impregnation method and (b) (NH4)2CO3 deposition–precipitation (DP) method; reaction conditions: GHSV = 460,000 h−1; feeding = 24.7 vol.% CO, 15.91 vol.% N2, 59.37 vol.% H2O, adapted from [30], with permission from publisher Elsevier, 2015.
Tang et al. [31] investigated the use of Pt/CeO2 nanofibers, with a diameter of 80–120 nm, obtained by electrospinning method; after calcining the nanofibers at 400 °C, 5–10 nm particles were obtained (Figure 4). The activity of the Pt/CeO2 nanofiber catalyst was compared with that of CeO2 nanofibers, with that of Pt/CeO2 nanofiber prepared without the use of electrospinning and with that of Pt/CeO2 powder. The results showed superior catalytic activity of the Pt-CeO2 nanofiber at low temperatures.

Figure 4.
SEM (a–d) and HR-TEM images (e,f) of CeO2 (a,b) and Pt/CeO2 (c–f) nanofibers, adapted from [31], with permission from Elsevier, 2012.
2.1.2. Effect of the Preparation Method of the Ceria-Based Solid Solutions and Mixed Oxide Support
Jeong et al. [32] carried out a comparative study on nano-sized Pt/Ce0.8Zr0.2O2 and Pt/Ce0.2Zr0.8O2 catalysts to evaluate the effect of ceria cubic structure and zirconia tetragonal structure. The nano-sized supports were prepared by one-step co-precipitation/digestion method, while platinum was loaded by incipient wetness impregnation method. Higher CO conversion value was obtained with the Pt/Ce0.8Zr0.2O2 catalyst, due to higher Pt dispersion, easier reducibility and higher OSC (oxygen storage capacity) of the cubic Ce0.8Zr0.2O2 support. Palma et al. [33] compared the performance of platinum-based catalysts supported on commercial CeO2 and CeO2/ZrO2 and on a prepared CeO2/γ-Al2O3 oxide (14% w/w of Ce). The Pt/CeO2/ZrO2 catalyst showed better activity and selectivity than the other catalysts for all the investigated temperatures; the stability test showed a deactivation in the first 40 h of reaction, attributed to sintering phenomena, and a stable trend in the further 40 h of reaction.
Castano et al. [34] compared the catalytic behavior of gold and platinum-based catalysts supported on commercial γ-alumina, CeO2/Al2O3 and Ce0.8Fe0.2/Al2O3 prepared by incipient wetness impregnation. The results showed a stronger influence of the support nature in the Au-based catalysts than in the Pt ones; the authors ascribed this effect to the water dissociation, which might take place only on the support for Au catalysts, while it can occur both on the support and on the metal particle in the case of Pt catalysts.
Zhang et al. [35] investigated the effect of a dopamine treatment on the catalyst preparation. The support was obtained by wet impregnation while the Pt/Ce0.4Ti0.6O2 catalyst was immersed in a dopamine solution, then heated at 700 °C in N2 for 2 h, and finally calcined at 500 °C for further 2 h (Figure 5). The activity tests were carried out on both the treated catalyst and the untreated one, showing that below 250 and above 350 °C the CO conversion values were almost similar, while in this temperature range, the treated catalyst showed better catalytic activity. The stability tests evidenced that the untreated catalyst deactivated during 12 h of reaction, while the treated catalyst did not show any deactivation, due to its strong resistance to Pt nanoparticles sintering, also due to the thermal treatment in N2 at 700 °C, which leaded to a Pt nanoparticles structure re-arrangement.
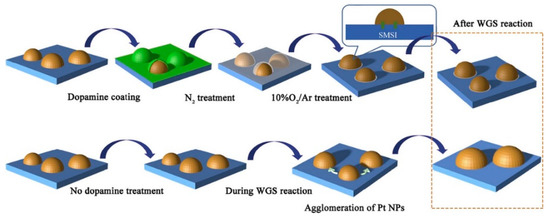
Figure 5.
Schematic representation for the synthesis of doped and undoped of platinum-based catalysts supported on ceria-titania, reproduced from [35], with permission from Elsevier, 2019.
Wan et al. [36] compared the activity of a core-shell Pt/CeO2 nanotube catalyst encapsulated in porous SiO2 (Pt/CeO2 nanotube@ SiO2), with a Pt/CeO2 catalyst, obtained by loading commercial ceria, in time on stream tests. The CeO2 nanotubes were synthesized by hydrothermal method, the platinum was loaded by deposition method, while the encapsulation was realized by using a modified Stöber method [37]. The Pt/CeO2 nanotube@ SiO2 catalyst showed higher activity than the other catalysts due to the high surface area, which increases the contact interface between the metal nanoparticles and transition-metal oxide supports, moreover after the aging time a serious deactivation of the not SiO2 encapsulated catalysts occurred. This result highlighted the ability of SiO2 encapsulation to prevent the Pt nanoparticle sintering. Roh et al. [38] compared the activity of Pt-based catalysts, prepared by incipient wetness impregnation of CeO2 and ZrO2 supports obtained by precipitation method, MgO prepared by calcination of magnesium nitrate at 500 °C, and commercial Al2O3 and MgO-Al2O3 (MgO = 30 wt%). The results evidenced the dependency of the catalytic activity from the catalyst reduction property and the platinum dispersion; the Pt/CeO2 catalyst showed the highest CO conversion and hydrogen selectivity in the whole investigated temperature range, due to the better reducibility of Pt/CeO2 and, above all, to the higher oxygen storage capacity of CeO2.
2.1.3. Effect of Preparation Method of Supports Different from Ceria
Yati et al. [39] compared the WGS activity of Pt@Al2O3 nanorod catalyst, obtained by NaBH4 reduction of trimethyl(tetra-decyl) ammonium bromide (TTAB)–stabilized Pt NPs precursor solution, on polymerized alumina, with that of Pt/Al2O3-A and Pt/Al2O3-S catalysts, obtained by impregnation aerogel alumina and sol-gel alumina respectively, with the TTAB-stabilized solution. The Pt@Al2O3 catalyst showed higher CO conversion than the other ones in all the studied conditions. The time on stream tests highlighted the higher stability of Pt@Al2O3 catalyst compared to the other catalysts, which showed a much faster deactivation due to the sintering phenomena. The TEM images of Pt@Al2O3 suggested a stabilizing effect of the tiny alumina nanorods which surrounded the Pt nanoparticles, producing an unusually larger metal-support interface compared to Pt supported on the bulk alumina surface.
Subramanian et al. [40] investigated the use of nano-sized Pt/ZnO catalyst in the WGS reaction, by using of two different ZnO support, spherical (ZRT) and rod type (Z70), obtained by precipitation method at ambient temperature and 70 °C respectively. The ZRT type catalysts showed higher TOF and reaction rate; the best performance was attributed to a better interfacial contact between Pt and ZnO. Martinelli et al. [41] investigated the effect of yttrium in 0.5wt%Pt/Zr(1−x)Y(x)Oδ (x = 0.1, 0.25, 0.5, 0.75, 0.9) catalysts for the LTS. The supports were prepared by homogeneous precipitation of zirconia and yttrium nitrate using sodium hydroxide as the precipitating agent. The catalytic activity of the mixed oxide-based catalyst was compared to those of 0.5wt%Pt/ZrO2 and 0.5wt%Pt/Y2O3.
The yttria supported Pt catalyst showed the worst performance, while the best performance was obtained with 0.5%Pt/Zr0.9Y0.1O1.95 catalyst, which had the higher concentration of surface defects, as confirmed by DRIFT of adsorbed CO, and a higher surface formates mobility, as suggested by the formates decomposition experiments. The presence of yttrium deformed the zirconia lattice, improving the oxygen mobility, the high number of defects in the crystal structures of the mixed oxide system increased the amount of bridging OH groups or oxygen vacancy sites on the surface of the support, thus increasing the active sites for associative or redox reaction mechanisms.
Kwon et al. [42] investigated the use of 1wt%Pt/TiO2/PRGO catalysts (PRGO = partially reduced graphite oxide), with different TiO2 content. The Pt/TirGO-n catalysts were prepared by adding the appropriate amount of Ti(OnBu)4 (n = 1, 3, 5, 7 mL) to PRGO followed by impregnation with the platinum precursor, moreover, for comparison, Pt/TiO2 was also prepared without adding PRGO. The activity tests results demonstrated that the incorporation of PRGO in the TiO2 support improved the catalytic activity; moreover, the best catalytic activity was provided by Pt/TirGO-5 (Figure 6). The functional groups on the PRGO surface behaved as anchors, strongly binding the titanium ions; an optimal TiO2/PRGO ratio induced a uniform dispersion of nanosized TiO2 on the PRGO surface, by balancing the number of functional groups, moreover, it modulated the number of surface PRGO defects, maximizing the reactivity of the functional groups with the interacting TiO2 precursor.
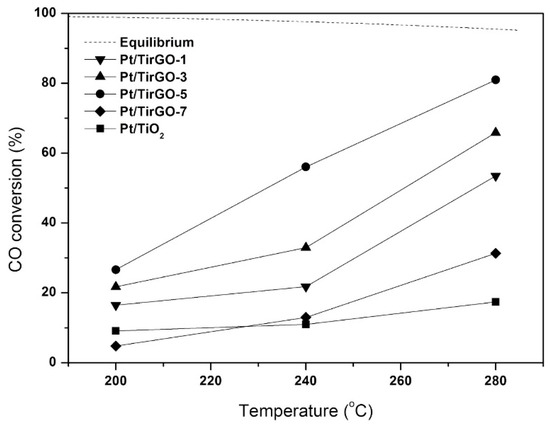
Figure 6.
CO conversions in water gas shift (WGS) reaction with Pt/TirGO catalysts and Pt/TiO2 (S/C ratio = 3.3, GHSV = 47,700 h−1), reproduced from [42], with permission from Elsevier, 2017.
2.1.4. Commercial Supports
Franchini et al. [43] studied the influence of zirconia polymorphism in Pt/ZrO2 catalyst for the WGS reaction, by using monoclinic zirconia (m-ZrO2) and tetragonal zirconia (t-ZrO2). The activity tests highlighted the higher performance, in terms of CO conversion, of the Pt/m-ZrO2 catalyst, due to the higher concentration of coordinatively unsaturated sites on the monoclinic structure which allowed a more extensive CO adsorption on the support surface. In addition, the catalysts deactivation was also investigated through the analysis of metal dispersion after the WGS reaction evaluating the cyclohexane dehydrogenation rate. The results showed a gradual drop in platinum dispersion for both catalysts, with a stronger reduction in the Pt/m-ZrO2 catalyst, due to sintering phenomena.
2.1.5. Conclusions
Summarizing, the supports play a crucial role in regulating the catalytic activity; the particle size and the porosimetric characteristics of the support can modulate the dispersion of the active phases, furthermore the interactions between active phases and support can modify the electronic characteristics of the catalytic systems, regulating both the activity and the selectivity. The platinum-based catalysts supported on reducible oxides, such as ceria and ceria/zirconia, certainly represent a significant example, where the redox properties of the supports can tune the catalytic activity and determining the reaction mechanism. The use of specific shapes such as nanorods and nanofibers can provide better performance, in preventing the particle sintering and enhancing the catalytic activity.

Table 1.
Selected catalysts per article reported in the Section 2.1: Monometallic Catalysts: the supports.
Table 1.
Selected catalysts per article reported in the Section 2.1: Monometallic Catalysts: the supports.
| Selected Catalyst (Particle/Crystallite Size) | Preparation Procedure | Operative Condition WGS | CO Conversion (XCO) (Temperature) | Ref. |
|---|---|---|---|---|
| 1wt%Pt/CeO2 (12 nm) | Pt loading by wet impregnation; CeO2 by microwave-assisted hydrothermal synthesis | WHSV = 40,000 mL·g−1·h−1; H2O/CO = 6 | XCO ≈ 97% (T = 360 °C) | [23] |
| 1wt%Pt/CeO2 (not specified) | Wet impregnation + Ar plasma treatment | WHSV = 40,000 mL·g−1·h−1; CO/H2O = 6 | XCO ≈ 97% (T = 280 °C) | [24] |
| 1wt%Pt/CeO2 (14.0 nm) | Pt loading by wet impregnation; CeO2 by supercritical antisolvent process | GHSV = 5000 h−1; H2O/CO = 3 | XCO ≈ 99% (T = 287 °C) | [25] |
| 1wt%Pt/CeO2 (5.8 nm) | Pt loading by wet impregnation; CeO2 by supercritical antisolvent process | WHSV = 1.13 gCO·gcat−1·h−1; H2O/CO = 3 | XCO ≈ 98% (T = 280 °C) | [26] |
| 1wt%Pt/CeO2 (7.9 nm) | Incipient wetness impregnation | GHSV = 45,625 h−1; H2O/(CH4 + CO + CO2) = 2 | XCO ≈ 87% (T = 320 °C) | [27] |
| 1wt%Pt/CeO2 (1.4 nm) | Incipient wetness impregnation | GHSV = 45,625 h−1; H2O/(CH4 + CO + CO2) = 2 | XCO ≈ 82% (T = 320 °C) | [28] |
| 1wt%Pt/CeO2 nanorods (1.6 nm) | Pt loading by incipient wetness impregnation; CeO2 by hydrothermal process for 12 h | GHSV = 95,541 h−1; H2O/(CH4 + CO + CO2) = 2 | XCO ≈ 77% (T = 360 °C) | [29] |
| 3wt%Pt/CeO2-nanorod (<2 nm) | Pt by impregnation method | GHSV = 4.6 × 105 h−1; H2O/CO = 2.4 | XCO ≈ 92% (T = 600 °C) | [30] |
| 1wt%Pt/CeO2 nanofibers (4.5 nm) (dnanofibers = 80–120 nm) | Electrospinning technology | WHSV = 60,000 mL·gcat−1·h−1; H2O/CO = 5.3 | XCO ≈ 98% (T = 350 °C) | [31] |
| 1wt% Pt/Ce0.8Zr0.2O2 (1.69 nm) | Incipient wetness impregnation | GHSV = 45,515 h−1; H2O/(CH4 + CO + CO2) = 2 | XCO ≈ 86% (T = 320 °C) | [32] |
| 1Wt%Pt/CeO2/ZrO2 (7.22 nm) | Wet impregnation | GHSV = 5000 h−1; H2O/CO = 5 | Equilibrium CO conversion at 200 °C | [33] |
| 2wt%Pt/Ce0.8Fe0.2/Al2O3 (2.2 nm) | Wet impregnation | GHSV = 4000 h−1; H2O/CO = 6.9 | Equilibrium CO conversion at 280 °C | [34] |
| 1.63wt%Pt/Ce0.4Ti0.6O2 (7.25 nm) | Wet impregnation | GHSV = 3600 h−1; H2O/(CO + CO2) = 4.8 | XCO ≈ 91% (T = 400 °C) | [35] |
| 0.9wt%Pt/CeO2@SiO2-nanotube (3.1 nm) | CeO2 by hydrothermal synthesis method SiO2 shell by modified Stober method | WHSV = 36,000 mL·gcat−1·h−1; H2O/CO = 3 | XCO ≈ 30% (T = 250 °C) | [36] |
| 1wt%Pt/CeO2 (3 nm) | Incipient wetness impregnation | GHSV = 45,515 h−1; H2O/(CH4 + CO + CO2) = 2 | XCO ≈ 88% (T = 320 °C) | [38] |
| 1wt%Pt@Al2O3-nanorods (10.4 nm) | Pt loading by NaBH4 reduction Al2O3 by polymerization | GHSV = 22,500 h−1; H2O/CO = 2 | XCO ≈ 96% (T = 450 °C) | [39] |
| 1Wt%Pt/sZnOspherical morphology (1.5 nm) | Incipient wetness impregnation | GHSV = 9583 h−1; H2O/(CH4 + CO + CO2) = 2 | XCO ≈ 92% (T = 240 °C) | [40] |
| 0.5wt%Pt/Zr0.9Y0.1O1.95 (0.5–2 nm) | Incipient wetness impregnation | GHSV = 120,220 h−1; H2O/CO = 8.7 | XCO ≈ 74% (T = 300 °C) | [41] |
| 1wt%Pt/TiO2/PRGO-5 (11.3 nm) | Incipient wetness impregnation | GHSV = 47,770 h−1; H2O/(CH4 + CO + CO2) = 3.3 | XCO ≈ 81% (T = 280 °C) | [42] |
| 1wt%Pt/ZrO2-monoclinic (10.1 nm) | Incipient wetness impregnation | WHSV = 43,200 mL·gcat−1·h−1; H2O/(CO + CO2) = 3.2 | XCO ≈ 65% (T = 300 °C) | [43] |
2.2. Monometallic Catalysts: The Active Phase
This section is devoted to the Pt-based monometallic catalysts for the WGS reaction, focusing on the effect of the active phase. The preparation method is reviewed in Section 2.2.1; the effect of the platinum loading is reviewed in Section 2.2.2; the comparative studies between platinum and rhodium are reviewed in Section 2.2.3; finally a brief conclusion is also provided. At the end of the section Table 2 reports the performance of one selected catalyst per article.
2.2.1. Effect of the Preparation Method
Jain et al. [44] investigated the use of reactive spray deposition technology (RSDT) in the preparation of nano Pt/CeO2 catalysts. The catalytic activity was compared to those reported in literature for Pt/CeO2 catalysts prepared with conventional processes (sol-gel, co-precipitation, and incipient wetness impregnation), under the same operating conditions. The results highlighted the superior activity of the catalyst prepared by RSDT, due to the uniform distribution of Pt nanoparticles on the ceria surface and to the non-agglomeration between the particles. Lu et al. [45] investigated the effect of using a photochemical method, through UV irradiation in the range 200–400 nm generated by a 500 W quartz mercury vapor arc lamp, in preparing three catalysts. The Pta/CeO2 catalyst was synthesized by irradiation of the precursor suspension, the Ptb/CeO2 was obtained by adding poly(vinylpyrrolidone) to the precursor suspension, while the Ptc/CeO2 was obtained by adding poly(vinylpyrrolidone) and 4-benzyolbenzoic to the precursor suspension. The latter catalyst showed both the best catalytic activity, attributable to the smaller particle Pt size (1.7 nm) and to the uniform distribution on the surface of the porous CeO2 nanofibers, and the best stability (no notable deactivation over 12 h of reaction).
Rajesh et al. [46] investigated the effect of the incorporation of platinum, in cationic form, into the stable perovskite lattice BaCeO3. Three types of catalysts were prepared, of general formula BaCe1−xPtxO3−δ, (x = 2, 4 and 6 mol%) by the citrate-gel method. The catalytic activity and the evolution of active species were studied in two reaction cycles, discovering that the catalytic activities in the second cycle were much higher than those in the first cycle; the authors attributed this behavior to the enrichment of the cationic platinum on the surface, since it was observed that the overall Pt surface concentration increased after the first cycle, with a prevalence of Pt(IV), and after the second cycle, the amount of Pt(II) species on the surface increases at the expense of Pt(IV).
2.2.2. Effect of the Platinum Loading
Tiwari et al. [47] studied the effect of loading and particles size in platinum-based catalysts supported on nanocrystalline ceria. Three catalysts, Xwt%Pt/CeO2 (X = 0.9, 1.9, 4.7), were prepared by hydrothermal method, with Pt particle sizes equal to 0.8, 3.7, 7.8 nm respectively. The stability of the catalyst was strongly dependent from the platinum loading and reaction temperature; the 0.9wt%Pt/CeO2 catalyst was stable during 30 h of reaction at 140 °C, on the contrary the CO conversion decreased with the time for 1.9wt%Pt-CeO2 and 4.7wt%Pt-CeO2 at the same reaction conditions. On the other hand, the 0.9wt%Pt-CeO2 catalyst showed a decrease in the CO conversion with time at 180 °C, that the authors related to the Pt2+ reduction to metallic Pt, which occurred at 151 °C. The XPS studies, in fact, revealed that the fresh catalyst contained Pt2+ as the active specie for the WGS reaction.
Shim et al. [48] investigated Pt-loaded Ce0.75Zr0.25O2 yolk−shell microspheres (Figure 7). The catalysts were prepared by a spray pyrolysis method with different platinum loadings (0.5 wt%, 2 wt% and 4 wt%). The 2wt%Pt/Ce0.75Zr0.25O2 catalyst was the most active in WGS reaction, a further increase of the Pt loading was detrimental. The time on stream tests, performed at 320 °C for in a 20 h of reaction, highlighted the excellent stability of all of the yolk-shell catalysts, demonstrating the ability of the Ce0.75Zr0.25O2 yolk-shell-structure to prevent the Pt sintering, thereby maintaining a high catalyst activity.
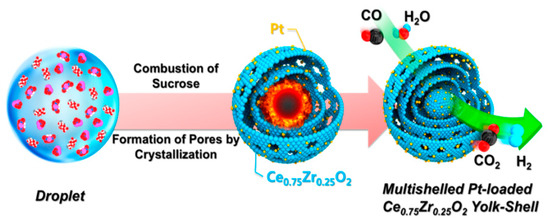
Figure 7.
Schematic diagram of the formation mechanism of Pt-doped Ce0.75Zr0.25O2 powders with yolk−shell structure, reproduced from [48], with permission from American Chemical Society, 2016.
Cornaglia et al. [49] studied the platinum loading on Xwt%Pt/La2O3·SiO2 (X = 0.02, 0.1, 0.2, 0.6, 1.2%). All the Pt catalysts showed good stability, but the 0.6 and 0.1 wt% Pt-based formulations showed superior stability even under severe conditions of start-up and shut-down cycles. The 1wt%Pt/La2O3·SiO2 catalyst showed the highest activity per gram of platinum, due to the complete reduction of surface metallic platinum; in fact, for higher loadings, the surface platinum was incompletely reduced at the same temperature, negatively effecting the catalytic activity. The 0.1wt%Pt/La2O3·SiO2 catalyst was also tested as a catalyst in a Pd-Ag membrane reactor (Pd/Ag ratio 77/23), obtaining a CO conversion higher than those of equilibrium obtainable in a conventional reactor. On the contrary, an increase in the GHSV significantly dropped the hydrogen recovery, as the reactants residence time in the reactor decreased.
The effect of the platinum loading on Pt(x)@TiO2 (x = 1.0, 1.2, 1.7, 2.2, 2.6, 2.6 nm) yolk/shell nanosphere was also investigated by Zhao et al. [50]. The yolk/shell nanostructure allowed sufficient exposure of the active sites to the reactants because of the presence of void spaces in the shell, maintaining at the same time the protective effect against the Pt nanoparticles sintering. Furthermore, the particle size had a significant influence on the catalytic activity; the Pt(1.0)@TiO2 catalyst showed the highest CO conversion values among the tested catalysts, reaching the thermodynamic equilibrium at a lower temperature than the other catalysts. The Pt(1.0)@TiO2 catalyst showed a much lower non-reactive metallic Pt fraction, confirming an increased average oxidation valence and hence more electron-deficient metal centers.
Galeano et al. [51] investigated the effect of the active metal loading in Pt encapsulated NaA zeolite. The catalysts were prepared by hydrothermal method, using two heating sources, conventional Pt(x)_NaA_CH (x = 2.2, 1.6, 0.6, 0.2 wt%) and microwave radiation (Pt(0.5)_NaA_MW). The microwave method allowed to significantly reduce the synthesis time. For comparison, a catalysts prepared by ionic exchange was also prepared (Pt(0.6)_NaA_IEx). The Pt(2.2)_NaA_CH catalyst showed the highest CO conversions at both the studied temperatures, moreover, the time on stream tests, carried out on the Pt(0.6)_NaA_CH and Pt(0.6)_NaA_MW catalysts, highlighted the excellent stability provided by the encapsulation method, which limited the sintering phenomena.
2.2.3. Comparative Studies between Pt and Rh
Roberts et al. [52] performed a study on the long-term stability tests (i.e., >600 h) on platinum and rhodium-based catalysts, supported both on titania and on ceria, using a typical reformate fuel processing stream. Both Pt-based and Rh-based catalysts were prepared by wet impregnation technique using the Chemspeed Isynth robot [53]. All the catalysts deactivated overtime, however Pt/CeO2 exhibited the best performance; the Rh/TiO2 catalyst showed a significant deactivation after 500 h of use, partially due to the increase in space velocity, the Pt/TiO2 catalyst showed very low catalytic activity, probably due to the support redox properties.
Cavusoglu et al. [54] compared the activity of Xwt%Pt/CeO2 and Ywt%Rh/CeO2 in HTS reaction, at different metal loading (Y = 3.1 and 1.9, X = 3.7 and 1.7). The catalysts were prepared by flames spray pyrolysis, for comparison a 0.74wt%Pt/CeO2 catalyst was prepared by incipient wetness impregnation method. The Pt-based catalysts showed higher activity than the Rh-based catalysts, moreover, the Pt-based flame made catalysts showed higher activity than the Pt/CeO2 catalyst prepared by impregnation. Summarizing Pt-based catalysts were more active than the Rh-based samples, moreover the latter showed methane formation starting from 300 °C.
The use of Rh/La2O3·SiO2 and Pt/La2O3·SiO2 catalysts in a membrane reactor for the WGS reaction, evaluating the influence of the main operating and design variables on the CO conversion and H2 recovery, was investigated by Cornaglia et al. [55]. The catalytic behavior was preliminary studied in a conventional fixed bed reactor, showing similar catalytic activity, however Pt/La2O3·SiO2 provided a higher hydrogen selectivity. Subsequently the activity tests were carried out at 400 °C, under atmospheric pressure, in a double tubular palladium membrane reactor. The time on stream tests highlighted the excellent stability of the catalysts, no significant deactivation occurred in 100 h of reaction. The effect of the H2O/CO ratio was also investigated employing a sweep gas flow rate, showing an increase in H2 recovery with the decrease of the H2O/CO ratio, due to the H2 partial pressure increase.
2.2.4. Conclusions
Summarizing, the catalytic activity is strongly related to the active phase loading, the best performance was reported for Pt loading ≈ 1 wt%, moreover an enhanced stability can be reached by incorporating the active phase, the best stability was reported for particle sizes < 1 nm. Finally, the comparative studies between Pt and Rh demonstrate a superior performance of the Pt-based catalysts, both in terms of CO conversion and hydrogen yield.
Table 2.
Selected catalysts per article reported in Section 2.2: Monometallic Catalysts: the Active Phase.
Table 2.
Selected catalysts per article reported in Section 2.2: Monometallic Catalysts: the Active Phase.
| Selected Catalyst (Particle/Crystallite Size) | Preparation Procedure | Operative Condition WGS | CO Conversion/Rate (XCO)/(rCO) (Temperature) | Ref. |
|---|---|---|---|---|
| 1wt%Pt/CeO2 (1.5–2 nm) | Reactive Spray Deposition Technology | GHSV = 8622 h−1; H2O/CO = 3 | Equilibrium CO (T = 350 °C) | [44] |
| 3wt%Pt/CeO2 (1.7 nm) | Pt loading by photochemical method adding PVP and 4-benzyolbenzoic acid; CeO2 by co-electrospinning | WHSV = 1.2 × 105 mL·g−1·h−1; H2O/CO = 5 | XCO ≈ 95% (T = 450 °C) | [45] |
| BaCe0.96Pt0.04O(3-δ) (<100 nm) | Citrate-gel method | GHSV = 5000 h−1; H2O/CO = 4.5 | XCO ≈ 86% (T = 400 °C) | [46] |
| 0.9wt%Pt/CeO2 (0.8 nm) | Hydrothermal method | GHSV = 5000 h−1; H2O/CO = 1 | XCO ≈ 97% (T = 140 °C) | [47] |
| 2wt%Pt/Ce0.75Zr0.25O2 (7.45 nm) | Yolk−shell microspheres formation by a spray pyrolysis process | GHSV = 18,193 h−1; H2O/(CH4 + CO + CO2) = 2 | XCO ≈ 89% (T = 320 °C) | [48] |
| 1.2wt%Pt/La2O3·SiO2 | Incipient wetness impregnation | GHSV = 2.8 × 106 h−1; H2O/CO = 3 | rCO = 350 mol·g−1·min−1 (T = 400 °C) | [49] |
| Pt@TiO2 (1.0 nm) | Yolk-shell nanospheres by a reverse micelle system | WHSV = 40,000 mL·gcat−1·h−1; H2O/CO = 5 | XCO ≈ 99%, T = 260 °C) | [50] |
| 2.2wt%Pt_NaA (not specified) | NaA zeolite by hydrothermal syntesis with conventional heating; Pt loading by encapsulation | GHSV = 6421 h−1; H2O/CO = 2 | XCO ≈ 96% (T = 400 °C) | [51] |
| 0.5wt%Pt/CeO2 (1.3 nm) | By wet impregnation | H2O/(CO + CO2) = 2.5 | XCO ≈ 45% (T = 275 °C) | [52] |
| 3.7wt% Pt/CeO2 (2.3 nm) | Flamespray pyrolisis method | WHSV = 5 × 104 mL·g−1·h−1; H2O/CO = 4 | XCO ≈ 97% (T = 250 °C) | [54] |
| 0.6wt%Pt/La2O3·SiO2 (not specified) | By Incipient wetness impregnation | WHSV = 6–24 × 103 mL·g−1·h−1; H2O/CO = 3 | XCO ≈ 95% (T = 400 °C) | [55] |
2.3. Polymetallic Catalysts and Addition of Promoters: The Active Phase
The addition of a second metal or a promoter is a widely used strategy to improve the performance of the catalysts, both in terms of catalytic activity and selectivity. In this section, the effect of the addition of promoters to the active species and to the support is reviewed, and the description is divided into three different paragraphs: Section 2.3.1 (the addition of Na, Re, Mo, V and Ni), Section 2.3.2 (comparative studies between multiple metals) and Section 2.3.3 (the addition of promoters to the support). Two tables are provided, Table 3 and Table 4, reporting the performance of one catalyst per reviewed article, moreover, at the end of the section a brief summary is also provided.
2.3.1. The Addition of Na, Re, Mo, V and Ni
Jeong et al. [56] investigated the addition of Na to Pt/CeO2 catalysts, by varying the Na amounts in the range 1–3 wt%. The best performance, in terms of catalytic activity and stability, at a temperature of 240 °C, were obtained with the 1wt%Pt-2wt%Na/CeO2 catalyst. The authors attributed this better behavior to the easier reducibility and higher oxygen storage capacity (OSC) of the catalyst.
Azzam et al. [57] investigated the influence of preparation strategies, the Pt/Re molar ratios, and the metals contents on the catalytic activity and stability of Pt–Re/TiO2 catalysts. The results indicated that the most significant effect of Re on the catalyst performance was obtained if the Re precursor was impregnated prior to the Pt precursor without intermediate drying and calcination, if a Pt/Re molar ratio of unity was used, and if a 0.5 wt% content of each metal was applied. The authors, by means of H2-TPR and in situ FTIR spectroscopy, deeply investigating the catalyst behavior, and they reported that Pt and Pt–Re catalysts exhibited identical CO vibration peaks, indicating that the Re did not interact with the Pt particles under WGS reaction conditions. However, similar experiments on reduced catalysts in the presence of CO showed that Re was partially reduced and interacted significantly with CO, thus enhancing the water activation, since the latter was able to oxidize metallic Re under WGS reaction conditions, resulting in new types of OH groups.
