
Structural and Biochemical Characterization of the Nucleosome Containing Variants H3.3 and H2A.Z
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Material and Methods
2.1. Protein Production
2.2. DNAs
2.3. Nucleosome Reconstitution
2.4. HinfI Endonuclease Accessibility Assay
2.5. MNase Accessibility Assay
2.6. Nucleosome-Sliding Assay
2.7. Vitrification
2.8. Cryo-EM Data Collection
2.9. Image Processing
2.10. Model Building and Refinement
2.11. Quantification and Statistical Analyses
3. Results
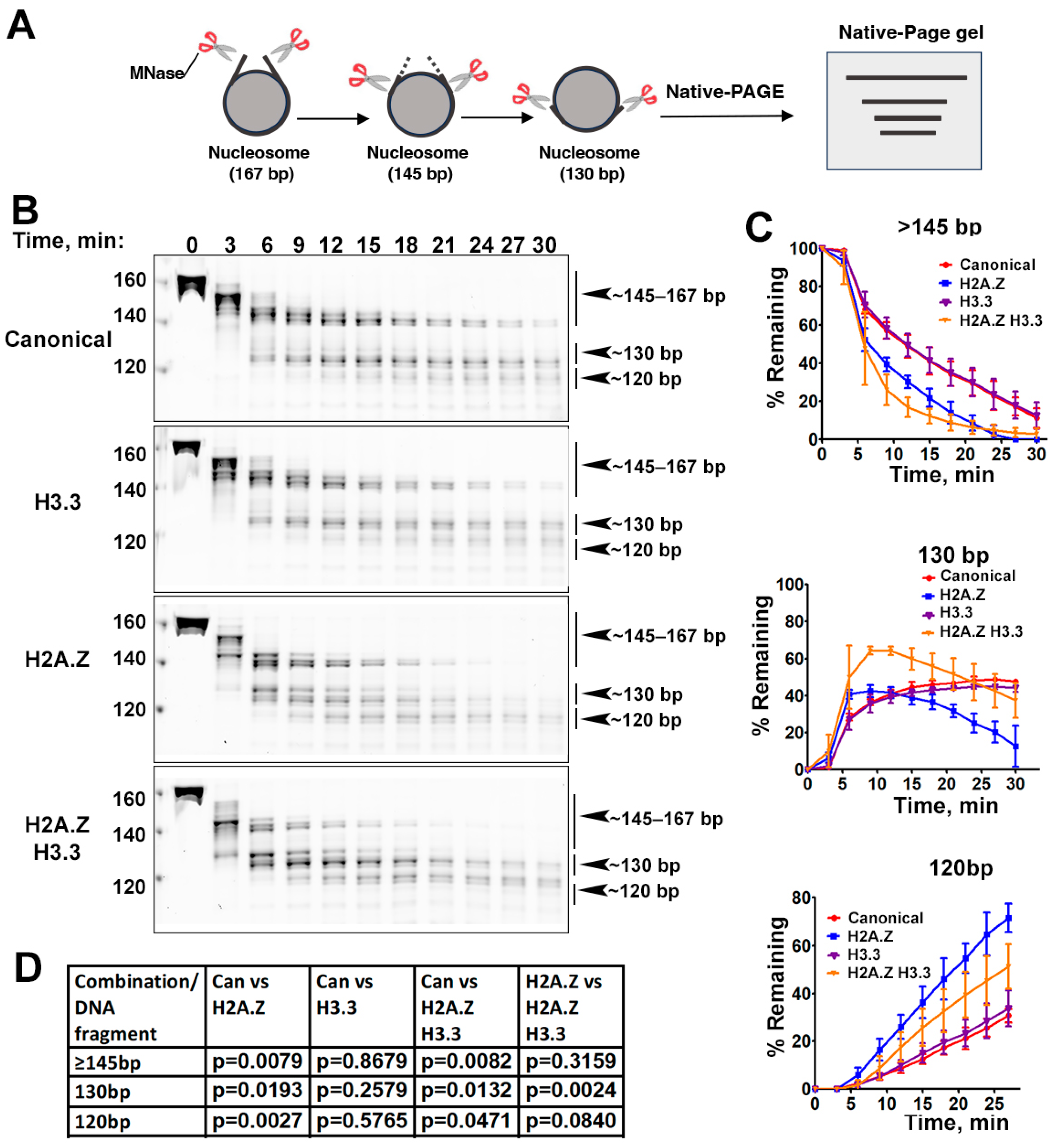
3.1. The Incorporation of Double Variant H2A.Z-H3.3 Enhances the Terminal DNA Accessibility on Nucleosomes
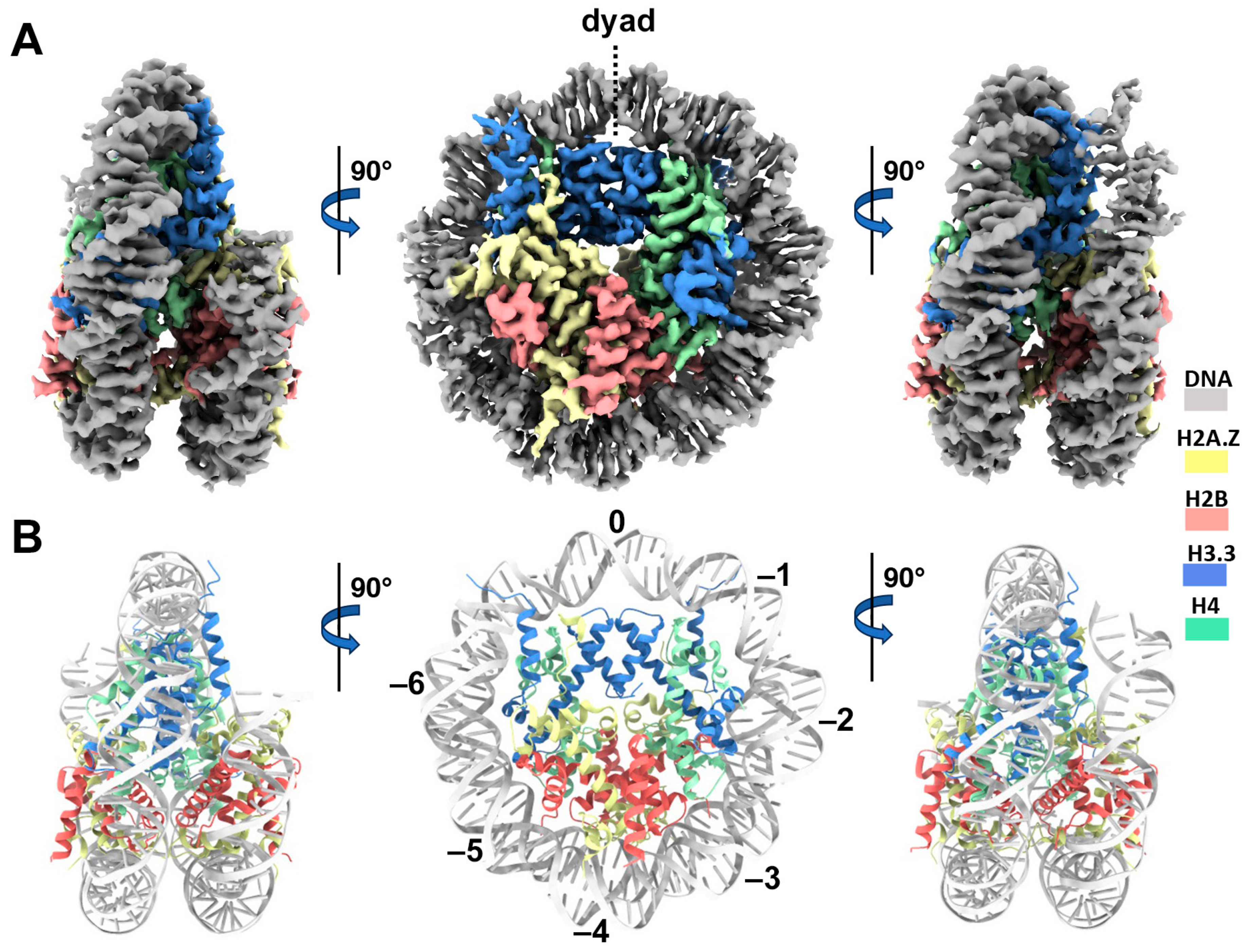
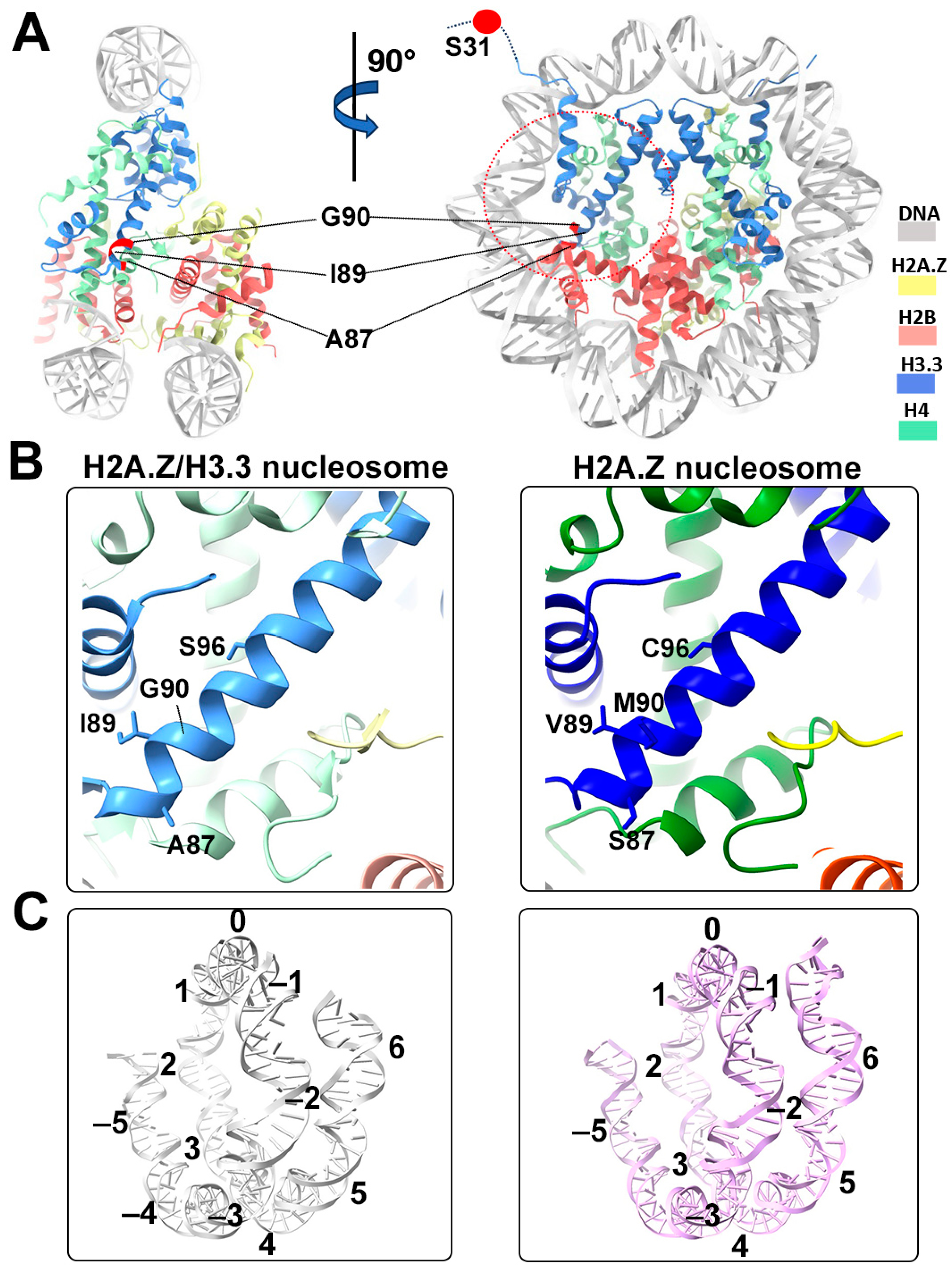
3.2. Cryo-EM Structure of the H2A.Z-H3.3 Double-Variant Nucleosome
3.3. INO80-Mediated Nucleosome Sliding on H2A.Z- H3.3 Double-Variant Nucleosome
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tagami, H.; Ray-Gallet, D.; Almouzni, G.; Nakatani, Y. Histone H3.1 and H3.3 complexes mediate nucleosome assembly pathways dependent or independent of DNA synthesis. Cell 2004, 116, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Brush, D.; Dodgson, J.B.; Choi, O.R.; Stevens, P.W.; Engel, J.D. Replacement variant histone genes contain intervening sequences. Mol. Cell. Biol. 1985, 5, 1307–1317. [Google Scholar] [PubMed]
