Integrating Flotation and Pyrometallurgy for Recovering Graphite and Valuable Metals from Battery Scrap

 , , , and
, , , and 
Abstract
1. Introduction
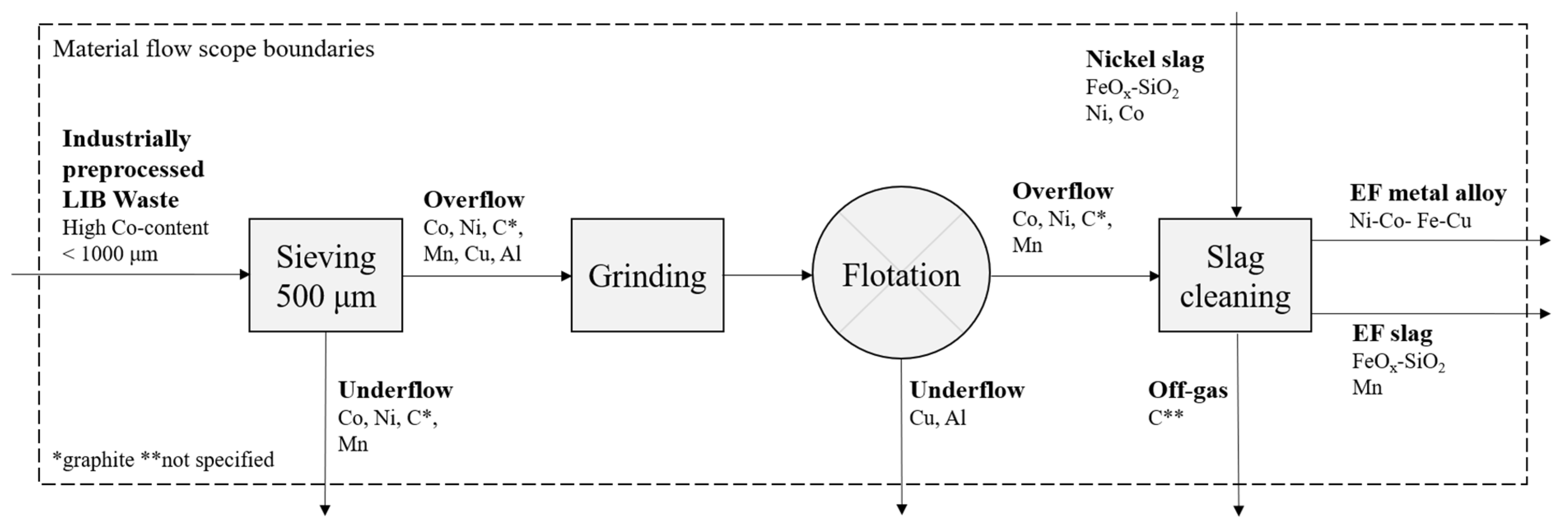
2. Materials and Methods
2.1. Froth Flotation
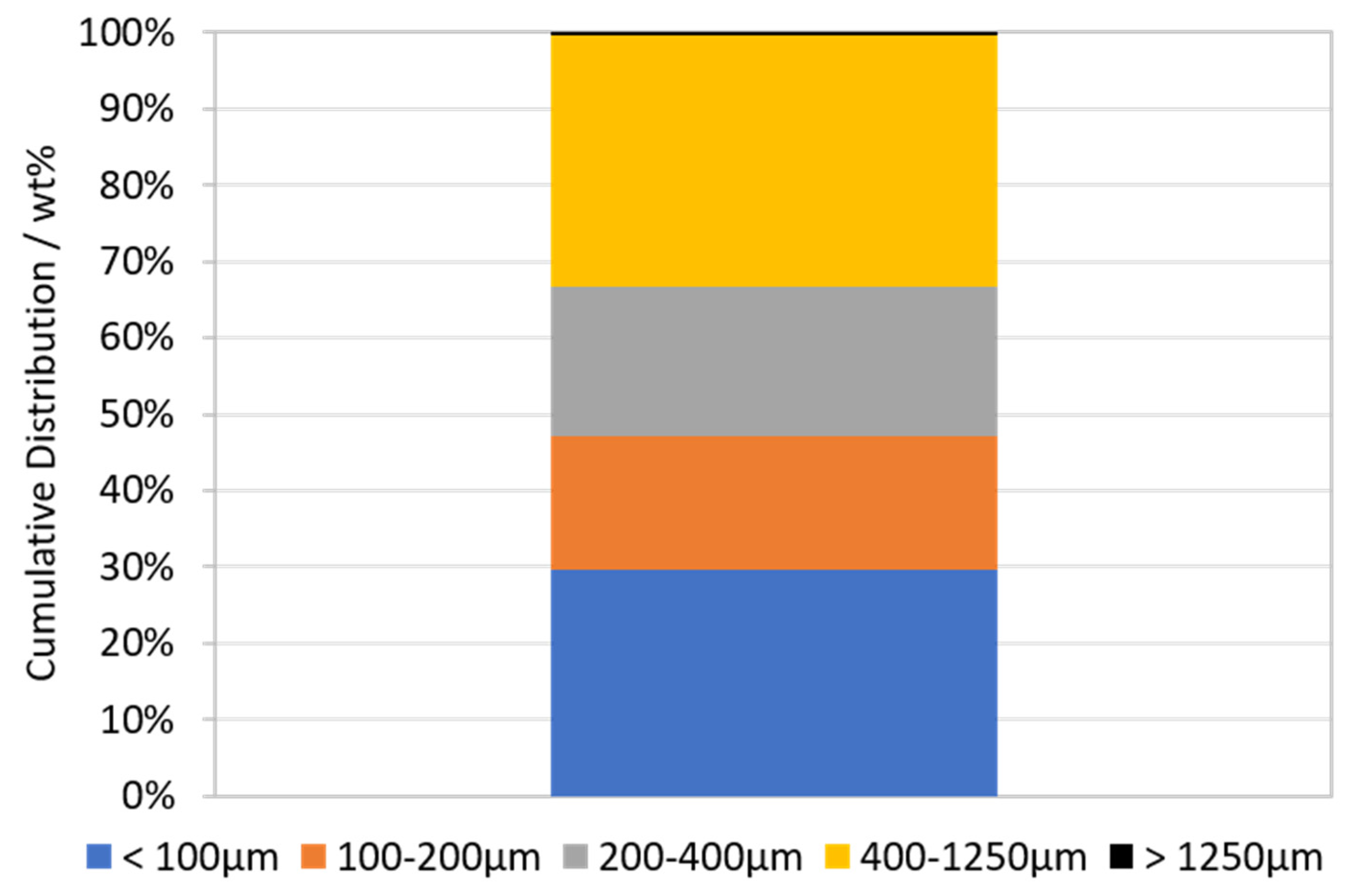
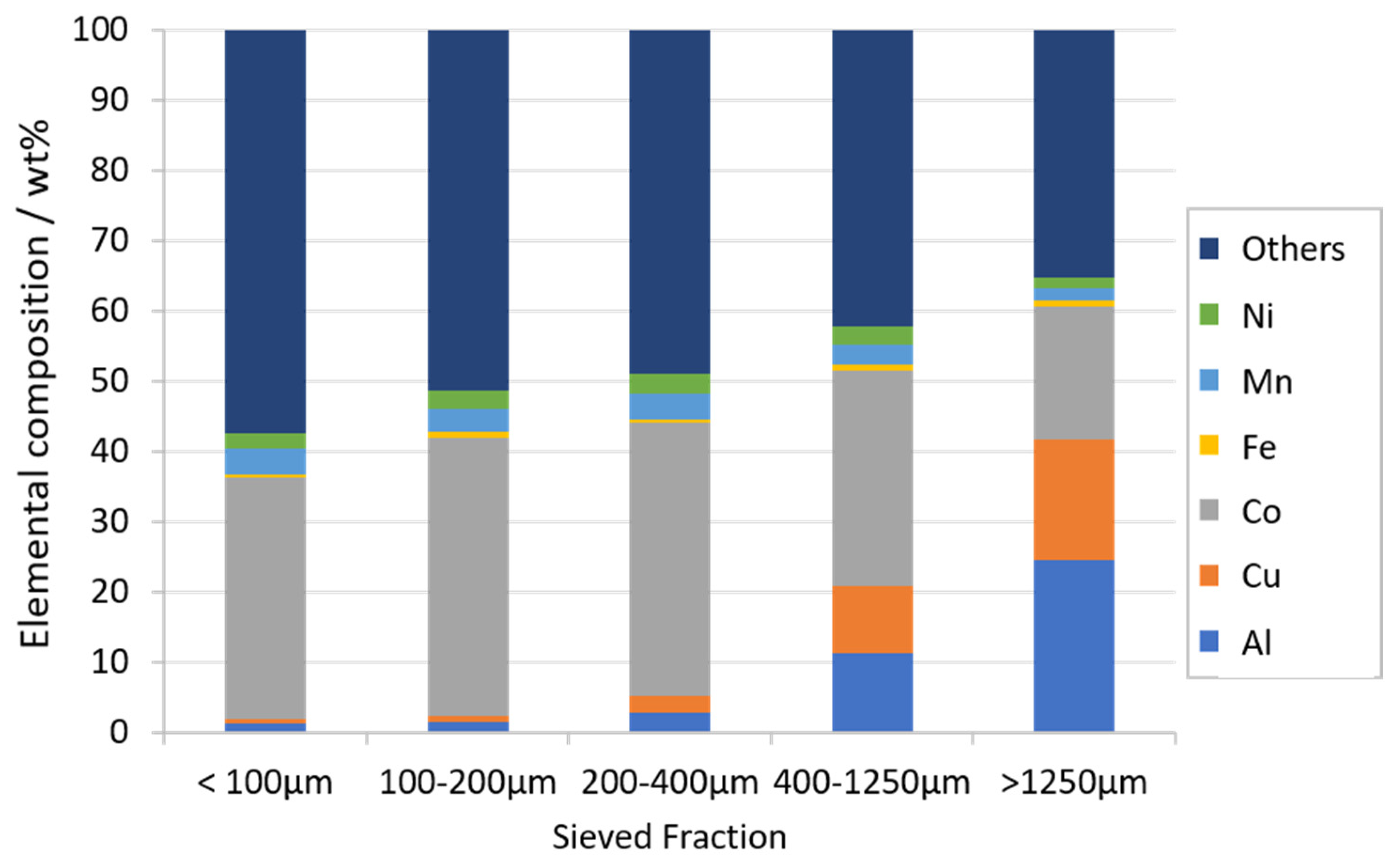
2.1.1. Sample Preparation and Characterization of the Waste Lithium Ion Battery Feed
2.1.2. Experimental Procedure of Froth Flotation
2.1.3. Characterization of the Flotation Products
- = Fraction of feed weight reporting to the froth at time t (1, 10, 25 min) [wt%]
- = Metal content of Cu in the mineral [wt%]
- = Fraction of Cu reporting to the froth at time t (1, 20, 25 min) [wt%]
- = Fraction of Cu reporting to the feed at time t (1, 10, 25min) [wt%].
