Exercise-Induced Mitohormesis for the Maintenance of Skeletal Muscle and Healthspan Extension


{kind=link}
{kind=link}
Abstract
1. Introduction
2. Age-Related Decline in Mitochondrial Function
3. Sarcopenia and Its Effect on Overall Health
4. Skeletal Muscle Mitochondrial Dysfunction and Sarcopenia
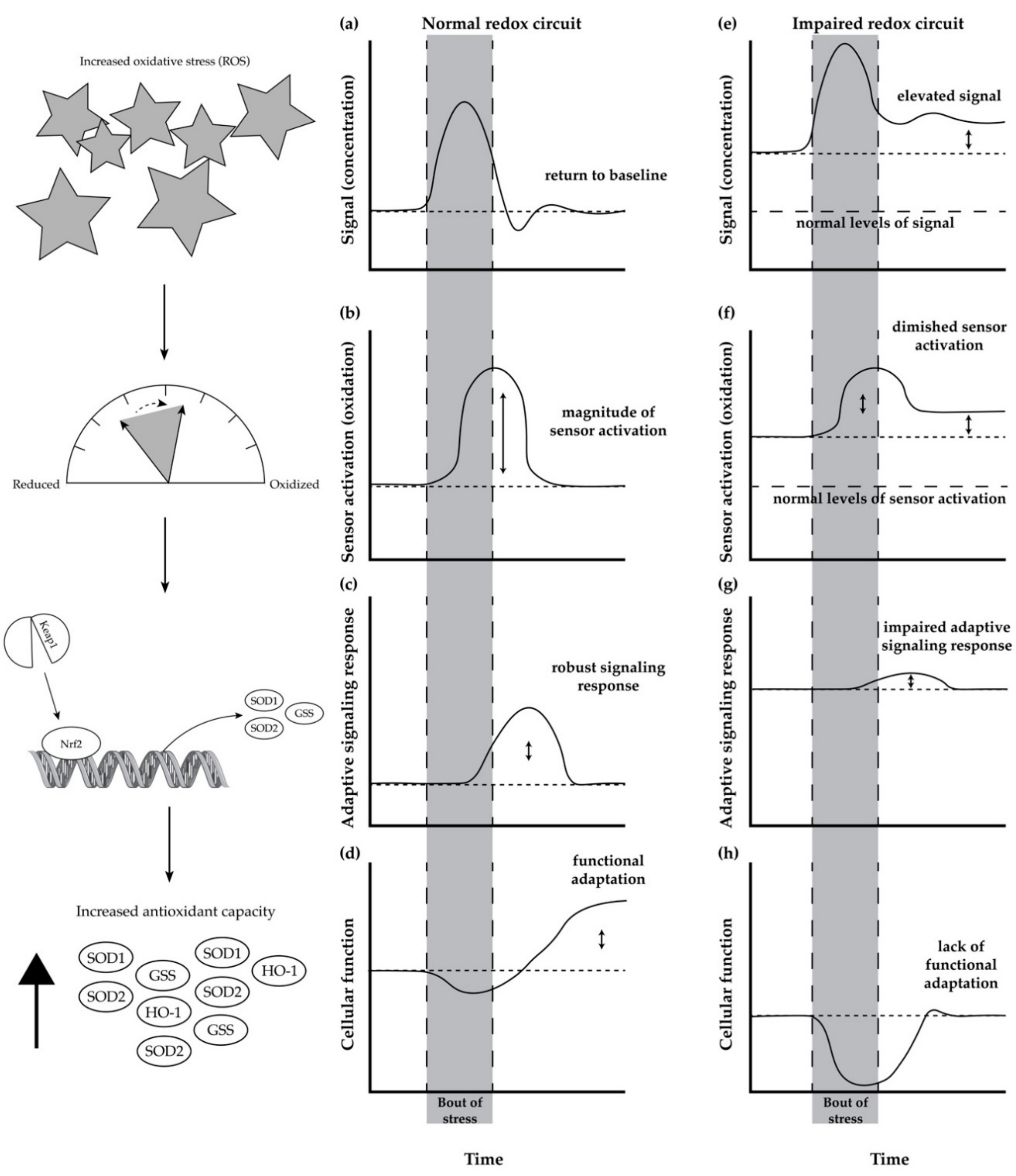
5. Redox Circuits and Redox Homeostasis
Age-Related Impairment in Redox Homeostasis and Its Consequence in Skeletal Muscle
6. Mitohormesis as a Mechanism to Restore Redox Homeostasis
6.1. Mitohormesis, Aerobic Exercise, and Healthspan Extension
6.2. Mitohormetic Effects of a Bout of Aerobic Exercise
6.3. Mitohormetic Adaptations from Aerobic Exercise Training
7. Targeting Nrf2 as a Complementary or Alternative Approach to Restore Redox Homeostasis
7.1. Directly Scavenging ROS with Exogenous Antioxidants
7.2. Upregulation of Endogenous Antioxidants
8. Gaps and Future Directions
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Beard, J.R.; Officer, A.M.; Cassels, A.K. The World Report on Ageing and Health. Gerontologist 2016, 56 (Suppl. 2), S163–S166. [Google Scholar] [CrossRef]
- European Commission. The 2015 Ageing Report: Economic and Budgetary Projections for the 28 EU Member States (2013–2060); European Commission Economic and Financial Affairs European Economy; European Commission: Brussels, Belgium, 2015; pp. 1–424. [Google Scholar]
- Olshansky, S.J.; Goldman, D.P.; Zheng, Y.; Rowe, J.W. Aging in America in the twenty-first century: Demographic forecasts from the MacArthur Foundation Research Network on an Aging Society. Milbank Q. 2009, 87, 842–862. [Google Scholar] [CrossRef] [PubMed]
- Kaeberlein, M. How healthy is the healthspan concept? Geroscience 2018, 40, 361–364. [Google Scholar] [CrossRef] [PubMed]
- Seals, D.R.; Justice, J.N.; LaRocca, T.J. Physiological geroscience: Targeting function to increase healthspan and achieve optimal longevity. J. Physiol. 2016, 594, 2001–2024. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, B.K.; Berger, S.L.; Brunet, A.; Campisi, J.; Cuervo, A.M.; Epel, E.S.; Franceschi, C.; Lithgow, G.J.; Morimoto, R.I.; Pessin, J.E.; et al. Geroscience: Linking Aging to Chronic Disease. Cell 2014, 159, 709–713. [Google Scholar] [CrossRef]
- Rebelo-Marques, A.; De Sousa Lages, A.; Andrade, R.; Ribeiro, C.F.; Mota-Pinto, A.; Carrilho, F.; Espregueira-Mendes, J. Aging Hallmarks: The Benefits of Physical Exercise. Front. Endocrinol. 2018, 9, 258. [Google Scholar] [CrossRef]
- Yun, J.; Finkel, T. Mitohormesis. Cell Metab. 2014, 19, 757–766. [Google Scholar] [CrossRef]
- Holloway, G.P.; Holwerda, A.M.; Miotto, P.M.; Dirks, M.L.; Verdijk, L.B.; Van Loon, L.J.C. Age-Associated Impairments in Mitochondrial ADP Sensitivity Contribute to Redox Stress in Senescent Human Skeletal Muscle. Cell Rep. 2018, 22, 2837–2848. [Google Scholar] [CrossRef]
- Drake, J.C.; Yan, Z. Precision remodeling: How exercise improves mitochondrial quality in myofibers. Curr. Opin. Psychol. 2019, 10, 96–101. [Google Scholar] [CrossRef]
- Seo, A.Y.; Joseph, A.M.; Dutta, D.; Hwang, J.C.Y.; Aris, J.P.; Leeuwenburgh, C. New insights into the role of mitochondria in aging: Mitochondrial dynamics and more. J. Cell Sci. 2010, 123, 2533–2542. [Google Scholar] [CrossRef] [PubMed]
- Ureshino, R.P.; Rocha, K.K.; Lopes, G.S.; Bincoletto, C.; Smaili, S.S. Calcium signaling alterations, oxidative stress, and autophagy in aging. Antioxid. Redox Signal. 2014, 21, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Short, K.R.; Bigelow, M.L.; Kahl, J.; Singh, R.; Coenen-Schimke, J.; Raghavakaimal, S.; Nair, K.S. Decline in skeletal muscle mitochondrial function with aging in humans. Proc. Natl. Acad. Sci. USA 2005, 102, 5618–5623. [Google Scholar] [CrossRef] [PubMed]
- Lanza, I.R.; Short, D.K.; Short, K.R.; Raghavakaimal, S.; Basu, R.; Joyner, M.J.; McConnell, J.P.; Nair, K.S. Endurance Exercise as a Countermeasure for Aging. Diabetes 2008, 57, 2933–2942. [Google Scholar] [CrossRef] [PubMed]
- Porter, C.; Hurren, N.M.; Cotter, M.V.; Bhattarai, N.; Reidy, P.T.; Dillon, E.L.; Durham, W.J.; Tuvdendorj, D.; Sheffield-Moore, M.; Volpi, E.; et al. Mitochondrial respiratory capacity and coupling control decline with age in human skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E224–E232. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, U.F.; Krustrup, P.; Kjaer, M.; Rasmussen, H.N. Experimental evidence against the mitochondrial theory of aging a study of isolated human skeletal muscle mitochondria. Exp. Gerontol. 2003, 38, 877–886. [Google Scholar] [CrossRef]