Ciftci et al. [58] studied the activity of Re addition to carbon-supported Pt catalysts in aqueous-phase reforming (APR) of glycerol and the WGS reaction, and the results were compared with the ones of carbon-supported alone Pt and Re. The experimental tests showed that the Re addition resulted in a higher conversion, due to the increase of the dehydration (C–O cleavage) reactions with respect to the decarbonylation (C–C cleavage) ones. This preference was attributed to the presence of acidic sites generated in the steam treated PtRe alloys.
Sener et al. [59] tested at 270 °C PtMo bimetallic catalysts supported on carbon and silica. The carbon supported catalysts, in which the Pt nanoparticles were saturated with Mo species at a Mo:Pt atomic ratio of 0.32, showed a strong promotional effect of Mo, which increased the TOF more than 2000 folds. On the contrary, the silica-supported catalysts, even if more active, showed a smaller promotional effect of Mo. This higher promoting effect in the carbon-supported catalysts was attributed to the formation of active sites for WGS at the interface between Pt atoms and Mo moieties that were possibly in an oxidized form.
The addition of Mo was also investigated by Osman et al. [60], in Pt and Cu supported on Mo2C/η-Al2O3, Mo2C/γ-Al2O3 or Mo2C catalysts. The activity tests results showed the superior activity of 4wt%Pt/Mo2C/η-Al2O3 catalyst, highlighting the synergistic effect between the two most active phases of Pt metal and Mo2C.
Kokumai et al. [61] studied the effect of vanadium addition to Pt/Al2O3 catalysts for the WGS reaction. The results showed an enhance catalyst activity, however, the increase of V content did not clearly affect the WGS activity, which might be related to the formation of polymeric VOx species interacting with alumina surface instead of new interfacial Pt-VOx sites that would favour the reaction. The in-situ characterization of the catalysts showed that metallic Pt and V3+/V4+ species were present during WGS reaction regardless of V loading.
Chein et al. [62] compared the performance of mono-(Pt) and bimetallic (Pt-Ni) catalysts supported by CeO2 and Al2O3. The results showed that the CeO2 addition to the support improved the activity of the Pt/Al2O3 catalyst only for the high S/C ratio cases and enhanced the activity of the bimetallic Pt-Ni catalyst in terms of CO conversion, H2 yield and stability.
Pt-Ni bimetallic catalysts, supported on γ-Al2O3, SiO2, TiO2, CeO2, HSA-ZrO2 and LSA-ZrO2, were investigated also by Wang et al. [63]. The results showed that the bimetallic catalyst had a higher activity than the monometallic one, due to the change in CO binding energy as result of the synergistic effect of the two metals. Moreover, the reducibility of the support had a positive effect on the catalytic activity, that was higher for the catalysts supported on reducible or partially reducible supports (CeO2, TiO2 and HSA-ZrO2) if compared with the one of the catalysts supported on non-reducible supports (γ-Al2O3, SiO2 and LSA-ZrO2). A deep investigation of the results indicated that the Pt-Ni bimetallic catalysts follow the activity trend of CeO2 > HSA-ZrO2 > TiO2∼ γ-Al2O3 > SiO2 > LSA-ZrO2, moreover, at similar CO conversion, the trend for the production of the undesirable CH4 was SiO2 > CeO2∼ γ-Al2O3 > TiO2∼ HSA-ZrO2.
2.3.2. Comparative Studies between Multiple Metals
Gunay et al. [64] investigated the effects of adding a promoter (K, Co and Ni) to Pt-CeO2/Al2O3 catalyst by means of both experimental tests and computational analysis through modular neural networks (Figure 8). The catalyst without promoters (1wt%Pt-1.25wt%Ce/Al2O3) showed the best performance, when a mixture of 5% CO and 10% H2O was feed, so clearing that the promoters had no effect on the catalyst activity. On the contrary, when the feed composition was changed by adding CO2 and H2, K addition (1wt%Pt-1.25wt%K-1.25wt%Ce/Al2O3) had the effect to significantly increase the catalytic activity, if compared to the other modified catalysts and to the not modified Pt-Ce.
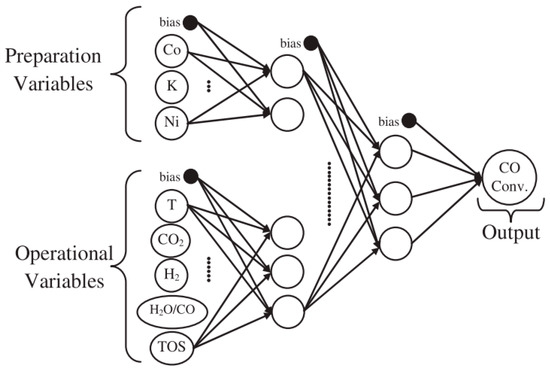
Figure 8.
Topology of the optimum neural network model, reproduced from [61], with permission from Elsevier, 2017.
Palma et al. [65] investigated the catalytic activity in low temperature WGS reaction of different bimetallic 1%Pt/1%M/CeZrO4 catalysts (M = Na, Mo, Sn, Cu, Zn). The results indicated that in the temperature range of 200–400 °C the 1Pt/1Sn/CeZrO4 catalyst showed the higher both CO conversion and H2-selectivity, while at lower temperatures the monometallic 1Pt/CeZrO4 catalyst showed the best performance. Further investigations were performed by studying the effect of the addition of Re, La and Rh) to PtM/CeZrO4 catalysts [66]. The results indicated that the 1Pt/1Re/CeZrO4 showed the best performance in terms of both CO conversion and H2 selectivity even at low temperatures and if compared to the 1Pt/CeZrO4 catalyst. Moreover, the effect of the sequence of impregnation of the two active metals was also evaluated, and the best performance was related to the catalyst in which was firstly loaded the rhenium.
Izquierdo et al. [67] prepared different Pt based mono and bimetallic catalysts, which were deposited into a microchannel reactor and tested in the temperature range of 300–450 °C. All the catalysts contained 25% of CeO2, the bimetallic ones contained 2.5% Pt and 2.5% of Me where Me = Ni, Co, Mo, Pd, Fe, Re, Y, Cu or Zn. The results showed that even if the tested catalysts, except the ones with Cu and Zn, reached the CO equilibrium conversion at 400 °C, all suffered of deactivation problems. Only the 2.1Pt–2.1Re catalyst showed stable activity for a duration of more than 100 h.
Rajesh et al. [68] prepared different Pt doped in the B site of LaBO3 (B = Mn, Fe, Co) perovskite catalysts, and this was found to be successful only in the case of Fe and Co and not in case of Mn, due to the sintering of Pt in the latter, so showing a distinct resistance to lattice incorporation and an intolerance to oxygen vacancies which would be created consequent to Pt substitution. Therefore, LaMn1−xPtxO3−d showed an extremely poor activity in WGS reaction. On the contrary, Pt doped Co and Fe perovskite catalysts showed a CO conversion of about 90% above 300 °C [69]. The authors attributed this enhanced catalytic activity to the multiple oxidation states (+4 and +2) of Pt in these compounds: in this way, a reduction of the B ions from the ideal state of +3 was observed for compensating the charge of Pt(IV), which was in turn found to enhance the oxygen vacancies on the surface. The ionic Pt species in tandem with oxygen vacancies capable of acting as water adsorption sites may be enhancing their WGS activity suppressing CO or CO2 hydrogenation.
Table 3.
Summary of the main catalytic results from the articles reported in Section 2.3.1 and Section 2.3.2.
Table 3.
Summary of the main catalytic results from the articles reported in Section 2.3.1 and Section 2.3.2.
| Selected Catalyst (Particle/Crystallite Size) | Preparation Procedure | Operative Condition WGS | CO Conversion/Rate (XCO)/(r) (Temperature) | Ref. |
|---|---|---|---|---|
| 1wt%Pt-2wt%Na/CeO2 (2.2 nm) | By incipient wetness impregnation | GHSV = 45,515 h−1; H2O/(CH4 +CO + CO2) = 2.0 | Equilibrium CO conversion (T = 310 °C) | [56] |
| 0.5wt%Pt–0.5wt%Re/TiO2 (not specified) | By co-impregnation | GHSV = 410,000 h−1; H2O/CO = 2.5 | XCO ≈ 90% (T = 300 °C) | [57] |
| Pt0.25-Mo0.75/C (1.27 nm) | By controlled surface reaction | WHSV = 240,000 mL·gcat−1·min−1; H2O/CO = 2 | r ≈ 10 μmol/gcat·s (T = 300 °C) | [58] |
| 4.3wt%Pt/64.6wt%Mo2C (not specified) | By wet impregnation | GHSV = 125,000 h−1 H2O/(CO + CO2) = 1.23 | XCO = 70% (T = 250 °C) | [60] |
| 4.79wt%VOx-0.49wt%Pt/Al2O3 (not specified) | By wet impregnation | WHSV = 80,000 mL·gcat−1·h−1; H2O/CO = 3 | XCO ≈ 60% (T = 450 °C) | [61] |
| 2.5wt%Pt-2.5wt%Ni/5wt%CeO2/Al2O3 (not specified) | By wetness incipient impregnation | W/FCOin = 20.37 gcat·h/molCO; H2O/CO = 5 | XCO ≈ 80% (T = 750 °C) | [62] |
| 1wt%Pt-1.25wt%K-1.25wt%CeO2/Al2O3 (not specified) | By incipient wetness co-impregnation | WHSV = 24,000 mL·gcat−1·h−1; H2O/(CO + CO2) = 0.67 | XCO ≈ 60% (T = 300 °C) | [64] |
| 1wt%Pt/1wt%Sn/CeZrO4 (9.3 nm) | By wet impregnation | GHSV = 10,000 h−1; H2O/CO = 3.75 | Equilibrium CO conversion (T = 230 °C) | [65] |
| 1wt%Pt-1wt%Re/CeZrO4 (9.4 nm) | By wet impregnation | GHSV = 10,000 h−1; H2O/CO = 3.75 | Equilibrium CO conversion (T = 200 °C) | [66] |
| 2.1wt%Pt–2.1wt%Re/25wt%CeO2/Al2O3 (1.4–5.0 nm) | By the incipient wetness impregnation | WHSV = 400,000 mL·gcat−1·h−1 H2O/(CO + CO2)= 2.2 | XCO ≈ 74% (T = 400 °C) | [67] |
| LaCo0.94Pt0.04O3-δ (75.5 nm) | By pyrolysis | GHSV = 5000 h−1; H2O/(CO + CO2) = 1.8 | XCO ≈ 90% (T = 325 °C) | [69] |
2.3.3. The Addition of Promoters to the Support
Mohamed et al. [70] investigated the effect of different loadings of Pt–Au/FSM-16 catalysts. The authors presented a non-conventional preparation technique, by synthesizing nanowire Pt–Au clusters introducing Au+ into in situ designed Pt carbonyl clusters. The Au addition provided positive effects in decreasing the platinum reduction temperatures as well as stabilizing the carbonyl clusters formed, moreover, the 2.5Au-2.5Pt/FSM16) catalyst showed higher activity than that of 2.5Pt/FSM-16.
Buitrago et al. [71] studied the performance of carbon-supported platinum catalysts promoted by ceria. The catalysts were prepared by first dispersing CeO2 over an activated carbon support at different loadings (20, 30 and 40 wt%), to obtain a high ceria surface area, and then by incorporating platinum by impregnation with three different solutions (acetonic, aqueous and ethanolic) of [Pt(NH3)4](NO3)2 to investigate the effect of the solvent in the final catalyst. The results of the activity tests were compared with the ones obtained by using Pt/CeO2 and Pt/C catalysts as references. The results showed that the best performance was obtained by the catalysts with 40 wt% CeO2 prepared by aqueous impregnation of the platinum precursor, that achieved conversions higher than 70% at 300 °C, and showed no deactivation under reaction at this temperature for 120 h.
Zugic et al. [72] investigated the controlled addition of Na, through Na ion-exchange by means of nitric acid oxidation of the carbon nanotubes (so allowing the creation of anchoring sites for platinum) in the activation of multi-walled carbon nanotube-supported platinum catalysts (Pt/MWNT) for the WGS reaction (Figure 9).
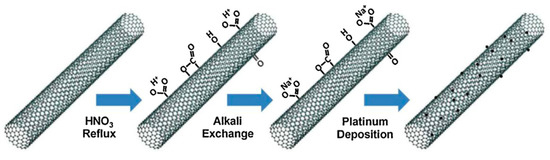
Figure 9.
Schematic of the preparation of alkali-promoted Pt/MWNT catalysts, reproduced from [72], with permission from Elsevier, 2014.
The results showed that the Na addition increased the catalyst activity, by altering the surface oxygen distribution through the stabilization of an oxidized platinum state. Meira et al. [73] studied the effect of ceria addition (12 and 20 wt%) to Pt/Al2O3 catalysts. The supports were prepared by adding cerium precursor during the sol-gel synthesis of Al2O3 or by impregnation of Al2O3. The results showed that CeO2 addition increased the WGS specific reaction rates up to seven times, suggesting that the preparation method had a significant impact on the structural and surface properties of ceria, and that the participation of interfacial Pt-O-Ce sites gave a crucial contribution to the significant increases of specific reaction rate.
Mao et al. [74] prepared Pt based catalysts by using as supports LaCoO3 films deposited onto MgAl2O4 powders by atomic layer deposition (ALD). The prepared catalysts were characterized by a 0.5 nm thickness of films and exhibited a perovskite structure after redox cycling at 800 °C; in addition, the films covered the substrate uniformly, as well as the Pt on the support. The reduced forms of the LaCoO3-containing (with 3 wt% Pt), compared with conventional Pt on MgAl2O4, were highly active for the WGS reaction, while the oxidized catalysts showed much lower activities.
Brandt et al. [75] studied the activity of Pt-Re surfaces, prepared with different methods. In one case Pt and Re surfaces were prepared by annealing Re films on Pt (111) to form Pt-Re surface alloys, in another case Pt was deposited on Re/Pt (111), finally Pt was deposited on Re clusters supported on highly oriented pyrolytic graphite (HOPG) surfaces. In all cases, the turnover frequency (TOF) was higher for Pt on Re surface compared to pure Pt, while the lower TOFs for PtRe alloy surface and PtRe clusters on HOPG, compared to PtRe/TiO2 suggested a promotional effect of the support.
2.3.4. Conclusions
Summarizing, the results of the comparative studies highlighted the positive effect of rhenium and alkali metals addition, both in the CO conversion and on the stability of the catalysts. In the case of rhenium, the optimal loading was found to be 0.5 wt% Re with a Pt/Re ratio equal to 1 and an impregnation sequence in which rhenium was the first metal loaded. Sodium promotion has been successful used, through the surface modification of fascinating multi-walled carbon nanotube, which modify the surface oxygen distribution, thus stabilizing an oxidized platinum state.
Table 4.
Summary of the main catalytic results from the articles reported in Section 2.3.3 (the addition of promoters to the support).
Table 4.
Summary of the main catalytic results from the articles reported in Section 2.3.3 (the addition of promoters to the support).
| Selected Catalyst (Particle/Crystallite Size) | Preparation Procedure | Operative Condition WGS | CO Conversion/H2 Formation Rate (XCO)/(rH2) (Temperature) | Ref. |
|---|---|---|---|---|
| 2.5wt%Au-2.5wt%Pt/FSM16 (7 nm) | Pt and Au loading by co-impregnation; FSM by surfactant templation | H2O/CO = 0.08 | rH2 = 5 mmol·gcat−1 (T = 77 °C) | [70] |
| 1wt%Pt–40wt%CeO2/C (not specified) | Pt and Ce loading by impregnation under vacuum with acetone | WHSV = 60,000 mL·gcat−1·h−1 H2O/CO = 20.5 | XCO > 70% (T = 300 °C) | [72] |
| 20wt%CeO2/1wt%Pt/Al2O3 (5.7 nm) | By sol-gel synthesis | WHSV = 130,000 mL·gcat−1·h−1 H2O/CO = 10 | XCO ≈ 95% (T = 350 °C) | [73] |
3. Kinetics of Water-Gas Shift Reaction
The study and the evaluation of the kinetic parameters for water-gas shift reaction has been carried out over both monometallic as well as bimetallic and promoted platinum-based catalysts. In the following two paragraphs, a review of the approach followed by various authors regarding WGS kinetics is reported.
3.1. Monometallic Catalysts
In this section, the kinetic studies of Pt-based monometallic catalysts are reviewed. The section has been divided into three paragraphs based on the type of support: Section 3.1.1 reports the studies of the articles on Pt/Mo2C-based catalysts; Section 3.1.2 reports the studies of the article on Pt/CeO2-based catalysts; and Section 3.1.3 report the studies of the article on Pt/MnO2-based and Pt/strontium hydroxy and fluorapatite catalysts. A brief summary concludes the section.
3.1.1. Pt/Mo2C-Based Catalysts
Schweitzer et al. [76] employed monometallic Pt catalysts supported on nanostructured molybdenum carbide (Mo2C) for water gas shift reaction. They compared the activation energies of the 4wt%Pt/Mo2C catalysts with those reported for other catalytic systems finding slightly lessened values (53 kJ·mol−1 for 3.8wt%Pt/Mo2C, 80 kJ·mol−1 for 5wt%Pt/CeO2, 51 kJ·mol−1 for 2wt%Pt/TiO2 and 66 kJ·mol−1 for 0.5wt%Pt/TiO2), which demonstrate a higher activity of the sample supported on molybdenum carbide. The improved reaction rates for WGS recorded over the Pt/Mo2C catalyst were explained via TEM characterizations, which revealed a low contact angle between the active particles and the Mo2C and proved a strong interaction Pt-support. They also identified two different reaction rates for the active sites on the particles surface and on their perimeter, finding that those on the perimeter are the rate-determining steps for WGS. Highly dispersed raft-like Pt particles were identified on the Mo2C support and, by increasing the Pt loading, their diameter increased, without affecting the thickness. Such particles are characterized by high surface to volume and perimeter to volume ratios, which is a relevant benefit when the rate-determining step is linked to perimeter active centers.
The effect of passivation on the activity of the Pt/ Mo2C catalyst was also investigated [77]. After support (Mo2C) synthesis via temperature programmed reaction, the reactor was removed from the furnace and cooled at room temperature; the unpassivated material was exposed to inert atmosphere while the passivated sample was treated in air (referred as p-Mo2C). Thereafter, Pt deposition was carried out via wet impregnation in both cases. The specific surface area of Mo2C and p-Mo2C were very similar; however, Pt particles were small and uniformly dispersed onto the unpassivated support while very large agglomerates of platinum were detected over the Pt/p-Mo2C sample. As a result, WGS rates and turnover frequencies (TOFs) were lower for the passivated catalyst, proving that the formation of the passivation layer had a negative impact on the metal-support interactions. The same authors also tested a 4wt%Pt-22wt%Mo2C/Al2O3 catalyst [78], which displayed similar activation energies compared to the Pt/Mo2C sample, lower than those observed for the Mo2C-free catalyst (Pt/Al2O3). The WGS rates were measured between 380 and 420 °C with a GHSV = 125,000 h−1 and the kinetic results obtained for the two catalysts containing Mo2C were compared in terms of normalized reaction rates (measured at 240 °C and evaluated by subtracting to the rate of the catalysts those measured for the supports Mo2C and Mo2C/Al2O3 and by dividing for the platinum loading). The profile obtained in the two cases (Figure 10) showed a very similar trend, demonstrating that the type of active sites as well as the interaction between Pt and molybdenum carbide are almost unaffected by Al2O3 deposition. Additionally in this case, atypical morphologies were observed for the Pt particles in the sample containing Mo2C, better dispersed than the cubo-octahedral particles of the Pt/Al2O3 sample.
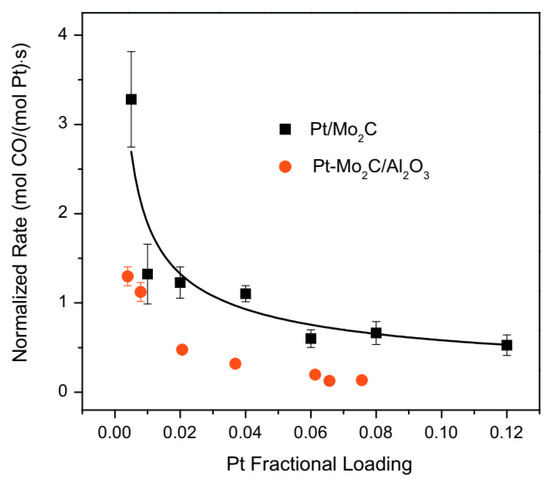
Figure 10.
Dependence of platinum normalized water–gas shift rates on the Pt loading for Pt/Mo2C and Pt-Mo2C/Al2O3 adapted from [78], with permission from Elsevier, 2013; P = 1 atm, T = 240 °C, 11% CO, 43% H2, 6% CO2, 21% H2O, 19% N2, WHSV = 70 h−1.
The presence of small platinum coverage on the carbide surface was also identified as the ground for the excellent activity of Pt/MoC catalysts [79], tested for WGS reaction under CO/H2O ratio of 2:1 between 135 and 195 °C. Small particles of platinum, in fact, assured a better and close contact with the MoC support, with the synergistic interactions Pt-MoC favouring a quick water dissociation: the WGS reaction occurs at Pt-carbide interface with the dissociation of water on the carbide side while CO is bounded to the Pt particles. The resulting activation energy (38 kJ·mol−1) was very low compared to the values reported for Pt(111) single crystals; a similar result was also recorded over a Pt/TiC catalyst. For both the supports (MoC and TiC), the superior performance of the 1:1 carbide was observed. In fact, for these carbides, the number of Mo or Ti atoms exposed is lower while, due to the positive charge of the metals, the Mo2C and Ti2C supports are responsible for stronger interactions with OH and O, thus causing slower WGS reaction rates.
3.1.2. Pt/CeO2-Based Catalysts
Wen et al. [80] synthetized platinum nanoclusters encapsulated on the internal concave surface of mesoporous CeO2 (Pt@mp-CeO2) or impregnated on the external surface of ceria nanoroads (Pt/rod-CeO2). Despite the particle sizes of Pt nanoclusters are similar in the two cases (3–6 nm), the activation energy was 60 kJ·mol−1 for the Pt@mp-CeO2, lower than the value of 78 kJ·mol−1 recorded for the Pt/rod-CeO2 (150 °C < T < 240 °C, 5.6% CO, 22.44% H2O, 71.96% Ar, WHSV = 4.2 h−1). The interaction between Pt and the local interface for the two samples is similar; however, the density of oxygen vacancies on the internal concave surface is higher compared to those measured for the Pt/rod-CeO2, which is related to a reduced adsorption energy of the OH groups and an improved reaction rate.
The synthesis of nano-sized CeO2 having high surface area and the strong interaction between Pt and CeO2 resulted in a highly active 1wt%Pt/CeO2 catalyst, as shown by Jeong et al. [81]. The support was synthetized through the thermal decomposition of crystalline cerium hydroxy carbonate, which was prepared via a novel precipitation/digestion method. The effect of pre-calcination temperature (400–700 °C) and aging time (0–8 h) on the kinetics was investigated (Figure 11) and the best results were recorded for the sample prepared at 400 °C and for an aging time of 4 h, which displayed the lowest activation energy (55 kJ·mol−1), ascribable to the relevant presence of stabilized Ce-PtOx species, which are highly active for the WGS reaction.
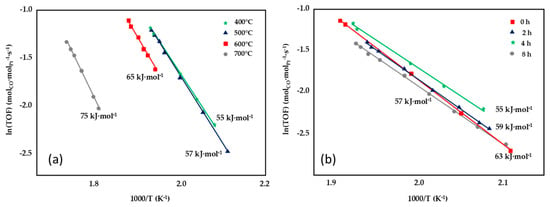
Figure 11.
Effect of pre-calcination temperature (a) and aging time (b) on the Arrhenius plots of the turnover frequency, adapted from [81], with permission from Elsevier, 2015; CO conversion obtained over Pt/CeO2 catalysts tested at P = 1 atm, 6.5% CO, 7.1% CO2, 0.7% CH4, 42.4% H2, 28.7% H2O, 14.5% N2, GHSV = 45,515 h−1.
The same authors [82] compared the performance of 1wt%Pt/CeO2, 1wt%Pt/ZrO2 and 1wt%Pt/Ce(1−x)ZrxO2 catalysts (x = 0.2, 0.8) and the different catalyst activity was correlated to their reduction profiles. The Pt/CeO2 catalyst, despite showing the worst Pt dispersion (37.6% compared to 66.9% of the Pt/Ce0.8Zr0.2O2, for example), displayed the lowest activation energy and the highest turnover frequency (Table 5). The Pt/CeO2 showed very high reducibility compared to the other samples, with the first peak of hydrogen consumption at 70 °C: due to the temporary reduction of Ce4+ to Ce3+, the support is able to supply active oxygen to oxidize CO in CO2; then is able to be re-oxidized by taking oxygen form water. Thus, beside dispersion, catalyst reducibility is another key factor affecting the activity for WGS reaction.
Table 5.
Turnover frequency (TOF) and activation energy (Ea) for WGS reaction over Pt/CeO2, PtZrO2 and Pt/Ce(1−x)ZrxO2 catalysts, date from [82]; at P = 1 atm, 6.5% CO, 7.1% CO2, 0.7% CH4, 42.4% H2, 28.7% H2O, 14.5% N2, GHSV = 45515 h−1.
Torrente-Murciano and Garcia-Garcia [83] synthetized two different nanostructured ceria supports (nanorods and nanocubes) by changing the operative conditions during hydrothermal treatment and compared their performance for WGS with a catalyst supported on ceria commercial nanoparticles; 1.5 wt% platinum was deposited by incipient wetness over the three supports. Despite the Pt particles size was slightly lower for the samples supported on CeO2 nanotubes and nanoparticles, the catalyst prepared from nanorods assured the best performance for WGS. In fact, catalyst activity and selectivity are mainly affected by platinum-ceria support interactions compared to the Pt crystallites dimension. The authors found that, for ceria nanorods, the selective exposure of the (100) and (110) crystal plane resulted in an optimum Pt-CeO2 interaction, thus strongly increasing WGS reaction rate and catalyst selectivity. In fact, methane formation was very low over the latter catalyst, proving that methanation is suppressed. Under a reacting stream of 5% CO, 5% H2O, 90% Ar and a GHSV = 4520 h−1, the 3 catalysts displayed similar activation energies (in the range 45–50 kJ·mol−1) while the reaction rates increased in the following order Pt/CeO2 cubes < Pt/CeO2 particles < Pt/CeO2 rods (43.1 < 49.9 < 189.8 molCO·kgcatalyst·h−1).
In the attempt of improving metal-support interaction, Deal et al. [84] prepared three alumina supported Pt catalysts by using the supercritical fluid deposition of an organometallic precursor (platinum acetylacetonate) for Pt deposition. Three samples were synthetized and tested for WGS reaction (1.25wt%Pt/Al2O3, 1.25wt%Pt-5wt%CeOx/Al2O3 and pre-treated Pt-CeOx/Al2O3, where the pre-treatment of the bare support was carried out in a H2/N2 stream at 300 °C for 4 h). As depicted in Figure 12, the addition of ceria to the Pt/Al2O3 catalyst significantly reduced (of almost 50%) the activation energy for the WGS reaction while similar values were recorded for the two Pt-CeOx/Al2O3 (71 kJ·mol−1); however, the pre-reduction did not allow a promotion of the reaction rate and this result is ascribable to the formation of both Pt cluster and crystalline nanoparticles upon the treatment. On the contrary, for the not pre-treated sample, a more uniform particle size was observed with a migration of ceria to form a crystalline shell around the Pt nanoparticles, thus increasing the number of interfacial sites which promotes CO oxidation.
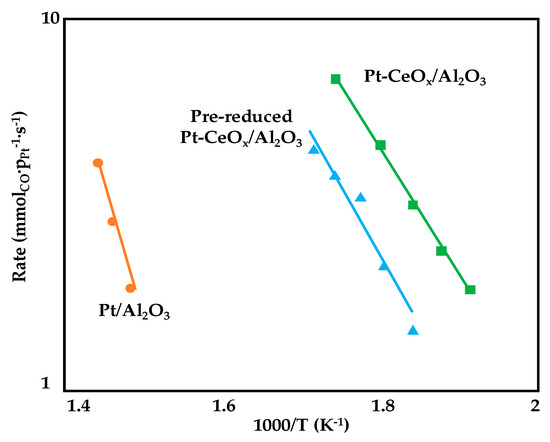
Figure 12.
Arrhenius plots used to calculate apparent activation energies for WGS reaction on supported Pt catalysts, adapted from [84], with permission from Elsevier, 2017; P = 1 atm, 16% CO, 32% H2, 32% H2O, 20% N2, GHSV = 75,000 mL·gcat−1·h−1.
3.1.3. Pt/MnO2-Based and Pt/strontium Hydroxy and Fluorapatite Catalysts
Shan et al. [85] investigated the performance of Pt-based catalysts supported on MnO2 nanoroads prepared via hydrothermal method (1wt%Pt/MnO2). Kinetic measurements were performed under an 8%CO, 24% H2O, 68% Ar stream and in a temperature range of 150–350 °C. The activation energy of WGS reaction calculated from the Arrhenius plot was 56 kJ·mol−1, which demonstrated the comparable activity of the MnO2 supported sample with Pt/CeO2 catalysts described in the literature. The authors, in fact, proved that, during WGS reaction, non-stoichiometric MnO1−x were formed and that, similarly to the results reported for ceria, oxygen vacancies participate into the reaction. Kuai et al. [86] applied a synchronous pyrolysis−deposition route for the addition of platinum to mesoporous MnOx. This technique allowed preparing atomically dispersed catalysts. By comparing the activity of as-prepared catalyst with a sample synthetized via conventional route (impregnation), the first one catalyst displayed a WGS activity more than two times higher and this result is ascribable to the lower number of single-atom active sites on the impregnated catalyst. The activation energy for the sample prepared by the pyrolysis-deposition route was 78 kJ·mol−1, which well agreed with the values reported for Pt single-atom catalysts with various supports. As depicted in Figure 13, by changing the platinum loading from 0.1 to 0.5 at%, the WGS activity displayed a linear increase, proving that the single Pt atom bounded to the support is active for the water gas shift reaction and that monodispersed single-atom active sites are present in the samples. Pt catalysts supported on a binary γ-Al2O3-α-Ga2O3 oxide, prepared via a simple-step precipitation and characterized by a good dispersion of the active phase, have also been tested for WGS reaction. Catalyst activity was investigated under a 2.5% CO, 10% H2O and 87.5% N2 mixture with a GHSV = 18,000 h−1. The 3wt%Pt/γ-Al2O3/25wt%α-Ga2O3 displayed a light-off reaction temperature of 280 °C with an apparent activation energy of 49.4 kJ·mol−1. For these catalysts, it was found that the presence of isolated basal or peripheral Pt atom species, rather than platinum dispersion, strongly contributed to the catalysts performance [87].