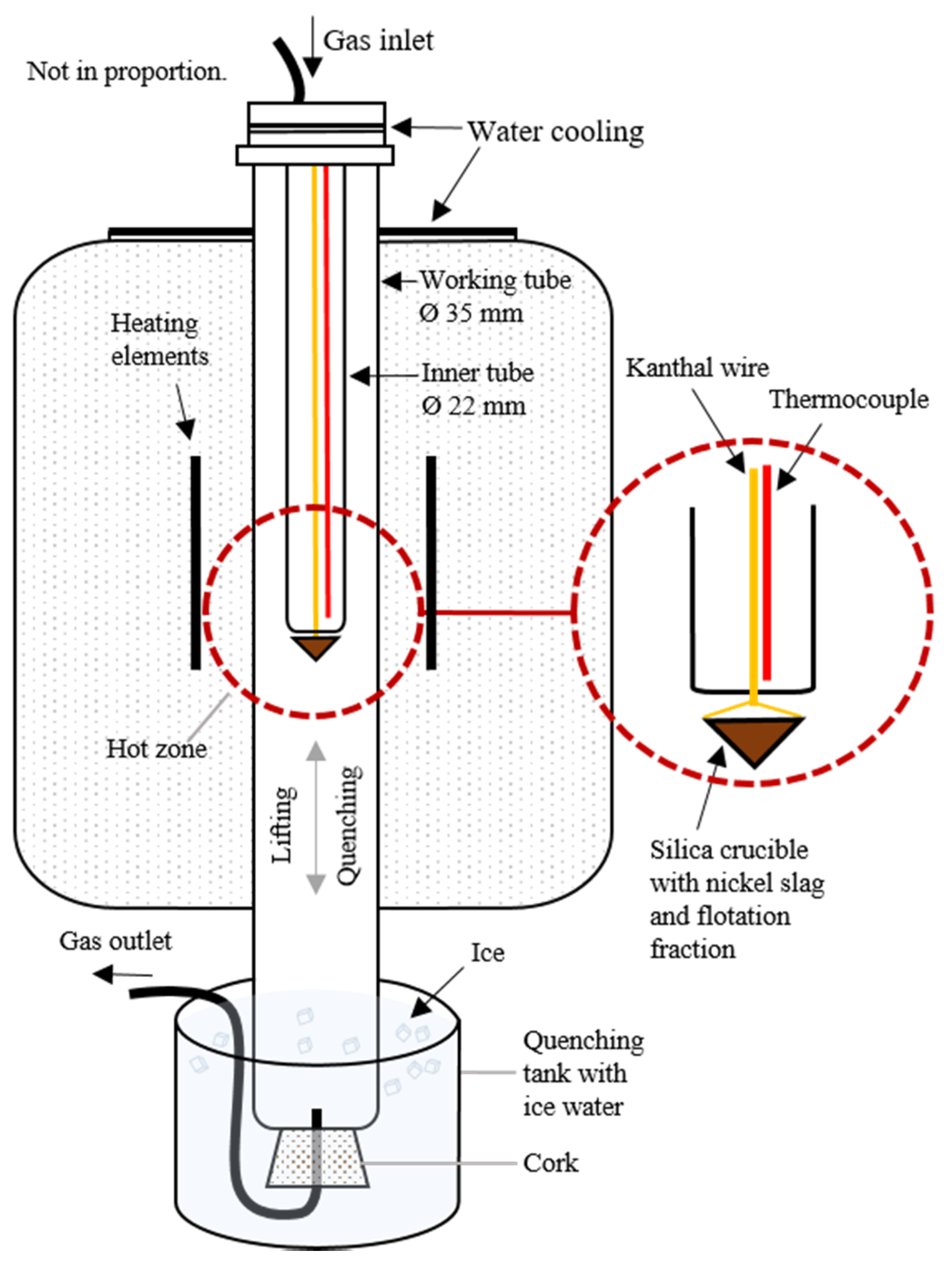
2.2. Slag Cleaning
2.2.1. Materials
2.2.2. Experimental Procedure
3. Results & Discussion
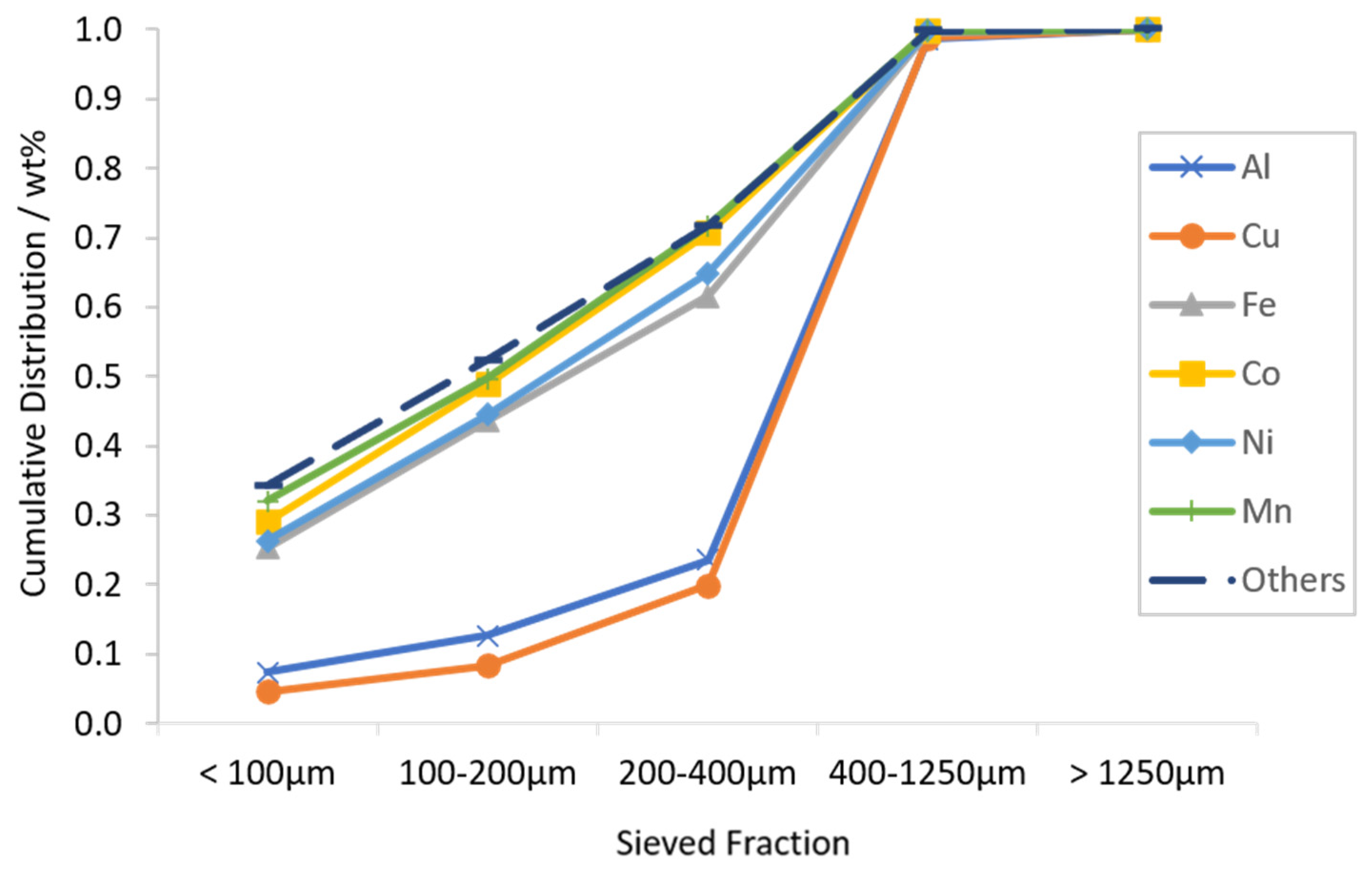
3.1. Characterization of LIB Waste
3.2. Flotation Experiments
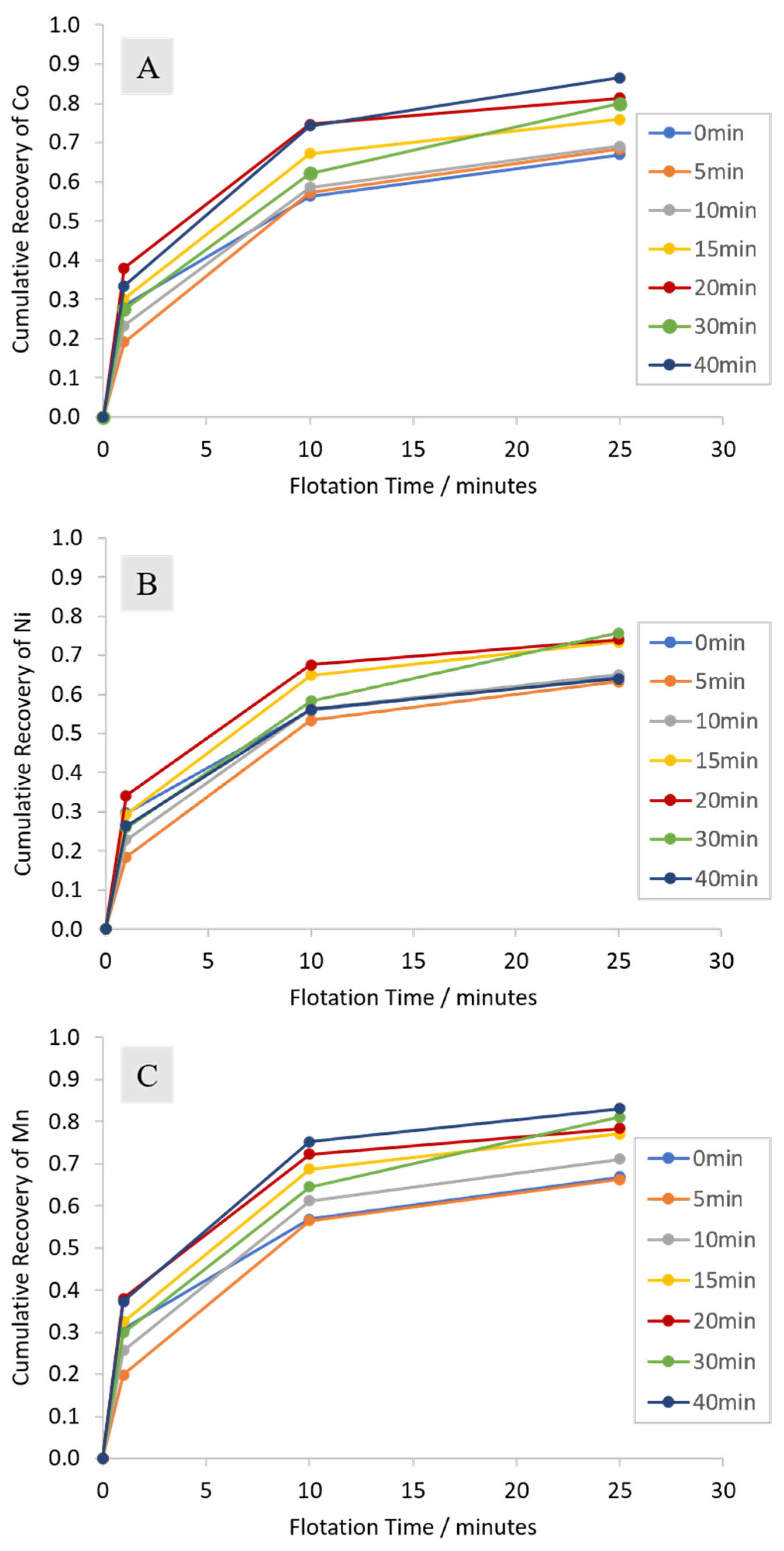
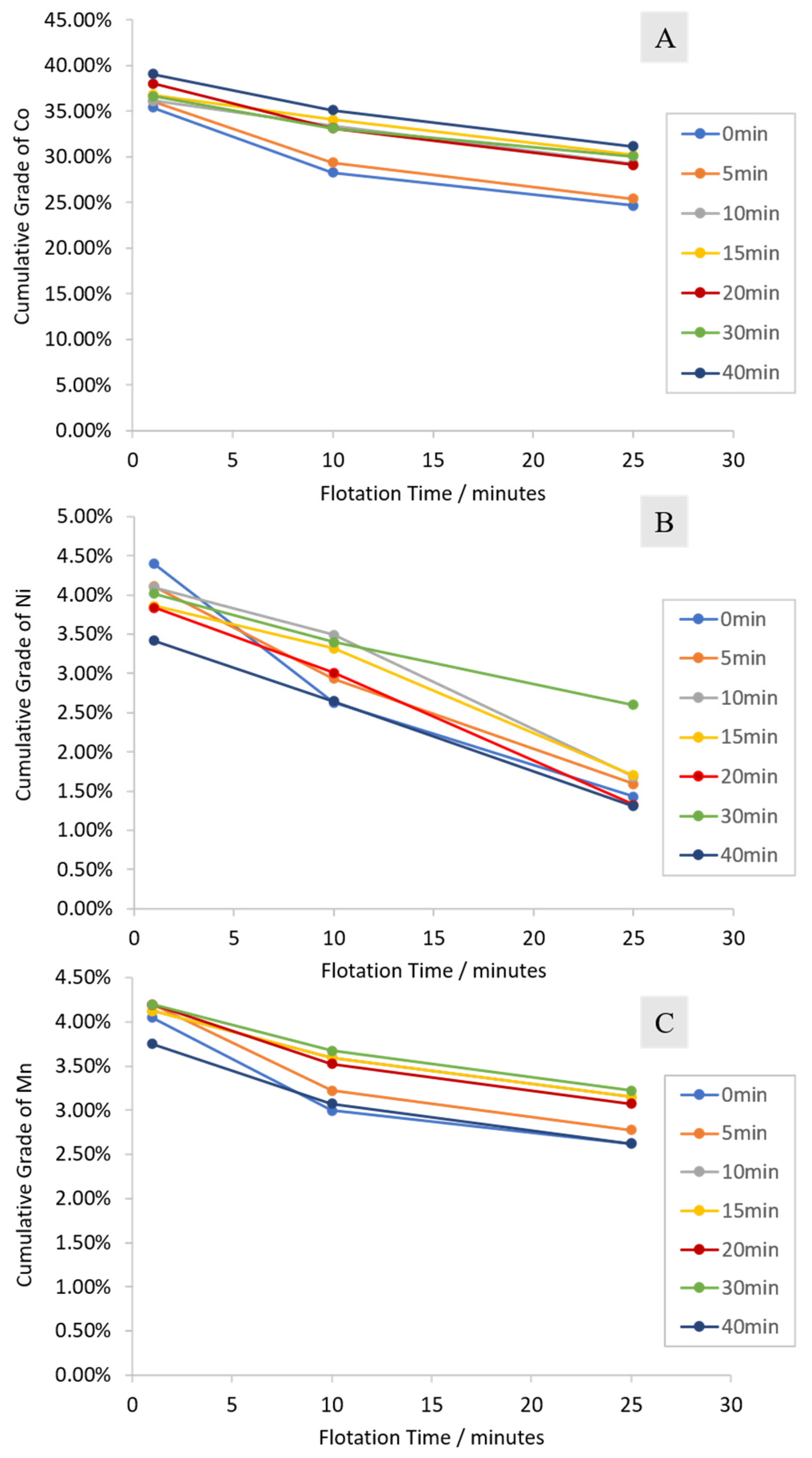
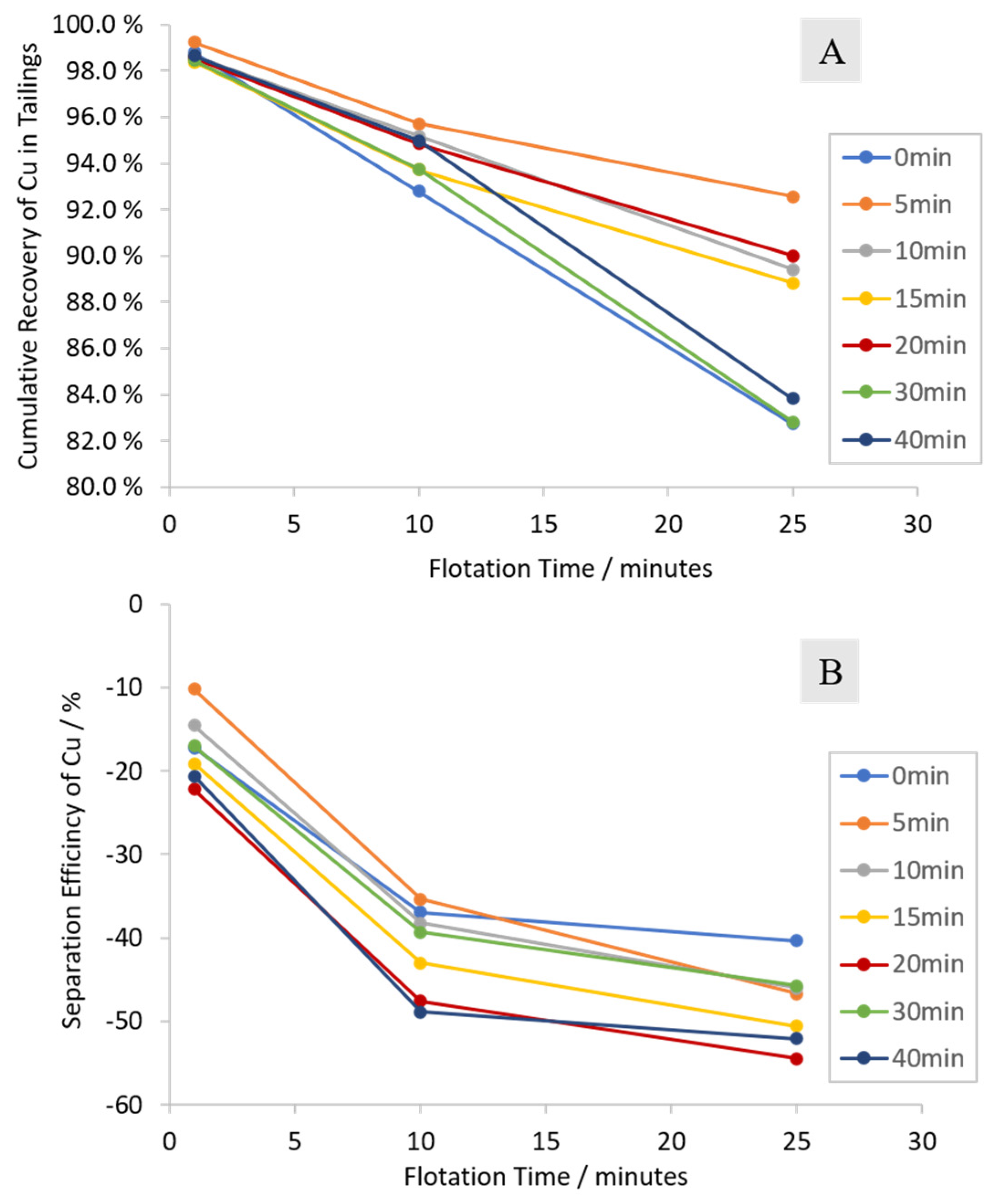
3.2.1. Cumulative Grades and Recoveries
3.2.2. Characterization of the Froth Fraction Used for the Nickel Slag Cleaning
3.3. Reduction of Metals
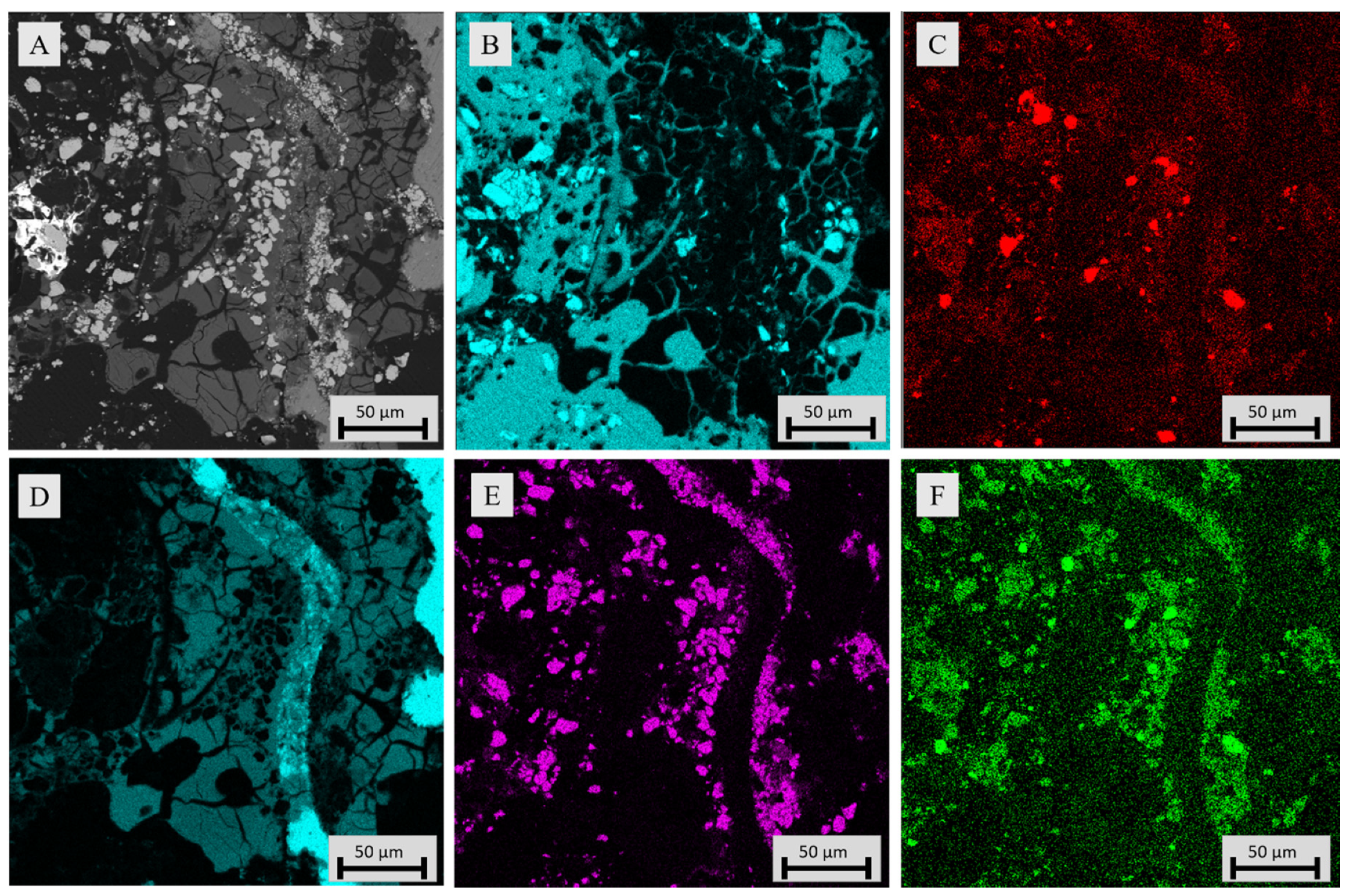
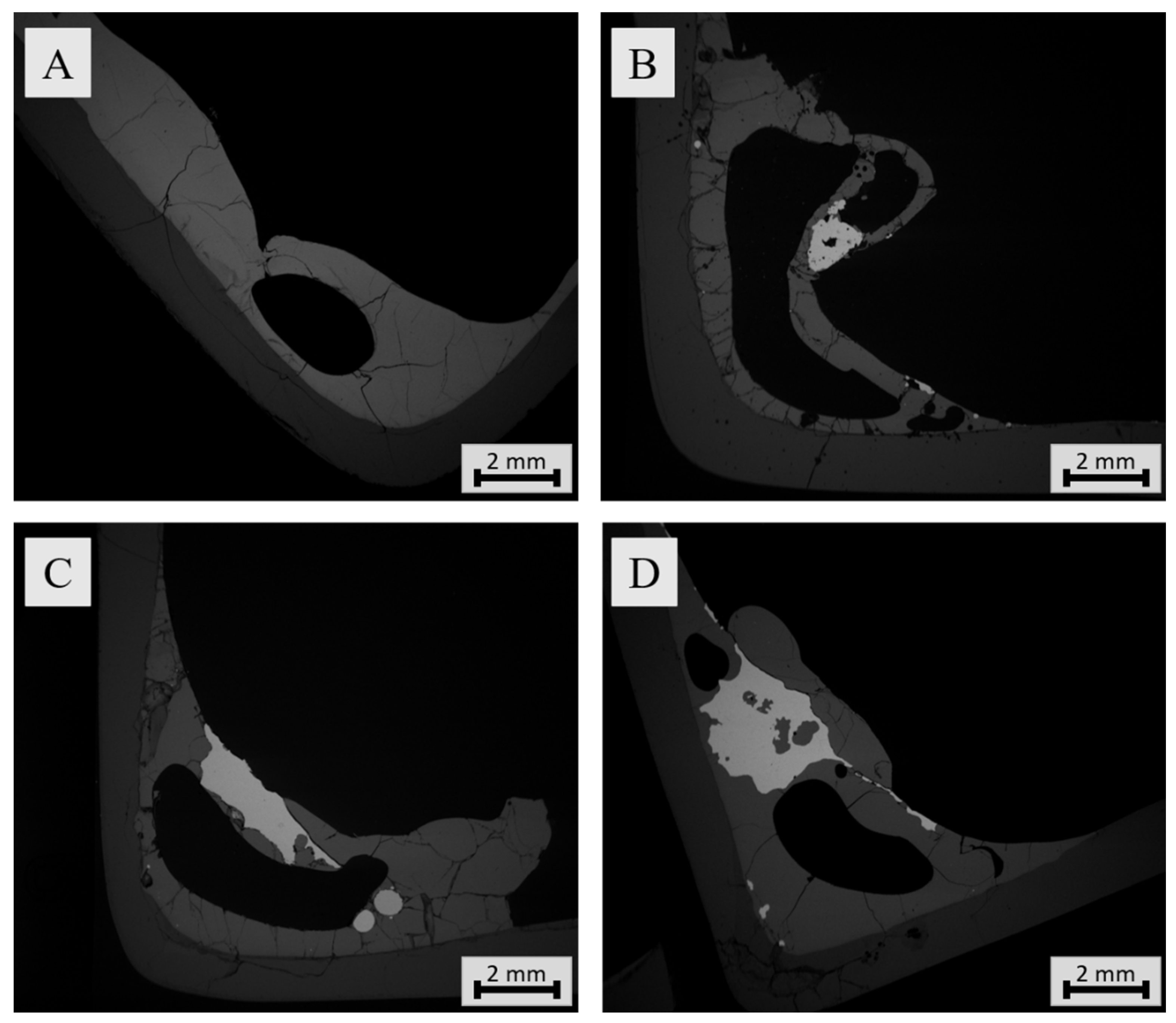
3.3.1. Sample Microstructure
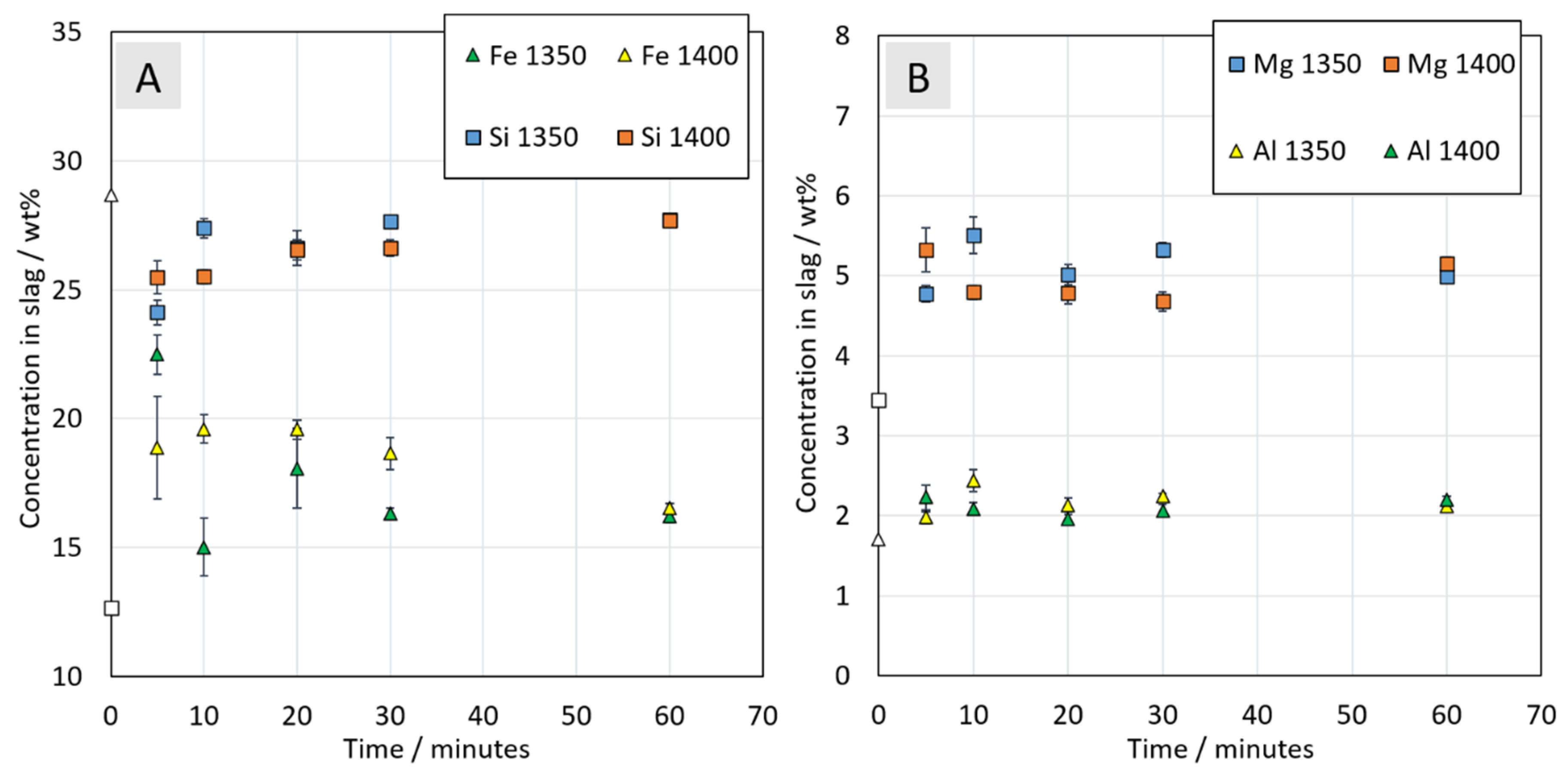
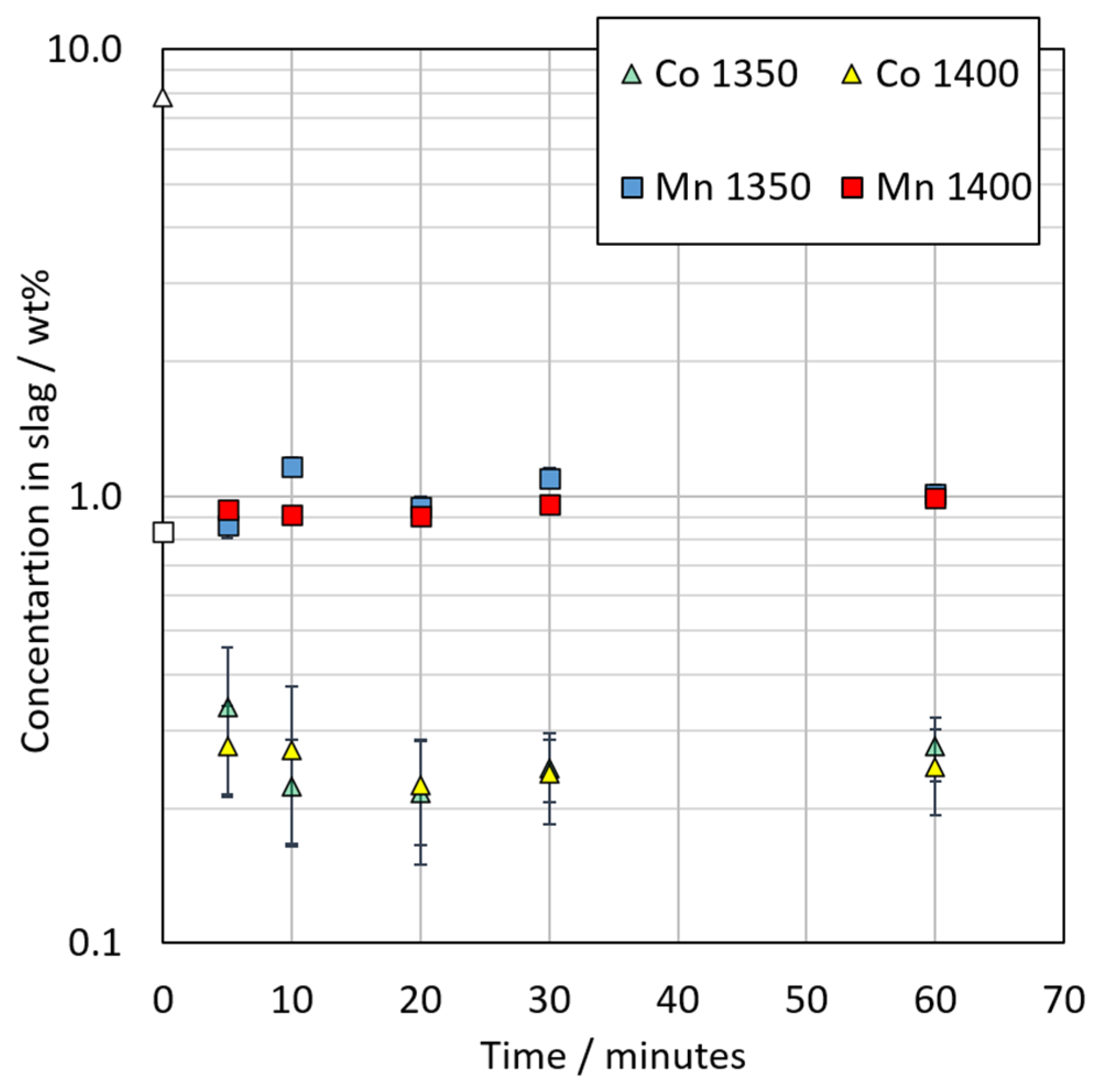
3.3.2. Chemical Composition of Slag
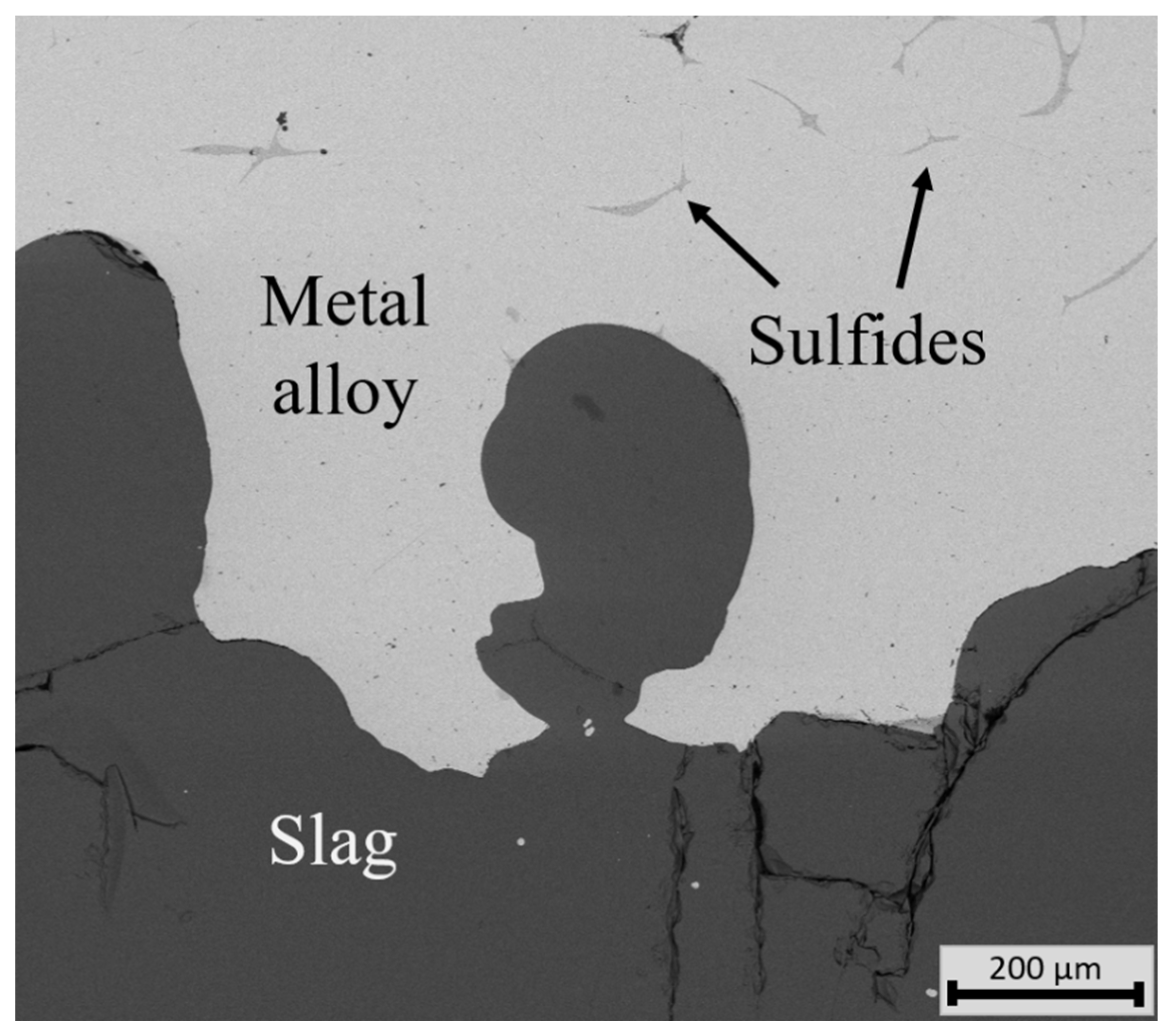
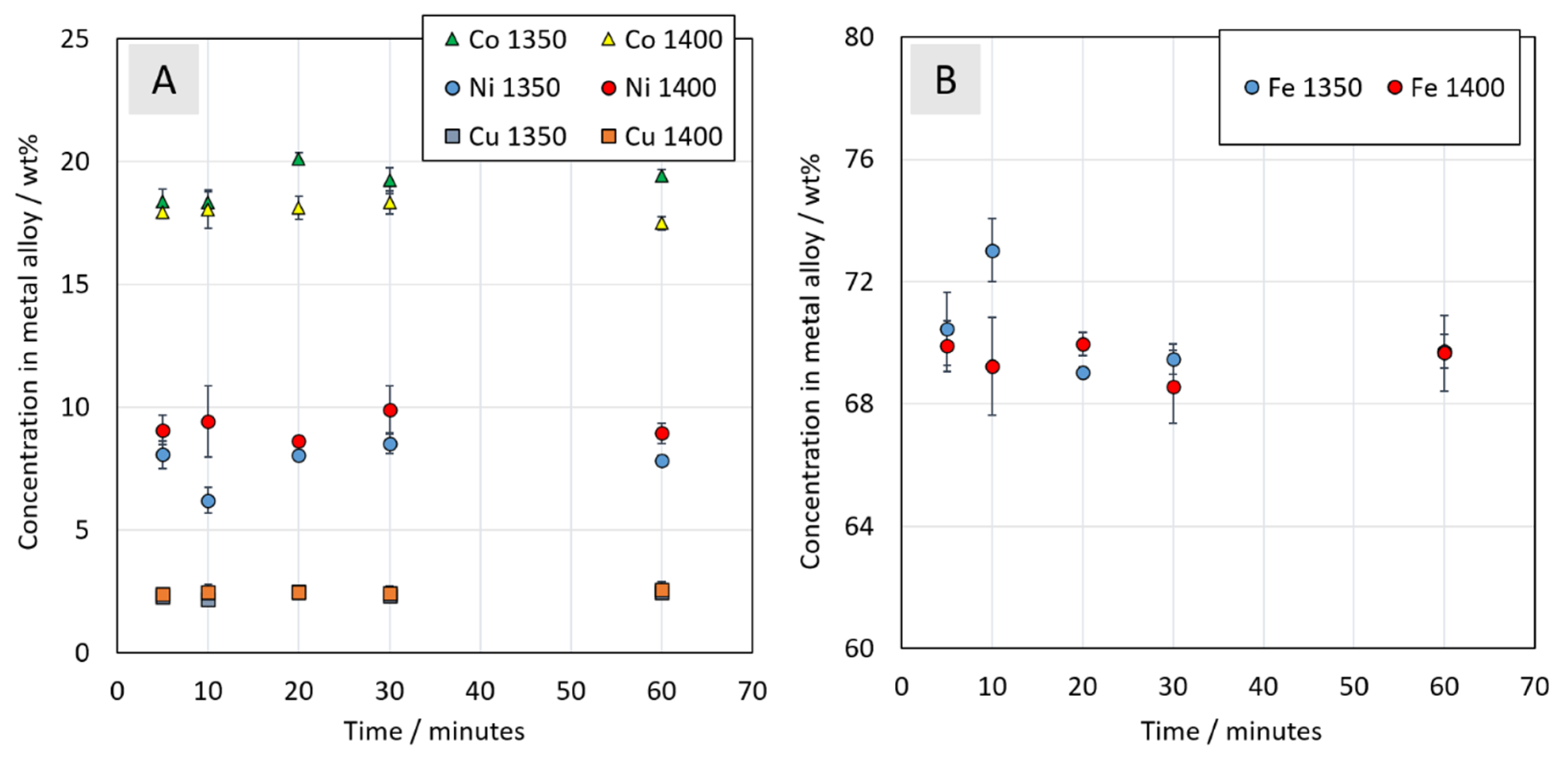
3.3.3. Chemical Composition of Metal Alloy
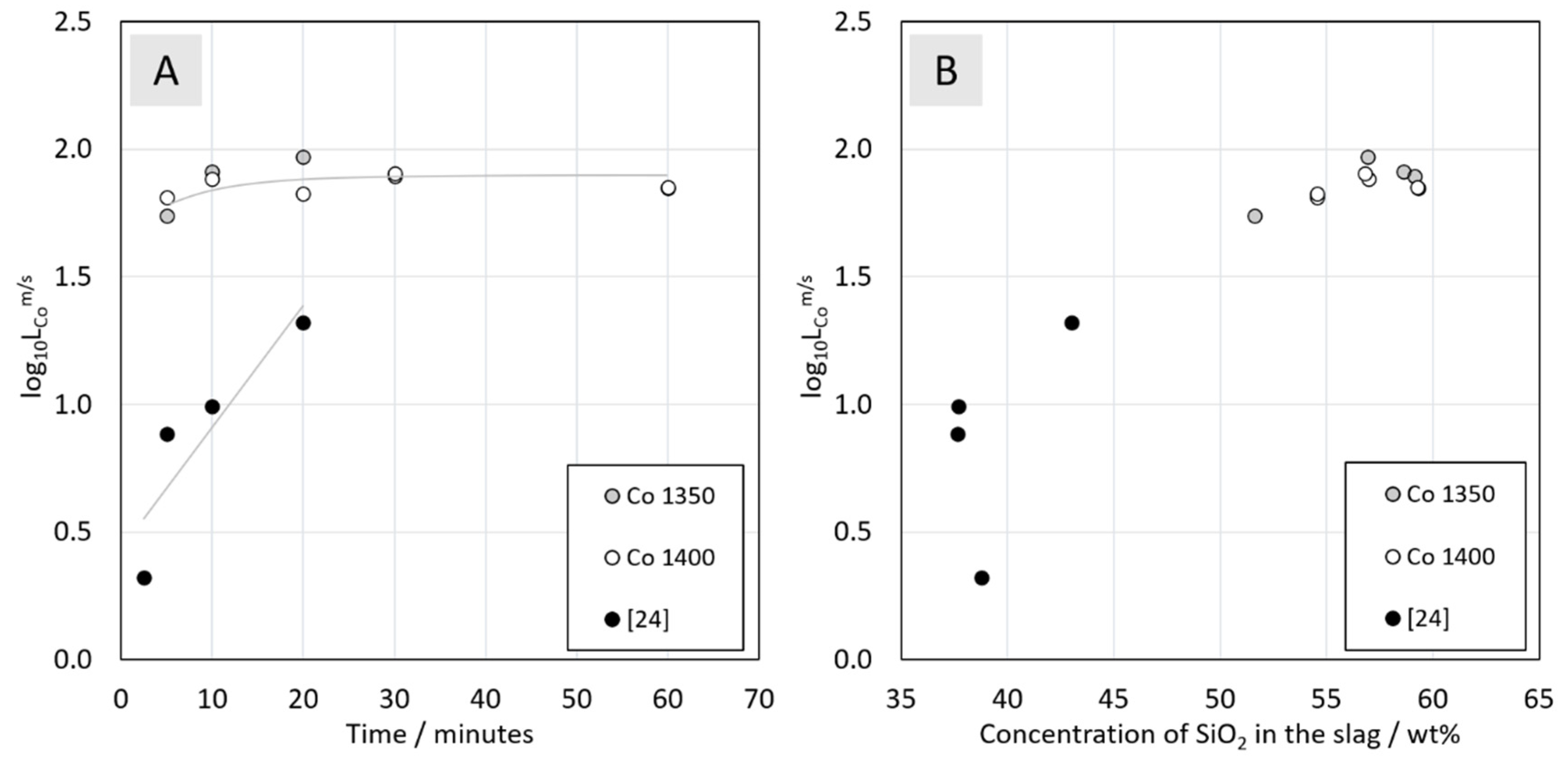
3.3.4. Distribution of Cobalt between Metal Alloy and the Slag
4. Conclusions
- Froth flotation is introduced as a mechanical pre-treatment for pyrometallurgical battery recycling
- Graphite in the flotation fraction is utilized to replace coke in the nickel slag cleaning.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Huang, B.; Pan, Z.; Su, X.; An, L. Recycling of lithium-ion batteries: Recent advances and perspectives. J. Power Sources 2018, 399, 274–286. [Google Scholar] [CrossRef]
- A Vision for a Sustainable Battery Value Chain in 2030, Insight Report, September 2019. Unlocking the Full Potential to Power Sustainable Development and Climate Change Mitigation. Available online: http://www3.weforum.org/docs/WEF_A_Vision_for_a_Sustainable_Battery_Value_Chain_in_2030_Report.pdf (accessed on 4 March 2020).
- Wentker, M.; Greenwood, M.; Leker, J. A bottom-up approach to lithium-ion battery cost modeling with a focus on cathode active materials. Energies 2019, 12, 504. [Google Scholar] [CrossRef]