- Gouspillou, G.; Bourdel-Marchasson, I.; Rouland, R.; Calmettes, G.; Biran, M.; Deschodt-Arsac, V.; Miraux, S.; Thiaudiere, E.; Pasdois, P.; Detaille, D.; et al. Mitochondrial energetics is impaired in vivo in aged skeletal muscle. Aging Cell 2013, 13, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Larsen, S.; Hey-Mogensen, M.; Rabøl, R.; Stride, N.; Helge, J.W.; Dela, F. The influence of age and aerobic fitness: Effects on mitochondrial respiration in skeletal muscle. Acta Physiol. (Oxf.) 2012, 205, 423–432. [Google Scholar] [CrossRef]
- Distefano, G.; Standley, R.A.; Dubé, J.J.; Carnero, E.A.; Ritov, V.B.; Stefanovic-Racic, M.; Toledo, F.G.S.; Piva, S.R.; Goodpaster, B.H.; Coen, P.M. Chronological Age Does not Influence Ex-vivo Mitochondrial Respiration and Quality Control in Skeletal Muscle. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2015, 72, 535–542. [Google Scholar]
- Rosenberg, I.H. Summary comments. Am. J. Clin. Nutr. 1989, 50, 1231–1233. [Google Scholar] [CrossRef]
- Cruz-Jentoft, A.J.; Bahat, G.; Bauer, J.; Boirie, Y.; Bruyère, O.; Cederholm, T.; Cooper, C.; Landi, F.; Rolland, Y.; Sayer, A.A.; et al. Writing Group for the European Working Group on Sarcopenia in Older People 2 (EWGSOP2), and the Extended Group for EWGSOP2 Sarcopenia: Revised European consensus on definition and diagnosis. Age Ageing 2019, 48, 16–31. [Google Scholar] [CrossRef] [PubMed]
- Bulow, J.; Ulijaszek, S.J.; Holm, L. Rejuvenation of the term Sarcopenia. J. Appl. Physiol. 2018, 134, 512. [Google Scholar] [CrossRef] [PubMed]
- Langer, H.T.; Mossakowski, A.A.; Baar, K.; Alcazar, J.; Martin-Rincon, M.; Alegre, L.M.; Ara, I.; Calbet, J.A.L.; Hinkley, J.M.; Coen, P.M.; et al. Commentaries on Viewpoint: Rejuvenation of the term sarcopenia. J. Appl. Physiol. 2019, 126, 257–262. [Google Scholar] [PubMed]
- Anker, S.D.; Morley, J.E.; von Haehling, S. Welcome to the ICD-10 code for sarcopenia. J. Cachexia Sarcopenia Muscle 2016, 7, 512–514. [Google Scholar] [CrossRef] [PubMed]
- Janssen, I.; Heymsfield, S.B.; Ross, R. Low relative skeletal muscle mass (sarcopenia) in older persons is associated with functional impairment and physical disability. J. Am. Geriatr. Soc. 2002, 50, 889–896. [Google Scholar] [CrossRef] [PubMed]
- Visser, M.; Goodpaster, B.H.; Kritchevsky, S.B.; Newman, A.B.; Nevitt, M.; Rubin, S.M.; Simonsick, E.M.; Harris, T.B. Muscle mass, muscle strength, and muscle fat infiltration as predictors of incident mobility limitations in well-functioning older persons. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2005, 60, 324–333. [Google Scholar] [CrossRef]
- Goodpaster, B.H.; Park, S.W.; Harris, T.B.; Kritchevsky, S.B.; Nevitt, M.; Schwartz, A.V.; Simonsick, E.M.; Tylavsky, F.A.; Visser, M.; Newman, A.B. The loss of skeletal muscle strength, mass, and quality in older adults: The health, aging and body composition study. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2006, 61, 1059–1064. [Google Scholar] [CrossRef]
- Landi, F.; Cruz-Jentoft, A.J.; Liperoti, R.; Russo, A.; Giovannini, S.; Tosato, M.; Capoluongo, E.; Bernabei, R.; Onder, G. Sarcopenia and mortality risk in frail older persons aged 80 years and older: Results from ilSIRENTE study. Age Ageing 2013, 42, 203–209. [Google Scholar] [CrossRef]
- Peterson, S.J.; Braunschweig, C.A. Prevalence of Sarcopenia and Associated Outcomes in the Clinical Setting. Nutr. Clin. Pr. 2016, 31, 40–48. [Google Scholar] [CrossRef]
- Pedersen, B.K.; Febbraio, M.A. Muscles, Exercise and Obesity: Skeletal Muscle as a Secretory Organ; Nature Publishing Group: London, UK, 2012; pp. 1–9. [Google Scholar]
- Scott, D.; de Courten, B.; Ebeling, R. Sarcopenia: A potential cause and consequence of type 2 diabetes in Australia’s ageing population? Med. J. Aust. 2016, 205, 329–333. [Google Scholar] [CrossRef]
- Wang, Y.; Lee, D.-C.; Brellenthin, A.G.; Sui, X.; Church, T.S.; Lavie, C.J.; Blair, S.N. Association of Muscular Strength and Incidence of Type 2 Diabetes. Mayo Clin. Proc. 2019, 94, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Maliszewska, K.; Adamska-Patruno, E.; Goscik, J.; Lipinska, D.; Citko, A.; Krahel, A.; Miniewska, K.; Fiedorczuk, J.; Moroz, M.; Gorska, M.; et al. The Role of Muscle Decline in Type 2 Diabetes Development: A 5-Year Prospective Observational Cohort Study. Nutrients 2019, 11, 834. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-R.; Jung, S.M.; Bang, H.; Kim, H.S.; Kim, Y.B. Association between muscle strength and type 2 diabetes mellitus in adults in Korea. Medicine 2018, 97, e10984. [Google Scholar] [CrossRef] [PubMed]