- Maze, I.; Noh, K.M.; Soshnev, A.A.; Allis, C.D. Every amino acid matters: Essential contributions of histone variants to mammalian development and disease. Nat. Rev. Genet. 2014, 15, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Clarkson, M.J.; Wells, J.R.; Gibson, F.; Saint, R.; Tremethick, D.J. Regions of variant histone His2AvD required for Drosophila development. Nature 1999, 399, 694–697. [Google Scholar] [CrossRef] [PubMed]
- Faast, R.; Thonglairoam, V.; Schulz, T.C.; Beall, J.; Wells, J.R.; Taylor, H.; Matthaei, K.; Rathjen, P.D.; Tremethick, D.J.; Lyons, I. Histone variant H2A.Z is required for early mammalian development. Curr. Biol. 2001, 11, 1183–1187. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, B.; Gorovsky, M.A. Essential and nonessential histone H2A variants in Tetrahymena thermophila. Mol. Cell. Biol. 1996, 16, 4305–4311. [Google Scholar] [CrossRef] [PubMed]
- Ridgway, P.; Brown, K.D.; Rangasamy, D.; Svensson, U.; Tremethick, D.J. Unique residues on the H2A.Z containing nucleosome surface are important for Xenopus laevis development. J. Biol. Chem. 2004, 279, 43815–43820. [Google Scholar] [CrossRef] [PubMed]
- Hardy, S.; Jacques, P.E.; Gévry, N.; Forest, A.; Fortin, M.E.; Laflamme, L.; Gaudreau, L.; Robert, F. The euchromatic and heterochromatic landscapes are shaped by antagonizing effects of transcription on H2A.Z deposition. PLoS Genet. 2009, 5, e1000687. [Google Scholar] [CrossRef] [PubMed]
- Raisner, R.M.; Hartley, P.D.; Meneghini, M.D.; Bao, M.Z.; Liu, C.L.; Schreiber, S.L.; Rando, O.J.; Madhani, H.D. Histone variant H2A.Z marks the 5′ ends of both active and inactive genes in euchromatin. Cell 2005, 123, 233–248. [Google Scholar] [CrossRef]