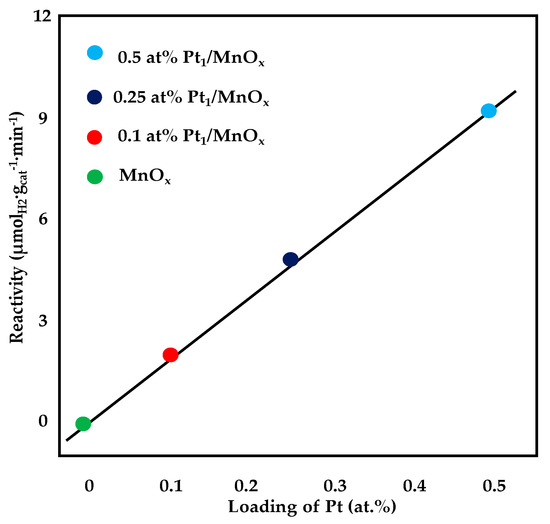
Figure 13.
Reactivity of WGS as a function of platinum loading, adapted from [86], with permission from American Chemical Society, 2018; T = 200 °C, 10% CO, 3% H2O, 87% He (linear fitting with R2 = 0.9988).
Miao et al. [88] deposited Pt nanoparticles (1 wt%) on strontium hydroxy and fluorapatite. WGS activity was investigated between 250 and 450 °C, with a reacting mixture of 5% CO, 20% H2O, 75% He under a GHSV = 150,000 mL·gcat−1·h−1. The prepared catalysts displayed high reducibility of the Pt oxidized species, comparable to the data reported for supports such as ceria: the interactions between the ionic support and the platinum particles allowed the formation of small Pt cluster on the apatite, characterized by improved reducibility and enhanced activity for WGS reaction. In particular, these supports are able to facilitate the activation of water: due to the presence of the apatite cations, the adsorption of water molecules in a strongly polarized fashion on the apatite surface assures an easier H2O dissociation. While CO is activated on the noble metal, water links to the ionic phosphates and the formate species are the main reaction intermediates. The comparison with other works found in the literature showed an increase of the reaction rate on the Pt/Sr apatite of almost 30% with respect to the previously described Pt/Sr apatite, with activation energies being slightly lower (70–72 kJ·mol−1). The reaction rate increased linearly with Pt loading up to 1 wt%, reaching the value of 0.68 molCO·molPt−1·s−1 at 300 °C.
3.1.4. Conclusions
Summarizing, in this section, among the described samples, the most advantageous catalyst, which displayed very low activation energy for the investigated reaction, was the Pt-MoC, with an Ea of 38 kJ·mol−1, while the activation energy of the much more used PtCeO2-based catalysts was in the range 55–80 kJ·mol−1.
3.2. Polymetallic and Promoted Catalysts
Various metals, including molybdenum and alkali, have been used as promoters for WGS reaction. In addition, the effect of support doping by the introduction of various metals has also been investigated. This section has been divided into three paragraphs based on the type of promoter: Section 3.2.1 reports the studies of the articles on Mo-promoted catalysts; Section 3.2.2 reports the studies of the article on alkali-promoted catalysts; and Section 3.2.3 reports the studies of the article on the rare earth and transition metals-promoted catalysts. A brief summary concludes the section.
3.2.1. Mo-Promoted Catalysts
Williams et al. [89] reported an increase in WGS rate upon Mo promotion for Pt/SiO2 and Pt/Al2O3 catalysts. Characterization studies revealed that Mo was in close proximity to Pt: the formation of Pt-Mo bimetallic particles decreased the carbon monoxide coverage over platinum and increased the CO reaction order, which is normally close to zero for Mo-free catalysts. Moreover, molybdenum provided a support-type effect by increasing the ability of the support to dissociate water in a similar manner over both Al2O3 and SiO2. Thus, a reduction in apparent activation energies of 20–40 kJ·mol−1 was observed for the Mo-promoted catalysts. However, reaction rates increased up to a molybdenum content of 3.4 wt% over alumina and 4.2% on silica: the highest turnover frequency can be reached for Pt-Pt to Pt-Mo ratios ranging between 4 and 5, with the best results over 3.4wt%Mo/Al2O3 and 1.2wt%Mo/SiO2. The reaction orders, the activation energies the TOFs values obtained over the alumina and silica-based catalysts are summarized in Table 6.
Table 6.
Results of kinetic measurements over doped and un-doped Pt/SiO2 and Pt/Al2O3 catalysts; P = 1 atm, 4–21% CO, 5–25% CO2, 11–34% H2O, and 14–55% H2, data from [89].
Similarly, a loading of 1.7 wt% of Mo on a 3.5 wt% Pt/SiO2 catalysts assured a reduction in the activation energy from 67 to 44 kJ·mol−1 (kinetic measurements were performed between 150 and 300 °C under a CO/H2O ratio of 3:10 and a GHSV = 18,000 mL·gcat−1·h−1). The authors found that the active sites for WGS reaction lied on the interface between MoOx patches and the Pt particles and that the loading of 1.7% (Mo/Pt atomic ratio ) assured the optimal coverage of molybdenum oxides on platinum. In such conditions, a bifunctional reaction mechanism was identified: water can be activated on the interfacial sites and react with the carbon monoxide adsorbed on the nearby Pt nanoparticles [90].
A bifunctional reaction mechanism with CO activated on Pt-Mo nanoparticles and water dissociating on the Mo2C support was also described by Sabnis et al [91]. They deposited platinum nanoparticles on multiwalled carbon nanotubes (MCNTs) and evaluated the effect of Mo addition by incipient wetness impregnation; during the reduction process of the dried material (carried out in the presence of pure H2 at 600 °C for 3 h), Mo reacted with the carbon of the support to form Mo2C domains. Additionally in this case, the addition of Mo to the Pt/MCNT catalysts resulted in a reduction of the apparent activation energy (from 83 kJ·mol−1 measured for the 4wt%Pt/MCNT to 48 kJ·mol−1 recorded over the 5wt%Pt-10wt%Mo/MCNT). Various Pt/Mo2C/MCNT samples were prepared changing the Mo lading at a fixed Pt content of 1.5 wt% and varying the Pt loading at a fixed Mo content of 10 wt%. Very close apparent activation energies, apparent reaction order and reaction rate per total mole of Pt were measured over the above samples, which demonstrates that the chemical nature of the active sites is similar, independently form Pt as well as Mo loadings. However, as depicted in Figure 14, higher Pt loadings result in a linear increase of the WGS rate per gram of catalyst while a stabilization in the curve obtained by increasing the Mo loading was observed above 10 wt%. These results suggest that higher Mo contents favour the formation of less active sites while the number of the most active (Pt-Mo) sites increase with Pt loading. In fact, by varying the platinum content, it is possible to enhance the number of Pt-Mo alloy bimetallic particles and the dominant active sites for WGS reaction are located both on the Pt-Mo bimetallic particles and on the interface between Pt-Mo particles and the Mo2C domain. Thus, a linear increase of WGS rate per gram of catalyst was observed with the increase of support surface area covered by the Pt-Mo nanoparticles.
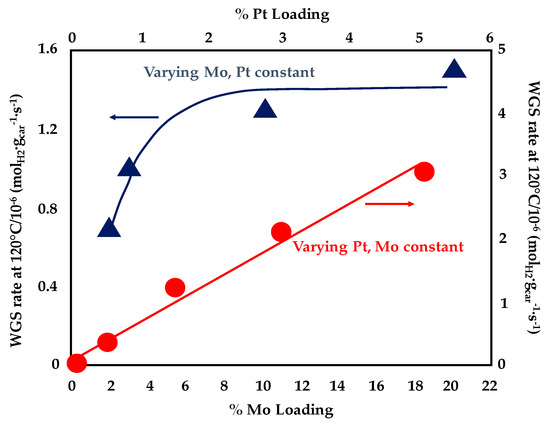
Figure 14.
WGS rate per gram of catalyst at 120 °C as a function of Pt and Mo lading for Pt/Mo2C/MCNT adapted from [91], with permission from Elsevier, 2015; P = 1 atm, 7% CO, 22% H2O, 8.5%, CO2, 37.5% H2.
3.2.2. Alkali-Promoted Catalysts
For WGS reaction, it was shown that the deposition of single-atom Pt catalytic centers (via different techniques, including incipient wetness impregnation and reverse microemulsion method) on numerous reducible, non-reducible and non-oxide supports (titania, silica, zeolites, alumina and carbon) rather than mixed metal structures (nanoparticles, clusters and atoms) is beneficial to enhance WGS activity [92]. In fact, it was found that catalytically active -OH species are associated with the isolated Pt atom sites. It was also described that the addition of alkali ions (i.e., sodium) can stabilize the atomic Pt catalytic sites and strongly increase the amount of such hydroxyls, independently from the selected support. The catalysts were prepared by wet impregnation; the Pt nominal content was 0.5 wt% while the Pt:Na atomic ratio was fixed to 1:10. The Na-containing samples displayed similar TOF (Figure 15) as well as apparent activation energies (70 ± 10 kJ·mol−1). This result can be explained considering that alkali ions are able to form a shell around each Pt site, able to supply the -OH for the reaction with CO and acting as a support itself. This structure is equally active whatever the selected support.
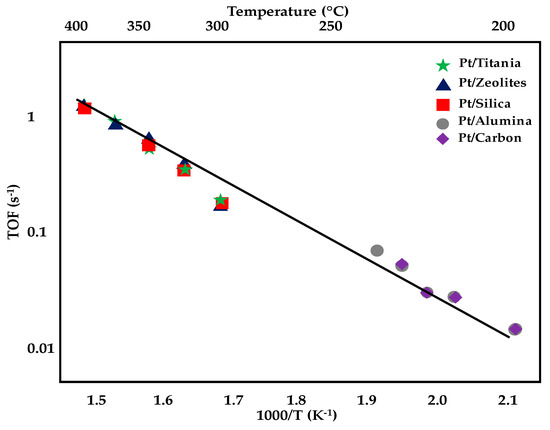
Figure 15.
TOF of the WGS reaction on Na-containing Pt catalysts on various supports, adapted from [92], with permission from Elsevier, 2017; 11% CO, 26% H2O, 7% CO2, 26% H2, 30% He.
The formation of well-dispersed Pt-Ox species associated with Na ions, which are stabilized around them, was also reported by Wang et al. [93]. They prepared core-shell 1 wt%Pt@SiO2 catalysts by the reverse microemulsion method, with platinum species embedded in the silica shell, the resulting platinum cores are of the order of 7–10 nm while the thickness of silica shell was 60 nm. Na (1.5–6 wt%) or CeO2 (2 wt%) were added as promoters and a comparison with the Pt/SiO2 catalysts prepared by conventional routes were also provided. While the sodium-free Pt@SiO2 and Pt/SiO2 were inactive for WGS reaction at 300 °C under a 11% CO, 26% H2O, 26% H2, 7% CO2, 30% He, both the Na and Ce catalysts due to the formation of stable Pt-Ox(OH)–M species (where M = Na or Ce), assured conversion above 50%. In fact, Na and Ce ions stabilized the Pt ions, boosting the regeneration of the hydroxyl groups, thus providing OH to complete the reaction. The apparent activation energy for both the promoted and un-promoted catalysts ranged between 65 and 75 kJ·mol−1; however, the difference among them is related to the population and accessibility of the Pt-Ox(OH)–M species, with increased for the core-shell catalysts.
Zhu et al. [94] investigated the effect of sodium addition on the WGS activity of 1wt%Pt/TiO2 catalysts; the kinetic measurements were performed between 250 and 300 °C under a stream of 2.83% CO, 5.66% H2O, 37.74% H2 and 53.77% He. Na incorporation slightly increased the apparent activation energy of WGS reaction (from 74 to 80 kJ·mol−1 in the case of the 4 wt% loaded catalyst); they also found that the promotional effect of sodium over the co-impregnated catalysts is significantly higher compared to the samples prepared by sequential impregnation: co-impregnation, in fact, improved the metal-promoter interactions (through the stabilization of a Pt-O-Na phase) and assured a partial coverage of the Pt surface by NaOx. By increasing the Na loading in the interval 0–10 wt%, the maximum improvement in WGS activity was recorded at 3–4 wt%. Such content, in fact, assured the formation of 2–4 layers of NaOx on the TiO2 surface, corresponding to the strongest metal-promoter interactions; a further increase in the Na loading led to the formation of a NaOx multilayer, with a low fraction of exposed Pt and high surface basicity. The same authors described two possible reaction pathways for WGS over Pt-Na/TiO2 catalysts: at low temperature and high H2O/CO ratio, the associative pathway was favoured, involving the interaction of CO on Pt with −OH on the support leading to the generation of surface formate, which further decomposed to CO2 and H2; the redox mechanism was predominant at high temperatures and low H2O/CO ratio, where CO is oxidized on Pt by the oxygen coming from the support to form CO2 while the resulting oxygen vacancy is refilled by steam, thus releasing hydrogen.
The promotional effect of sodium on the WGS activity between 200 and 400 °C has also been reported for Pt catalysts supported on oxygen-free multiwalled carbon nanotubes [95]. Pt (1 wt%) was added to the carbon nanotubes by incipient wetness impregnation and Na was deposited via a further step to give a Na/Pt molar ratio of 6. High WGS activity was observed over the promoted catalyst, due to the capability of sodium (oxide) alone to provide sites for water activation. Thus, sodium oxide was able to activate Pt even over oxygen-free supports, acting as a “ceria analogue”. However, due to the high affinity of hydrogen with the catalyst surface, the hydrogen storage capacity of the carbon support in the presence of Pt and Na was high, which was responsible for an increase in the apparent activation energy of almost 30 kJ·mol−1 (from 75 to 105 kJ·mol−1) under a stream of 11% CO, 25% H2, 25% H2O, 7% CO2, 32% He: under such conditions, hydrogen desorption limited the water gas shift reaction rate.
The role of the support (TiO2, L-zeolites and mesoporous silica MCM-41) on the WGS activity has been investigated for different Na-promoted and un-promoted Pt catalysts [96]; platinum was deposited by a simple incipient wet impregnation procedure and Pt loading was fixed to 0.5 wt% while the Na was co-impregnated leading to a Pt:Na atomic ratio was 1:10. Kinetic measurements were performed between 250 and 300 °C under a 11% CO, 26% H2O, 26% H2, 7% CO2, 30% He stream. While the formation of nanoparticles was observed over the Na-free catalysts, no Pt nanoclusters were detected in the promoted samples, where 80% of the total platinum was present as isolated atoms without Pt-Pt bonding. No change in the apparent activation energy (75–80 kJ·mol−1) for the promoted and un-promoted catalysts over the various catalysts was observed, which is related to the presence of similar structured Pt-Ox sites. However, Na addition increased the number of such sites, thus resulting in an enhanced WGS rate. Among the selected support, silica and zeolite are considered inert under the point of view of supply capacity of active −OH species while TiO2 is regarded as an active support. Independently from the kind of support (active or inert oxide), single-atomic Pt-sites stabilized by Na through O- ligands were identified, which assured similar catalytic properties to the prepared samples and highlights the role of Pt−O(OH)x− species on the activity, indirectly influenced by the catalytic support.
Xie et al. [97] compared the activity for WGS reaction of a 2wt%Pt/ZrO2 catalyst prepared by supporting platinum (via atomic layer deposition) on tetragonal ZrO2 (t-ZrO2) synthetized via hydrothermal method (the use of NaOH as the mineralizer made available Na ions within the structure) with that recorded in the presence of commercial nonporous monoclinic zirconia (m-ZrO2). Na presence assured the stabilization of zirconia in the tetragonal phase and a WGS rate of 40 × 10−6 mol·gcat−1·s−1 was recorded (at 250 °C, 1 atm, 6.8% CO, 21.9% H2O, 8.5% CO2, 37% H2, 25.8% Ar, WHSV = 0.96 h−1), which is almost 5 times higher than that measured for the catalyst deposited on the monoclinic support.
The kinetics of low-temperature (200–250 °C) WGS reaction was studied over Pt/Al2O3 and Pt/TiO2 upon the addition of alkali additives (Na, Li and K) [98]. The catalysts were prepared by incipient wetness impregnation with a Pt loading of 0.8 wt%, while the alkali metal/Pt molar ratio ranged from 7 to 125. For the Al2O3 series, Na promotion resulted in an increase of the reaction order for water and hydrogen, while a lessened reaction order was observed for CO, independently from the sodium content. Similar results were also observed upon Li and K promotion. An increase in the apparent activation energy was induced by Na and Li addition, while potassium promotion did not affect the value of Ea. However, sodium addition resulted in the highest increase of TOF normalized by Pt on the surface. Thus, for the TiO2 series, only the effect of Na was investigated, finding similar results in terms of reaction order and apparent activation energy compared to the alumina-based catalysts. The increase in the water reaction order was ascribed to the lower coverage of hydroxyl and hydroxyl generating-intermediates determined by the alkali addition. In fact, Na, Li and K enhanced the decomposition rate of formates, with a faster consumption of OH groups, thus reducing their coverage. Alkali also affected the CO adsorption on platinum by boosting Pt-C bonds and lowering the CO orders; in addition, higher hydrogen orders indicated less negative adsorption energies. The WGS kinetics over the alumina and titania promoted catalysts were similar (Figure 16), indicating that the alkali metal created new active sites on the support while Pt remained in its metallic state.
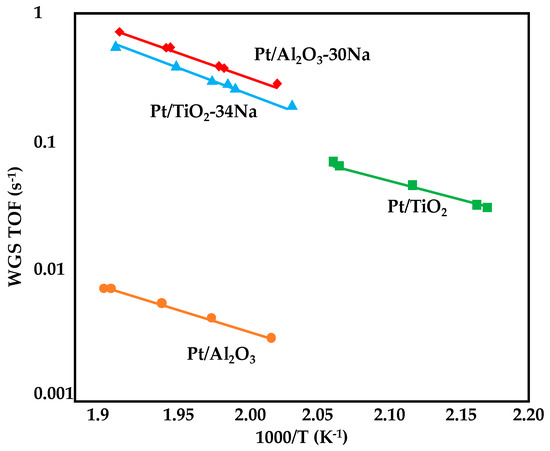
Figure 16.
Arrhenius plot for the promoted and un-promoted Pt/Al2O3 and Pt/TiO2 catalysts, reproduced or adapted from [98], with permission from Elsevier, 2012; P = 1 atm, 6.8% CO, 8.5% CO2, 21.9% H2O, 37.4% H2, 24.4% Ar.
Kusche et al. [99] studied the effect of the surface modification by a thin film of molten alkali salts for Pt/Al2O3 catalysts. A commercial Pt/Al2O3 catalyst (supplied by Alfa Aesar) was immersed into a solution of the salt and the alkali-hydroxide-coated samples were dried under vacuum at 150 °C for 4 h. Kinetic measurements were performed at 5 bar under a 1:1 ratio of CO/H2O and at a contact time of 0.5 s. By changing the KOH surface coating in the interval 0–30 wt%, the full activation of the surface of the Pt/Al2O3 catalyst was reached for a loading of 7.5 wt%, with an increase of TOF from 17 (K-free sample) to 95 h−1. A further increase of the coating loading resulted in a worsening of the WGS performance, ascribable to diffusional limitations (a too high degree of pore filling had negative effects on mass transfer). Moreover, a slight increase in the activation energy (from 54 to 63–67 kJ·mol−1) was observed, as a consequence of the different adsorption behavior of the reactants upon the thin film deposition. Salt coatings of LiOH, NaOH and CsOH were also deposited on the alumina catalysts, with the maximum increase in TOF over the 20 wt% NaOH coated sample (from 17 to 86 h−1). The enhanced WGS activity of the coated catalysts is ascribed to the presence of OH- groups on the catalysts surface, which accelerated all the steps involving hydroxyls. The dissociative adsorption of water on Al2O3 and its diffusion towards the interface between the active species and the support are highly enhanced by the hygroscopic nature of the alkali hydroxyls and their basicity. The potassium ions electronically modified the active Pt sites while the OH- groups altered the support, thus resulting in an increase of WGS activity. The influence of promoters addition (Mg, Ca, Sr, Ba) on the WGS activity of a 1wt%Pt/TiO2 catalyst was reported in the work of Panagiotopoulou and Kondaries [100].
Kinetic measurements were performed between 150 and 350 °C under a 3% CO, 10% H2O, 87% He stream at a GHSV of 73,000 h−1. The addition of all the selected alkali oxides (excluded MgO) shifted the CO conversion curve towards lower temperatures compared to the Pt/TiO2 sample; the promotional effect on the reaction rate decreased in the order Ca > Sr > Ba > unpromoted sample > Mg. Apparent activation energy was unaffected by alkali addition and changed between 65 and 73 kJ·mol−1; in fact, the promoters had no effect on the reaction mechanism but changed the pre-exponential factor through the variation in the population of active sites present at the metal-support interface. The increase in CaO loading from 0 to 2 wt% enhanced the WGS activity while a further growth to 4 wt% had a negative impact on catalyst performance. The catalytically active sites for WGS reaction (Pt-□s-Ti3+ sites), able to adsorb and activate carbon monoxide and water, are placed at the metal-support interface. Thus, small amounts of promoters are capable to weaken H2 adsorption strength and boost CO adsorption strength, with a consequent increase in the WGS rate. For higher alkali contents, the strength of CO chemisorption on the above sites underwent a further growth, with a consequent lower activity for WGS. In fact, such reaction is favoured under conditions of intermediate CO heat of adsorption. For the Pt/TiO2 catalyst loaded by 2 wt% CaO, the effect of calcination temperature was also investigated, finding a maximum in WGS activity for the sample treated at 600 °C. In fact, high calcination temperatures promote the formation of oxygen vacancies in the vicinity of the alkali atoms, due to the surface substitution of Ti atoms with Ca ones. However, a further increase of the temperature led to a decrease of specific surface area and a growth of TiO2 crystallite size, which resulted in lower conversions.
3.2.3. Rare Earth and Transition Metals-Promoted Catalysts
Kim et al. [101] investigated the Ce promotion in Pt-CeOx/CexZr1−xO2 catalyst was prepared by co-impregnation. The composition of the reacting mixture was 6.7% CO, 6.7% CO2, 33.2% H2O, 53.4% H2 and the kinetic experiments were performed between 170 and 320 °C. The effect of Ce doping on apparent activation energy was negligible with Ea around 92–94 kJ·mol−1 for the un-promoted sample and 97–102 kJ·mol−1 for the Pt-CeOx/CexZr1−xO2 catalysts. Moreover, for x = 0.05, 0.2 and 0.4, a growth in CO conversion was observed with respect to the Pt/CexZr1−xO2, while a further increase in the molar fraction of ceria in the support resulted in a worsening of the performances for WGS. In fact, low molar ratios of Ce/(Ce + Zr) in the support assured the best intimate contact between Pt and CeOx species, thus enhancing the WGS reaction rate.
Similarly, the effect of the addition of 10 and 20 wt% CeO2 to 1wt%Pt/SBA-16 catalysts was investigated by Carta et al. [102]. Pt-CeO2/SBA-16 were prepared both via the deposition–precipitation (DP) or the co-impregnation route (IMP). WGS activity was investigated between 150 and 350 °C under a 2% CO, 10% H2O, 88% Ar feeding at 50,000 mL·gcat−1·h−1. For the DP series, a CO conversion of 50% was reached at 258 °C (light-off temperature) over the 10% CeO2 loaded catalyst, while a double ceria content allowed a reduction of the light-off temperature to 224 °C. Likewise, the impregnated samples displayed a CO conversion of 50% at 203 and 190 °C for a CeO2 loading of 10 and 20%, respectively. The apparent activation energy for the ceria-promoted samples ranged between 50 and 80 kJ·mol−1. For the samples prepared by deposition-precipitation, highly dispersed nano-crystallites of CeO2 were deposited on the ordered mesoporous silica support, with a considerable surface area of the final catalyst and excellent accessibility of platinum. On the contrary, during impregnation, a confinement of ceria particles within the silica matrix occurred, with a relevant surface area reduction; however, despite the lower Pt accessibility, the IMP samples were characterized by a more efficient interaction between the Pt nanoparticles and ceria promoter. Thus, the catalysts prepared via the deposition-precipitation route, due to their anti-sintering behavior, are suitable for high temperature WGS (300–350 °C) while the impregnated catalysts are effective for low temperature operation.
WGS kinetics have also been investigated over Ce0.67Fe0.33O2-δ and Ce0.65Fe0.33Pt0.02O2-δ solid solutions prepared by the sonication method [103] under a 3.8% CO, 23% H2O, 73.3% N2 reaction mixture at GHSV = 55,900 h−1. The Pt-free catalyst displayed a good activity for WGS reaction at moderate temperatures, ascribable to the sufficient CO adsorption properties of iron. As a result, an almost complete CO conversion was recorded at 450 up to 550 °C. The Fe and Pt substituted ceria catalyst reached total conversion at 285 °C, with 100% values recorded until 550 °C. Pt presence enhanced the CO oxidation activity of the catalyst due to higher lattice oxygen removal by CO compared to the Ce0.67Fe0.33O2-δ. In fact, the first step of WGS reaction over the Ce0.65Fe0.33Pt0.02O2-δ sample involves CO oxidation to carbon dioxide by lattice oxygen. Thus, additional oxide ion vacancies are created in the catalyst lattice and such vacant lattice oxygen become sites for water adsorption. Finally, the release of oxygen from the lattice allows the site regeneration. A WGS rate of 2.8 µmol·g−1·s−1 at 450 °C was recorded over the Ce0.67Fe0.33O2-δ catalyst, with an apparent activation energy of 33.8 kJ·mol−1; an higher reaction rate (4.05 µmol·g−1·s−1 at 275 °C) with a lessened activation energy (12.1 kJ·mol−1) over the Pt ion substituted catalyst was observed, ascribable to the synergistic interaction of platinum with Ce and Fe ions.
The WGS activity of noble metals (Pt and Pd) as well as Fe ions substituted TiO2 was also studied [104]; Ti0.84Pt0.01Fe0.15O2-δ and Ti0.73Pd0.02Fe0.25O2-δ catalysts were synthetized by a sonochemical route and the activity tests were performed between 140 and 500 °C under a 1.3% CO, 35.5% H2O, 63.2% N2 stream at 48,000 h−1. The comparison with the performance of Fe-free catalysts (Pt/TiO2 and Ti0.99Pt0.01O2−δ samples) confirmed that ionic substitution of iron and Pt/Pd in the TiO2 structure enhanced the population of TiO2 surface oxygen/defects, thus improving WGS activity. The Pd-based catalyst assured almost total CO conversion from 260 to 500 °C while the Pt-substituted sample reached complete conversion at 300 °C; a lower apparent activation energy was also measured for the Ti0.73Pd0.02Fe0.25O2−δ catalyst (42 kJ·mol−1 against 63 kJ·mol−1 found over the Pt-based catalyst) with a rate of reaction of 4.36 µmol·g−1·s−1 at 240 °C. A lessened rate was also recorded for the Ti0.73Pd0.02Fe0.25O2−δ sample (2.74 µmol·g−1·s−1 at 280 °C). Despite the Pd-based catalyst displayed superior WGS activity under a H2 and CO2-free stream, by feeding a mixture of 2% CO, 10% CO2, 40% H2, 48% N2, equilibrium CO conversion was only reached in the presence of Pt, which appeared a more suitable catalyst to be selected under real WGS conditions. For the WGS reaction over the Pt and Pd-based catalysts, a hybrid mechanism was proposed (involving both the redox and formate pathway and accounting for the utilization of oxygen vacancies as well as OH groups), in which the redox step was dominant. Thus, CO can be either adsorbed on Pt/Pd, extracting the lattice oxygen from titania while H2O dissociates over the oxide vacancy sites of TiO2 producing H2 and oxygen, which re-oxidizes the support, or react with the OH groups originated by the H2O dissociation on TiO2, generating formate species, which decompose to CO2 and H2.
The addition of Si or Al to Ce0.98Pt0.02O2-δ catalysts prepared by the sonication method considerably improved their WGS activity between 150 and 440 °C [105]. A nearby complete CO conversion was reached at 260 °C by the Si-based catalyst and at 270 °C over the Ce0.88Al0.1Pt0.02O2-δ, with no decay until 440 °C over both the samples. As shown in Figure 17, the CO conversion rate was improved upon Si substitution instead of Al, with a consequently lower apparent activation energy (51 kJ·mol−1 over Ce0.88Al0.1Pt0.02O2-δ and 32 kJ·mol−1 for the Ce0.88Si0.1Pt0.02O2−δ catalyst).
Figure 17.
Variation of CO reaction rate with temperature over Ce0.88Si0.1Pt0.02O2-δ and Ce0.88Al0.1Pt0.02O2-δ catalysts adapted from [105], with permission from Wiley, 2012; P = 1 atm, 2% CO, 10% CO2, 10% H2, 55% H2O, 33% N2, GHSV = 48,000 h−1.
Such difference was ascribed to the fact that Si, despite being non-reducible, upon substitution into the Ce0.98Pt0.02O2−δ structure, is able to improve the reducibility of the final catalyst, with an enhancement of the oxygen storage capacity and a resulting higher catalyst activity compared to the Al substituted sample. However, both Al and Si addition increased the oxygen vacancies of the Ce0.98Pt0.02O2−δ catalyst, which are the active centers for water dissociation and are able to provide oxygen to oxidize CO. Moreover, the comparison with the performance of Pt-impregnated Ce0.9Si0.1O2−δ and Ce0.9Al0.1O2d catalysts revealed the higher activity of the samples having Pt ionically substituted within the structure. In fact, the metallic Pt is not capable to activate water dissociation while, in the catalysts prepared by sonication, the enhanced metal-support interactions made available two redox cycles (Ce3+-Ce4+ and Pt2+-Pt4+) during the reaction, instead of the only ceria one, able to both provide oxide ion vacancies. The effect of ceria modification by La addition was also investigated [106]. Pt nanoparticles (0.5 wt%) were deposited on Ce1−xLaxO2−δ(x = 0.0, 0.2, 05, 0.8 and 1.0) carriers prepared via citrate sol-gel method and the influence of Ce/La atomic ratio on the kinetic rates of the Pt supported catalysts was studied. WGS activity was investigated between 200 and 400 °C under a 3% CO, 10% H2O, 87% He with a GHSV = 40,000 h−1. By changing this ratio, a variation in the reactivity of the catalytic sites distributed along the Pt-support interface was observed. The largest specific kinetic rate of WGS reaction per length of perimeter of Pt-support interface was measured for x = 0.2 while the worst results were recorded for the ceria-free catalyst. The Ce0.8La0.2O2−δ support, in fact, displayed the highest reducibility at low temperatures and the highest oxygen storage capacity among the prepared catalysts, which assures the presence of weakly-bonded oxygen around the Pt nanoparticles, thus resulting in a higher rate of CO2 formation during WGS.