- Batsis, J.A.; Mackenzie, T.A.; Lopez-Jimenez, F.; Bartels, S.J. Sarcopenia, sarcopenic obesity, and functional impairments in older adults: National Health and Nutrition Examination Surveys 1999–2004. Nutr. Res. 2015, 35, 1031–1039. [Google Scholar] [CrossRef] [PubMed]
- Atkins, J.L.; Whincup, P.H.; Morris, R.W.; Lennon, L.T.; Papacosta, O.; Wannamethee, S.G. Sarcopenic Obesity and Risk of Cardiovascular Disease and Mortality: A Population-Based Cohort Study of Older Men. J. Am. Geriatr. Soc. 2014, 62, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Chin, S.O.; Rhee, S.Y.; Chon, S.; Hwang, Y.-C.; Jeong, I.-K.; Oh, S.; Ahn, K.J.; Chung, H.Y.; Woo, J.-T.; Kim, S.-W.; et al. Sarcopenia Is Independently Associated with Cardiovascular Disease in Older Korean Adults: The Korea National Health and Nutrition Examination Survey (KNHANES) from 2009. PLoS ONE 2013, 8, e60119. [Google Scholar] [CrossRef] [PubMed]
- McLeod, M.; Breen, L.; Hamilton, D.L.; Philp, A. Live strong and prosper: The importance of skeletal muscle strength for healthy ageing. Biogerontology 2016, 17, 497–510. [Google Scholar] [CrossRef]
- Koopman, R.; van Loon, L.J.C. Aging, exercise, and muscle protein metabolism. J. Appl. Physiol. 2009, 106, 2040–2048. [Google Scholar] [CrossRef]
- Haus, J.M.; Carrithers, J.A.; Trappe, S.W.; Trappe, T.A. Collagen, cross-linking, and advanced glycation end products in aging human skeletal muscle. J. Appl. Physiol. 2007, 103, 2068–2076. [Google Scholar] [CrossRef]
- Larsson, L.; Sjödin, B.; Karlsson, J. Histochemical and biochemical changes in human skeletal muscle with age in sedentary males, age 22–65 years. Acta Physiol. Scand. 1978, 103, 31–39. [Google Scholar] [CrossRef]
- Lauretani, F.; Russo, C.R.; Bandinelli, S.; Bartali, B.; Cavazzini, C.; Di Iorio, A.; Corsi, A.M.; Rantanen, T.; Guralnik, J.M.; Ferrucci, L. Age-associated changes in skeletal muscles and their effect on mobility: An operational diagnosis of sarcopenia. J. Appl. Physiol. 2003, 95, 1851–1860. [Google Scholar] [CrossRef] [PubMed]
- Schaap, L.A.; Pluijm, S.M.F.; Deeg, D.J.H.; Visser, M. Inflammatory markers and loss of muscle mass (sarcopenia) and strength. Am. J. Med. 2006, 119, 526. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Freire, M.; Scalzo, P.; D’Agostino, J.; Moore, Z.A.; Diaz-Ruiz, A.; Fabbri, E.; Zane, A.; Chen, B.; Becker, K.G.; Lehrmann, E.; et al. Skeletal muscle ex vivo mitochondrial respiration parallels decline in vivo oxidative capacity, cardiorespiratory fitness, and muscle strength: The Baltimore Longitudinal Study of Aging. Aging Cell 2018, 17, e12725. [Google Scholar] [CrossRef] [PubMed]
- Hepple, R.T.; Rice, C.L. Innervation and neuromuscular control in ageing skeletal muscle. J. Physiol. 2016, 594, 1965–1978. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.J. Reactive oxygen species in sarcopenia: Should we focus on excess oxidative damage or defective redox signalling? Mol. Asp. Med. 2016, 50, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Snow, L.M.; Fugere, N.A.; Thompson, L.V. Advanced glycation end-product accumulation and associated protein modification in type II skeletal muscle with aging. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2007, 62, 1204–1210. [Google Scholar] [CrossRef]
- Dos Santos, S.L.; Baraibar, M.A.; Lundberg, S.; Eeg-Olofsson, O.; Larsson, L.; Friguet, B. Oxidative proteome alterations during skeletal muscle ageing. Redox Biol. 2015, 5, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Vasilaki, A.; Richardson, A.; Van Remmen, H.; Brooks, S.V.; Larkin, L.; McArdle, A.; Jackson, M.J. Role of nerve-muscle interactions and reactive oxygen species in regulation of muscle proteostasis with ageing. J. Physiol. 2017, 595, 6409–6415. [Google Scholar] [CrossRef]
- Musci, R.V.; Hamilton, K.L.; Miller, B.F. Targeting mitochondrial function and proteostasis to mitigate dynapenia. Eur. J. Appl. Physiol. 2018, 118, 1–9. [Google Scholar] [CrossRef]
- Collins, Y.; Chouchani, E.T.; James, A.M.; Menger, K.E.; Cochemé, H.M.; Murphy, M.P. Mitochondrial redox signalling at a glance. J. Cell Sci. 2012, 125, 801–806. [Google Scholar] [CrossRef]
- Gaffney, C.J.; Pollard, A.; Barratt, T.F.; Constantin-Teodosiu, D.; Greenhaff, P.L.; Szewczyk, N.J. Greater loss of mitochondrial function with ageing is associated with earlier onset of sarcopenia in C. elegans. Aging 2018, 10, 3382. [Google Scholar] [CrossRef] [PubMed]
- Coen, P.M.; Musci, R.V.; Hinkley, J.M.; Miller, B.F. Mitochondria as a Target for Mitigating Sarcopenia. Front. Physiol. 2019, 9, 1883. [Google Scholar] [CrossRef] [PubMed]