- Soboleva, T.A.; Nekrasov, M.; Pahwa, A.; Williams, R.; Huttley, G.A.; Tremethick, D.J. A unique H2A histone variant occupies the transcriptional start site of active genes. Nat. Struct. Mol. Biol. 2011, 19, 25–30. [Google Scholar] [CrossRef]
- Weber, C.M.; Henikoff, J.G.; Henikoff, S. H2A.Z nucleosomes enriched over active genes are homotypic. Nat. Struct. Mol. Biol. 2010, 17, 1500–1507. [Google Scholar] [CrossRef]
- Zhang, H.; Roberts, D.N.; Cairns, B.R. Genome-wide dynamics of Htz1, a histone H2A variant that poises repressed/basal promoters for activation through histone loss. Cell 2005, 123, 219–231. [Google Scholar] [CrossRef] [PubMed]
- John, S.; Sabo, P.J.; Johnson, T.A.; Sung, M.H.; Biddie, S.C.; Lightman, S.L.; Voss, T.C.; Davis, S.R.; Meltzer, P.S.; Stamatoyannopoulos, J.A.; et al. Interaction of the glucocorticoid receptor with the chromatin landscape. Mol. Cell 2008, 29, 611–624. [Google Scholar] [CrossRef] [PubMed]
- Sutcliffe, E.L.; Parish, I.A.; He, Y.Q.; Juelich, T.; Tierney, M.L.; Rangasamy, D.; Milburn, P.J.; Parish, C.R.; Tremethick, D.J.; Rao, S. Dynamic histone variant exchange accompanies gene induction in T cells. Mol. Cell. Biol. 2009, 29, 1972–1986. [Google Scholar] [CrossRef]
- Wong, M.M.; Cox, L.K.; Chrivia, J.C. The chromatin remodeling protein, SRCAP, is critical for deposition of the histone variant H2A.Z at promoters. J. Biol. Chem. 2007, 282, 26132–26139. [Google Scholar] [CrossRef] [PubMed]