- Commission Staff Working Document on the Evaluation of the Directive 2006/66/EC on Batteries and Accumulators and Waste Batteries and Accumulatros and Repealing Directive 91/157/EEC. Available online: https://ec.europa.eu/environment/waste/batteries/pdf/evaluation_report_batteries_directive.pdf (accessed on 23 April 2020).
- Shi, J.; Chen, M.; Li, Y.; Eric, H.; Klemettinen, L.; Lundström, M.; Taskinen, P.; Jokilaakso, A. Sulfation roasting mechanism for spent lithium-ion battery metal oxides under SO2-O2-Ar atmosphere. JOM 2019, 71, 4473–4482. [Google Scholar] [CrossRef]
- Wang, X.; Gaustad, G.; Babbitt, C.W.; Richa, K. Economies of scale for future lithium-ion battery recycling infrastructure. Resour. Conserv. Recycl. 2014, 83, 53–62. [Google Scholar] [CrossRef]
- Bernhart, W. Recycling of lithium-ion batteries in the context of technology and price developments. Atzelectron. Worldw. 2019, 14, 38–43. [Google Scholar] [CrossRef]
- Gu, F.; Guo, J.; Yao, X.; Summers, P.A.; Widijatmoko, S.D.; Hall, P. An investigation of the current status of recycling spent lithium-ion batteries from consumer electronics in China. J. Clean. Prod. 2017, 161, 765–780. [Google Scholar] [CrossRef]
- Werner, D.; Peuker, U.A.; Mütze, T. Recycling chain for spent lithium-ion batteries. Metals 2020, 10, 316. [Google Scholar] [CrossRef]
- Zheng, X.; Zhu, Z.; Lin, X.; Zhang, Y.; He, y.; Cao, H.; Sun, Z. A mini-review on metal recycling from spent lithium ion batteries. Engineering 2018, 4, 361–370. [Google Scholar] [CrossRef]
- Rämä, M.; Nurmi, S.; Jokilaakso, A.; Klemettinen, L.; Taskinen, P.; Salminen, J. Thermal processing of jarosite leach residue for a safe disposable slag and valuable metals recovery. Metals 2018, 8, 744. [Google Scholar] [CrossRef]
- Directive 2006/66/EC of the European Parliament and of the Council of 6 September 2006 on Batteries and Accumulators and Waste Batteries and Accumulators and Repealing Directive 91/157/EEC. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:02006L0066-20131230&rid=1 (accessed on 30 March 2020).