- Martinez Guimera, A.; Welsh, C.M.; Proctor, C.J.; McArdle, A.; Shanley, D.P. “Molecular habituation” as a potential mechanism of gradual homeostatic loss with age. Mech. Ageing Dev. 2018, 169, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.P. Redefining oxidative stress. Antioxid. Redox Signal. 2006, 8, 1865–1879. [Google Scholar] [CrossRef] [PubMed]
- Lomeli, N.; Bota, D.A.; Davies, K.J.A. Diminished stress resistance and defective adaptive homeostasis in age-related diseases. Clin. Sci. 2017, 131, 2573–2599. [Google Scholar] [CrossRef] [PubMed]
- Baumann, C.W.; Kwak, D.; Liu, H.M.; Thompson, L.V. Age-induced oxidative stress: How does it influence skeletal muscle quantity and quality? J. Appl. Physiol. 2016, 121, 1047–1052. [Google Scholar] [CrossRef] [PubMed]
- Shigenaga, M.K.; Hagen, T.M.; Ames, B.N. Oxidative damage and mitochondrial decay in aging. Proc. Natl. Acad. Sci. USA 1994, 91, 10771–10778. [Google Scholar] [CrossRef]
- Hybertson, B.M.; Gao, B.; Bose, S.K.; McCord, J.M. Oxidative stress in health and disease: The therapeutic potential of Nrf2 activation. Mol. Asp. Med. 2011, 32, 234–246. [Google Scholar] [CrossRef]
- Pansarasa, O.; Bertorelli, L.; Vecchiet, J.; Felzani, G.; Marzatico, F. Age-dependent changes of antioxidant activities and markers of free radical damage in human skeletal muscle. Free Radic. Biol. Med. 1999, 27, 617–622. [Google Scholar] [CrossRef]
- Miller, C.J.; Gounder, S.S.; Kannan, S.; Goutam, K.; Muthusamy, V.R.; Firpo, M.A.; Symons, J.D.; Paine, R., III; Hoidal, J.R.; Rajasekaran, N.S. Disruption of Nrf2/ARE signaling impairs antioxidant mechanisms and promotes cell degradation pathways in aged skeletal muscle. BBA Mol. Basis Dis. 2012, 1822, 1038–1050. [Google Scholar] [CrossRef]
- Safdar, A.; deBeer, J.; Tarnopolsky, M.A. Dysfunctional Nrf2—Keap1 redox signaling in skeletal muscle of the sedentary old. Free Radic. Biol. Med. 2010, 49, 1487–1493. [Google Scholar] [CrossRef] [PubMed]
- Rattan, S.I.S.; Demirovic, D.; Nizard, C. A preliminary attempt to establish multiple stress response profiles of human skin fibroblasts exposed to mild or severe stress during ageing in vitro. Mech. Ageing Dev. 2017, 170, 92–97. [Google Scholar] [CrossRef] [PubMed]
- McArdle, A.; Pollock, N.; Staunton, C.A.; Jackson, M.J. Aberrant redox signalling and stress response in age-related muscle decline: Role in inter- and intra-cellular signalling. Free Radic. Biol. Med. 2019, 132, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Guimera, A.M.; Shanley, D.P.; Proctor, C.J. Modelling the role of redox-related mechanisms in musculoskeletal ageing. Free Radic. Biol. Med. 2019, 132, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Suh, J.H.; Shenvi, S.V.; Dixon, B.M.; Liu, H.; Jaiswal, A.K.; Liu, R.-M.; Hagen, T.M. Decline in transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis, which is reversible with lipoic acid. Proc. Natl. Acad. Sci. USA 2004, 101, 3381–3386. [Google Scholar] [CrossRef] [PubMed]
- Pluquet, O.; Pourtier, A.; Abbadie, C. The unfolded protein response and cellular senescence. A Review in the Theme: Cellular Mechanisms of Endoplasmic Reticulum Stress Signaling in Health and Disease. Am. J. Physiol. Cell Physiol. 2015, 308, C415–C425. [Google Scholar] [CrossRef] [PubMed]
- Wright, V.P.; Reiser, P.J.; Clanton, T.L. Redox modulation of global phosphatase activity and protein phosphorylation in intact skeletal muscle. J. Physiol. 2009, 587, 5767–5781. [Google Scholar] [CrossRef] [PubMed]
- Crilly, M.J.; Tryon, L.D.; Erlich, A.T.; Hood, D.A. The role of Nrf2 in skeletal muscle contractile and mitochondrial function. J. Appl. Physiol. 2016, 121, 730–740. [Google Scholar] [CrossRef] [PubMed]
- Drenth, H.; Zuidema, S.; Bunt, S.; Bautmans, I.; Van Der Schans, C.; Hobbelen, H. The Contribution of Advanced Glycation End product (AGE) accumulation to the decline in motor function. Eur. Rev. Aging Phys. Act. 2016, 13, 3. [Google Scholar] [CrossRef]
- Burkart, A.; Shi, X.; Chouinard, M.; Corvera, S. Adenylate kinase 2 links mitochondrial energy metabolism to the induction of the unfolded protein response. J. Biol. Chem. 2011, 286, 4081–4089. [Google Scholar] [CrossRef]
- Fernando, R.; Drescher, C.; Nowotny, K.; Grune, T.; Castro, J.P. Impaired proteostasis during skeletal muscle aging. Free Radic. Biol. Med. 2019, 132, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Ben-Zvi, A.; Miller, E.A.; Morimoto, R.I. Collapse of proteostasis represents an early molecular event in Caenorhabditis elegans aging. Proc. Natl. Acad. Sci. USA 2009, 106, 14914–14919. [Google Scholar] [CrossRef] [PubMed]