The kinetic parameters of Pt/Re/CeO2-based catalysts for WGS reaction were recently reported [107]. Three xPt/yRe/CeO2 powder catalysts (x/y = 1/2, 1/1 and 2/1; x + y ≈ 1 wt%) were prepared via dry impregnation, while the WGS activity was investigated between 230 and 330 °C, under a reacting mixture of 43 vol% H2, 7 vol% CO2, 7 vol% CO, X vol% H2O (X = 14, 20 or 24), with a contact time τ = 131 ms. The activity tests highlighted the best activity of the catalyst with the Pt/Re ratio equal to 2/1; the calculated kinetic parameters suggested that the reaction pathway involved not only the WGS reaction, but also CO as well as CO2 methanation. The calculated apparent activation energies increased with the decrease of the platinum content in the Pt/Re ratio, placing in the range 78–118 kJ·mol−1.
3.2.4. Conclusions
Summarizing, in this section the lowest activation energy was attained with solid solutions containing platinum as active species (Ce0.88Si0.1Pt0.02O2-δ and Ce0.65Fe0.33Pt0.02O2 showing Ea values of 32 of 12.1 kJ·mol−1, respectively). Thus, Pt-based solid solutions promoted by ceria displayed the highest activity for WGS reaction.
4. Reaction Mechanisms
The WGS reaction mechanism is currently widely discussed and investigated. A shared point of view is that it has a strict correlation with the nature of the active metal and the support, and of course it is also related to metal/support interaction; for this reason, several reaction mechanisms have been proposed in literature. In the next paragraphs a series of relevant published achievements, both experimental results and density functional theory (DFT) calculations, on the preferred WGS mechanism on monometallic, promoted and supported catalysts, are reported.
4.1. Monometallic Catalysts Supported on Single and Mixed Oxides
As a general point of view, WGS reaction operates in a bifunctional manner over supported metal catalysts, i.e., both the metal sites and the support take part to the reaction. Concerning Pt-based catalysts supported on reducible metal oxides, it is possible to individuate two main mechanisms: the redox and the associative mechanism [108]. The former, reported in Figure 18a, involves the support reduction and oxidation: CO adsorbs on the metallic site and diffuses to the metal/support interface, where the first redox reaction takes place through CO oxidation to CO2 and support reduction; the produced oxygen vacancy is now an active site for H2O adsorption, and so the second redox reaction occur, reducing water to H2 and re-oxidizing the support. It is convincing that this mechanism occurs in the high-temperature water-gas shift (HTS), while instead it has been questioned the possibility of decomposing water to deposit an oxygen atom in the vacancy of the support in the low-temperature WGS (LTS) conditions [109]. For this reason, the associative mechanism was proposed, and it is a little more complex because it involves the formation of a reaction intermediate, which can be either carbonate (CO3) (Figure 18b), formate (HCOO) (Figure 18c) or, as suggested by theoretical calculations, carboxylate (HOCO) (Figure 18d) [110]. This mechanism occurs when CO adsorption and diffusion to the metal/support interface is not able to reduce the support: in this mechanism, water interaction with support determines the presence of OH group on the support surface with whom the intermediate specie can react and form CO2 and H2. The type of mechanism that is prevalent, the kind of intermediate specie formed during the reaction and the path that the intermediate specie follows to convert itself in CO2 and H2 are all aspects which depend mainly on the nature of support and also on the platinum particles dimension [111]. Several studies have been conducted with Pt-based catalysts supported onto reducible metal oxides, because of their redox cycle, oxygen storage capacity, thermal and chemical stability. Among them, ceria was found to be the most promising, in terms of activity and stability, but it has been proved that its catalytic behavior could be enhanced by the addition of a second oxide, which modifies the structural properties of the lattice, thus changing also its chemical properties [112,113]. The section has been divided in five paragraphs based on the type of support: Section 4.1.1 (CeO2- and CeO2-TiO2-supported platinum catalysts), Section 4.1.2 (Ca and Si addition to CeO2-supported platinum catalysts), Section 4.1.3 (CeO2ZrO2-supported platinum catalysts), Section 4.1.4 (CeO2La2O3-supported platinum catalysts) and Section 4.1.5 (different-supported platinum catalysts). A brief summary concludes the section.
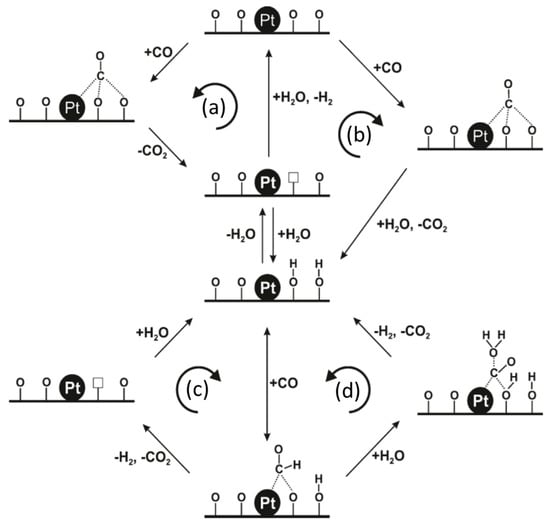
Figure 18.
WGS proposed mechanisms: (a) redox mechanism; associative mechanism with (b) carbonate (CO3) formation, (c) formate (HCOO) formation, (d) carboxylate (HOCO) formation. Reproduced from [116], with permission from American Chemical Society, 2011.
4.1.1. CeO2- and CeO2-TiO2-Supported Platinum Catalysts
Jain et al. [114] investigated the effect of ceria structural properties in a series of Pt/CeO2 catalysts where ceria was synthesized with three different methods. 5%wt nano-Pt (0.5–2 nm) was deposited by reacting spray deposition technology respectively on: C1, sol-gel method synthesized ceria; C2, combustion chemical vapor deposition prepared ceria; C3, commercial ceria provided by Sigma-Aldrich. The activity tests pointed out that the best formulation was obtained with mesoporous ceria C1 which presented the smaller crystallites size (5.8 nm) and the highest SSA (187 m2/g). The DRIFTS (diffuse reflectance infrared fourier transform spectroscopy) analysis showed the clear presence of formate band for C1 and C3, while only a small peak was detected for C2, concluding that formate mechanism dominates the reaction over C1 and C3 while C2 promotes the redox mechanism. The authors attributed the enhancement in the catalytic activity to the textural properties of the support, which are responsible of a different OH groups adsorption energy and of a higher number of more accessible active sites for the reaction intermediate.
An interesting dopant agent for ceria-based catalysts is yttria. A Pt/Ce0.6Y0.4O2 catalyst was studied in comparison with Pt/CeO2 and Pt/Y2O3 for the WGS reaction by Lee et al. [115] at 250 °C. The results of the study pointed out that the catalytic activity does not depend on the platinum particle size, whereas the increase in activity was found to be related to the enhancement in support reducibility. Moreover, the DRIFTS studies suggested that, for the Pt/Ce0.6Y0.4O2 catalyst, the adsorbed CO could react with oxygen from the support such as in a redox mechanism, while Pt/CeO2 and Pt/Y2O3 samples revealed a stronger CO adsorption, which made difficult to transport oxygen to the CO chemisorbed on Pt site. In fact, the high reducibility of Pt/Ce0.6Y0.4O2 catalyst promotes the existence of OH groups, which act as active sites and are related to the oxygen vacancies in the support: for this reason, the authors propose that the high oxygen mobility leads to a reduction in the CO adsorption strength.
Kalamaras et al. [116] reported a very interesting study above Pt-based catalysts supported on CeO2, TiO2 and a CeO2-TiO2 mixed oxide support, with the focus on the effect of temperature and support on the reaction mechanism. The authors found that in presence of CeO2 and CeO2-TiO2 and in low-temperature conditions (200 °C) the WGS reaction proceeded exclusively through the redox mechanism; at higher temperature (300 °C), instead, a very high concentration of OH species on the supports was detected, determining the competition of the associative mechanism with the redox mechanism. Nevertheless, the contribution of the former to the overall reaction rate was found to be small. On TiO2 support the only observed WGS mechanism was the redox one; moreover, TiO2-supported sample was the only one which showed a decrease in the reaction performances above 287 °C: this was ascribed to an excessive reduction of the support, leading to a self-poisoning of the catalyst. The authors also provided a differentiation of three possible associative mechanisms, one having a carbonate and two having a formate as reaction intermediate. At high temperature and in presence of CeO2 and CeO2-TiO2 supports the carbonate route could be excluded but it was not possible to identify which formate associative mechanism participated to the reaction.
The catalytic performances of Pt/Ce1−xTixO2−∂ formulations (x = 0, 0.2, 0.5, 0.8, 1) were also investigated by Petallidou et al. [111] showing that Pt/CeO2 and Pt/TiO2 catalysts have a very similar activity towards the WGS reaction, but the addition of Ti4+ ions in the ceria lattice offers the possibility of remarkably enhance the CO conversion. Moreover, the authors estimated the intrinsic reactivity (k, s−1) of 0.1%Pt/Ce0.8Ti0.2O2−∂ and 0.5%Pt/Ce0.8Ti0.2O2−∂ in order to correlate the activity to the Pt particle size, which was 1.1 and 1.7 nm, respectively. They found that the 0.5%-loaded sample showed better reaction performances, and explained this evidence considering that smaller Pt particles form Pt atoms with lower coordination number and different local electron density, which influences the electron density on adjacent support oxygen atoms. As in the redox mechanism the reaction occurs at the metal/support interface, the bond strength at the interface is a key aspect in determining WGS reaction rate.
Pt/CeO2-TiO2 formulations were also tested by Luo et al. [117] but the support was obtained, in this case, by impregnating CeO2 onto an anatase TiO2 support. The authors defined three different geometries depending on the CeO2 concentration: 1D, corresponding to ultra-fine dispersed clusters, smaller than 1 nm, 2D, corresponding to trapezoidal chains and three-dimensional hemispherical nanoparticles [118]. These possible structures were found capable of providing unique electronic and geometrical properties that led to different catalytic performances in WGS reaction, with low Ce-loaded samples giving the highest rate of CO2 production. This result was ascribed to the fact that Ce-rich samples contain more oxidized ceria (Ce4+) while 1D CeO2 particles (low CeO2 concentration) enclose mainly Ce3+ states, thus they can be easily reduced, leading to the highest WGS activity. Concerning the mechanism, instead, these different CeO2 structures did not affect the reaction pathway, which involved the formation of the same intermediate species on all the samples. Peaks related to the presence of strongly bound carbonyl species and of formate species were observed through the DRIFTS analysis, even if the authors suggested that formates act as spectators and not active intermediates for the WGS pathway, thus suggesting a prevalent redox mechanism [117].
4.1.2. Ca and Si Addition to CeO2-Supported Platinum Catalysts
A positive effect was observed also in the case of Ca addition to Pt/CeO2 catalysts. The mechanism on Pt-based doped CeO2-supported catalysts was studied by Linganiso et al. [119] on the basis of previous computational evaluations which claim the enhancement in O-mobility with the addition of a different element in the ceria lattice. The authors then evaluated the effect of Ca addition (as divalent element), observing that it has the ability of weakening the Ce-O bond, thus improving at the same time O-mobility and reducibility of support. The CexCa1−xOy (Ce:Ca equal to 100:0, 90:10, 75:25, 50:50 and 0:100) prepared catalysts showed that the best Ca:Ce ratio was 50:50 for the whole temperature range. The authors ascribed the positive result, given from Ca addition, to the enhancement of formate decomposition rate due to the higher O-mobility. This caused an increased accessibility of O-bound intermediates generated on the surface, resulting in higher LTS rates.
Ce doping was evaluated also from Hwang et al. [120] by modifying the support with Si addition. From a structural point of view, the surface area increased as a consequence of Si addition, and, at the same time, it was observed a decrease in both crystalline size and interplanar spacing, thus obtaining smaller particles with a lower crystallinity degree. Addition of Si enhanced the support reducibility, thus offering a promising potential to increase the number of superficial hydroxyl groups, which are an active specie for WGS reaction. The DRIFTS spectra observed on Pt/C100, Pt/S5C95 and Pt/S100 (respectively Si:Ce ratio equal to 0:100, 5:95, 100:0) showed that Pt supported on modified ceria had 2.5 times higher LTS rate than Pt/C100. Furthermore, while in CeO2 and Si-modified ceria supported catalysts the band related to formate was detected, it was found to be absent in the Pt/S100 catalyst, where CO was converted into CO2 by the mono-functional mechanism suggested by Vignatti et al. [121].
4.1.3. CeO2ZrO2-Supported Platinum Catalysts
CeO2-ZrO2 mixed oxide also represents a widely studied support for WGS reaction: this is because Pt/CeO2 catalysts deactivate easily in shutdown/startup operations, which are typical modes in fuel cells applications, while ZrO2 is attractive because its mechanical resistance and thermal stability but it presents less activity towards the reaction. For this reason, several studies report the investigation of Pt/CeO2-ZrO2 formulations with very promising results.
Vignatti et al. [122] focused on three CeO2-ZrO2 mixed oxides prepared by sol-gel method with different CeO2:ZrO2 ratios. The study revealed that there is not a linear trend of the performances with respect to the zirconia addition to ceria: in particular, Pt/CexZr1−xO2 catalysts with x ≥ 0.5 have a better activity than Pt/CeO2 while Zr-rich samples and Pt/ZrO2 are the least active catalysts. Concerning the reaction mechanism, the study confirmed the idea of formate associative mechanism proposed in literature. The formates formation rate clearly depends on the OH groups concentration on the support surface, and OH groups formation occurs mainly on the Ce3+ defect sites. This is the reason why Ce-rich samples showed a better catalytic activity: Zr4+ ions addition to CeO2 induces important lattice distortions that increase the reducibility of the support, thus resulting in a mixed oxide which can ensure higher OH groups formation. On the Zr-rich samples, DRIFTS analysis showed that the formates C-H stretching bands are shifted to high wavenumbers (Figure 19), suggesting their higher stability on these supports rather than the Ce-rich ones, resulting in a lower formates decomposition rate, which becomes the rate-limiting step in the WGS reaction mechanism.
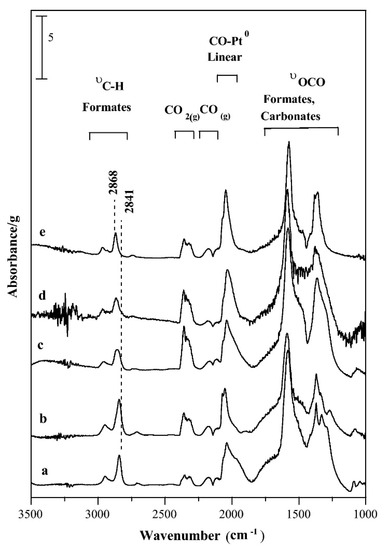
Figure 19.
In situ (diffuse reflectance infrared fourier transform spectroscopy) DRIFTS spectra at 250 °C on Pt/CeO2 (a); Pt/Ce0.75Zr0.25O2 (b); Pt/Ce0.50Zr0.50 (c); Pt/Ce0.25Zr0.75O2 (d); Pt/ZrO2 (e) (5% CO + 10% H2O in N2; Wcat = 50 mg; P = 101.3 kPa; Fv = 60 mL·min−1), reproduced from [122], with permission from Elsevier, 2011.
A different mechanism for almost the same formulation was proposed by Kalamaras et al. [123], who reported the effect of CeO2 doping with Zr4+ ions and the Pt crystallites dimension on the WGS reaction performances and mechanism. In agreement with what reported above, Ce-rich samples gave the best activity results, because of the increasing in Ce3+ sites concentration, but the authors found that the reaction proceeded mainly through the redox mechanism, with carbonate species formed as reaction intermediates and with a determining dependence on the temperature. In particular, at low temperature the reaction was found to proceed only through the redox mechanism, while at high temperature also a parallel associative-formate mechanism was observed, even if with lesser extent. Furthermore, the addition of Zr4+ ions to the ceria lattice induced a change in the structure of the reaction intermediate: in CeO2 support, CO adsorption on Pt site and its diffusion at the metal/support interface led to a carbonate that decomposes into CO2; in the mixed oxides supports, instead, CO adsorbs on Pt sites and then diffuses completely on the support, leading to a modified C-containing intermediate.
Pinaeva et al. [124] focused on the catalytic properties of lanthana-doped CeO2-ZrO2 both with and without platinum. They proposed a WGS mechanism on the basis of a detailed transient kinetic study, where the reactants interaction with the support and the active metal can be summarized as reported in Table 7. The study revealed that the reaction rate is mainly related to the surface/bulk oxygen diffusion and to the support ability of accumulating oxygen that can oxidize the adsorbed CO. The presence of Pt sites induced a preferential adsorption of CO onto the metallic sites, which led to a higher overall reaction rate. The addition of La to the support lattice resulted in a higher dispersion of platinum, thus achieving a higher concentration of Pt2+ sites located at the Pt/support interface.
Table 7.
WGS mechanism on Pt/CeO2–ZrO2(-La2O3).
4.1.4. CeO2La2O3-Supported Platinum Catalysts
Petallidou et al. [125] investigated the ceria modification with lanthana, in a series of 0.5%Pt/Ce1−xLaxO2−∂ formulations prepared by different methods, in the temperature range 250–350 °C. Among the investigated preparation procedures, the urea co-precipitation (U-method) gave the best results in terms of CO conversion and kinetic rate: the authors ascribed this evidence to the different support composition that this method determined compared to the others and also to the highest surface acidity and basicity showed by this sample. U-method was found to promote the formation of a two-phase system, made by La2O3 and a solid solution Ce1−xLaxO2−∂, while the others preparation procedures gave only the latter; furthermore, the obtained support showed the least extent of La3+ doping, the lowest concentration of oxygen vacancies and the highest size of support crystals and platinum particles. Considering the main occurrence of the associative mechanism on the basis of formates band observation, the authors reported that Pt supported on La2O3 does not promote the decomposition of the adsorbed formate towards H2 and CO2. Nevertheless, because of the two-phase nature of the support, it is reasonable to suggest that in the sample prepared via U-method the Ce1−xLaxO2−∂ phase has x close to 0.2 which was found to be the optimal La loading in a previous study [126]. On the basis of the results obtained from this study, U-method was adopted for the preparation of Pt/(100−x)wt%Ce1−xLaxO2−∂-xwt%CNT with the aim of investigate about the effect of Ce1−xLaxO2−∂/CNT ratio on LTS (300 °C) catalytic activity [127].
The support containing 44.1% CNT showed the highest kinetic rate and CO conversion, and this result was correlated to several properties, mainly related to the support composition: (i) Pt-CO and Pt-H bond strength was found to be the highest among all the different samples; (ii) the 44.1%CNT-sample exhibited the best Ce1−xLaxO2−∂ dispersion, thus the best Pt dispersion; (iii) the 44.1%CNT-sample exposed the highest concentration of Ce3+ species; (iv) the SSITKA studies revealed the highest concentration of active carbon-containing intermediates. The reaction mechanism study showed that, independently from the CNT wt%, the “redox” and the “associative with −OH group regeneration” mechanisms occur in parallel. In particular, concerning the C-path, COads species are formed on Pt sites while formate species (HCOO−) at the metal/support interface, while for the H-path atomic hydrogen is formed on Pt and O/−OH species at the metal/support interface, together with the formates.
In a subsequent work, the effect of Ti4+, La3+ and Zr4+ doping of ceria supported platinum nanoparticles catalysts was evaluated [128]; in particular Pt/CeO2, Pt/TiO2, Pt/Ce0.8Ti0.2O2−δ, Pt/Ce0.8La0.2O2−δ and Pt/Ce0.8Zr0.5O2−δ have been tested by the authors under WGS reaction at 200–300 °C in order to investigate the mechanistic and kinetic aspects of the process. Among all the tested samples, the Ti4+-doped catalyst exhibited higher WGS activity, result that has been addressed by the authors to a dual motivation: first, Ti4 doping has been found to enhance the concentration of labile oxygen and its surface mobility on ceria supported catalysts, as the Ce4+ → Ce3+ reduction energy is lowered, thus increasing the concentration of Ce3+ defect sites and, secondly, the existence of a higher site reactivity and a larger concentration for active “H-containing” and “C-containing” intermediates formed within a reactive zone around each Pt nanoparticle has been detected in the study; this latter aspect in another important intrinsic kinetic parameter that is considered to strongly influence the WGS rate of reaction. Focus on the involved mechanisms in the WGS reactions for the Pt supported ceria-doped samples revealed that both redox and associative mechanisms were involved and the extent of contribution of each reaction path to the overall kinetic rate depended on the dopant used; indeed with Pt/Ce0.8Zr0.5O2−δ the absence of formate species indicated that the redox mechanisms was controlling the WGS reaction, while on Pt/Ce0.8Ti0.2O2−δ, Pt/Ce0.8La0.2O2−δ both associative and redox mechanism were reported by the authors.
Linganiso et al. [129] investigated the effect of different metals as dopants in Pt-based catalysts supported on cerium dioxide. Their study focused on the impact that specific cations (i.e., Ba, La, Y, Hf and Zn) may have on the catalytic behavior of each sample, when added to the ceria matrix. In particular, the attention was paid to the catalyst reducibility and on the mobility of formates, as O-bound associated species are proposed to be intermediates in the LTS reaction [129]. The results pointed out that the dopants addition resulted in an enhanced catalyst reduction, with a remarkable increase in the extents of both CeO2 surface shell and bulk reduction; furthermore, ceria doping allowed the occurrence of bridging OH groups formation at lower temperature. The DRIFTS measurements indicated that the formate decomposition rate (strictly related to the O-mobility of O-bound associated intermediates) had the following trend: Ba > Y > Hf > La > undoped ceria > Zn. The easier decomposition of O-bound surface species resulted in an increased O-mobility, and consequently in a higher low temperature water gas shift activity, with a decreasing activity order which was the same observed via DRIFT measurements for formates decomposition.
A different study on Pt/Ce1−xLaxO2−∂ (x = 0, 0.2, 1) formulations was performed by Kalamaras et al. [130] with the aim of identifying the active and spectators reaction intermediates and of revealing the prevailing reaction path, among the redox and associative mechanisms proposed in literature. The authors found that the reaction follows both the mechanisms for all the catalysts, but the extent of participation of each to the overall reaction rate depends on the support composition, according to the results obtained in previous studies. Furthermore, three kinds of linear adsorbed CO were considered as active intermediates, among which the LF-linear CO was the most active towards water.
4.1.5. Different-Supported Platinum Catalysts
The associative mechanism was found to occur alone in the case of Pt/HfO2 in presence of steam at 130 °C: a DRIFTS study conducted by Ribeiro et al. revealed the presence of formates during CO adsorption, formed by its reaction with bridging OH group on the support surface [109]. The authors also suggested that the RDS of the LTS could be the breaking of the formate C-H bond; furthermore, they reported that a correlation exists between the formate C-H bond frequency and the ionic radius of the metal in the partially reduced metal oxide, in particular: the larger the ionic radius, the lower is the bond frequency, leading to a weaker C-H bond. Alumina is a typical support with a large variety of applications: even if reducible supports appear to be more active towards the WGS reaction, it still finds a wide interest.
Busca et al. [131] reported the study of a Pt/Al2O3 catalyst in the CO-WGS with temperatures up to 400 °C. As alumina is a non-reducible support, the redox mechanism cannot occur; nevertheless, IR studies revealed that even if the greatest part of Pt on the surface is present in reduced form, there were some Ptn+ dispersed cations located in correspondence of the defects of alumina crystals, such as corners and edges. These cations strongly interact with the support and are extremely active in CO oxidation, more than Pt0 sites. Transition metal carbides (TMCs) have demonstrated in recent studies to possess interesting catalytic activity; in particular, molybdenum carbide (Mo2C) was found to be suitable for the WGS reaction: the addition of metals could even enhance these catalytic properties. Sabnis et al. [132] reported a comparative study in which several admetals (Pt, Au, Pd and Ni) were supported over Mo2C and tested in WGS conditions at 120 °C. The authors correlated the reaction rate to the surface coverage of a specie: Mo2C alone showed a remarkably lower H2O reaction order than CO, resulting in very low CO coverage, thus slight catalytic activity; promoted Mo2C revealed instead an increased CO reaction order, corresponding to a high surface coverage. These observations led to the conclusion that the metal nanoparticle represent the CO activation site, while H2O adsorbs and activates on Mo2C, with the reaction taking place at the metal/support interface.
Apatite-supported Pt catalysts were discovered to lead to very high specific reaction rates at 300 °C, exceeding some Pt/CeO2 formulations by up to 50% [133]. The study conducted by Miao et al. highlights that H2O activation (on apatite) is even more challenging than CO (on Pt sites), and it can be enhanced by changing the Ca/P ratio in the apatite. In particular, the authors reported that H2O adsorbs molecularly on the (001) surface, while it spontaneously dissociates on (010) surface: this produces new superficial groups that are involved in the reaction, Ca-OH and PO-H. Considering the non-reducible nature of apatite, the redox mechanism widely proposed for the WGS reaction is, in this case, non-applicable; the suggested reaction path follows an associative mechanism, with formates as reaction intermediates.
WGS reaction on non-reducible support was studied in presence of zeolite-supported catalysts by Ding et al. [134]. The aim of the study was the identification and characterization of the active sites, in particular at low temperature. The authors reported that Pt/HZSM-5 (Si/Al ratio equal to 62) gave two possible adsorption sites: Pt single atoms (Pt-SA) and Pt nanoparticles (Pt-NPs). Below 100 °C, it was observed that only CO molecules adsorbed onto Pt-NPs could be oxidized to CO2, while CO adsorbed onto Pt-SA remained unchanged under the reaction conditions. As possible explanations, the authors stated that: (i) O2 activation is strictly dependent on the Pt clusters size, i.e., small clusters present less electron back-donation to the antibonding O2 orbital, thus less efficient O2 activation; (ii) the reactivity is related to the binding strength of the Pt-CO bonds. In the latter case, CO-(Pt-SA) bond was found to be stronger than CO-(Pt-NPs), resulting in a lower catalytic activity for the CO oxidation.
Rivero-Crespo et al. [135] reported a very interesting study of the WGS reaction in presence of Pt catalysts supported onto metal-organic frameworks (MOF). These are crystalline porous materials that allow the stabilization of ultra-small metal clusters in reduced form: the authors obtained, through gram-scale synthesis, a MOF-supported Pt11+ single-atom catalyst (SAC) with an exceptional activity towards the LTS. The proposed mechanism can be summarized as follows: the more reactive coordinated H2O molecule (Figure 20a), far from the MOF wall, transfers a proton first to the neighbour H2O (Figure 20b, arrow a) and then to the H2O molecule in the second coordination sphere (Figure 20b, arrow b), leading to an -OH group on Pt11+ and a H3O+ cation on the water cluster (Figure 20c). The -OH group is the responsible for the CO activation on Pt1; then CO undergoes OH and then H2O addition, giving two possible intermediates in equilibrium, orthoformate Pt-C(OH)3 and formate Pt-COOH.
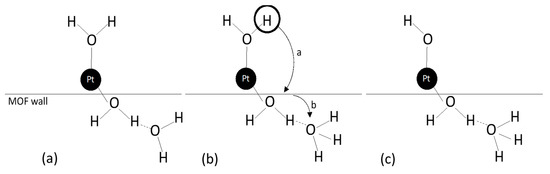
Figure 20.
CO activation mechanism on Pt/MOF catalysts: (a) coordinated H2O molecule, (b) transferring of a proton first to the neighbour H2O molecule, (c) -OH group on Pt11+ and a H3O+ cation on the water cluster.
Chen et al. [136] reported a comparative study of Pt/FeOx SAC and Pt/FeOx with Pt-NPs for the LTS. As an opposite result with respect to the work of Ding et al. [134] on HZSM-5 supported catalysts previously reported, the authors found that Pt-NPs exhibit higher CO adsorption strength than Pt-SAC. On Pt-NPs adsorbed CO reacts with the OH groups present on the surface and forms intermediate formates following the associative mechanism; formates then decompose to produce CO2 and H2. Pt sites in Pt-SAC induce the formation of oxygen vacancies which dissociate water to H2; the adsorbed O interacts with the weakly-adsorbed CO to produce CO2, such as in the redox mechanism. The catalyzed reaction activation energy is lower in the case of Pt-SAC and, as result, this sample exhibited the highest specific activity. Unsupported Pt(111) single crystals were also studied to provide a more detailed knowledge on the WGS reaction mechanism.
The work proposed by Flaherty et al. [137] reports the behavior of monofunctional platinum Pt(111) for the WGS reaction at 339 °C, 26 Torr CO and 10 Torr H2O. The authors individuated a reaction intermediate, I*, which could be a carboxylate (OCOH) or a formate (HCOO): the proposed mechanism suggests I* formation from the oxidation of CO* with OH*, and the subsequent observations of I* transformation in CO2 and H2 revealed that I* decomposes by linear transition state, thus ensuring I* is a carboxylate. Table 8 reports a summary of the above-discussed studies, including the proposed mechanism, the catalyst formulation and, where possible, the Pt particle size.
Table 8.
Summary of the mechanism reported in Section 4.1: Monometallic Catalysts Supported on Single and Mixed Oxides).
4.1.6. Conclusions
Summarizing, it is possible to point out that ceria promotes one mechanism or another depending on the preparation method: in fact, both the associative and redox mechanisms were found to occur on Pt/CeO2 catalysts. In presence of yttria doped ceria the redox mechanism was found to be preferential, while for mixed oxides, instead, the preferential mechanism was found to be the associative one. Furthermore, both for Ca-doped and Si-doped ceria, the reaction was found to follow the associative path. Concerning the CeO2ZrO2-supported catalysts, the reported studies are not according in the observed mechanism: in fact, Vignatti et al. [122] proposed the associative mechanism because of the presence of formate in the DRIFTS study, while Kalamaras et al. [123] reported the occurrence of the redox mechanism, with carbonate species as intermediates. Lantana addition to ceria-supported catalysts led to the occurrence of both the associative and redox mechanisms, as reported by the majority of the reviewed studies. In general, the associative mechanism was found to be the easiest to occur, also in presence of other supports, which present very different characteristics from ceria and ceria-based mixed oxides.
4.2. Polymetallic and Promoted Catalysts
As previously discussed in the monometallic catalysts mechanisms paragraph, the two general mechanistic schemes proposed for WGS reaction over metal oxide-supported noble metal catalysts are the (i) “redox or regenerative” mechanism and (ii) the “adsorptive or associative” mechanism. However, an extensive research effort is still devoted to the deep comprehension of some kinetic and mechanistic aspects of the WGS over supported metal catalysts, such as the controlling mechanistic path, the rate-determining step, the chemical nature of the active “carbon-containing” and “hydrogen-containing” intermediates, and their location (e.g. support, metal-support interface, metal surface or metal and support surfaces); indeed, awareness of molecular mechanisms in the WGS reaction would represent a powerful way to improve the catalysts activity, selectivity and stability, thus optimizing the industrial conditions of the WGS process [128].