- Harper, G.; Sommerville, R.; Kendrick, E.; Driscoll, L.; Slater, P.; Stolkin, R.; Walton, A.; Christensen, P.; Heidrich, O.; Lambert, S.; et al. Recycling lithium-ion batteries from electric vehicles. Nature 2019, 575, 75–86. [Google Scholar] [CrossRef]
- Velázquez-Martínez, O.; Valio, J.; Santasalo-Aarnio, A.; Reuter, M.; Serna-Guerrero, R. A critical review of lithium-ion battery recycling processes from a circular economy perspective. Batteries 2019, 5, 68. [Google Scholar] [CrossRef]
- Nitta, N.; Wu, F.; Lee, J.T.; Yushin, G. Li-ion battery materials: Present and future. Mater. Today 2015, 18, 252–264. [Google Scholar] [CrossRef]
- Lv, W.; Wang, Z.; Cao, H.; Sun, Y.; Zhang, Y.; Sun, Z. A critical review and analysis on the recycling of spent lithium-ion batteries. ACS Sustain. Chem. Eng. 2018, 6, 1504–1521. [Google Scholar]
- Or, T.; Gourley, S.W.D.; Kaliyappan, K.; Yu, A.; Chen, Z. Recycling of mixed cathode lithium-ion batteries for electric vehicles: Current status and future outlook. Carbon Energy 2020, 2, 6–43. [Google Scholar] [CrossRef]
- Porvali, A.; Aaltonen, M.; Ojanen, S.; Velazquez-Martinez, O.; Eronen, E.; Liu, F.; Wilson, B.P.; Serna-Guerrero, R.; Lundström, M. Mechanical and hydrometallurgical processes in HCl media for the recycling of valuable metals from Li-ion battery waste. Resour. Conserv. Recycl. 2019, 142, 257–266. [Google Scholar] [CrossRef]
- Mayyas, A.; Steward, D.; Mann, M. The case for recycling: Overview and challenges in the material supply chain for automotive li-ion batteries. SMT 2018, 19. [Google Scholar] [CrossRef]
- Chen, M.; Ma, X.; Chen, B.; Arsenault, R.; Karlson, P.; Simon, N.; Wang, Y. Recycling end-of-life electric vehicle lithium-ion batteries. Joule 2019, 3, 2622–2646. [Google Scholar] [CrossRef]
- Dańczak, A.; Klemettinen, L.; Kurhila, M.; Taskinen, P.; Lindberg, D.; Jokilaakso, A. Behavior of battery metals lithium, cobalt, manganese and lanthanum in black copper smelting. Batteries 2020, 6, 16. [Google Scholar] [CrossRef]