- Narasimhan, M.; Hong, J.; Atieno, N.; Muthusamy, V.R.; Davidson, C.J.; Abu-Rmaileh, N.; Richardson, R.S.; Gomes, A.V.; Hoidal, J.R.; Rajasekaran, N.S. Nrf2 deficiency promotes apoptosis and impairs PAX7/MyoD expression in aging skeletal muscle cells. Free Radic. Biol. Med. 2014, 71, 402–414. [Google Scholar] [CrossRef] [PubMed]
- Tapia, P.C. Sublethal mitochondrial stress with an attendant stoichiometric augmentation of reactive oxygen species may precipitate many of the beneficial alterations in cellular physiology produced by caloric restriction, intermittent fasting, exercise and dietary phytonutrients: “Mitohormesis” for health and vitality. Med. Hypotheses 2006, 66, 832–843. [Google Scholar] [PubMed]
- Ristow, M.; Schmeisser, K. Mitohormesis: Promoting Health and Lifespan by Increased Levels of Reactive Oxygen Species (ROS). Dose-Response 2014, 12, 288–341. [Google Scholar] [CrossRef]
- Miller, B.F.; Robinson, M.M.; Bruss, M.D.; Hellerstein, M.; Hamilton, K.L. A comprehensive assessment of mitochondrial protein synthesis and cellular proliferation with age and caloric restriction. Aging Cell 2012, 11, 150–161. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, K.L.; Miller, B.F. Mitochondrial proteostasis as a shared characteristic of slowed aging: The importance of considering cell proliferation. J. Physiol. 2017, 595, 6401–6407. [Google Scholar] [CrossRef]
- Wolff, C.A.; Reid, J.J.; Musci, R.V.; Linden, M.A.; Konopka, A.R.; Peelor, F.F.; Miller, B.F.; Hamilton, K.L. Differential Effects of Rapamycin and Metformin in Combination with Rapamycin on Mechanisms of Proteostasis in Cultured Skeletal Myotubes. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2019, 1, glz058. [Google Scholar] [CrossRef]
- Cox, C.S.; McKay, S.E.; Holmbeck, M.A.; Christian, B.E.; Scortea, A.C.; Tsay, A.J.; Newman, L.E.; Shadel, G.S. Mitohormesis in Mice via Sustained Basal Activation of Mitochondrial and Antioxidant Signaling. Cell Metab. 2018, 28, 776–786. [Google Scholar] [CrossRef]
- Schulz, T.J.; Zarse, K.; Voigt, A.; Urban, N.; Birringer, M.; Ristow, M. Glucose Restriction Extends Caenorhabditis elegans Life Span by Inducing Mitochondrial Respiration and Increasing Oxidative Stress. Cell Metab. 2007, 6, 280–293. [Google Scholar] [CrossRef]
- Merry, T.L.; Ristow, M. Mitohormesis in exercise training. Free Radic. Biol. Med. 2016, 98, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Penedo, F.J.; Dahn, J.R. Exercise and well-being: A review of mental and physical health benefits associated with physical activity. Curr. Opin. Psychiatry 2005, 18, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Blair, S.N.; Morris, J.N. Healthy Hearts—And the Universal Benefits of Being Physically Active: Physical Activity and Health. Ann. Epidemiol. 2009, 19, 253–256. [Google Scholar] [CrossRef] [PubMed]
- Hillman, C.H.; Erickson, K.I.; Kramer, A.F. Be smart, exercise your heart: Exercise effects on brain and cognition. Nat. Rev. Neurosci. 2008, 9, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Warburton, D.E.R. Health benefits of physical activity: The evidence. Can. Med Assoc. J. 2006, 174, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Yusuf, S.; Hawken, S.; Ôunpuu, S.; Dans, T.; Avezum, A.; Lanas, F.; McQueen, M.; Budaj, A.; Pais, P.; Varigos, J.; et al. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): Case-control study. Lancet 2004, 364, 937–952. [Google Scholar] [CrossRef]
- Kiens, B.; Richter, E.A.; Wojtaszewski, J.F. Exercise physiology: From performance studies to muscle physiology and cardiovascular adaptations. J. Appl. Physiol. 2014, 117, 943–944. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Clausen, J.S.R.; Marott, J.L.; Holtermann, A.; Gyntelberg, F.; Jensen, M.T. Midlife Cardiorespiratory Fitness and the Long-Term Risk of Mortality: 46 Years of Follow-Up. J. Am. Coll. Cardiol. 2018, 72, 987–995. [Google Scholar] [CrossRef] [PubMed]
- Imboden, M.T.; Harber, M.P.; Whaley, M.H.; Finch, W.H.; Bishop, D.L.; Kaminsky, L.A. Cardiorespiratory Fitness and Mortality in Healthy Men and Women. J. Am. Coll. Cardiol. 2018, 72, 2283–2292. [Google Scholar] [CrossRef]
- Strasser, B.; Burtscher, M. Survival of the fittest: VO2max, a key predictor of longevity? Front. Biosci. (Landmark Ed) 2018, 23, 1505–1516. [Google Scholar] [CrossRef]