It is widely known that polymetallic catalysts and promoters addition could result in an improvement of the catalytic performances. For instance, bimetallic catalysts have shown, in many studies, superior activity, stability and selectivity in comparison with the monometallic counterparts, showing chemical and physical behaviors that are more than just the sum of the pure metals; these enhanced properties have been attributed to electronic effects from metal-metal interactions, modifications in the surface structure, or the creation of new bifunctional active sites [138,139]. The section has been divided in three paragraphs: Section 4.2.1 (Pt-based bimetallic catalysts supported on different oxides); Section 4.2.2 (alkali metals promotion of Pt-based catalysts); and Section 4.2.3 (Other metals promotion of Pt-based bimetallic catalysts). A brief summary concludes the section.
4.2.1. Pt-Based Bimetallic Catalysts Supported on Different Oxides
Concerning the inspection of Pt-based bimetallic catalysts for the WGS reaction and their influence on the reaction mechanisms, Aragao et al. [140] evaluated the effect of selective Fe deposition on Pt/SiO2 catalysts, synthesized by controlled surface reactions (CSR) of (cyclohexadiene)iron tricarbonyl with hydrogen-treated supported Pt nanoparticles. Fe addition to the monometallic sample led to 5 times increase of the WGS turnover frequency (TOF), as a result of the Fe redox properties, indeed oxidation of Fe2+ into Fe3+ ensues water activation. Investigation concerning the mechanism of the reaction pointed out that the FeOx species were highly dispersed and Fe was characterized by an oxidation state < +2, thus suggesting the capability of FeOx to activate H2O and to promote the reaction with the adsorbed CO on a nearby Pt site.
Xu et al. [141] focused their attention to ruthenium, that is characterized by high WGS activity and cost less than Pt but, in the reaction conditions typical of WGS can lead to methane production; for this purpose three catalysts were prepared: Pt/CeO2, Ru/CeO2, and Pt-Ru/CeO2, in order to evaluate if alloying Ru with Pt could lower the Ru tendency to form methane. The catalytic investigation pointed out similar activities for Pt/CeO2 and Pt-Ru/CeO2 while, Ru/CeO2 showed worse performances. Moreover, it has been verified the possibility to inhibit the methanation reaction by alloying Ru with Pt, since extremely low CH4 production has been obtained on Pt-Ru/CeO2. The methanation reaction inhibition has been addressed by the authors to the capacity of the alloy to prevent the cleavage of C-O bonds, thus avoiding the subsequent hydrogenation of C to CH4. DRIFTS studies were performed to identify the predominant mechanism; during which, the presence of substantial amounts of formate (HCOO-) has been detected on Pt/CeO2, while lower amounts of formate were encountered with Ru/CeO2 and Pt-Ru/CeO2 but, for all the catalysts, formates completely disappeared at 350 °C. Furthermore, since the inhibition of formates did not influence the catalytic activity, it has been pointed out their non-involvement in the main reaction pathway.
A study devoted to understanding the mechanism that leads to obtain higher WGS performances on bimetallic Pt-Re catalyst in comparison with the monometallic Pt has been carried out by Duke and co-authors [139]. Indeed, prior studies pointed out that higher stability and superior activity were achieved on Pt-Re catalysts, attributing these results to two possible phenomenon: (i) ReOx could provide the active sites for water dissociation, thus enhancing the WGS activity in the hypothesis that the O-H bond represents the rate-limiting step in the mechanism and (ii) the CO poisoning could be reduced on Pt-Re surfaces due to the weaker binding of CO compared to that obtained on pure Pt catalysts. On this purpose, Pt-Re clusters supported on titania were prepared with different Re loadings 0-2 monolayer (ML), where ML is defined as the packaging density of Pt(111) (1.52 × 1015 atoms/cm2) or Re(0001) (1.52 × 1015 atoms/cm2), and moreover, Re oxidation states were also be determined after WGS reactions. The study pointed out that Pt-Re bimetallic clusters resulted more active than the pure Pt cluster in the WGS reaction, and in particular higher performances were exhibited by 2 ML Pt on 2 ML Re and 0.5 ML Re on 2 ML Pt. Moreover, investigations towards the oxidation states influence showed that preoxidized Pt-Re clusters were characterized by lower activity in comparison with both pure Pt and the unoxidized Pt-Re clusters, thus suggesting that ReOx does not provide active sites during the WGS reaction. Furthermore, it has been proved by the authors that the CO poisoning could be the main factor affecting the WGS activity, indeed, CO coverage after the reactions was lower on Pt-Re alloy.
Anil et al. [142] prepared noble metal (Pt, Ru and Pd) substituted Mn3O4 catalysts and, among them, Pt substituted Mn3O4 showed the best performances (99.9% conversion at 260 °C). The authors proposed a redox mechanism, which was subsequently validated using the experimental data; in particular, three elementary steps were considered: (1) the reversible adsorption of CO on active metal, (2) the dissociation of water at oxide vacancy on the support and retention of intermediate oxygen species and (3) the formation of CO2.
where CO_Pt express the CO adsorption on Pt surface, ν′ represents the oxide vacancy of support and O′ the support oxygen species. Moreover, DRIFTS studies evidenced that CO was linearly adsorbed on the Pt sites and then it reacted with carbonate species forming CO2 and with the OH groups of the support to product bicarbonates. These latter may also react with OH groups resulting in water and carbonates formation.
Rajesh et al. [143] developed platinum and yttrium doped BaCeO3 perovskites aiming to a deep comprehension of the oxygen vacancies role in the WGS reaction. Oxygen vacancies are, in fact, hypothesized to play a crucial role in the process, for instance, with ceria-supported catalysts they are supposed to represent the sites responsible for the water splitting step; however, their role is still not completely explained since the difficulties related to the fine-tuning of the oxygen-deficient sites with the conventional catalysts. Whereas, the authors developed BaCe1−xPtxO3−δ catalysts, fixing the Pt concentrations to 2% mol and progressively increasing Y from 2 to 30% in the B site, so that the oxygen vacancies were systematically enhanced to maintain electrical neutrality. It has been seen that the 6% Y-substituted catalyst showed the best activity, and further investigations pointed also out that the compound was characterized by the most symmetric B site coordination, thus highlighting that is not the extent of oxygen vacancies that play the crucial role but their structural characteristics. The proposed mechanism involved the water adsorption in the vacancy sites and its dissociation, whose energetics also depend on the coordination environment, thus demonstrating that the better activity achieved by the 6% Y-substituted compound could be attributed to the symmetric environment around B ion, energetically advantageous for the adsorption and desorption of water molecules.
4.2.2. Alkali Metals Promotion of Pt-Based Catalysts
Moreover, several studies have been devoted in the last years, to the effect of promoters’ addition to Pt-based catalysts in the WGS mechanism and, in particular, alkali metals adoption has attracted major attention in this field. Alkali metals have been found to play crucial roles in the reaction pathway, indeed, their presence can (i) modify the support properties and ease the formation of hydroxyl groups, thus originating sites in close contact with the dispersed metal, (ii) affect the strength of the formate C-H bonds, resulting in an enhancement of the formate decomposition rate and, moreover, (iii) provide a film of alkali-OH on the surface, and increase the number of H2O activation sites [144]. Faust et al. [145] performed deeper investigations in this field on a Pt/TiO2 catalyst, indeed in their study a first evaluation of the support particle size and its surface chemistry (hydroxyl groups) has been carried out, followed by an investigation towards the modification of the support material obtained by Na deposition. Via chemical vapor deposition (CVD), the authors prepared Pt nanoparticles catalysts on two TiO2 support with different specified OH-densities, WGS experiments and catalysts characterization showed that higher amount of hydroxyl groups on the support surface positively influenced the performances of the catalysts. Moreover, Pt/NaTiO2 nanoparticles with very narrow Pt particle size distribution and high Pt surface area were developed in this study; the additional Na coating resulted in an increase of the catalytic activity by a factor of 3, mainly attributed to the Na influence on the adsorption of CO. Na doping investigation, performed by Martinelli et al. [146] for Pt/YSZ catalysts, resulted in higher conversions and reactions rates for WGS reaction carried out between 250 and 400 °C (Low temperature WGS). Further analysis on the involved mechanism pointed out that the presence of Na allowed a weakening of the C-H bond of the formate species, and the water-assisted formate decomposition has been identified as the rate limiting step of the process. The proposed mechanism, presented in Figure 21, involved the support promotion of the formate formation via the reaction between CO and the defect-associated bridging OH groups, while Pt was involved in the formate dehydrogenation.
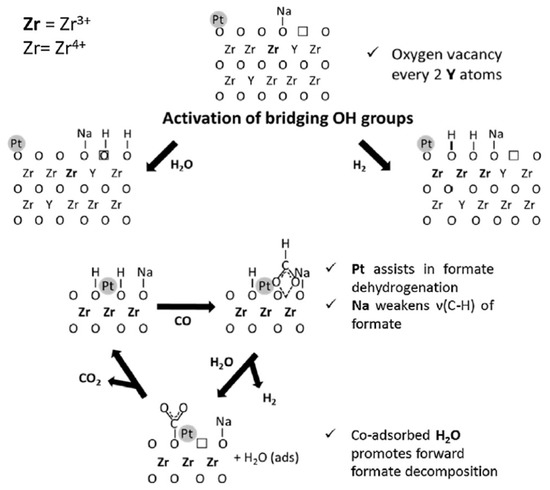
Figure 21.
LT WGS mechanism proposed by Martinelli et al. on Na doped Pt/YSZ catalysts, reproduced from [146], with permission from Elsevier, 2017.
Analogous outcomes have been obtained by Gao et al. [147] in the inspection of alkali-doped silica supported Pt catalysts; indeed, their results suggested that the formate C-H bond breaking could be the rate limiting step in the associative mechanism, and Na doping led to an electronically weakening of the C-H bond. In their study, the authors performed a first comparison between Pt/CeO2 and Pt/SiO2, finding higher WGS activity on ceria due to its capability to generate associated formate species; furthermore, the effect of both light and heavy alkali addition has been evaluated adopting Li, Na, K, Rb and Cs doped catalysts. Li, Na and K addition resulted in an improvement of the catalysts performances, deriving by the enhanced breakability of the C-H bond while, with higher alkali metals, the obtained activity was lower, maybe attributable to the major difficulty in the decomposition of the second intermediate (i.e., carbonate). Conclusions of this study pointed out that the adopted catalyst should provide three important characteristics: (i) a stable high surface area of the support that would allow to obtain a good dispersion of the active phase, (ii) an alkali species that would promote the formation of reactive OH groups, thus favouring the generation of formate species upon addition of CO and (iii) a metal species in close contact with the alkali metal that would assist the formate dehydrogenation during its forward decomposition. Moreover, light alkali metals have been identified by the authors as the most suitable dopant, since they do not result in the production of stable carbonates that may result in the suppression of the catalytic cycle, as resulted, on the other hand, with the adoption of Rb or Cs.
Within alkali metals, potassium has also attracted attention as a dopant for the Pt-based catalysts in WGS reactions; in particular, Watson et al. [148] evaluated the potassium effect on Pt/ZrO2 catalysts, analysing the influence of the K loading on the reaction mechanism. The study pointed out that K addition resulted in an improvement of the LT-WGS activity, achieved by the acceleration in the rate of the formate decomposition; indeed, in the associative mechanism proposed by the authors the formate decomposition represented the rate limiting step and hence, the weakening of the C-H bond would result in the enhancement of the catalyst performances. Under this purpose, K loading between the 0.85 wt% and the 3.4 wt% have been tested under DRIFTS experiments, and the best outcomes were reached with the 2.6 wt% of potassium, that resulted in a faster decomposition of formate, ensuring in the meantime that the Pt nanoparticles surface remained largely uncovered. Indeed, with higher potassium concentrations, 3.4 wt%, the platinum nanoparticles were completely covered by the alkali, thus resulting in the inhibition of the Pt activity towards the formate dehydrogenation. On the other hand, with lower K loadings, e.g. 0.85 wt%, there was no improvement in the WGS activity.
Potassium doping has also been investigated by Kaftan et al. [149] through the development of a KOH-coated Pt/Al2O3 catalyst. The KOH coating resulted in an improvement of the WGS activity, leading to a lowering of the onset reaction temperature of approximately 40 °C. Moreover, the introduction of the KOH coating modified the involved mechanism indeed, while on the uncoated catalyst the reaction mechanism was predominantly formate driven, adding the KOH coating led to a more selective mechanism, which involved mainly carbonates as the most abundant surface species. The authors associated the increased WGS activity with the enhanced availability of OH groups on the metal, achieved with the addition of the coating.
4.2.3. Other Metals Promotion of Pt-Based Bimetallic Catalysts
Apart from alkali metals, other metals have also been studied as dopant for WGS Pt-based catalysts, for instance Hwang et al. [150] investigated the effect of Ti addition to Pt/ZrO2 catalyst with different Ti loadings. DRIFT studies performed on the catalyst containing the 20 wt% of Ti, Pt/Ti[20]/ZrO2 highlighted that a faster decomposition of the formate species was achieved, indeed Ti addition facilitated the Pt particles dispersion and increased the density of OH groups, thus resulting in a positive effect on the WGS reaction rate. Moreover, the authors proposed for the investigated catalyst an associative mechanism with red-ox regeneration, characterized by the steps (4) to (7).
where S and Sv are the support surface site and oxygen vacancy site on the support, respectively. Table 9 reports a summary of the above-discussed studies, including the proposed mechanism, the catalyst formulation and, where possible, the Pt particle size.
Table 9.
Summary of the mechanism reported in Section 4.2: Polymetallic and Promoted Catalysts.
4.2.4. Conclusions
An associative mechanism has been encountered in the case of Fe addition to Pt/SiO2 catalysts and also in the case of Pt/CeO2, Ru/CeO2, and Pt-Ru/CeO2 catalysts, while in the case of Pt, Ru and Pd substituted Mn3O4 the occurrence of a redox mechanism has been observed. In the case of alkali metals promotion to Pt-based catalysts, the occurrence of the associative mechanism has been encountered for all the investigated catalytic formulations.
4.3. DFT and Theoretical Studies
As already underlined, the WGS reaction mechanism on Pt-based catalysts is still a matter of debate, however standard DFT methods and multistep approaches, in the case of particularly complex systems [151], such as reducible oxides, can provide fundamental information to elucidate the results of experimental studies. In this section, the main results of a series of theoretical-based studies, concerning the reaction kinetics, the reaction mechanisms and surface interactions, of platinum catalytic systems for WGS reaction, are reported. The first two paragraphs are devoted to the studies on unsupported monometallic (Section 4.3.1) and polymetallic surface models (Section 4.3.2), while the following paragraphs report the studies on TiO2-supported Pt models (Section 4.3.3), CeO2-supported Pt models (Section 4.3.4) and MgO-supported Pt and bimetallic supported models (Section 4.3.5).
4.3.1. Unsupported Monometallic Pt Surface Models
The structure sensitivity of the WGS reaction on Pt, using a multiscale modelling approach that integrates density functional theory (DFT) calculations and kinetic Monte Carlo (KMC) simulation, was investigated by Stamatakis et al. [152]. The DFT calculations were used to calculate the reaction barriers and the pre-exponential factors of the elementary steps of the WGS mechanism (adsorption-desorption events, water and hydroxyl decomposition, carboxyl and formate intermediates formation, CO2 production), involving combinations of step and terrace sites. The obtained values, of total energy and of vibrational frequencies of the stable species and activated complexes, were then used in KMC framework (Figure 22) to calculate the turnover frequency for Pt(111), Pt(211), and Pt(322) as function of the CO/H2O ratios and of the temperatures. The results showed that on Pt(111), for all the PCO studied the reaction proceeded through the carboxyl pathway, and the rate-determining step was the carboxyl formation, but under H2O excess the redox mechanism assumed a non-negligible rate, anyway it was still slower than the carboxyl pathway. The Pt(211) case was more complicated, because of the different involvement of the sites in the elementary events. At low CO/H2O ratios, the H2O dissociation was active on both step and terrace sites, as well as, at the interface, the adsorbed oxygen reacted with CO, and CO2 and H2 desorbed. The rate-determining step appeared to be the desorption; the carboxyl formation was still observed but the CO oxidation became more important as the temperature increased.
Figure 22.
(a) Model of the Pt(211) surface used in the DFT calculations. (b) Lattice for the Pt(211) used in the KMC simulations, adapted from [152], with permission from American Chemical Society, 2011.
Fajín and Cordeiro [153] studied, by periodic DFT calculations, the performance of Pt(5,3) nanotubes in WGS reaction. The results suggested that, on Pt(5,3) nanotubes, the associative mechanism, through the carboxyl intermediate followed by the carboxyl dehydrogenation, was active for WGS reaction. Comparison between the calculated activation energies and the adsorption energies demonstrated that the desorption processes were not competitive, moreover, the study revealed the low tendency to the sintering of the Pt(5,3) nanotubes. Lin et al. [154] investigated the WGS reaction mechanism, on the fcc(111) surfaces of the close-packed transition metals, such as Co, Ni, and Cu (from the 3d row), Rh, Pd, and Ag (from the 4d row) and Ir, Pt, and Au (from the 5d row), by means of DFT calculations. The results showed that three main reaction pathways were involved in the mechanism: redox, carboxyl, and formate (Table 10). The formation steps, in the three mechanisms, were energetically competitive, showing a similar decreasing trend both for ΔH and Ea from left to right and from up to down across the periodic table, supporting the experimental evidence, which show the best performance on the bottom-right d-block metals of Cu, Pt, and Au. Comparing the reaction pathways on these three metals, it turns out that on Au(111) surface, the redox pathway was dominant, while on Cu(111) and Pt(111) surfaces the three pathways contributed similarly in CO consumption.
Table 10.
Reaction mechanisms.
Clay et al. [155] reported the results of a DFT comparative study on five competing WGS reaction pathways on Pd and Pt(111) surface models. The computational results demonstrated that the reaction barriers on Pt were lower than on Pd, thus a higher intrinsic WGS reaction rate was predicted for on Pt. The kinetic models showed that the reaction proceeds via carboxyl intermediate, and the identification of the RDS was not simple: the progressive exothermicity of the CO binding, i.e., the decreasing of the CO binding energy with the increase of the coverage, suggested a shift of the RDS from water dissociation to carboxyl formation.
4.3.2. Unsupported Polymetallic Surface Models
Lian et al. [156] studied a series of promoted Pt-based catalysts with Pt3M formula (M = Pt, Cu, Mo, Ni, Rh) for WGS reaction, by DFT calculations. For all the Pt3M catalytic systems the preferred reaction pathway was the associative carboxylate-formate mechanism; three sequential elementary steps were involved: the O-H bond cleavage, the OCOH formation and the formate dissociation. The bimetallic clusters promoted the water decomposition more than the monometallic Pt4, moreover, Pt3Cu and Pt3Rh showed the best performance in WGS reaction. Guo et al. [157] investigated the reaction mechanism operating on TM@Cu12 core-shell nanoclusters (TM = Co, Rh, Ir, Ni, Pd, Pt, Cu, Ag, Au) for WGS reaction by DFT calculations, demonstrating that the carboxyl one was preferred. In the case of Pt@Cu12 clusters the rate-determining step was the formation of a complex between TM@Cu12 with CO2*(ads) and H2*(ads). Finally, the TOF obtained with the transition metals of the group 9 (Co, Rh and Ir) were higher than those obtained with the metals of group 10 (Ni, Pd and Pt) and 11 (Ag and Au).
Fang et al. [158] investigated the mechanism of the WGS reaction on Au32M6 (M = Cu, Pt, Pd, Rh, Ir) core-shell nanoclusters by DFT calculations. Based on the first-principles calculations of the geometry structure, the Au38 cluster was selected as the model sample to obtain the core-shell nanoclusters Au32M6. The results showed a larger charge transfer in the core-shell structure than in the Au38 nanocluster, moreover the calculated binding energies highlighted the higher stability of the Au32M6 nanoclusters. The d-orbital electrons of Au32Pd6 nanocluster were the closest to the Fermi level; the energy of the d-band center of Au32Pd6 was −2.93 eV, while those of Au32Pt6 and Au38 were −3.11 eV and −3.19 respectively. In all the nanoclusters the Au atom acted as electron acceptor, while the other metal as electron donor, in the adsorption of the reaction intermediates. The Au32Pd6 nanocluster exhibited the better electronic activity and the best catalytic activity, however for all the core-shell nanoclusters the reaction pathway was the same of that followed by Au32Pt6 (Table 11).
Table 11.
Activation energy (Ea) and reaction energy (ΔE) of elementary reactions on the Au32Pt6 nanocluster, data from [158].
Cao et al. [159] investigated the WGS reaction mechanism on Cu6TM clusters (TM = Co, Ni, Cu, Rh, Pd, Ag, Ir, Pt, Au). Carboxyl was the predominant mechanism in the case of TM = Co, Ni, Rh, Pd, Ir, and Pt, while formate mechanism was predominant in the case of TM = Cu, Ag and Au. The WGS reaction mechanism on a Cu-Pt-Au ternary alloy and the binary Cu-Au and Pt-Au catalytic systems were studied by Xue et al. [160], by means of DFT calculations. The results showed that Pty-Au(111) was more stable and exhibited a d-band center closer to Fermi level than Cux-Au(111). The ternary alloy Cu3-Pt3-Au(111) was more stable than the binary systems, showed the d-band center closest to Fermi level (−3.05 eV) and exhibited the one of the higher adsorption energy for CO and H2O. The RDS, the energy barriers, the reaction heat and the rate constant for the three investigated reaction mechanisms are reported in Table 12., showing that the lowest energy barrier and reaction heat were associated to formate mechanism, while the largest rate constant was associated to the redox mechanism.
Table 12.
Activation energy (Ea), reaction energy (ΔE) and rate constant (k) of rate determining steps (RDS) on Cu3-Pt3-Au(111), data from [160].
Really interesting is a recent published article in which AuM (M = Ni, Cu, Pt)-promoted LDHs catalysts were investigated, with both DRIFT and DFT studies, in WGS reaction. The in-situ DRIFTS study and DFT calculations elucidated the atomistic reaction mechanism, suggesting a formate-mediated reaction pathway coupled with a redox route, which proceeded by bypassing a direct *O-H bond breakage step [161].
4.3.3. TiO2-Supported Pt Models
The supported platinum catalysts are extremely interesting systems, as they combine the oxygen adsorption properties of platinum nanoparticles with the ability of the support to easily dissociate water [162].
One of the most studied catalytic system is platinum supported on TiO2; Heyden and co-workers published a series of article in which the interaction between the metal and the support and the behavior of the WGS reaction mechanism on Pt/TiO2 model, were studied. The nature of the interface between the electronic structure of Ptn (n = 1–8) clusters on the partially reduced TiO2(110) rutile surface in the WGS reaction was investigated by ab initio thermodynamic simulations [163]. Fully spin-polarized periodic DFT calculations, using the frozen-core all-electron projector-augmented-wave (PAW) method of Blöchl [164], as implemented in the VASP (Vienna ab-initio molecular-dynamics package) program, were performed. Choosing the PBE functional [165], within the GGA, to describe exchange and correlation effects. The results showed that the Pt atoms bound to the TiO2(110) surface, both stochiometric and partially reduced, through covalent interactions.
The clustering of Pt atoms, on the oxygen vacancies of rutile surface, gave a close-packed structure with (111) facet, and a less dense structure, with (100) facet away from oxygen vacancies. The rutile reducibility was promoted by Pt clusters, the smaller the Pt cluster size the greater the effect. Under WGS reaction conditions, the formation of surface oxygen vacancies at the interface was thermodynamically favoured, the Pt clusters (noninterfacial) were in the reduced state and probably covered/poisoned by CO, while only the interfacial Pt atoms were available sites for the reaction. The hydrogen adsorption was weak both on the interfacial Pt and oxygen atoms, suggesting that hydrogen spillover from the Pt to the rutile surface and the reverse process were thermodynamically possible. On the contrary, the formation of stable hydride species on the Pt atoms neighbouring oxygen vacancies of TiO2(110), which displace the interfacial OH groups, suggested a negative effect on the reaction rate, as supposed in previous work on similar catalytic systems [166].
In furthers studies the role of the TPB in determining the activity and selectivity of Pt8/TiO2(110) was investigated [167]. The results showed that the CO-promoted redox mechanism was dominant in the low temperature range 200–350 °C, while the redox mechanism was dominant above 400 °C. Based on the Campbell’s degree of rate control analysis [168], the overall reaction rate on TiO2 support was significantly determined by H2O dissociation and H-diffusion, while the CO adsorption on the Pt clusters appeared as a less important contributor. The calculated TOFs at the TPB for Pt/TiO2(110) were two orders of magnitude higher than the reported ones for P(111) [169], probably because TPB acted as unique site. The improved activity was explained as the result of a reduced CO adsorption strength on the Pt sites, an increased number of oxygen vacancies and an improved water activation and dissociation, at the TPB.
The redox and associative carboxyl pathways were also investigated at a corner Pt site of the Pt/TiO2 (110) interface [170], suggesting that the redox pathway was preferred in the temperature range 200–400 °C, in fact the calculated rates were six order of magnitude higher, suggesting that a promotion effect of the oxygen vacancies in the H2O adsorption and dissociation, was active. Pt atoms strongly adsorbed the CO thus the CO2 desorption resulted as RDS, moreover the corner Pt were less active than the edge Pt sites below 300 °C, however above 300 °C the activity was the opposite. A UQ framework was applied to WGS reaction, on a Pt8 cluster supported on a rutile TiO2 (110) surface, in order to quantify the uncertainties of various QoIs, obtained by microkinetic models developed from first principles, such as TOF, apparent activation barriers and reaction orders [171]. The free energy probabilistic model, was obtained by three different classes of functionals, such as GGA, meta-GGA, and hybrid functionals; the functionals used to obtain a correlations for the covariance matrix were PBE [165], the revised PBE functional [172], the Heyd−Scuseria functional [173] and the Minnesota functional (M06L) [174].
A Dirichlet distribution has been used to perform a thermodynamics correction [175], while the dominant catalytic cycle was identified by using Kullback−Leibler [176]; the results showed that the CO-promoted cycle was dominant in the temperature range 200–327 °C, the redox cycle was equivalent to CO-promoted above 327 °C, while formate and carboxyl pathway did not play any role. The UQ was also applied to investigate the most active site among Pt(111) terrace model and edge and corner interface model, in WGS reaction, for Pt supported on titatina catalysts [177]. The results showed that the edges were the active sites, and the CO-promoted redox mechanism was preferred below 300 °C, while above 300 °C the classical redox mechanism contributed the overall reaction rate. The water and the surface O-H bond dissociation were the RDS, however at low temperatures (230 °C) the CO adsorption became partially RDS, while at 300 °C the interface TiO2 oxygen vacancies formation became partially RDS in the redox pathway.
The WGS activity was studied also on atomically dispersed supported platinum in comparison to metallic platinum clusters on TiO2(110) [178]. The DFT calculations showed that the reaction rates and the apparent energy barriers for the Pt2+ sites were similar to those at the interface edge of Pt, while the RDS were similar to that at the corner Pt interface. The Pt2+ sites exhibited ligands characteristics similar to those of homogeneous catalysts, moreover the interface edge Pt sites and the Pt2+ sites were more active at low temperature, where the oxygen vacancies formation played a significant role, while the corner Pt sites were the most active above 300 °C. These results were confirmed in further studies, in which CO-adsorbed Pt atom supported on undoped-TiO2 surface showed lower activity than the Pt2+ supported on cation-doped TiO2, due to the higher oxygen vacancies formation energy calculated for the former [179].
4.3.4. CeO2-Supported Pt Models
Pt/ceria model is extremely interesting catalytic system for the WGS reaction, due to the ability of ceria to react and dissociate water on the oxygen vacancies or Ce3+ sites [180]. However, the role of oxygen vacancies in ceria-supported platinum catalysts, for the WGS reaction, is object of debate. Vecchietti et al. [181] studied the role of oxygen vacancies by doping ceria with gallium. The doping allowed to tune the ceria oxygen vacancies without affecting the metal dispersion. The higher activity of Pt/CeO2 with respect Pt/Ce80Ga20, showed an inverse relationship between the catalytic activity and the amount of oxygen vacancies. The DFT calculations results suggested that the water activation was not the RDS in these systems; moreover, the carboxyl mechanism, activated at the oxide-metal interface, was the preferred route (Figure 23). Bruix et al. [182] investigated Pt nanoparticles on CeO2(111) and CeOx/TiO2(110) model catalysts for WGS reaction, by valence photoemission experiments and DFT calculations. The UPS spectra showed a density of Pt 5d states close the Fermi level for Pt/CeO2(111) and Pt/CeOx/TiO2(110), which was much smaller than the expected for metallic platinum.
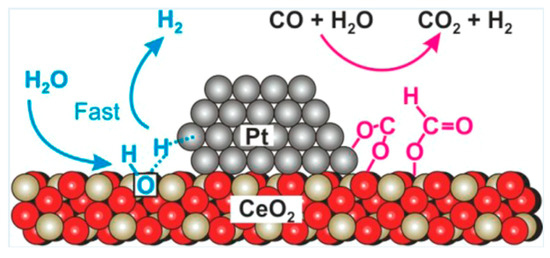
Figure 23.
Representation of the proposed mechanism, reproduced from [181], with permission from American Chemical Society, 2014.
The interaction between platinum and ceria enhanced the ability of the metal to adsorb water and dissociate the O–H bond. Comparing the water dissociation reaction on Pt(111) and Pt8/CeO2(111) an increase in the exothermicity was observed, due to a different geometry and charge distribution, which better accommodated the adsorbates. The catalytic activity in Pt8/CeO2(111) increased with the Pt coverage until ≈ 0.25 ML (Figure 24), and decreased for higher coverage, the XPS studies showed that during the WGS reaction the ceria support was partially reduced to CeO1.90–1.93, comparing these data with STM results reported for the Pt/CeO2(111) and Pt/CeO1.88(111) systems [183], the authors concluded that the highest activity corresponded to Pt particles size with a diameter in the range 0.4–1.7 nm.
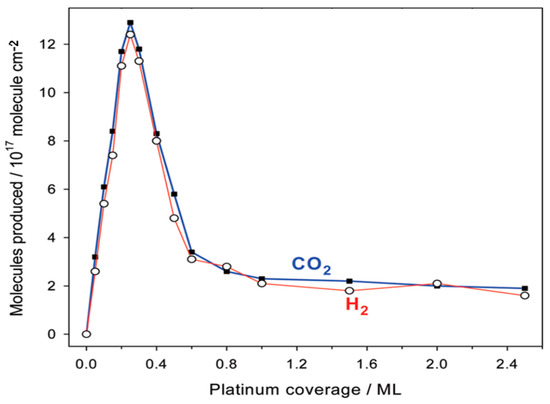
Figure 24.