- Ren, G.; Xiao, S.; Xie, M.; Pan, B.; Chen, J.; Wang, F.; Xia, X. Recovery of valuable metals from spent lithium ion batteries by smelting reduction process based on FeO-SiO2-Al2O3 slag system. Trans. Nonferr. Metals Soc. 2017, 27, 450–456. [Google Scholar] [CrossRef]
- Ren, G.; Xiao, S.; Xie, M.; Pan, B.; Fan, Y.; Wang, F.; Xia, X. Recovery of valuable metals from spent lithium-ion batteries by smelting reduction process based on MnO-SiO2-Al2O3 slag system. In Advances in Molten Slags, Fluxes, and Salts, Proceedings of the 10th International Conference on Molten Slags, Fluxes and Salts (MOLTEN16), 22–25 May 2016; Reddy, R.G., Chaubal, P., Pistorius, P.C., Pal, U., Eds.; TMS (The Minerals, Metals & Materials Society): Pittsburgh, PA, USA, 2016. [Google Scholar]
- Ruismäki, R.; Dańczak, A.; Klemettinen, L.; Taskinen, P.; Lindberg, D.; Jokilaakso, A. Integrated battery scrap recycling and nickel slag cleaning with methane reduction. Minerals 2020, 10, 435. [Google Scholar] [CrossRef]
- Yu, J.; He, Y.; Ge, Z.; Li, H.; Xie, W.; Wang, S. A promising physical method for recovery of LiCoO2 and graphite from spent lithium-ion batteries: Grinding flotation. Sep. Purif. Technol. 2018, 190, 45–52. [Google Scholar] [CrossRef]
- Wang, F.; Zhang, T.; He, Y.; Zhao, Y.; Wang, S.; Zhang, G.W.; Zhang, Y.; Feng, Y. Recovery of valuable materials from spent lithium-ion batteries by mechanical separation and thermal treatment. J. Clean. Prod. 2018, 185, 646–652. [Google Scholar] [CrossRef]
- Liu, J.; Wang, H.; Hu, T.; Bai, X.; Wang, S.; Xie, W.; Juan, H.J.; He, Y. Recovery of LiCoO2 and graphite from spent lithium-ion batteries by cryogenic grinding and froth flotation. Miner. Eng. 2020, 148, 106223. [Google Scholar] [CrossRef]
- Zhang, G.; He, Y.; Feng, Y.; Wang, H.; Zhu, X. Pyrolysis-ultrasonic-assisted flotation technology for recovering graphite and LiCoO2 from spent lithium-ion batteries. ACS Sustain. Chem. Eng. 2018, 6, 10896–10904. [Google Scholar]