- Konopka, A.R.; Suer, M.K.; Wolff, C.A.; Harber, M.P. Markers of Human Skeletal Muscle Mitochondrial Biogenesis and Quality Control: Effects of Age and Aerobic Exercise Training. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2014, 69, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Holloszy, J.O.; Booth, F.W. Biochemical adaptations to endurance exercise in muscle. Annu. Rev. Physiol. 1976, 38, 273–291. [Google Scholar] [CrossRef] [PubMed]
- Harber, M.P.; Konopka, A.R.; Undem, M.K.; Hinkley, J.M.; Minchev, K.; Kaminsky, L.A.; Trappe, T.A.; Trappe, S. Aerobic exercise training induces skeletal muscle hypertrophy and age-dependent adaptations in myofiber function in young and older men. J. Appl. Physiol. 2012, 113, 1495–1504. [Google Scholar] [CrossRef] [PubMed]
- Harber, M.P.; Konopka, A.R.; Douglass, M.D.; Minchev, K.; Kaminsky, L.A.; Trappe, T.A.; Trappe, S. Aerobic exercise training improves whole muscle and single myofiber size and function in older women. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 297, R1452–R1459. [Google Scholar] [CrossRef] [PubMed]
- Hojman, P.; Gehl, J.; Christensen, J.F.; Pedersen, B.K. Molecular Mechanisms Linking Exercise to Cancer Prevention and Treatment. Cell Metab. 2018, 27, 10–21. [Google Scholar] [CrossRef]
- Gries, K.J.; Raue, U.; Perkins, R.K.; Lavin, K.M.; Overstreet, B.S.; D’Acquisto, L.J.; Graham, B.; Finch, W.H.; Kaminsky, L.A.; Trappe, T.A.; et al. Cardiovascular and skeletal muscle health with lifelong exercise. J. Appl. Physiol. 2018, 125, 1636–1645. [Google Scholar] [CrossRef]
- Short, K.R.; Vittone, J.L.; Bigelow, M.L.; Proctor, D.N.; Rizza, R.A.; Coenen-Schimke, J.M.; Nair, K.S. Impact of aerobic exercise training on age-related changes in insulin sensitivity and muscle oxidative capacity. Diabetes 2003, 52, 1888–1896. [Google Scholar] [CrossRef]
- Powers, S.K.; Jackson, M.J. Exercise-Induced Oxidative Stress: Cellular Mechanisms and Impact on Muscle Force Production. Physiol. Rev. 2008, 88, 1243–1276. [Google Scholar] [CrossRef]
- Reid, M.B. Invited Review: Redox modulation of skeletal muscle contraction: What we know and what we don’t. J. Appl. Physiol. 2001, 90, 724–731. [Google Scholar] [CrossRef]
- Miotto, P.M.; Holloway, G.P. Exercise-induced reductions in mitochondrial ADP sensitivity contribute to the induction of gene expression and mitochondrial biogenesis through enhanced mitochondrial H2O2 emission. Mitochondrion 2019, 46, 116–122. [Google Scholar] [CrossRef]
- Jackson, M.J.; McArdle, A. Age-related changes in skeletal muscle reactive oxygen species generation and adaptive responses to reactive oxygen species. J. Physiol. 2011, 589, 2139–2145. [Google Scholar] [CrossRef] [PubMed]
- Vasilaki, A.; McArdle, F.; Iwanejko, L.M.; McArdle, A. Adaptive responses of mouse skeletal muscle to contractile activity: The effect of age. Mech. Ageing Dev. 2006, 127, 830–839. [Google Scholar] [CrossRef] [PubMed]
- Webb, R.; Hughes, M.G.; Thomas, A.W.; Morris, K. The Ability of Exercise-Associated Oxidative Stress to Trigger Redox-Sensitive Signalling Responses. Antioxidants 2017, 6, 63. [Google Scholar] [CrossRef] [PubMed]
- Richter, E.A.; Ruderman, N.B. AMPK and the biochemistry of exercise: Implications for human health and disease. Biochem. J. 2009, 418, 261–275. [Google Scholar] [CrossRef] [PubMed]
- Aronson, D.; Violan, M.A.; Dufresne, S.D.; Zangen, D.; Fielding, R.A.; Goodyear, L.J. Exercise stimulates the mitogen-activated protein kinase pathway in human skeletal muscle. J. Clin. Investig. 1997, 99, 1251–1257. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.L.; Gomez-Cabrera, M.C.; Steinhafel, N.; Vina, J. Acute exercise activates nuclear factor (NF)-κB signaling pathway in rat skeletal muscle. FASEB J. 2004, 18, 1499–1506. [Google Scholar] [CrossRef]
- Ogborn, D.I.; McKay, B.R.; Crane, J.D.; Parise, G.; Tarnopolsky, M.A. The unfolded protein response is triggered following a single, unaccustomed resistance-exercise bout. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2014, 307, R664–R669. [Google Scholar] [CrossRef]
- Estébanez, B.; de Paz, J.A.; Cuevas, M.J.; González-Gallego, J. Endoplasmic Reticulum Unfolded Protein Response, Aging and Exercise: An Update. Front. Physiol. 2018, 9, 1744. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2017, 19, 121–135. [Google Scholar] [CrossRef]
- O’Neill, H.M.; Maarbjerg, S.J.; Crane, J.D.; Jeppesen, J.; Jørgensen, S.B.; Schertzer, J.D.; Shyroka, O.; Kiens, B.; van Denderen, B.J.; Tarnopolsky, M.A.; et al. AMP-activated protein kinase (AMPK) β1β2 muscle null mice reveal an essential role for AMPK in maintaining mitochondrial content and glucose uptake during exercise. Proc. Natl. Acad. Sci. USA. 2011, 108, 16092–16097. [Google Scholar] [CrossRef]