WGS activities of model Pt/CeO2(111) catalysts as function of admetal coverage (CO/H2O = 2, T = 352 °C), adapted from [182], with permission from America Chemical Society, 2012.
Senanayake et al. [184] investigated the electronic metal support interaction of Ni and Pt nanoparticles on CeO2(111) surface, by core and valence photoemission. The results showed a large electronic perturbation for Pt nanoparticle in contact with ceria surface; the Pt/CeO2(111) system exhibited a density of the Pt 5d state, near the Fermi level, much smaller than the expected for metallic Pt. In particular, for Pt(111) a strong photoemission signal was observed in the range 0–1 eV, while for Pt/CeO2(111), at a coverage less than 0.25 ML, only a weak signal was observed. The electronic perturbation enhanced the ability of platinum nanoparticles to adsorb and to dissociate water, making it a highly active catalyst for WGS reaction. The exothermicity of the water dissociation increase from Pt(111) (ΔE = 0.65 eV) to Pt79 (ΔE = −0.03 eV) and even more to Pt8 (ΔE = −0.41 eV), thus the dissociation was favoured on Pt8, moreover the DFT calculations showed an enhanced ability of Pt8 to cleave the O-H bond, when the cluster was deposited on CeO2(111) or Ce40O80 surfaces. The Pt8/CeO2(111) can be considered a fluxional system which is able to change geometry and electronic distribution to accommodate the adsorbates, and which has a unique ability to dissociate water.
Additionally, in the case of Pt/CeO2 model catalysts Heyden and co-workers published a series of relevant articles. The growth pattern of small Ptn (n = 1–10) clusters on the stoichiometric and partially reduced ceria (111) surfaces, was investigated by DFT calculations [185]. The results of the constrained ab initio thermodynamic simulations showed that the clustering of Pt atoms, both on stoichiometric and partially reduced ceria, was a closed-packed structure with a (111) facet, and that the platinum promoted the reducibility of ceria surface. Under oxidizing conditions, the oxygen vacancies did not play a significant role, while under reducing conditions, both by CO and H2, oxygen vacancies and vacancy clusters were thermodynamically favoured. The non-interfacial Pt atoms were easily covered by CO, increasing the hydrogen adsorption energy and decreasing the CO adsorption at TPB, however the presence of CO did not change the ceria redox behavior. The hydrogen spillover to ceria surface was observed: the hydrogen changed the redox behavior of the ceria surface, making double vacancy clusters unstable in a H2/H2O environment. On the other hand, the redox cyclability of the hydroxylated ceria surface was improved by the presence of CO, which stabilized the double vacancy clusters of the Pt10/CeO2 catalyst. The CO-assisted associative carboxyl pathways of the WGS reaction, at the Pt/CeO2 (111) interface, where the creation of oxygen vacancies is not involved, was also investigated by periodic DFT calculations [186].
The microkinetic analysis, obtained from first principles, showed that the reaction rates were two orders of magnitude lower and the energy barriers were 0.2 eV higher than in the associative carboxyl pathway with redox regeneration, which involves the oxygen vacancies formation during the catalytic cycle. The reaction order towards H2O was 1.0, while the reaction order towards H2 was −1.0, moreover the Campbell’s degree of rate control analysis [168] suggested the −COOH dissociation as the RDS. Further studies [187] suggested that, contrary to what is normally hypothesizes about the role of the CO which could poison the metal surface, the CO adsorbed molecules were able to assist the WGS reaction at the neighbouring sites, by reducing the CO adsorption strength. Water dissociated preferentially at the oxygen vacancies by transfer of a H atom to the neighbouring surface oxygen, but when they were all covered the dissociation occurred to the metal-oxide interface. The microkinetic modelling suggested that two reaction pathways were operating, the redox and the associative carboxyl pathway with redox regeneration. Comparing the data of Pt/CeO2 with the data of Pt/TiO2, previously reported [167], it was possible to conclude that the stability of the oxygen vacancy increased the activation barrier and reduce the low-temperature activity.
4.3.5. MgO-Supported Pt and Bimetallic Supported Models
Carrasquillo-Flores et al. [188] published a mechanistic study on the Pt-Re/Vulcan XC-72 catalysts for WGS reaction. DFT analysis was used to obtain the parameters of the elementary steps on Pt(111) and Pt3Re(111) model surfaces, to build a comprehensive mean-field microkinetic model, to study the active sites. The comparison between the experimental observations and the DFT results, showed that the monometallic Pt(111) model was a good representation of the active site, in WGS reaction. More complicated was the case of the Pt-Re, for which the active site cannot be simply represented by Pt3Re (111) model surface, but more complex structures should be considered, such as mixed metal-metal oxides. Formate was identified as a by-product, while carboxyl as a key intermediate; although the OH-assisted decomposition of COOH in CO2 and H2 was favoured, the low OH coverage was a kinetic limitation, so the reaction proceeded by direct COOH decomposition.
Wang et al. [189] investigated the WGS reaction mechanism, by DFT calculations, on K-free and K-promoted Pt40 nanorod supported by ZrO2 model, as well as on K-free and K-modified Pt(111) surface. The calculation results demonstrated that carboxyl mechanism was preferred, and the H2O dissociation was the RDS. The Pt40/ZrO2 was more active than Pt(111) due to the presence of the support, which facilitated the H2O dissociation by strengthening the O-H bond at the transition and final state. The addition of K enhanced the WGS activity of Pt40/ZrO2 by reducing the apparent energy activation of the reaction as well as of the energy barriers of the single steps. The promotion effect of K was attributed to the stabilization at the transition state, by K-O bond formation, of the oxygenated species derived from the H2O and COOH dissociation. On the contrary the addition of K to Pt(111) was detrimental for the COOH dissociation, however, a promotion effect was active in the case of the H2O dissociation, despite less than in the case of Pt40/ZrO2, suggesting a structure sensitivity.
Ghanekar et al. [190] studied the WGS mechanism at the interface between a “quasi-one dimensional” platinum nanowire and an irreducible MgO support, by using a combination of periodic DFT calculations and microkinetic modelling. The results showed that the reaction proceeded almost exclusively at the Pt/MgO interface. Moreover, the CO coverage was able to change the reaction pathway, which switched from redox to carboxyl mechanism, when this effect is considered.
4.3.6. Conclusions
Summarizing, although the studies reviewed in Section 4.3.1 suggested a strong correlation between the reaction conditions and the operating mechanism on Pt surface models, the carboxyl route seems to be preferred, however, on Pt(111) surface under H2O excess the redox mechanism showed a non-negligible rate. Moreover, in the case of the bimetallic surface models, the carboxyl route is preferred, while in the case of the Pt-Au systems the redox mechanism become not negligible. The studies on TiO2-supported Pt models, showed a correlation between the temperature and the operating reaction mechanism, in particular at high temperature the classical redox mechanism is preferred, while at low temperature the CO-promoted redox mechanism is operating. On the other hand, in the case of CeO2-supported Pt models, the operating reaction mechanism seems to be strongly affected by the presence of ceria, this catalytic system can be considered a “fluxional system”, which is able to accommodate the adsorbates, by adapting the geometry and the electronic distribution. The optimal Pt particle size distribution (0.4–1.7 nm), as well as the Pt coverage (0.25 ML), was identified, to reach the best catalytic activity. The redox and the associative carboxyl pathway with redox regeneration seems to be contemporary active at the neighbouring sites, while the carboxyl mechanism at the oxide-metal interface. However, since this catalytic system is of great interest, further studies seem to be necessary to better clarify the reaction mechanisms. Finally, in the case of MgO-supported Pt and bimetallic supported, the carboxyl route seems to be preferred; the presence of a second metal such as rhenium complicate the identification of the active site model surface, while the doping with potassium allows to reduce the apparent energy activation.
5. Deactivation Studies
As discussed before, standard WGS processes require two separate CO conversion steps: HTS, usually operated in the range 310–450 °C with a Fe/Cr catalyst, and LTS, within the temperature range 200–250 °C and in presence of a more active Cu/ZnO catalyst. These conventional formulations have many drawbacks, including their pyrophoricity, low thermal stability, suffering to prolonged exposures to condensed water, which are related both to the frequent start-up and shut-down procedures and to the reaction environment. Furthermore, of course they undergo poisoning if exposed to sulphur-containing streams, and this condition easily occurs when a WGS step is associated to an IGCC (integrated gasification combined cycle) process. All these factors cause severe deactivation of the WGS catalysts, which is one of the main issues related to the WGS process.
Various deactivation mechanisms have been suggested over the years, including formation of carbonates or thiols (in the case of sour conditions) on the catalyst surface, which inhibit the activity of the metallic sites, and sintering of the active specie, which reduces the number of the active sites for the reaction [191]. While in the first case the nature of the active specie and of the support and their interaction plays a key role, in the latter the catalyst structure acquires relevance and a recent solution for improving the thermal stability of metal nanoparticles is represented by their encapsulation within an oxide lattice, such as silica or alumina [192].
Concerning the commercial catalysts, none of the conventional formulations seems to be suitable in sour condition WGS, and in the past ten years noble metal catalysts started to be considered as a promising solution [193]. High temperature WGS in sour conditions was studied by Liu et al. with a low-content Pt/CeO2 catalyst. The authors reported that when increasing the Pt content from 0.38% to 2% there is both an increase in activity and a decrease in selectivity; furthermore, the higher the Pt loading, the higher was the catalyst resistance to H2S deactivation in the whole investigated temperature range. Nevertheless, because of the strong drop in selectivity observed with the 2% Pt/CeO2 sample, the authors performed a 300 h stability test over 1% Pt/CeO2 catalyst: the study pointed out that, after a fast decrease in CO conversion in the first 10 h and a further slight reduction for the following 90 h, the sample reached a value of 73.3% that was then kept stable for the last 200 h (Figure 25). The authors suggested that Pt sites strongly promote OH groups formation in their neighbourhood, thus reducing the H2S dehydrogenation in the proximity of the active sites, simplifying its desorption before thiol groups could be formed. Thus, the proposed sulphur tolerance mechanism is a competitive H2S and H2O adsorption on the ceria surface that led to the formation of OH and SH groups; the hydrogen that adsorbs on Pt spills over to the support surface, promoting SH species desorption and H2S re-formation: in this way, the catalytic activity of Pt/CeO2 catalyst is preserved [193].
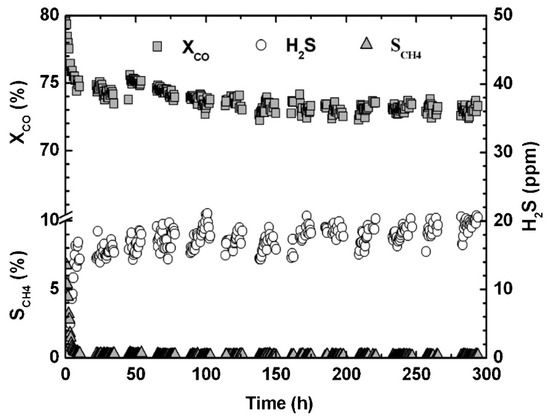
Figure 25.
300 h stability test over 1% Pt/CeO2 catalyst in the presence of 20 ppm H2S at 400 °C and S/CO = 2, reproduced or adapted from [193], with permission from Elsevier, 2011.
Further related evaluations were conducted in a following study, where the authors focused on core-shell structured catalysts Pt@CeO2 and Pt@CeO0.67Zr0.33O2: the samples were prepared in order to obtain platinum encapsulated in a ceria shell, in order to avoid a direct contact between Pt and H2S. The catalysts were found to be highly selective and with a near to equilibrium conversion at 450 °C: Pt@CeO2 showed the best activity results in non-sour conditions, while Pt@CeO0.67Zr0.33O2 displayed the best performances in sour conditions. The authors ascribed this result to the larger pore size of ceria, which was beneficial for reactants but also for H2S to pass through to platinum; the smaller pores of Pt@CeO0.67Zr0.33O2 determined instead its stronger ability to resist H2S [194].
The stability of a commercial formulation provided by Johnson Matthey was investigated in sour conditions by Neto et al. [195] The work showed that 1 ppm of H2S is not sufficient to deteriorate the catalyst for 100 h, in none of the tested operating conditions; the further increase in H2S concentration to 300 ppm did not cause deactivation as well, at least for the evaluated time of 150 min. Silva et al. [196] studied several Pt supported catalysts suitable for high temperature WGS, reporting that while in clean WGS conditions the best results were achieved by the samples with highly reducible supports (ceria and ceria-zirconia mixed oxides), Pt/CeO2 formulation seemed to be the most affected by sulphur poisoning, demonstrating the highest sour deactivation and the minimum recovery of the catalytic activity. As the deactivation increased in the order Ce0.25Zr0.75O2 < γ-Al2O3 < ZrO2 < Ce0.75Zr0.25O2 < CeO2, Pt/Ce0.25Zr0.75O2 was selected as the sample with the greatest potential to be applied to sour WGS.
WGS in sour condition has been recently investigated by Silva et al. [197] also with Nb2O5 supported catalysts: in a first study, they reported the absence of deactivation phenomena for a Pt/Nb2O5 in presence of 50 ppm of H2S in a 2.5 h stability test; a subsequent investigation reports instead a comparison of different active species (Pt, Cu and Au) supported on Nb2O5. Pt/Nb2O5 exhibited an intermediate activity during the clean WGS test (with Au giving the best activity results) but while in the case of the Au sample the activity dropped to zero in the presence of H2S, the Pt catalysts presented no deactivation for the whole 4 h test. Because of the high sulphur resistance, the catalyst was tested also with an H2S concentration of 1000 ppm and the activity still remained unvaried; with the introduction of a clean stream after the sulphur resistance test was, instead, observed an increase in the activity that did not decrease for the following 20 h. While the recovery of the initial activity could be explained by the low stability of the S-Pt bond, the enhancement in the catalytic performance after sulphur exposure remains not clarified: a possible reason could be the formation of niobium sulphide in the catalyst surface, which could act as new active sites [198]. As discussed in the first part of this paragraph, sulphur exposure is not the only driving factor for a WGS catalyst deactivation. Frequent stop-start operations are another aspect which strongly affects the lifetime of a WGS catalyst, so several studies were conducted also in sour component-free atmosphere.
Lombardo et al. [199] tested in HTS conditions (400 °C) different La-containing formulations: Rh(0.6)/La2O3(27)SiO2, Rh(0.6)/La2O3 and Pt(0.6)/La2O3(27)SiO2. A stability test performed for 50 h showed that the former formulation did not exhibit any change in CO rate, while the latter, which presented a higher initial CO rate, suffered of a strong deactivation. The Pt(0.6)/La2O3(27)SiO2 was confirmed as the most active and selective catalyst, showing a remarkably high resistance to deactivation; in fact it was tested under three different steam/CO ratios and six cycles of start-stop operations and it did not show any change in CO conversion. Rajesh et al. reported a study on the stability of a BaCe0.98Pt0.2O3−∂ catalyst in which Pt is present in +2 oxidation state and it is incorporated into the lattice. The authors performed three sets of experiments, testing three different shut-down conditions: each test was composed of two cycle of activity tests in the range of 300–400 °C, with each test taking approximately 2.5 h: the two cycles were separated by one of the selected shut-down procedures. The catalyst was found to remain unvaried in ionic form under reaction conditions and under reducing start-up/shut-down operations. The authors stressed the catalyst by applying shut-down conditions selected on the basis of several reported deactivation mechanisms, including the sintering of active species, carbonate formation which leads to inhibition of the active sites, and Ce over reduction. They concluded that the presence of Ba in the lattice may have helped with the carbonate formation and that the peculiar perovskite structure seemed to tolerate Ce reduction, preventing the structural issues occurring in CeO2 supported catalysts [191]. A summary of the reviewed studies treated in this paragraph is given in Table 13.
Table 13.
Summary of the evaluated deactivation conditions reported in Section 5: Deactivation Studies.
6. Electrochemical Promotion
Electrochemical promotion of catalysis (EPOC), also named NEMCA (non-Faradaic electrochemical modification of catalytic activity) effect, is a very promising concept for boosting catalytic processes [200]. This general, well-established phenomenon in catalysis aims at controlling in-situ both the activity and the selectivity of a catalyst through application of electric stimuli [201]. EPOC is observed in presence of solid electrolyte materials and porous catalysts. Ions contained in these electrolytes (O2−, H+, Na+ OH−, etc.) are electrochemically pumped to the catalyst surface, where they act as promoting species, so modifying the catalyst’s electronic properties and consequently its catalytic activity and selectivity. In more detail, the application of an anodic polarization has the consequence of strengthening electron-donor adsorbates and weakening the binding strength of electron-accepting adsorbates [201]. The resulting electrochemical activation magnitude is much higher than that predicted by Faraday’s law. The apparent Faradaic efficiency, Λ, is expressed by Equation (8).
where: ∆r is the increase in the catalytic rate, I is the current, F is the Faraday’s constant and n is the electrons number. A process is considered non-Faradaic when |Λ| is higher than 1. Another parameter commonly used to quantify EPOC is the rate enhancement ratio, ρ, expressed as the ratio between the promoted closed-circuit catalytic rate, r, and the unpromoted open-circuit catalytic rate, ro. Compared to classical promotion, the polarization can control the in-situ coverage of the promoting species, and it allows the in-situ modification of the promoter amount under working reaction conditions. On the contrary, in the case of the classical promotion, the control of the promoter coverage on the active phase cannot be exactly performed, as part of the promoter is deposited on catalytically inert support areas. Moreover, the amount of promoter is fixed before catalyst exposure to reaction conditions and it cannot be modified afterwards [202]. Nevertheless, EPOC has not yet been directly applied in the industry despite recent developments on electrochemical reactors [200]. The main problem is that this technology presents a far lower activity per catalyst mass than that of nano-dispersed powdered catalysts, which exhibit much higher specific surface areas and metallic dispersions. Therefore, many research efforts have been focused on the use of EPOC to intensify several catalytic reactions, including the most important H2 production reactions [202]. In the case of the WGS reaction, different researches were performed by using porous Pt catalysts.
Souentie et al. [203], studied the effect of EPOC by using a closed-end tube of 8 mol% Y2O3-stabilized ZrO2 (YSZ) (13 mm outer diameter and 2 mm thickness) as solid electrolyte. Both on the inner and the outer side of the tube, a thin coating of Pt organometallic paste (followed by calcination in air for 12 h at 800 °C) was deposited, so realizing, respectively, the Pt electrode serving as the counter (and/or reference) electrode, and the Pt electrode serving as the working electrode. The final metal loading was 0.83 mg Pt/cm2, while the geometrical area of the electrode was about 3 cm2, and the catalytically active surface area was about 1 mmol Pt. The authors used a fuel cell type electrochemical reactor, and the reaction was performed in the temperature range 300–400 °C, and at values of the PH2O/PCO ratio in the range 2.85–31. The results evidenced that under open-circuit and polarization conditions the order dependence of the catalytic reaction rate was negative on PCO and positive on PH2O. Moreover, the results highlighted that a negative potential application (−1.5 V) caused the sensible increase (up to 200%) of the catalytic reaction rate, with apparent Faradaic efficiency values up to 110. The authors attributed this positive effect of the cathodic polarization to (i) the weakening of the Pt–CO bond strength and (ii) the increase in surface concentration of O2 ion vacancies near the Pt-gas-support three-phase boundaries.
Sekine et al. [204], investigated catalytic WGS reaction with and without an electric field, in order to elucidate the effect of the electric field to the catalytic reaction. The catalyst was 1wt%Pt/10 mol%La–ZrO2. The results showed that the electric field had a positive effect on the reaction, since a drastic kinetic promotion was observed, with the decrease of apparent activation energy from 98.3 kJ·mol−1 for the reaction without the electric field to 50.9 kJ·mol−1 for the reaction in the electric field. The authors attribute this important result to the change of reaction mechanism, since in the electric field, CO adsorption weakened and redox mechanism using surface lattice oxygen proceeded. More recently, Cui et al. [205] studied a room-temperature electrochemical water–gas shift process, where the water is reduced to H2 at the cathode and the CO is oxidized at the anode. The authors reported the production of high purity hydrogen (over 99.99%) with a Faradaic efficiency of approximately 100%. The authors combined experiments with theoretical calculations for the optimization of the anode structure to facilitate CO diffusion and of the PtCu catalyst to optimize CO adsorption. The authors prepared the catalyst by supporting Pt nanoparticles on well-graphitized carbon nanotubes (Pt@CNTs, 40 wt%), previously treated in H2 at 400 °C to remove the oxygen containing groups from the CNTs surface and improve hydrophobicity. Subsequently, the catalyst was decorated with 1.5 μg/cm2, aiming to build water-free compartments at the interface, of PTFE and Pt, to promote CO diffusion and facilitate CO colliding with the surface sites. The results showed that the process can be successfully performed with an anodic onset potential of about 0 volts (versus the reversible hydrogen electrode) at room temperature and atmospheric pressure. Moreover, the optimized PtCu catalyst achieved a current density of 70.0 mA/cm2 at 0.6 volts (12 times higher than that of commercial Pt/C (40 wt%) catalyst), and remained stable for even more than 475 h. The main results are summarized in Table 14.
Table 14.
Summary of preparation procedures and operative conditions used in electrochemical promotion of catalysis (EPOC) applied to WGS reaction.
7. Pt-Based Catalysts for Medium Temperature Single Stage WGS Process
An extremely relevant aspect in the evaluation of platinum-based catalysts for the WGS reaction is the performance comparison with the conventional catalysts used for the HTS and LTS processes, as a prerequisite for replacement in the production processes. In this field, the catalyst cost is considered a critical issue: in 2003 Ladebeck and Wagner [206] reported an estimation about the relative costs of precious metals with respect to the conventional catalysts. The estimated cost of conventional catalysts, e.g., Fe/Cr/Cu and Cu/Zn catalysts were respectively 10.6 $ and 17.3 $ per liter; while, for a noble metal-based catalyst, assuming a metal loading of 2 wt% of the washcoat, the noble metal cost was the 40% of the total cost of the catalyst. Hence, considering a noble metal cost of 600 $ troy/ounce, on an equal volume basis the noble metal was about 14 times of the conventional catalysts; thus, the metal catalysts activity should be increased 10 times to be competitive. In the last 15 years the price of the metals has considerably fluctuated, however from 2006 up to now the price of copper, zinc, iron and platinum decreased of 20–30% each [207]; therefore, it can be assumed a negligible relative costs variation. Therefore, the problem of the high cost of the platinum-based catalysts has remained substantially unchanged. On the other hand, the catalytic activity represents another important issue that lead to remarkable differences between the double-stage and the single stage process, so the evaluation of the catalytic performances is more complex. Indeed, as highlighted in the introduction, one of the critical issues of the conventional WGS process is the double stage configuration, which can be considered not suitable for some production processes, such as the distributed hydrogen production. The design of a single-stage WGS process clashes with the problem of identifying new catalytic formulations, active in a wider temperatures range than conventional catalysts. Platinum-based catalysts meet the desired characteristics, being highly active in the medium-temperature range (250–400 °C). Another important aspect is the platinum loading, recent published work has studied the optimization of the catalysts [182], showing that good performance can be reached with low platinum loading (≈1 wt%) [46,107]. Furthermore, the exothermicity of the reaction might be managed to drive down the outlet temperatures, through the use of micro-channel reactors [208] and highly conductive structured catalysts [107]. With particular attention to the latter solution, although with a higher cost and a lower activity, in some temperature ranges the platinum-based catalytic formulations seem to be the only viable route [22].
8. Conclusions
In this short review, a comprehensive literature survey of the last decade on Pt-based catalysts in the WGS reaction has been provided. The effect of the catalyst’s preparation on the catalytic behavior in terms of platinum loading, support characteristics, preparation technique and promotion effect have been accounted for in the first part of the review article. The preparation technique influences the catalysts activity and stability in several ways, such as preventing the particles sintering, strengthening the platinum/support surface interaction, and allowing a high contact surface between the catalyst and the reactant mixture. The finding of the optimal platinum loading allows us to tune the particle size and distribution: a too low loading could provide an insufficient amount of active phase on the support surface; on the contrary, a too high loading could lead to an excessive reduction of the cationic platinum and to particle aggregation, which are detrimental for activity in the WGS reaction, locating the optimal loading to ≈ 1 wt%. The supports also play a fundamental role, as the oxygen mobility is strictly involved in the WGS reaction mechanism. The effect of promoters in boosting the catalytic activity has also been shown, with particular emphasis to electrochemical promotion, also named the NEMCA (non-Faradaic electrochemical modification of catalytic activity) effect, in which an in-situ control of the activity and selectivity is obtained through the application of electric stimuli. Compared to classical promotion, the polarization can control the in-situ coverage of the promoting species, by allowing the in-situ modification of the promoter amount under working reaction conditions. On the contrary, in the case of the classical promotion, the control of the promoter coverage on the active phase cannot be exactly performed, as part of the promoter is deposited on catalytically inert support areas. Moreover, the amount of promoter is fixed before catalyst exposure to reaction conditions and it cannot be modified afterwards.
A series of relevant articles has been reviewed, focusing on the kinetic behavior, finding that the Pt/Mo2C-based catalytic systems have the lowest activation energy (Ea of 38 kJ·mol−1). As widely reported, WGS reaction takes place on the Pt-support sites at the periphery of the interface; thus, increasing the Pt dispersion with a consequent enhancement of the Pt-support interfacial sites is a crucial strategy to improve reaction rates. In this regard, the synthesis of catalysts with atomic Pt sites increased the number of hydroxyls on the catalyst surface, which act as oxidizing agents for carbon monoxide. The catalyst reducibility and population of oxide vacancy centers (active centers for water adsorption) was also shown to affect CO conversion rate. Thus, the addition of promoters or second metals to Pt-supported catalysts was mainly addressed to increase the number of active sites at the metal-support interface for CO adsorption. Articles on the reaction mechanisms were also reviewed, reporting both experimental and DFT studies. The WGS reaction operates in a bifunctional manner over the supported metal catalysts: both the metal sites and the support take part in the reaction, with the metal always adsorbing CO and the metal/support behavior giving rise to mainly two different mechanisms, the redox and the associative mechanisms. What emerges from the review is that the redox mechanism can only occur in the presence of highly reducible supports, such as ceria, while it is impossible to state a clear correlation between the catalyst formulation and the kind of associative mechanism, which can offer very different reaction paths. Polymetallic formulations and metals doping represent widely adopted routes in the improvement of the WGS catalysts performances, mainly achieved as a consequence of the reaction mechanisms modifications obtained.
A series of computational studies on Pt surfaces and Pt clusters supported on TiO2 and CeO2 have also been reviewed, to identify the active site and the reaction mechanism and clarify the role of the support. In the case of metal surfaces, a preference for the carboxyl mechanism was found, while the case of supported models was more complicated. In presence of titania as support, the redox mechanism seems to be preferred at high temperatures, while the CO-promoted redox mechanism is preferred at low temperature. The case of ceria is very intriguing, where the reducibility of the support could play a crucial role; the reported studies suggested an inverse relationship between the catalytic activity and the amount of oxygen vacancies; moreover, the preferred WGS mechanism seems to be the carboxyl one, however the redox pathway also seems to play a role. Finally, a series of deactivation studies were also discussed. Conventional WGS catalysts, both for HTS and LTS, suffer of severe deactivation when exposed to sulphur atmospheres and being subjected to frequent start-up/shut-down operations. The substitutions of the most common formulation with platinum-based catalysts offers a concrete possibility to obtain catalysts with high activity and a remarkable resistance to deactivation.
The reviewed works reported interesting results also for stability test time up to 300 h, and very stressful reaction conditions (H2S concentration up to 1000 ppm), identifying Pt as a promising active speciefor WGS durable catalysts. Although the duration of 300 h is not comparable to the characteristic times of industrial applications, based on a predictive model, the reported trend would suggest stability for long time periods, however further studies would be necessary. Finally, a short paragraph (Pt-Based Catalysts for Medium Temperature Single Stage WGS Process) was devoted to underline the future prospective in design the WGS single-stage process, to which the platinum-based catalysts are addressed.