- Zhang, G.; He, J.; Wang, H.; Feng, Y.; Xie, W.; Zhu, X. Application of mechanical crushing combined with pyrolysis-enhanced flotation technology to recover graphite and LiCoO2 from spent lithium-ion batteries. J. Clean. Prod. 2019, 231, 1418–1427. [Google Scholar] [CrossRef]
- He, Y.; Zhang, T.; Wang, F.; Zhang, G.; Zhang, W.; Wang, J. Recovery of LiCoO2 and graphite from spent lithium-ion batteries by Fenton reagent-assisted flotation. J. Clean. Prod. 2017, 143, 319–325. [Google Scholar] [CrossRef]
- Wills, B.A.; Finch, J. Wills’ Mineral Processing Technology: An Introduction to the Practical Aspects of Ore Treatment and Mineral Recovery; Butterworth-Heinemann: Oxford, UK, 2015; ISBN 978-0-08-097053-0. [Google Scholar]
- Wan, X.; Jokilaakso, A.; Fellman, J.; Klemettinen, L.; Marjakoski, M. Behavior of Waste Printed Circuit Board (WPCB) materials in the copper matte smelting process. Metals 2018, 8, 887. [Google Scholar] [CrossRef]
- Wan, X.; Jokilaakso, A.; Iduozee, I.; Eric, H.; Latostenmaa, P. Experimental research on the behavior of WEEE scrap in flash smelting settler with copper concentrate and synthetic slag. In Proceedings of the EMC 2019, Düsseldorf, Germany, 23–26 June 2019; Volume 3, pp. 1137–1150. [Google Scholar]
- Semykina, A.; Seetharaman, S. Recovery of manganese ferrite in nanoform from the metallurgical slags. Metals Mater. Trans. B 2011, 42, 2–4. [Google Scholar] [CrossRef]
- Sukhomlinov, D.; Klemettinen, L.; Avarmaa, K.; O’Brien, H.; Taskinen, P.; Jokilaakso, A. Distribution of Ni, Co, precious and platinum group metals in copper making process. Metall. Mater. Trans. B 2019, 50, 1752–1765. [Google Scholar] [CrossRef]
- Piskunen, P.; Avarmaa, K.; O’Bren, H.; Klemettinen, L.; Johto, H.; Taskinen, P. Precious metal distributions in direct nickel matte smelting with low-Cu mattes. Metall. Mater. Trans. B 2017, 49, 98–112. [Google Scholar] [CrossRef]