- Choi, S.L.; Kim, S.J.; Lee, K.T.; Kim, J.; Mu, J.; Birnbaum, M.J.; Soo Kim, S.; Ha, J. The regulation of AMP-activated protein kinase by H2O2. Biochem. Biophys. Res. Commun. 2001, 287, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, D.T.; Kaufman, R.J. That which does not kill me makes me stronger: Adapting to chronic ER stress. Trends Biochem. Sci. 2007, 32, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Muthusamy, V.R.; Kannan, S.; Sadhaasivam, K.; Gounder, S.S.; Davidson, C.J.; Boeheme, C.; Hoidal, J.R.; Wang, L.; Rajasekaran, N.S. Acute exercise stress activates Nrf2/ARE signaling and promotes antioxidant mechanisms in the myocardium. Free Radic. Biol. Med. 2012, 52, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Done, A.J.; Traustadóttir, T. Nrf2 mediates redox adaptations to exercise. Redox Biol. 2016, 10, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Piantadosi, C.A.; Carraway, M.S.; Babiker, A.; Suliman, H.B. Heme oxygenase-1 regulates cardiac mitochondrial biogenesis via Nrf2-mediated transcriptional control of nuclear respiratory factor-1. Circ. Res. 2008, 103, 1232–1240. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-C.; Yin, P.-H.; Lu, C.-Y.; Chi, C.-W.; Wei, Y.-H. Increase of mitochondria and mitochondrial DNA in response to oxidative stress in human cells. Biochem. J. 2000, 348, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Handy, D.E.; Loscalzo, J. Redox Regulation of Mitochondrial Function. Antioxid. Redox Signal. 2012, 16, 1323–1367. [Google Scholar] [CrossRef] [PubMed]
- Merry, T.L.; Ristow, M. Nuclear factor erythroid-derived 2-like 2 (NFE2L2, Nrf2) mediates exercise-induced mitochondrial biogenesis and the anti-oxidant response in mice. J. Physiol. 2016, 594, 5195–5207. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.M.; Dasari, S.; Konopka, A.R.; Johnson, M.L.; Manjunatha, S.; Esponda, R.R.; Carter, R.E.; Lanza, I.R.; Nair, K.S. Enhanced Protein Translation Underlies Improved Metabolic and Physical Adaptations to Different Exercise Training Modes in Young and Old Humans. Cell Metab. 2017, 25, 581–592. [Google Scholar] [CrossRef]
- Tucker, J.M.; Welk, G.J.; Beyler, N.K. Physical activity in U.S.: Adults compliance with the Physical Activity Guidelines for Americans. Am. J. Prev. Med. 2011, 40, 454–461. [Google Scholar]
- Marsaux, C.F.M.; Celis-Morales, C.; Hoonhout, J.; Claassen, A.; Goris, A.; Forster, H.; Fallaize, R.; Macready, A.L.; Navas-Carretero, S.; Kolossa, S.; et al. Objectively Measured Physical Activity in European Adults: Cross-Sectional Findings from the Food4Me Study. PLoS ONE 2016, 11, e0150902. [Google Scholar] [CrossRef]
- Poljsak, B.; Šuput, D.; Milisav, I. Achieving the Balance between ROS and Antioxidants: When to Use the Synthetic Antioxidants. Oxid. Med. Cell. Longev. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Cabrera, M.-C.; Domenech, E.; Romagnoli, M.; Arduini, A.; Borras, C.; Pallardo, F.V.; Sastre, J.; Viña, J. Oral administration of vitamin C decreases muscle mitochondrial biogenesis and hampers training-induced adaptations in endurance performance. Am. J. Clin. Nutr. 2008, 87, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Morrison, D.; Hughes, J.; Della, Gatta, P.A.; Mason, S.; Lamon, S.; Russell, A.P.; Wadley, G.D. Vitamin C and E supplementation prevents some of the cellular adaptations to endurance-training in humans. Free Radic. Biol. Med. 2015, 89, 852–862. [Google Scholar] [CrossRef] [PubMed]
- Bjelakovic, G.; Nikolova, D.; Gluud, L.L.; Simonetti, R.G.; Gluud, C. Antioxidant supplements for prevention of mortality in healthy participants and patients with various diseases. Cochrane Database Syst. Rev. 2012, 336, 1245. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Cabrera, M.-C.; Ristow, M.; Viña, J. Antioxidant supplements in exercise: Worse than useless? Am. J. Physiol. Endocrinol. Metab. 2012, 302, E476–E477. [Google Scholar] [CrossRef]
- Oh, S.; Komine, S.; Warabi, E.; Akiyama, K.; Ishii, A.; Ishige, K.; Mizokami, Y.; Kuga, K.; Horie, M.; Miwa, Y.; et al. Nuclear factor (erythroid derived 2)-like 2 activation increases exercise endurance capacity via redox modulation in skeletal muscles. Sci. Rep. 2017, 7, 12902. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-Y.; Choi, C.S.; Birkenfeld, A.L.; Alves, T.C.; Jornayvaz, F.R.; Jurczak, M.J.; Zhang, D.; Woo, D.K.; Shadel, G.S.; Ladiges, W.; et al. Targeted Expression of Catalase to Mitochondria Prevents Age-Associated Reductions in Mitochondrial Function and Insulin Resistance. Cell Metab. 2010, 12, 668–674. [Google Scholar] [CrossRef]