Author Contributions
All authors equally contributed to the conceptualization, methodology, software, validation, formal analysis, investigation, resources, data curation, writing-original draft preparation, writing-review and editing, visualization, supervision, project administration and funding acquisition of this manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work has received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 734561.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| Acronym | Extended term |
| ALD | Atomic layer deposition |
| BET | Brunauer–Emmett–Teller |
| CNTs | Carbon nanotubes |
| DFT | Density functional theory |
| DRIFTS | Diffuse reflection infrared Fourier transform spectroscopy |
| EPOC | Electrochemical promotion of catalysis |
| GGA | Generalized gradient approximation |
| GHSV | Gas Hourly Space Velocity |
| HOPG | Highly oriented pyrolytic graphite |
| HTS | High-temperature water-gas shift |
| IGCC | Integrated gasification combined cycle |
| KMC | Kinetic Monte Carlo |
| LDHs | Layered double hydroxides |
| LTS | Low-temperature water-gas shift |
| MCNTs | Multiwalled carbon nanotubes |
| MOF | Metal-organic frameworks |
| NEMCA | Non-Faradaic Electrochemical Modification of Catalytic Activity |
| NPs | Nanoparticles |
| OSC | Oxygen storage capacity |
| PBE | Perdew–Burke–Ernzerhof functional |
| PRGO | Partially reduced graphite oxide |
| PRO | Partially reducible oxide |
| QoIs | Quantities of interest |
| RDS | Rate determining step |
| RSDT | Reactive spray deposition technology |
| SAC | Single-atom catalyst |
| SSA | Specific Surface Area |
| TEM | Transmission Electron Microscopy |
| TMCs | Transition metal carbides |
| TOF | Turnover frequency |
| TPB | The three-phase boundary |
| UPS | UV photoelectron spectroscopy |
| UQ | Uncertainty quantification |
| WGS | Water gas shift |
References
- Jacobs, G.; Davis, B.H. Low Temperature Water-Gas Shift Catalysts; Royal Society of Chemistry: London, UK, 2008; pp. 122–285. [Google Scholar]
- Abe, M.; Iriki, T.; Yoshida, N. Ammonia Production from Soluble-N Fraction of Various Feedstuffs When Incubated with Rumen Fluid. Nihon Chikusan Gakkaiho 1992, 63, 721–727. [Google Scholar] [CrossRef]
- Vu, M.-H.; Sakar, M.; Do, T.-O. Insights into the Recent Progress and Advanced Materials for Photocatalytic Nitrogen Fixation for Ammonia (NH3) Production. Catalysts 2018, 8, 621. [Google Scholar] [CrossRef]
- Patart, G. Procédé de Production d’Alcools, d’Aldéhydes et d’Acides à Partir de Mélanges Gazeux Maintenus sous Pression et Soumis à l’Action d’Agents Catalytiques ou de l’Électricité’. Fr. Pat. Appl. 1922, 540, 543. [Google Scholar]
- Dawood, F.; Anda, M.; Shafiullah, G.M. Hydrogen production for energy: An overview. Int. J. Hydrog. Energy 2020, 45, 3847–3869. [Google Scholar] [CrossRef]
- LeValley, T.L.; Richard, A.R.; Fan, M. The progress in water gas shift and steam reforming hydrogen production technologies—A review. Int. J. Hydrog. Energy 2014, 39, 16983–17000. [Google Scholar] [CrossRef]
- Eigenberger, G. Catalytic Fixed-Bed Reactors. Handb. Heterog. Catal. 2008, 10, 2075–2106. [Google Scholar] [CrossRef]
- Bhaskar, T.; Balagurumurthy, B.; Singh, R.; Poddar, M.K. Thermochemical Route for Biohydrogen Production. In Biohydrogen; Pandey, A., Chang, J.-S., Hallenbeck, P.C., Larroche, C., Eds.; Elsevier: San Diego, CA, USA, 2013; pp. 285–316. [Google Scholar]
- Odabaşı, Ç.; Günay, M.E.; Yıldırım, R. Knowledge extraction for water gas shift reaction over noble metal catalysts from publications in the literature between 2002 and 2012. Int. J. Hydrog. Energy 2014, 39, 5733–5746. [Google Scholar] [CrossRef]
- Bosch, C.; Wild, W. Hydrogen Production. U.S. Patent 1,115,776, 3 November 1914. [Google Scholar]
- Zhu, M.; Wachs, I.E. Iron-Based Catalysts for the High-Temperature Water–Gas Shift (HT-WGS) Reaction: A Review. ACS Catal. 2015, 6, 722–732. [Google Scholar] [CrossRef]
- Dienes, E.K. Low Temperature Shift Reaction Involving a Zinc Oxide-Copper Catalyst. U.S. Patent No. 3,303,001, 7 February 1967. [Google Scholar]
- Gradisher, L.; Dutcher, B.; Fan, M. Catalytic hydrogen production from fossil fuels via the water gas shift reaction. Appl. Energy 2015, 139, 335–349. [Google Scholar] [CrossRef]
- Kam, R.; Scotta, J.; Amal, R.; Selomulya, C. Pyrophoricity and stability of copper and platinum based water-gas shift catalysts during oxidative shut-down/start-up operation. Chem. Eng. Sci. 2010, 65, 6461–6470. [Google Scholar] [CrossRef]
- Pal, D.; Chand, R.; Upadhyay, S.; Mishra, P.K. Performance of water gas shift reaction catalysts: A review. Renew. Sustain. Energy Rev. 2018, 93, 549–565. [Google Scholar] [CrossRef]
- Bunluesin, T.; Gorte, R.; Graham, G. Studies of the water-gas-shift reaction on ceria-supported Pt, Pd, and Rh: Implications for oxygen-storage properties. Appl. Catal. B Environ. 1998, 15, 107–114. [Google Scholar] [CrossRef]
- Palma, V.; Pisano, D.; Martino, M. Comparative Study Between Aluminum Monolith and Foam as Carriers for The Intensification of The CO Water Gas Shift Process. Catalysts 2018, 8, 489. [Google Scholar] [CrossRef]
- Nguyen-Phan, T.-D.; Baber, A.E.; Rodriguez, J.A.; Senanayake, S.D. Au and Pt nanoparticle supported catalysts tailored for H2 production: From models to powder catalysts. Appl. Catal. A Gen. 2016, 518, 18–47. [Google Scholar] [CrossRef]
- Flytzani-Stephanopoulos, M. Supported metal catalysts at the single-atom limit—A viewpoint. Chin. J. Catal. 2017, 38, 1432–1442. [Google Scholar] [CrossRef]
- Hwang, K.-R.; Lee, S.-W.; Lee, D.-W.; Lee, C.-B.; Ji, S.-M.; Park, J.-S. Bi-functional hydrogen membrane for simultaneous chemical reaction and hydrogen separation. Int. J. Hydrog. Energy 2014, 39, 2614–2620. [Google Scholar] [CrossRef]
- Moncada, N.G.; Castaño, M.G.; Ivanova, S.; Centeno, M.Á.; Romero-Sarria, F.; Odriozola, J. New concept for old reaction: Novel WGS catalyst design. Appl. Catal. B Environ. 2018, 238, 1–5. [Google Scholar] [CrossRef]
- Palma, V.; Ruocco, C.; Cortese, M.; Martino, M. Recent Advances in Structured Catalysts Preparation and Use in Water-Gas Shift Reaction. Catalysts 2019, 9, 991. [Google Scholar] [CrossRef]
- Pastor-Pérez, L.; Ramos-Fernández, E.; Sepúlveda-Escribano, A. Effect of the CeO2 synthesis method on the behaviour of Pt/CeO2 catalysis for the water-gas shift reaction. Int. J. Hydrog. Energy 2019, 44, 21837–21846. [Google Scholar] [CrossRef]
- Pastor-Pérez, L.; Belda-Alcázar, V.; Marini, C.; Pastor-Blas, M.M.; Sepúlveda-Escribano, A.; Ramos-Fernandez, E.V. Effect of cold Ar plasma treatment on the catalytic performance of Pt/CeO2 in water-gas shift reaction (WGS). Appl. Catal. B Environ. 2018, 225, 121–127. [Google Scholar] [CrossRef]
- Palma, V.; Pietrosanto, A.; Martino, M.; Reverchon, E.; De Marco, I. Supercritical Antisolvent Process to Produce Cerium Oxide Nanoparticles as a Support for High-Activity Platinum Catalysts. Chem. Eng. Trans. 2017, 57, 967–972. [Google Scholar] [CrossRef]
- Franco, P.; Martino, M.; Palma, V.; Scarpellini, A.; De Marco, I. Pt on SAS-CeO2 nanopowder as catalyst for the CO-WGS reaction. Int. J. Hydrog. Energy 2018, 43, 19965–19975. [Google Scholar] [CrossRef]
- Potdar, H.S.; Jeong, D.-W.; Kim, K.-S.; Roh, H.-S. Synthesis of Highly Active Nano-Sized Pt/CeO2 Catalyst via a Cerium Hydroxy Carbonate Precursor for Water Gas Shift Reaction. Catal. Lett. 2011, 141, 1268–1274. [Google Scholar] [CrossRef]
- Roh, H.-S.; Potdar, H.S.; Jeong, D.-W.; Kim, K.-S.; Shim, J.-O.; Jang, W.-J.; Koo, K.Y.; Yoon, W.L. Synthesis of highly active nano-sized (1 wt.% Pt/CeO2) catalyst for water gas shift reaction in medium temperature application. Catal. Today 2012, 185, 113–118. [Google Scholar] [CrossRef]
- Im, H.B.; Kwon, S.J.; Byun, C.K.; Ahn, H.S.; Koo, K.Y.; Yoon, W.L.; Yi, K.B. Effect of Support Geometry on Catalytic Activity of Pt/CeO2 Nanorods in Water Gas Shift Reaction. Trans. Korean Hydrog. New Energy Soc. 2014, 25, 577–585. [Google Scholar] [CrossRef]
- Mei, Z.; Li, Y.; Fan, M.; Zhao, L.; Zhao, J. Effect of the interactions between Pt species and ceria on Pt/ceria catalysts for water gas shift: The XPS studies. Chem. Eng. J. 2015, 259, 293–302. [Google Scholar] [CrossRef]
- Tang, H.; Sun, H.; Chen, D.; Jiao, X. Fabrication of Pt/CeO2 nanofibers for use in water–gas shift reaction. Mater. Lett. 2012, 77, 7–9. [Google Scholar] [CrossRef]
- Jeong, D.-W.; Potdar, H.S.; Roh, H.-S. Comparative Study on Nano-Sized 1 wt% Pt/Ce0.8Zr0.2O2 and 1 wt% Pt/Ce0.2Zr0.8O2 Catalysts for a Single Stage Water Gas Shift Reaction. Catal. Lett. 2012, 142, 439–444. [Google Scholar] [CrossRef]
- Palma, V.; Pisano, D.; Martino, M.; Ricca, A.; Ciambelli, P. Comparative studies of low temperature water gas shift reaction over platinum based catalysts. Chem. Eng. Trans. 2014, 39, 31–36. [Google Scholar] [CrossRef]
- Castaño, M.G.; Reina, T.R.; Ivanova, S.; Centeno, M.Á.; Odriozola, J. Pt vs. Au in water–gas shift reaction. J. Catal. 2014, 314, 1–9. [Google Scholar] [CrossRef]
- Zhang, Y.; Cai, J.; Liu, Y.; Wang, X.; Au, C.-T.; Jiang, L. Preparation of sintering-resistant Pt nanocatalysts by dopamine mediation for water-gas shift reaction. Appl. Surf. Sci. 2019, 496, 1–8. [Google Scholar] [CrossRef]
- Wan, C.; Cheng, D.; Chen, F.; Zhan, X. Fabrication of CeO2 nanotube supported Pt catalyst encapsulated with silica for high and stable performance. Chem. Commun. 2015, 51, 9785–9788. [Google Scholar] [CrossRef] [PubMed]
- Stöber, W.; Fink, A.; Bohn, E. Controlled growth of monodisperse silica spheres in the micron size range. J. Colloid Interface Sci. 1968, 26, 62–69. [Google Scholar] [CrossRef]
- Roh, H.-S.; Jeong, D.-W.; Kim, K.-S.; Eum, I.-H.; Koo, K.Y.; Yoon, W.L. Single Stage Water–Gas Shift Reaction Over Supported Pt Catalysts. Catal. Lett. 2010, 141, 95–99. [Google Scholar] [CrossRef]
- Yati, I.; Ridwan, M.; Jeong, G.E.; Lee, Y.; Choi, J.-W.; Yoon, C.W.; Suh, D.J.; Ha, J.-M. Effects of sintering-resistance and large metal-support interface of alumina nanorod-stabilized Pt nanoparticle catalysts on the improved high temperature water gas shift reaction activity. Catal. Commun. 2014, 56, 11–16. [Google Scholar] [CrossRef]
- Subramanian, V.; Potdar, H.S.; Jeong, D.-W.; Shim, J.-O.; Jang, W.-J.; Roh, H.-S.; Jung, U.H.; Yoon, W.L. Synthesis of a Novel Nano-Sized Pt/ZnO Catalyst for Water Gas Shift Reaction in Medium Temperature Application. Catal. Lett. 2012, 142, 1075–1081. [Google Scholar] [CrossRef]
- Martinelli, M.; Jacobs, G.; Graham, U.M.; Shafer, W.D.; Cronauer, D.C.; Kropf, A.J.; Marshall, C.L.; Khalid, S.; Visconti, C.G.; Lietti, L.; et al. Water-gas shift: Characterization and testing of nanoscale YSZ supported Pt catalysts. Appl. Catal. A Gen. 2015, 497, 184–197. [Google Scholar] [CrossRef]
- Kwon, S.J.; Park, J.H.; Koo, K.Y.; Yoon, W.L.; Yi, K.B. Enhanced catalytic performance of Pt/TiO2 catalyst in water gas shift reaction by incorporation of PRGO. Catal. Today 2017, 113–121. [Google Scholar] [CrossRef]
- Franchini, C.A.; De Farias, A.M.D.; Albuquerque, E.; Dos Santos, R.; Fraga, M. Single-stage medium temperature water-gas shift reaction over Pt/ZrO2—Support structural polymorphism and catalyst deactivation. Appl. Catal. B Environ. 2012, 117, 302–309. [Google Scholar] [CrossRef]
- Jain, R.; Maric, R. Synthesis of nano-Pt onto ceria support as catalyst for water–gas shift reaction by Reactive Spray Deposition Technology. Appl. Catal. A Gen. 2014, 475, 461–468. [Google Scholar] [CrossRef]
- Lu, P.; Qiao, B.; Lu, N.; Hyun, D.C.; Wang, J.; Kim, M.J.; Liu, J.; Xia, Y. Photochemical Deposition of Highly Dispersed Pt Nanoparticles on Porous CeO2Nanofibers for the Water-Gas Shift Reaction. Adv. Funct. Mater. 2015, 25, 4153–4162. [Google Scholar] [CrossRef]
- Rajesh, T.; Rajarajan, A.K.; Gopinath, C.S.; Devi, R.N. Evidence of Cationic Pt Active for Water–Gas Shift Reaction: Pt-Doped BaCeO3 Perovskite. J. Phys. Chem. C 2012, 116, 9526–9532. [Google Scholar] [CrossRef]
- Tiwari, R.; Sarkar, B.; Tiwari, R.; Pendem, C.; Sasaki, T.; Saran, S.; Bal, R. Pt nanoparticles with tuneable size supported on nanocrystalline ceria for the low temperature water-gas-shift (WGS) reaction. J. Mol. Catal. A Chem. 2014, 395, 117–123. [Google Scholar] [CrossRef]
- Shim, J.-O.; Hong, Y.J.; Na, H.-S.; Jang, W.-J.; Kang, Y.C.; Roh, H.-S. Highly Active and Stable Pt-Loaded Ce0.75Zr0.25O2 Yolk–Shell Catalyst for Water–Gas Shift Reaction. ACS Appl. Mater. Interfaces 2016, 8, 17239–17244. [Google Scholar] [CrossRef]
- Cornaglia, C.A.; Tosti, S.; Múnera, J.F.; Lombardo, E.A. Optimal Pt load of a Pt/La2O3·SiO2 highly selective WGS catalyst used in a Pd-membrane reactor. Appl. Catal. A Gen. 2014, 486, 85–93. [Google Scholar] [CrossRef]
- Zhao, H.; Yao, S.; Zhang, M.; Huang, F.; Fan, Q.; Zhang, S.; Liu, H.; Ma, D.; Gao, C. Ultra-Small Platinum Nanoparticles Encapsulated in Sub-50 nm Hollow Titania Nanospheres for Low-Temperature Water–Gas Shift Reaction. ACS Appl. Mater. Interfaces 2018, 10, 36954–36960. [Google Scholar] [CrossRef]
- Galeano, Y.M.; Negri, F.; Moreno, M.S.; Múnera, J.; Cornaglia, L.; Tarditi, A.M. Pt encapsulated into NaA zeolite as catalyst for the WGS reaction. Appl. Catal. A Gen. 2019, 572, 176–184. [Google Scholar] [CrossRef]
- Roberts, S.J.; Fletcher, J.V.; Zhou, Y.; Luchters, N.T.; Fletcher, J.C. Water-gas shift of reformate streams over mono-metallic PGM catalysts. Int. J. Hydrog. Energy 2018, 43, 6150–6157. [Google Scholar] [CrossRef]
- Luchters, N.T.; Fletcher, J.; Roberts, S. Variability of Data in High Throughput Experimentation for Catalyst Studies in Fuel Processing. Bull. Chem. React. Eng. Catal. 2017, 12, 106. [Google Scholar] [CrossRef]
- Cavusoglu, G.; Miao, D.; Lichtenberg, H.; Carvalho, H.W.; Xu, H.; Goldbach, A.; Grunwaldt, J.-D. Structure and activity of flame made ceria supported Rh and Pt water gas shift catalysts. Appl. Catal. A Gen. 2015, 504, 381–390. [Google Scholar] [CrossRef]
- Cornaglia, C.A.; Adrover, M.E.; Múnera, J.F.; Pedernera, M.N.; Borio, D.O.; Lombardo, E.A. Production of ultrapure hydrogen in a Pd–Ag membrane reactor using noble metals supported on La–Si oxides. Heterogeneous modeling for the water gas shift reaction. Int. J. Hydrog. Energy 2013, 38, 10485–10493. [Google Scholar] [CrossRef]
- Jeong, D.-W.; Shim, J.-O.; Jang, W.-J.; Roh, H.-S. A Study on Pt-Na/CeO2 Catalysts for Single Stage Water Gas Shift Reaction. Trans. Korean Hydrog. New Energy Soc. 2012, 23, 111–116. [Google Scholar] [CrossRef]
- Azzam, K.G.; Babich, I.V.; Seshan, K.; Mojet, B.L.; Lefferts, L. Stable and Efficient Pt-Re/TiO2 catalysts for Water-Gas-Shift: On the Effect of Rhenium. ChemCatChem 2013, 5, 557–564. [Google Scholar] [CrossRef]
- Ciftci, A.; Ligthart, D.M.; Sen, A.O.; Van Hoof, A.; Friedrich, H.; Hensen, E.J. Pt-Re synergy in aqueous-phase reforming of glycerol and the water–gas shift reaction. J. Catal. 2014, 311, 88–101. [Google Scholar] [CrossRef]
- Sener, C.; Wesley, T.; Alba-Rubio, A.C.; Kumbhalkar, M.D.; Hakim, S.H.; Ribeiro, F.H.; Miller, J.T.; Dumesic, J.A. PtMo Bimetallic Catalysts Synthesized by Controlled Surface Reactions for Water Gas Shift. ACS Catal. 2016, 6, 1334–1344. [Google Scholar] [CrossRef]
- Osman, A.I.; Abu-Dahrieh, J.K.; Cherkasov, N.; Fernández-García, J.; Walker, D.; Walton, R.I.; Rooney, D.W.; Rebrov, E.V. A highly active and synergistic Pt/Mo2C/Al2O3 catalyst for water-gas shift reaction. Mol. Catal. 2018, 455, 38–47. [Google Scholar] [CrossRef]
- Kokumai, T.M.; Cantane, D.A.; Melo, G.T.; Paulucci, L.B.; Zanchet, D. VO x -Pt/Al2O3 catalysts for hydrogen production. Catal. Today 2017, 289, 249–257. [Google Scholar] [CrossRef]
- Chein, R.; Lin, Y.; Chen, Y.; Chyou, Y.; Chung, J. Study on water–gas shift reaction performance using Pt-based catalysts at high temperatures. Int. J. Hydrog. Energy 2014, 39, 18854–18862. [Google Scholar] [CrossRef]
- Wang, T.; Porosoff, M.D.; Chen, J. Effects of oxide supports on the water-gas shift reaction over PtNi bimetallic catalysts: Activity and methanation inhibition. Catal. Today 2014, 233, 61–69. [Google Scholar] [CrossRef]
- Günay, M.E.; Akpinar, F.; Önsan, Z.I.; Yıldırım, R.; Yıldırım, R. Investigation of water gas-shift activity of Pt–MOx–CeO2/Al2O3 (M = K, Ni, Co) using modular artificial neural networks. Int. J. Hydrog. Energy 2012, 37, 2094–2102. [Google Scholar] [CrossRef]
- Palma, V.; Martino, M.; Pisano, D.; Ciambelli, P. Catalytic Activities of Bimetallic Catalysts for Low Temperature Water Gas Shift Reaction. Chem. Eng. Trans. 2016, 52, 481–486. [Google Scholar] [CrossRef]
- Palma, V.; Martino, M. Pt-Re Based Catalysts for the Realization of a Single Stage Water Gas Shift Process. Chem. Eng. Trans. 2017, 57, 1657–1662. [Google Scholar] [CrossRef]
- Izquierdo, U.; Neuberg, S.; Pecov, S.; Pennemann, H.; Zapf, R.; Wichert, M.; Barrio, V.; Cambra, J.F.; Kolb, G. Hydrogen production with a microchannel heat-exchanger reactor by single stage water-gas shift; catalyst development. Chem. Eng. J. 2017, 313, 1494–1508. [Google Scholar] [CrossRef]
- Rajesh, T.; Devi, R.N. Resistance to ionic Pt insertion in oxygen excess LaMnO3 perovskite lattices and its effect in water gas shift reaction. J. Mol. Catal. A Chem. 2014, 395, 534–542. [Google Scholar] [CrossRef]
- Rajesh, T.; Upadhyay, A.; Sinha, A.K.; Deb, S.K.; Devi, R.N. Effect of Pt incorporation in LaBO3 (B = Mn, Fe, Co) perovskites on water gas shift activity. J. Mol. Catal. A Chem. 2014, 395, 506–513. [Google Scholar] [CrossRef]
- Mohamed, M.M.; Khairou, K. Fabrication and characterization of bimetallic Pt–Au nanowires supported on FSM-16 and their catalytic activities toward water–gas shift reaction. J. Colloid Interface Sci. 2011, 354, 100–108. [Google Scholar] [CrossRef]
- Buitrago-Sierra, R.; Ruiz-Martinez, J.; Silvestre-Albero, J.; Sepúlveda-Escribano, A.; Rodriguez-Reinoso, F. Water gas shift reaction on carbon-supported Pt catalysts promoted by CeO2. Catal. Today 2012, 180, 19–24. [Google Scholar] [CrossRef]
- Zugic, B.; Bell, D.C.; Flytzani-Stephanopoulos, M. Activation of carbon-supported platinum catalysts by sodium for the low-temperature water-gas shift reaction. Appl. Catal. B Environ. 2014, 144, 243–251. [Google Scholar] [CrossRef]
- Meira, D.; Ribeiro, R.; Mathon, O.; Pascarelli, S.; Bueno, J.; Zanchet, D. Complex interplay of structural and surface properties of ceria on platinum supported catalyst under water gas shift reaction. Appl. Catal. B Environ. 2016, 197, 73–85. [Google Scholar] [CrossRef]
- Mao, X.; Foucher, A.C.; Stach, E.A.; Gorte, R.J. “Intelligent” Pt Catalysts Based on Thin LaCoO3 Films Prepared by Atomic Layer Deposition. Inorganics 2019, 7, 113. [Google Scholar] [CrossRef]
- Brandt, A.J.; Maddumapatabandi, T.D.; Shakya, D.M.; Xie, K.; Seuser, G.S.; Farzandh, S.; Chen, D.A. Water-gas shift activity on Pt-Re surfaces and the role of the support. J. Chem. Phys. 2019, 151, 234714. [Google Scholar] [CrossRef]
- Schweitzer, N.M.; Schaidle, J.A.; Ezekoye, O.K.; Pan, X.; Linic, S.; Thompson, L.T. High Activity Carbide Supported Catalysts for Water Gas Shift. J. Am. Chem. Soc. 2011, 133, 2378–2381. [Google Scholar] [CrossRef] [PubMed]
- Wyvratt, B.M.; Gaudet, J.R.; Thompson, L.T. Effects of passivation on synthesis, structure and composition of molybdenum carbide supported platinum water–gas shift catalysts. J. Catal. 2015, 330, 280–287. [Google Scholar] [CrossRef]
- Wang, G.; Schaidle, J.A.; Katz, M.B.; Li, Y.; Pan, X.Q.; Thompson, L. Alumina supported Pt–Mo2C catalysts for the water–gas shift reaction. J. Catal. 2013, 304, 92–99. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Ramírez, P.J.; Gutierrez, R.A. Highly active Pt/MoC and Pt/TiC catalysts for the low-temperature water-gas shift reaction: Effects of the carbide metal/carbon ratio on the catalyst performance. Catal. Today 2017, 289, 47–52. [Google Scholar] [CrossRef]
- Wen, C.; Zhu, Y.; Ye, Y.; Zhang, S.; Cheng, F.; Liu, Y.; Wang, P.; Tao, D.F. (Feng) Water–Gas Shift Reaction on Metal Nanoclusters Encapsulated in Mesoporous Ceria Studied with Ambient-Pressure X-ray Photoelectron Spectroscopy. ACS Nano 2012, 6, 9305–9313. [Google Scholar] [CrossRef]
- Jeong, D.-W.; Jang, W.-J.; Shim, J.-O.; Han, W.-B.; Kim, H.-M.; Lee, Y.-L.; Bae, J.W.; Roh, H.-S. Optimization of a highly active nano-sized Pt/CeO2 catalyst via Ce(OH)CO3 for the water-gas shift reaction. Renew. Energy 2015, 79, 78–84. [Google Scholar] [CrossRef]
- Jeong, D.-W.; Potdar, H.S.; Shim, J.-O.; Jang, W.-J.; Roh, H.-S. H2 production from a single stage water–gas shift reaction over Pt/CeO2, Pt/ZrO2, and Pt/Ce(1−x)Zr(x)O2 catalysts. Int. J. Hydrog. Energy 2013, 38, 4502–4507. [Google Scholar] [CrossRef]
- Torrente-Murciano, L.; García-García, F. Effect of nanostructured support on the WGSR activity of Pt/CeO2 catalysts. Catal. Commun. 2015, 71, 1–6. [Google Scholar] [CrossRef]
- Deal, J.W.; Le, P.; Corey, C.B.; More, K.L.; West, C.W. Water-gas shift reaction on alumina-supported Pt-CeO catalysts prepared by supercritical fluid deposition. J. Supercrit. Fluids 2017, 119, 113–121. [Google Scholar] [CrossRef]
- Shan, J.; Nguyen, L.; Zhang, S.; Tao, F.-F. Water–Gas Shift on Pd/α-MnO2 and Pt/α-MnO2. Catal. Lett. 2015, 145, 1571–1580. [Google Scholar] [CrossRef]
- Kuai, L.; Liu, S.; Cao, S.; Ren, Y.; Kan, E.; Zhao, Y.; Yu, N.; Li, F.; Li, X.; Wu, Z.; et al. Atomically Dispersed Pt/Metal Oxide Mesoporous Catalysts from Synchronous Pyrolysis–Deposition Route for Water–Gas Shift Reaction. Chem. Mater. 2018, 30, 5534–5538. [Google Scholar] [CrossRef]
- De León, J.D.; Loera-Serna, S.; Zepeda, T.; Domínguez, D.; Pawelec, B.; Venezia, A.; Fuentes-Moyado, S. Noble metals supported on binary γ-Al2O3-α-Ga2O3 oxide as potential low-temperature water-gas shift catalysts. Fuel 2020, 266, 117031. [Google Scholar] [CrossRef]
- Miao, D.; Cavusoglu, G.; Lichtenberg, H.; Yu, J.; Xu, H.; Grunwaldt, J.-D.; Goldbach, A. Water-gas shift reaction over platinum/strontium apatite catalysts. Appl. Catal. B Environ. 2017, 202, 587–596. [Google Scholar] [CrossRef]
- Williams, W.D.; Bollmann, L.; Miller, J.T.; Delgass, W.N.; Ribeiro, F.H. Effect of molybdenum addition on supported platinum catalysts for the water–gas shift reaction. Appl. Catal. B Environ. 2012, 125, 206–214. [Google Scholar] [CrossRef]
- Xu, X.; Fu, Q.; Bao, X. MoOx-promoted Pt catalysts for the water gas shift reaction at low temperatures. Chin. J. Catal. 2015, 36, 750–756. [Google Scholar] [CrossRef]
- Sabnis, K.D.; Akatay, M.C.; Cui, Y.; Sollberger, F.G.; Stach, E.A.; Miller, J.T.; Delgass, W.N.; Ribeiro, F.H. Probing the active sites for water–gas shift over Pt/molybdenum carbide using multi-walled carbon nanotubes. J. Catal. 2015, 330, 442–451. [Google Scholar] [CrossRef]
- Yang, M.; Flytzani-Stephanopoulos, M. Design of single-atom metal catalysts on various supports for the low-temperature water-gas shift reaction. Catal. Today 2017, 298, 216–225. [Google Scholar] [CrossRef]
- Wang, Y.; Zhai, Y.; Pierre, D.; Flytzani-Stephanopoulos, M. Silica-encapsulated platinum catalysts for the low-temperature water-gas shift reaction. Appl. Catal. B Environ. 2012, 127, 342–350. [Google Scholar] [CrossRef]
- Zhu, X.; Shen, M.; Lobban, L.L.; Mallinson, R.G. Structural effects of Na promotion for high water gas shift activity on Pt–Na/TiO2. J. Catal. 2011, 278, 123–132. [Google Scholar] [CrossRef]
- Zugic, B.; Zhang, S.; Bell, D.C.; Tao, F.; Flytzani-Stephanopoulos, M. Probing the Low-Temperature Water–Gas Shift Activity of Alkali-Promoted Platinum Catalysts Stabilized on Carbon Supports. J. Am. Chem. Soc. 2014, 136, 3238–3245. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Liu, J.; Lee, S.; Zugic, B.; Huang, J.; Allard, L.F.; Flytzani-Stephanopoulos, M. A Common Single-Site Pt(II)–O(OH)x–Species Stabilized by Sodium on “Active” and “Inert” Supports Catalyzes the Water-Gas Shift Reaction. J. Am. Chem. Soc. 2015, 137, 3470–3473. [Google Scholar] [CrossRef]
- Xie, H.; Lu, J.; Shekhar, M.; Elam, J.W.; Delgass, W.N.; Ribeiro, F.H.; Weitz, E.; Poeppelmeier, K.R. Synthesis of Na-Stabilized Nonporous t-ZrO2 Supports and Pt/t-ZrO2 Catalysts and Application to Water-Gas-Shift Reaction. ACS Catal. 2012, 3, 61–73. [Google Scholar] [CrossRef]
- Pazmiño, J.H.; Shekhar, M.; Williams, W.D.; Akatay, M.C.; Miller, J.T.; Delgass, W.N.; Ribeiro, F.H. Metallic Pt as active sites for the water–gas shift reaction on alkali-promoted supported catalysts. J. Catal. 2012, 286, 279–286. [Google Scholar] [CrossRef]
- Kusche, M.; Bustillo, K.; Agel, F.; Wasserscheid, P. Highly Effective Pt-Based Water–Gas Shift Catalysts by Surface Modification with Alkali Hydroxide Salts. ChemCatChem 2015, 7, 766–775. [Google Scholar] [CrossRef] [PubMed]