- Hellstén, N.; Klemettinen, L.; Sukhomlinov, D.; O’Brien, H.; Taskinen, P.; Jokilaakso, A.; Salminen, J. Slag cleaning equilibria in iron silicate slag-copper systems. J. Sustain. Metall. 2019, 5, 463–473. [Google Scholar] [CrossRef]
- Shlesinger, M.E.; King, M.J.; Sole, K.C.T.G.; Davenport, W.G. Extractive Metallurgy of Copper—Chapter 5: Matte Smelting Fundamentals; Elsevier: Oxford, UK, 2011; pp. 73–88. [Google Scholar] [CrossRef]
- Stefanova, V.P.; Iliev, P.K.; Stefanov, B.S. Copper, nickel and cobalt extraction from FeCuNiCoMn alloy obtained after pyrometallurgical processing of deep sea nodules. In Proceedings of the Tenth ISOPE Ocean Mining and Gas Hydrates Symposium, Szczecin, Poland, 22–26 September 2013. [Google Scholar]
- Riekkola-Vanhanen, M. Finnish Expert Report on Best Available Techniques in Nickel Production; Finnish Environment Institute: Helsinki, Finland, 1999; ISBN 952-11-0507-0. Available online: https://helda.helsinki.fi/bitstream/handle/10138/40684/FE_317.pdf?sequence=1&isAllowed=y (accessed on 13 May 2020).
- Jones, R.T.; Denton, G.M.; Reynolds, Q.G.; Parker, J.A.L.; van Tonder, G.J.J. Recovery of cobalt, nickel, and copper from slags, using DC-arc furnace technology. In Proceedings of the International Symposium on Challenges of Process Intensification 35th Annual Conference of Metallurgists, Montreal, QC, Canada, 25–29 August 1996; Available online: http://www.mintek.co.za/Pyromet/Cobalt/Cobalt.htm (accessed on 30 March 2020).
- Li, N.; Guo, J.; Chang, Z.; Dang, H.; Zhao, X.; Ali, S.; Li, W.; Zhou, H.; Sun, C. Aqueous leaching of lithium from simulated pyrometallurgical slag by sodium sulfate roasting. RSC Adv. 2019, 9, 23908–23915. [Google Scholar] [CrossRef]
- Ayala, J.; Fernández, B. Recovery of manganese from silicomanganese slag by means of a hydrometallurgical process. Hydrometallurgy 2015, 158, 68–73. [Google Scholar] [CrossRef]