- Strong, R.; Miller, R.A.; Antebi, A.; Astle, C.M.; Bogue, M.; Denzel, M.S.; Fernandez, E.; Flurkey, K.; Hamilton, K.L.; Lamming, D.W.; et al. Longer lifespan in male mice treated with a weakly estrogenic agonist, an antioxidant, an α-glucosidase inhibitor or a Nrf2-inducer. Aging Cell 2016, 15, 872–884. [Google Scholar] [CrossRef]
- Donovan, E.L.; McCord, J.M.; Reuland, D.J.; Miller, B.F.; Hamilton, K.L. Phytochemical Activation of Nrf2 Protects Human Coronary Artery Endothelial Cells against an Oxidative Challenge. Oxid. Med. Cell. Longev. 2012, 2012. [Google Scholar] [CrossRef]
- Reuland, D.J.; Khademi, S.; Castle, C.J.; Irwin, D.C.; McCord, J.M.; Miller, B.F.; Hamilton, K.L. Upregulation of phase II enzymes through phytochemical activation of Nrf2 protects cardiomyocytes against oxidant stress. Free Radic. Biol. Med. 2013, 56, 102–111. [Google Scholar] [CrossRef]
- Bruns, D.R.; Ehrlicher, S.E.; Khademi, S.; Biela, L.M.; Peelor, F.F.; Miller, B.F.; Hamilton, K.L. Differential Effects of Vitamin C or Protandim on Skeletal Muscle Adaptation to Exercise. J. Appl. Physiol. 2018, 509, 565. [Google Scholar] [CrossRef] [PubMed]
- Konopka, A.R.; Laurin, J.L.; Musci, R.V.; Wolff, C.A.; Reid, J.J.; Biela, L.M.; Zhang, Q.; Peelor, F.F.; Melby, C.L.; Hamilton, K.L.; et al. Influence of Nrf2 activators on subcellular skeletal muscle protein and DNA synthesis rates after 6 weeks of milk protein feeding in older adults. Geroscience 2017, 29, 175–186. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kubo, E.; Chhunchha, B.; Singh, P.; Sasaki, H.; Singh, D.P. Sulforaphane reactivates cellular antioxidant defense by inducing Nrf2/ARE/Prdx6 activity during aging and oxidative stress. Sci. Rep. 2017, 7, 14130. [Google Scholar] [CrossRef] [PubMed]
- Al-Sawaf, O.; Fragoulis, A.; Rosen, C.; Kan, Y.W.; Sönmez, T.T.; Pufe, T.; Wruck, C.J. Nrf2 Protects Against TWEAK-mediated Skeletal Muscle Wasting. Sci. Rep. 2014, 4, 3625. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Waltz, T.B.; Kassahun, H.; Lu, Q.; Kerr, J.S.; Morevati, M.; Fivenson, E.M.; Wollman, B.N.; Marosi, K.; Wilson, M.A.; et al. Tomatidine enhances lifespan and healthspan in C. elegans through mitophagy induction via the SKN-1/ Nrf2 pathway. Sci. Rep. 2017, 7, 46208. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.D.; Duan, J.; Samuelson, A.T.; Gaffrey, M.J.; Wang, L.; Bammler, T.K.; Moore, R.J.; White, C.C.; Kavanagh, T.J.; Voss, J.G.; et al. Improving mitochondrial function with SS-31 reverses age-related redox stress and improves exercise tolerance in aged mice. Free Radic. Biol. Med. 2019, 134, 268–281. [Google Scholar] [CrossRef] [PubMed]
- Siegel, M.P.; Kruse, S.E.; Percival, J.M.; Goh, J.; White, C.C.; Hopkins, H.C.; Kavanagh, T.J.; Szeto, H.H.; Rabinovitch, P.S.; Marcinek, D.J. Mitochondrial-targeted peptide rapidly improves mitochondrial energetics and skeletal muscle performance in aged mice. Aging Cell 2013, 12, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, E.; Agostini, D.; Polidori, E.; Potenza, L.; Guescini, M.; Lucertini, F.; Annibalini, G.; Stocchi, L.; De Santi, M.; Stocchi, V. The Pleiotropic Effect of Physical Exercise on Mitochondrial Dynamics in Aging Skeletal Muscle. Oxid. Med. Cell. Longev. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Kramer, P.A.; Duan, J.; Qian, W.-J.; Marcinek, D.J. The Measurement of Reversible Redox Dependent Post-translational Modifications and Their Regulation of Mitochondrial and Skeletal Muscle Function. Front. Physiol. 2015, 6, 347. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Musci, R.V.; Hamilton, K.L.; Linden, M.A. Exercise-Induced Mitohormesis for the Maintenance of Skeletal Muscle and Healthspan Extension. Sports 2019, 7, 170. https://doi.org/10.3390/sports7070170
Musci RV, Hamilton KL, Linden MA. Exercise-Induced Mitohormesis for the Maintenance of Skeletal Muscle and Healthspan Extension. Sports. 2019; 7(7):170. https://doi.org/10.3390/sports7070170
Chicago/Turabian StyleMusci, Robert V., Karyn L. Hamilton, and Melissa A. Linden. 2019. "Exercise-Induced Mitohormesis for the Maintenance of Skeletal Muscle and Healthspan Extension" Sports 7, no. 7: 170. https://doi.org/10.3390/sports7070170
APA StyleMusci, R. V., Hamilton, K. L., & Linden, M. A. (2019). Exercise-Induced Mitohormesis for the Maintenance of Skeletal Muscle and Healthspan Extension. Sports, 7(7), 170. https://doi.org/10.3390/sports7070170