- Panagiotopoulou, P.; Kondarides, D.I. Effects of promotion of TiO2 with alkaline earth metals on the chemisorptive properties and water–gas shift activity of supported platinum catalysts. Appl. Catal. B Environ. 2011, 101, 738–746. [Google Scholar] [CrossRef]
- Kim, Y.-T.; You, S.J.; Park, E.D. Water–gas shift reaction over Pt and Pt–CeOx supported on CexZr1−xO2. Int. J. Hydrog. Energy 2012, 37, 1465–1474. [Google Scholar] [CrossRef]
- Carta, D.; Montini, T.; Casula, M.F.; Monai, M.; Bullita, S.; Fornasiero, P.; Corrias, A. The water gas shift reaction over Pt–CeO2 nanoparticles confined within mesoporous SBA-16. J. Mater. Chem. A 2017, 5, 20024–20034. [Google Scholar] [CrossRef]
- Mahadevaiah, N.; Singh, P.; Mukri, B.D.; Parida, S.K.; Hegde, M.S. Ce0.67Fe0.33O2−δ and Ce0.65Fe0.33Pt0.02O2−δ: New water gas shift (WGS) catalysts. Appl. Catal. B Environ. 2011, 108, 117–126. [Google Scholar] [CrossRef]
- Shinde, V.M.; Madras, G. Low temperature CO oxidation and water gas shift reaction over Pt/Pd substituted in Fe/TiO2 catalysts. Int. J. Hydrog. Energy 2012, 37, 18798–18814. [Google Scholar] [CrossRef]
- Shinde, V.M.; Madras, G. A Single-Stage Water-Gas Shift Reaction over Highly Active and Stable Si- and Al-Substituted Pt/CeO2 Catalysts. ChemCatChem 2012, 4, 1968–1978. [Google Scholar] [CrossRef]
- Petallidou, K.C.; Efstathiou, A.M. Low-temperature water-gas shift on Pt/Ce1−xLaxO2−δ: Effect of Ce/La ratio. Appl. Catal. B Environ. 2013, 140, 333–347. [Google Scholar] [CrossRef]
- Palma, V.; Gallucci, F.; Pullumbi, P.; Ruocco, C.; Meloni, E.; Martino, M. Pt/Re/CeO2 Based Catalysts for CO-Water–Gas Shift Reaction: From Powders to Structured Catalyst. Catalysts 2020, 10, 564. [Google Scholar] [CrossRef]
- Ratnasamy, C.; Wagner, J.P. Water Gas Shift Catalysis. Catal. Rev. 2009, 51, 325–440. [Google Scholar] [CrossRef]
- Ribeiro, M.; Jacobs, G.; Linganiso, L.; Azzam, K.G.; Graham, U.M.; Crocker, M. Low Temperature Water Gas Shift: Evaluation of Pt/HfO2 and Correlation between Reaction Mechanism and Periodic Trends in Tetravalent (Ti, Zr, Hf, Ce, Th) Metal Oxides. ACS Catal. 2011, 1, 1375–1383. [Google Scholar] [CrossRef]
- Rodríguez, J.A.; Hanson, J.C.; Stacchiola, D.J.; Senanayake, S.D. In situ/operando studies for the production of hydrogen through the water-gas shift on metal oxide catalysts. Phys. Chem. Chem. Phys. 2013, 15, 12004. [Google Scholar] [CrossRef]
- Petallidou, K.C.; Polychronopoulou, K.; Boghosian, S.; García-Rodríguez, S.; Efstathiou, A.M. Water–Gas Shift Reaction on Pt/Ce1–xTixO2−δ: The Effect of Ce/Ti Ratio. J. Phys. Chem. C 2013, 117, 25467–25477. [Google Scholar] [CrossRef]
- Yao, S.Y.; Xu, W.Q.; Johnston-Peck, A.C.; Zhao, F.Z.; Liu, Z.; Luo, S.; Senanayake, S.D.; Martínez-Arias, A.; Liu, W.; Rodriguez, J.A. Morphological effects of the nanostructured ceria support on the activity and stability of CuO/CeO2 catalysts for the water-gas shift reaction. Phys. Chem. Chem. Phys. 2014, 16, 17183–17195. [Google Scholar] [CrossRef]
- Barrio, L.; Zhou, G.; González, I.D.; Estrella, M.A.; Hanson, J.; Rodriguez, J.A.; Yerga, R.M.N.; Fierro, J.L.G. In situ characterization of Ptcatalysts supported on ceria modified TiO2 for the WGS reaction: Influence of ceria loading. Phys. Chem. Chem. Phys. 2012, 14, 2192–2202. [Google Scholar] [CrossRef]
- Jain, R.; Poyraz, A.S.; Gamliel, D.P.; Valla, J.; Suib, S.L.; Maric, R. Comparative study for low temperature water-gas shift reaction on Pt/ceria catalysts: Role of different ceria supports. Appl. Catal. A Gen. 2015, 507, 1–13. [Google Scholar] [CrossRef]
- Lee, S.M.; Kim, G.J.; Lee, S.H.; Hwang, I.H.; Hong, S.C.; Kim, S.S. Catalytic Performance of Ce0.6Y0.4O2-Supported Platinum Catalyst for Low-Temperature Water-Gas Shift Reaction. ACS Omega 2018, 3, 3156–3163. [Google Scholar] [CrossRef] [PubMed]
- Kalamaras, C.; Gonzalez, I.D.; Navarro, R.M.; Fierro, J.L.G.; Efstathiou, A.M. Effects of Reaction Temperature and Support Composition on the Mechanism of Water–Gas Shift Reaction over Supported-Pt Catalysts. J. Phys. Chem. C 2011, 115, 11595–11610. [Google Scholar] [CrossRef]
- Luo, S.; Barrio, L.; Nguyen-Phan, T.-D.; Vovchok, D.; Johnston-Peck, A.C.; Xu, W.; Stach, E.A.; Rodriguez, J.A.; Senanayake, S.D. Importance of Low Dimensional CeOx Nanostructures in Pt/CeOx–TiO2 Catalysts for the Water–Gas Shift Reaction. J. Phys. Chem. C 2017, 121, 6635–6642. [Google Scholar] [CrossRef]
- Luo, S.; Nguyen-Phan, T.-D.; Johnston-Peck, A.C.; Barrio, L.; Sallis, S.; Arena, D.A.; Kundu, S.; Xu, W.; Piper, L.F.; Stach, E.A.; et al. Hierarchical Heterogeneity at the CeOx–TiO2Interface: Electronic and Geometric Structural Influence on the Photocatalytic Activity of Oxide on Oxide Nanostructures. J. Phys. Chem. C 2015, 119, 2669–2679. [Google Scholar] [CrossRef]
- Linganiso, L.Z.; Jacobs, G.; Azzam, K.G.; Graham, U.M.; Davis, B.H.; Cronauer, D.; Kropf, A.J.; Marshall, C.L. Low-temperature water–gas shift: Strategy to lower Pt loading by doping ceria with Ca2+ improves formate mobility/WGS rate by increasing surface O-mobility. Appl. Catal. A Gen. 2011, 394, 105–116. [Google Scholar] [CrossRef]
- Hwang, K.-R.; Park, J.-S.; Ihm, S.-K. Si-modified Pt/CeO2 catalyst for a single-stage water-gas shift reaction. Int. J. Hydrog. Energy 2011, 36, 9685–9693. [Google Scholar] [CrossRef]
- Vignatti, C.; Avila, M.; Apesteguia, C.; Garetto, T. Catalytic and DRIFTS study of the WGS reaction on Pt-based catalysts. Int. J. Hydrog. Energy 2010, 35, 7302–7312. [Google Scholar] [CrossRef]
- Vignatti, C.; Avila, M.; Apesteguia, C.; Garetto, T. Study of the water-gas shift reaction over Pt supported on CeO2–ZrO2 mixed oxides. Catal. Today 2011, 171, 297–303. [Google Scholar] [CrossRef]
- Kalamaras, C.; Dionysiou, D.D.; Efstathiou, A.M. Mechanistic Studies of the Water–Gas Shift Reaction over Pt/CexZr1–xO2 Catalysts: The Effect of Pt Particle Size and Zr Dopant. ACS Catal. 2012, 2, 2729–2742. [Google Scholar] [CrossRef]
- Pinaeva, L.; Sadovskaya, E.; Ivanova, Y.; Kuznetsova, T.; Prosvirin, I.P.; Sadykov, V.; Schuurman, Y.; Van Veen, A.; Mirodatos, C. Water gas shift and partial oxidation of CH4 over CeO2–ZrO2(–La2O3) and Pt/CeO2–ZrO2(–La2O3): Performance under transient conditions. Chem. Eng. J. 2014, 257, 281–291. [Google Scholar] [CrossRef]
- Petallidou, K.C.; Boghosian, S.; Efstathiou, A.M. Low-temperature water–gas shift on Pt/Ce0.5La0.5O2−δ: Effect of support synthesis method. Catal. Today 2015, 242, 153–167. [Google Scholar] [CrossRef]
- Leet, J.S.; Kim, S.; Society, T.A.C. Synthesis and Characterization of Ce1−xGdxO2-δ Nanorods. J. Am. Ceram. Soc. 2007, 90, 661–663. [Google Scholar]
- Petallidou, K.; Polychronopoulou, K.; Fierro, J.; Efstathiou, A.M. Low-temperature water-gas shift on Pt/Ce0.8La0.2O2−δ–CNT: The effect of Ce0.8La0.2O2−δ/CNT ratio. Appl. Catal. A Gen. 2015, 504, 585–598. [Google Scholar] [CrossRef]
- Petallidou, K.C.; Kalamaras, C.; Efstathiou, A.M. The effect of La3+, Ti4+ and Zr4+ dopants on the mechanism of WGS on ceria-doped supported Pt catalysts. Catal. Today 2014, 228, 183–193. [Google Scholar] [CrossRef]
- Linganiso, L.Z.; Pendyala, V.R.R.; Jacobs, G.; Crocker, M.; Cronauer, D.C.; Kropf, A.J.; Marshall, C.L. Low-Temperature Water–Gas Shift: Doping Ceria Improves Reducibility and Mobility of O-Bound Species and Catalyst Activity. Catal. Lett. 2011, 141, 1723–1731. [Google Scholar] [CrossRef]
- Kalamaras, C.; Petallidou, K.C.; Efstathiou, A.M. The effect of La3+-doping of CeO2 support on the water-gas shift reaction mechanism and kinetics over Pt/Ce1−xLaxO2−δ. Appl. Catal. B Environ. 2013, 136, 225–238. [Google Scholar] [CrossRef]
- Busca, G.; Finocchio, E.; Escribano, V.S. Infrared studies of CO oxidation by oxygen and by water over Pt/Al2O3 and Pd/Al2O3 catalysts. Appl. Catal. B Environ. 2012, 113, 172–179. [Google Scholar] [CrossRef]
- Sabnis, K.D.; Cui, Y.; Akatay, M.C.; Shekhar, M.; Lee, W.-S.; Miller, J.T.; Delgass, W.N.; Ribeiro, F.H. Water–gas shift catalysis over transition metals supported on molybdenum carbide. J. Catal. 2015, 331, 162–171. [Google Scholar] [CrossRef]
- Miao, D.; Goldbach, A.; Xu, H. Platinum/Apatite Water-Gas Shift Catalysts. ACS Catal. 2016, 6, 775–783. [Google Scholar] [CrossRef]
- Ding, K.; Gulec, A.; Johnson, A.M.; Schweitzer, N.M.; Stucky, G.D.; Marks, L.D.; Stair, P.C. Identification of active sites in CO oxidation and water-gas shift over supported Pt catalysts. Science 2015, 350, 189–192. [Google Scholar] [CrossRef]
- Rivero-Crespo, M.; Mon, M.; Ferrando-Soria, J.; Lopes, C.W.; Boronat, M.; Leyva–Pérez, A.; Corma, A.; Hernandez-Garrido, J.C.; López-Haro, M.; Calvino, J.J.; et al. Confined Pt11+ Water Clusters in a MOF Catalyze the Low-Temperature Water-Gas Shift Reaction with both CO2 Oxygen Atoms Coming from Water. Angew. Chem. Int. Ed. 2018, 57, 17094–17099. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Lin, J.; Li, L.; Qiao, B.; Liu, J.; Su, Y.; Wang, X. Identifying Size Effects of Pt as Single Atoms and Nanoparticles Supported on FeOx for the Water-Gas Shift Reaction. ACS Catal. 2018, 8, 859–868. [Google Scholar] [CrossRef]
- Flaherty, D.W.; Yu, W.-Y.; Pozun, Z.; Henkelman, G.; Mullins, C.B. Mechanism for the water–gas shift reaction on monofunctional platinum and cause of catalyst deactivation. J. Catal. 2011, 282, 278–288. [Google Scholar] [CrossRef]
- Gerceker, D.; Motagamwala, A.H.; Rivera-Dones, K.R.; Miller, J.B.; Huber, G.W.; Mavrikakis, M.; Dumesic, J.A. Methane Conversion to Ethylene and Aromatics on PtSn Catalysts. ACS Catal. 2017, 7, 2088–2100. [Google Scholar] [CrossRef]
- Duke, A.S.; Xie, K.; Brandt, A.J.; Maddumapatabandi, T.D.; Ammal, S.C.; Heyden, A.; Monnier, J.R.; Chen, D.A. Understanding Active Sites in the Water–Gas Shift Reaction for Pt–Re Catalysts on Titania. ACS Catal. 2017, 7, 2597–2606. [Google Scholar] [CrossRef]
- Aragão, I.B.; Ro, I.; Liu, Y.; Ball, M.; Huber, G.W.; Zanchet, D.; Dumesic, J.A. Catalysts synthesized by selective deposition of Fe onto Pt for the water-gas shift reaction. Appl. Catal. B Environ. 2018, 222, 182–190. [Google Scholar] [CrossRef]
- Xu, W.; Si, R.; Senanayake, S.D.; Llorca, J.; Idriss, H.; Stacchiola, D.J.; Hanson, J.C.; Rodriguez, J.A. In situ studies of CeO2-supported Pt, Ru, and Pt–Ru alloy catalysts for the water–gas shift reaction: Active phases and reaction intermediates. J. Catal. 2012, 291, 117–126. [Google Scholar] [CrossRef]
- Anil, C.; Madras, G. Catalytic behaviour of Mn2.94M0.06O4-δ (M = Pt, Ru and Pd) catalysts for low temperature water gas shift (WGS) and CO oxidation. Int. J. Hydrog. Energy 2020, 45, 10461–10474. [Google Scholar] [CrossRef]
- Rajesh, T.; Devi, R.N. Role of Oxygen Vacancies in Water Gas Shift Reaction: Activity Study on BaCe0.98–xYxPt0.02O3−δ Perovskites. J. Phys. Chem. C 2014, 118, 20867–20874. [Google Scholar] [CrossRef]
- Cybulskis, V.J.; Wang, J.; Pazmiño, J.H.; Ribeiro, F.H.; Delgass, W.N. Isotopic transient studies of sodium promotion of Pt/Al2O3 for the water–gas shift reaction. J. Catal. 2016, 339, 163–172. [Google Scholar] [CrossRef]
- Faust, M.; Dinkel, M.; Bruns, M.; Bräse, S.; Seipenbusch, M.; Bräse, S. Support Effect on the Water Gas Shift Activity of Chemical Vapor Deposition-Tailored-Pt/TiO2 Catalysts. Ind. Eng. Chem. Res. 2017, 56, 3194–3203. [Google Scholar] [CrossRef]
- Martinelli, M.; Jacobs, G.; Shafer, W.D.; Crocker, M. Effect of alkali on C H bond scission over Pt/YSZ catalyst during water-gas-shift, steam-assisted formic acid decomposition and methanol steam reforming. Catal. Today 2017, 291, 29–35. [Google Scholar] [CrossRef]
- Gao, P.; Graham, U.M.; Shafer, W.D.; Linganiso, L.Z.; Jacobs, G.; Crocker, M. Nanostructure and kinetic isotope effect of alkali-doped Pt/silica catalysts for water-gas shift and steam-assisted formic acid decomposition. Catal. Today 2016, 272, 42–48. [Google Scholar] [CrossRef]
- Watson, C.D.; Martinelli, M.; Cronauer, D.C.; Kropf, A.J.; Marshall, C.L.; Jacobs, G. Low temperature water-gas shift: Optimization of K loading on Pt/m-ZrO2 for enhancing CO conversion. Appl. Catal. A Gen. 2020, 598, 117572. [Google Scholar] [CrossRef]
- Kaftan, A.; Kusche, M.; Laurin, M.; Wasserscheid, P.; Libuda, J.; Libuda, J. KOH-promoted Pt/Al2O3 catalysts for water gas shift and methanol steam reforming: An operando DRIFTS-MS study. Appl. Catal. B Environ. 2017, 201, 169–181. [Google Scholar] [CrossRef]
- Hwang, K.-R.; Ihm, S.-K.; Park, S.-C.; Park, J.S. Pt/ZrO2 catalyst for a single-stage water-gas shift reaction: Ti addition effect. Int. J. Hydrog. Energy 2013, 38, 6044–6051. [Google Scholar] [CrossRef]
- Ammal, S.C.; Heyden, A. Understanding the Nature and Activity of Supported Platinum Catalysts for the Water–Gas Shift Reaction: From Metallic Nanoclusters to Alkali-Stabilized Single-Atom Cations. ACS Catal. 2019, 9, 7721–7740. [Google Scholar] [CrossRef]
- Stamatakis, M.; Chen, Y.; Vlachos, D.G. First-Principles-Based Kinetic Monte Carlo Simulation of the Structure Sensitivity of the Water–Gas Shift Reaction on Platinum Surfaces. J. Phys. Chem. C 2011, 115, 24750–24762. [Google Scholar] [CrossRef]
- Fajín, J.L.; Cordeiro, M.N.D. Probing the efficiency of platinum nanotubes for the H2 production by water gas shift reaction: A DFT study. Appl. Catal. B Environ. 2020, 263, 118301. [Google Scholar] [CrossRef]
- Lin, C.-H.; Chen, C.-L.; Wang, J.-H. Mechanistic Studies of Water–Gas-Shift Reaction on Transition Metals. J. Phys. Chem. C 2011, 115, 18582–18588. [Google Scholar] [CrossRef]
- Clay, J.P.; Greeley, J.; Ribeiro, F.H.; Delgass, W.N.; Schneider, W.F. DFT comparison of intrinsic WGS kinetics over Pd and Pt. J. Catal. 2014, 320, 106–117. [Google Scholar] [CrossRef]
- Lian, X.; Guo, W.; Shu, J.; Zhang, X.; Liu, Z.; Zhang, Y.; Liu, R. A density functional theory study of the water–gas shift reaction promoted by Pt-based catalysts. Theor. Chem. Acc. 2015, 134. [Google Scholar] [CrossRef]
- Guo, L.; Li, A.; An, X.; Cao, Z.; Liu, N. Catalytic activity of TM@Cu12 core–shell nanoclusters for water gas shift reaction. Int. J. Hydrog. Energy 2015, 40, 8330–8340. [Google Scholar] [CrossRef]
- Fang, L.; Chen, T.; Meng, Y.; Wang, Y.; Xue, J.; Ni, Z.; Xia, S. Water-gas shift reaction catalyzed by Au32M6 (M = Cu, Pt, Pd, Rh, Ir) core-shell nanoclusters: A density functional theory study. Mol. Catal. 2020, 483, 110757. [Google Scholar] [CrossRef]
- Cao, Z.; Guo, L.; Liu, N. A Theoretical Study of the Water–Gas-Shift Reaction on Cu6TM (TM = Co, Ni, Cu, Rh, Pd, Ag, Ir, Pt, Au) Clusters. J. Clust. Sci. 2015, 27, 523–535. [Google Scholar] [CrossRef]
- Xue, J.-L.; Fang, L.; Luo, W.; Meng, Y.; Chen, T.; Xia, S.; Ni, Z.-M. Density functional study of water gas shift reaction catalyzed by Cu-Pt-Au ternary alloy. J. Fuel Chem. Technol. 2019, 47, 688–696. [Google Scholar] [CrossRef]
- Xia, S.; Fang, L.; Meng, Y.; Zhang, X.; Zhang, L.; Yang, C.; Ni, Z. Water-gas shift reaction catalyzed by layered double hydroxides supported Au-Ni/Cu/Pt bimetallic alloys. Appl. Catal. B Environ. 2020, 272, 118949. [Google Scholar] [CrossRef]
- Williams, W.D.; Greeley, J.; Delgass, W.N.; Ribeiro, F.H. Water activation and carbon monoxide coverage effects on maximum rates for low temperature water-gas shift catalysis. J. Catal. 2017, 347, 197–204. [Google Scholar] [CrossRef]
- Ammal, S.C.; Heyden, A. Nature of Ptn/TiO2(110) Interface under Water-Gas Shift Reaction Conditions: A Constrained ab Initio Thermodynamics Study. J. Phys. Chem. C 2011, 115, 19246–19259. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Azzam, K.; Babich, I.; Seshan, K.; Lefferts, L. Role of Re in Pt–Re/TiO2 catalyst for water gas shift reaction: A mechanistic and kinetic study. Appl. Catal. B Environ. 2008, 80, 129–140. [Google Scholar] [CrossRef]
- Ammal, S.C.; Heyden, A. Origin of the unique activity of Pt/TiO2 catalysts for the water–gas shift reaction. J. Catal. 2013, 306, 78–90. [Google Scholar] [CrossRef]
- Stegelmann, C.; Andreasen, A.; Campbell, C.T. Degree of Rate Control: How Much the Energies of Intermediates and Transition States Control Rates. J. Am. Chem. Soc. 2009, 131, 8077–8082. [Google Scholar] [CrossRef]
- Grabow, L.C.; Gokhale, A.A.; Evans, S.T.; Dumesic, J.A.; Mavrikakis, M. Mechanism of the Water Gas Shift Reaction on Pt: First Principles, Experiments, and Microkinetic Modeling. J. Phys. Chem. C 2008, 112, 4608–4617. [Google Scholar] [CrossRef]
- Ammal, S.C.; Heyden, A. Water–Gas Shift Catalysis at Corner Atoms of Pt Clusters in Contact with a TiO2 (110) Support Surface. ACS Catal. 2014, 4, 3654–3662. [Google Scholar] [CrossRef]
- Walker, E.; Ammal, S.C.; Terejanu, G.A.; Heyden, A. Uncertainty Quantification Framework Applied to the Water–Gas Shift Reaction over Pt-Based Catalysts. J. Phys. Chem. C 2016, 120, 10328–10339. [Google Scholar] [CrossRef]
- Hammer, B.; Hansen, L.B.; Nørskov, J.K. Improved adsorption energetics within density-functional theory using revised Perdew-Burke-Ernzerhof functionals. Phys. Rev. B 1999, 59, 7413–7421. [Google Scholar] [CrossRef]
- Heyd, J.; Scuseria, G.E.; Ernzerhof, M. Hybrid functionals based on a screened Coulomb potential. J. Chem. Phys. 2003, 118, 8207. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions. J. Chem. Phys. 2006, 125, 194101. [Google Scholar] [CrossRef]
- Plessis, S.; Carrasco, N.; Pernot, P. Knowledge-based probabilistic representations of branching ratios in chemical networks: The case of dissociative recombinations. J. Chem. Phys. 2010, 133, 134110. [Google Scholar] [CrossRef] [PubMed]
- Kullback, S.; Leibler, R.A. On Information and Sufficiency. Ann. Math. Stat. 1951, 22, 79–86. [Google Scholar] [CrossRef]
- Walker, E.; Mitchell, D.; Terejanu, G.A.; Heyden, A. Identifying Active Sites of the Water–Gas Shift Reaction over Titania Supported Platinum Catalysts under Uncertainty. ACS Catal. 2018, 8, 3990–3998. [Google Scholar] [CrossRef]
- Ammal, S.C.; Heyden, A. Water-Gas Shift Activity of Atomically Dispersed Cationic Platinum versus Metallic Platinum Clusters on Titania Supports. ACS Catal. 2016, 7, 301–309. [Google Scholar] [CrossRef]
- Ammal, S.C.; Heyden, A. Titania-Supported Single-Atom Platinum Catalyst for Water-Gas Shift Reaction. Chem. Ing. Tech. 2017, 89, 1343–1349. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Grinter, D.C.; Liu, Z.; Palomino, R.M.; Senanayake, S.D. Ceria-based model catalysts: Fundamental studies on the importance of the metal–ceria interface in CO oxidation, the water–gas shift, CO2 hydrogenation, and methane and alcohol reforming. Chem. Soc. Rev. 2017, 46, 1824–1841. [Google Scholar] [CrossRef]
- Vecchietti, J.; Bonivardi, A.; Xu, W.; Stacchiola, D.J.; Delgado, J.J.; Calatayud, M.; Collins, S.E. Understanding the Role of Oxygen Vacancies in the Water Gas Shift Reaction on Ceria-Supported Platinum Catalysts. ACS Catal. 2014, 4, 2088–2096. [Google Scholar] [CrossRef]
- Bruix, A.; Rodríguez, J.A.; Ramirez, P.J.; Senanayake, S.D.; Evans, J.; Park, J.B.; Stacchiola, D.J.; Liu, P.; Hrbek, J.; Illas, F. A New Type of Strong Metal–Support Interaction and the Production of H2 through the Transformation of Water on Pt/CeO2(111) and Pt/CeOx/TiO2(110) Catalysts. J. Am. Chem. Soc. 2012, 134, 8968–8974. [Google Scholar] [CrossRef]
- Zhou, Y.; Perket, J.M.; Zhou, J. Growth of Pt Nanoparticles on Reducible CeO2(111) Thin Films: Effect of Nanostructures and Redox Properties of Ceria. J. Phys. Chem. C 2010, 114, 11853–11860. [Google Scholar] [CrossRef]
- Senanayake, S.D.; Rodriguez, J.A.; Stacchiola, D.J. Electronic Metal–Support Interactions and the Production of Hydrogen through the Water-Gas Shift Reaction and Ethanol Steam Reforming: Fundamental Studies with Well-Defined Model Catalysts. Top. Catal. 2013, 56, 1488–1498. [Google Scholar] [CrossRef]
- Aranifard, S.; Ammal, S.C.; Heyden, A. Nature of Ptn/CeO2 (111) Surface under Water–Gas Shift Reaction Conditions: A Constrained ab Initio Thermodynamics Study. J. Phys. Chem. C 2012, 116, 9029–9042. [Google Scholar] [CrossRef]
- Aranifard, S.; Ammal, S.C.; Heyden, A. On the Importance of the Associative Carboxyl Mechanism for the Water-Gas Shift Reaction at Pt/CeO2 Interface Sites. J. Phys. Chem. C 2014, 118, 6314–6323. [Google Scholar] [CrossRef]
- Aranifard, S.; Ammal, S.C.; Heyden, A. On the importance of metal–oxide interface sites for the water–gas shift reaction over Pt/CeO2 catalysts. J. Catal. 2014, 309, 314–324. [Google Scholar] [CrossRef]
- Carrasquillo-Flores, R.; Gallo, J.M.R.; Hahn, K.R.; Dumesic, J.A.; Mavrikakis, M. Density Functional Theory and Reaction Kinetics Studies of the Water-Gas Shift Reaction on Pt-Re Catalysts. ChemCatChem 2013, 5, 3690–3699. [Google Scholar] [CrossRef]
- Wang, Y.-X.; Wang, G.-C. A systematic theoretical study of the water gas shift reaction on the Pt/ZrO2 interface and Pt(111) face: Key role of a potassium additive. Catal. Sci. Technol. 2020, 10, 876–892. [Google Scholar] [CrossRef]
- Ghanekar, P.; Kubal, J.; Cui, Y.; Mitchell, G.; Delgass, W.N.; Ribeiro, F.; Greeley, J. Catalysis at Metal/Oxide Interfaces: Density Functional Theory and Microkinetic Modeling of Water Gas Shift at Pt/MgO Boundaries. Top. Catal. 2020, 1–15. [Google Scholar] [CrossRef]
- Rajesh, T.; Devi, R.N. Pt States in BaCe0.98Pt0.02O3−δ during Start Up and Shut Down Operations Under Different Conditions: Stability and Activity of Ionic Pt in Water Gas Shift Reaction. Catal. Lett. 2014, 144, 2227–2232. [Google Scholar] [CrossRef]
- Ribeiro, R.; Meira, D.; Rodella, C.; Oliveira, D.; Bueno, J.M.C.; Zanchet, D. Probing the stability of Pt nanoparticles encapsulated in sol–gel Al2O3 using in situ and ex situ characterization techniques. Appl. Catal. A Gen. 2014, 485, 108–117. [Google Scholar] [CrossRef]
- Liu, B.; Goldbach, A.; Xu, H. Sour water–gas shift reaction over Pt/CeO2 catalysts. Catal. Today 2011, 171, 304–311. [Google Scholar] [CrossRef]
- Liu, B.; Xu, H.; Zhang, Z. Platinum based core–shell catalysts for sour water–gas shift reaction. Catal. Commun. 2012, 26, 159–163. [Google Scholar] [CrossRef]
- Neto, R.C.; Monteiro, I.; Maximino, R.; De Azevedo, J.T. Longevity test for a Water–Gas Shift catalyst. Int. J. Hydrog. Energy 2014, 39, 5242–5247. [Google Scholar] [CrossRef]
- Silva, L.P.; Terra, L.E.; Coutinho, A.C.; Passos, F.B. Sour water–gas shift reaction over Pt/CeZrO2 catalysts. J. Catal. 2016, 341, 1–12. [Google Scholar] [CrossRef]
- Silva, L.; Luis, E.; Ana, C. Sulphur-Tolerant Catalyst for Use in Water-Gas Shift Reactions, and Water-Gas Shift Process. Patent WO2016197211Al, 15 December 2016. [Google Scholar]
- Silva, L.P.; Freitas, M.M.; Santos, R.M.; Perez, G.; Terra, L.E.; Coutinho, A.C.; Passos, F.B. The effect of metal type on the sulfur tolerance of catalysts supported on niobia for sour water-gas shift reaction. Int. J. Hydrog. Energy 2018, 43, 3190–3202. [Google Scholar] [CrossRef]
- Lombardo, E.; Cornaglia, C.; Múnera, J. Development of an active, selective and durable water-gas shift catalyst for use in membrane reactors. Catal. Today 2016, 259, 165–176. [Google Scholar] [CrossRef]
- Kambolis, A.; Lizarraga, L.; Tsampas, M.N.; Burel, L.; Rieu, M.; Viricelle, J.-P.; Vernoux, P. Electrochemical promotion of catalysis with highly dispersed Pt nanoparticles. Electrochem. Commun. 2012, 19, 5–8. [Google Scholar] [CrossRef]
- Hajar, Y.M.; Venkatesh, B.; Baranova, E.A. Electrochemical Promotion of Nanostructured Palladium Catalyst for Complete Methane Oxidation. Catalysts 2019, 9, 48. [Google Scholar] [CrossRef]
- González-Cobos, J.; Valverde, J.L.; De Lucas-Consuegra, A. Electrochemical vs. chemical promotion in the H2 production catalytic reactions. Int. J. Hydrog. Energy 2017, 42, 13712–13723. [Google Scholar] [CrossRef]
- Souentie, S.; Lizarraga, L.; Kambolis, A.; Alves-Fortunato, M.; Valverde, J.L.; Vernoux, P. Electrochemical promotion of the water–gas shift reaction on Pt/YSZ. J. Catal. 2011, 283, 124–132. [Google Scholar] [CrossRef]
- Sekine, Y.; Yamagishi, K.; Nogami, Y.; Manabe, R.; Oshima, K.; Ogo, S. Low Temperature Catalytic Water Gas Shift in an Electric Field. Catal. Lett. 2016, 146, 1423–1428. [Google Scholar] [CrossRef]
- Cui, X.; Su, H.-Y.; Chen, R.; Yu, L.; Dong, J.; Mac, C.; Wang, S.; Li, J.-F.; Yang, F.; Xiao, J.; et al. Room-temperature electrochemical water–gas shift reaction for high purity hydrogen production. Nat. Commun. 2019, 10, 86. [Google Scholar] [CrossRef]
- Ladebeck, J.R.; Wagner, J.P. Catalyst development for water-gas shift. Handb. Fuel Cells 2010, 3, 190–201. [Google Scholar]
- Makets Insider. Available online: https://markets.businessinsider.com/commodities (accessed on 12 June 2020).
- Tonkovich, A.; Zilka, J.; Lamont, M.; Wang, Y.; Wegeng, R. Microchannel reactors for fuel processing applications. I. Water gas shift reactor. Chem. Eng. Sci. 1999, 54, 2947–2951. [Google Scholar] [CrossRef]

























© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).