- Baumgartner, S.J.; Groot, D.R. The recovery of manganese products from ferromanganese slag using a hydrometallurgical route. J. S. Afr. Inst. Min. Metall. 2014, 114, 2411–9717. Available online: http://www.scielo.org.za/scielo.php?script=sci_arttext&pid=S2225-62532014000400011&lng=en&nrm=iso (accessed on 14 May 2020).
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Fe | SiO2 | MgO | Al2O3 | Ni | CaO | Co | Cu | S | Zn |
|---|---|---|---|---|---|---|---|---|---|---|
| wt% | 35.86 | 33.86 | 7.14 | 3.12 | 3.46 | 1.65 | 0.46 | 0.52 | 0.15 | 0.06 |
| Grades | Co | Ni | Mn | Cu | O | Li | Other |
|---|---|---|---|---|---|---|---|
| Head Grade [wt%] | 19.00 | 2.26 | 2.39 | 24.29 | 10.32 | 2.24 | 39.50 |
| 0–1 min Grade [wt%] | 37.30 | 3.97 | 4.16 | 1.95 | 20.25 | 4.39 | 27.98 |
| Substance | Co | Cu | Li | Fe | Mn | Ni | Zn | S | SiO2 | MgO | Al2O3 | Graphite |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| wt% | 7.83 | 0.81 | 0.88 | 28.69 | 0.83 | 3.56 | 0.05 | 0.12 | 27.09 | 5.71 | 3.23 | 3.8–4.0 * |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruismäki, R.; Rinne, T.; Dańczak, A.; Taskinen, P.; Serna-Guerrero, R.; Jokilaakso, A. Integrating Flotation and Pyrometallurgy for Recovering Graphite and Valuable Metals from Battery Scrap. Metals 2020, 10, 680. https://doi.org/10.3390/met10050680
Ruismäki R, Rinne T, Dańczak A, Taskinen P, Serna-Guerrero R, Jokilaakso A. Integrating Flotation and Pyrometallurgy for Recovering Graphite and Valuable Metals from Battery Scrap. Metals. 2020; 10(5):680. https://doi.org/10.3390/met10050680
Chicago/Turabian StyleRuismäki, Ronja, Tommi Rinne, Anna Dańczak, Pekka Taskinen, Rodrigo Serna-Guerrero, and Ari Jokilaakso. 2020. "Integrating Flotation and Pyrometallurgy for Recovering Graphite and Valuable Metals from Battery Scrap" Metals 10, no. 5: 680. https://doi.org/10.3390/met10050680
APA StyleRuismäki, R., Rinne, T., Dańczak, A., Taskinen, P., Serna-Guerrero, R., & Jokilaakso, A. (2020). Integrating Flotation and Pyrometallurgy for Recovering Graphite and Valuable Metals from Battery Scrap. Metals, 10(5), 680. https://doi.org/10.3390/met10050680