- Greaves, I.K.; Rangasamy, D.; Ridgway, P.; Tremethick, D.J. H2A.Z contributes to the unique 3D structure of the centromere. Proc. Natl. Acad. Sci. USA 2007, 104, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Rangasamy, D.; Berven, L.; Ridgway, P.; Tremethick, D.J. Pericentric heterochromatin becomes enriched with H2A.Z during early mammalian development. EMBO J. 2003, 22, 1599–1607. [Google Scholar] [CrossRef] [PubMed]
- Farris, S.D.; Rubio, E.D.; Moon, J.J.; Gombert, W.M.; Nelson, B.H.; Krumm, A. Transcription-induced chromatin remodeling at the c-myc gene involves the local exchange of histone H2A.Z. J. Biol. Chem. 2005, 280, 25298–25303. [Google Scholar] [CrossRef]
- Gevry, N.; Chan, H.M.; Laflamme, L.; Livingston, D.M.; Gaudreau, L. p21 transcription is regulated by differential localization of histone H2A.Z. Genes Dev. 2007, 21, 1869–1881. [Google Scholar] [CrossRef]
- Kotekar, A.S.; Weissman, J.D.; Gegonne, A.; Cohen, H.; Singer, D.S. Histone modifications, but not nucleosomal positioning, correlate with major histocompatibility complex class I promoter activity in different tissues in vivo. Mol. Cell. Biol. 2008, 28, 7323–7336. [Google Scholar] [CrossRef]
- Latorre, I.; Chesney, M.A.; Garrigues, J.M.; Stempor, P.; Appert, A.; Francesconi, M.; Strome, S.; Ahringer, J. The DREAM complex promotes gene body H2A.Z for target repression. Genes Dev. 2015, 29, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Lashgari, A.; Millau, J.F.; Jacques, P.E.; Gaudreau, L. Global inhibition of transcription causes an increase in histone H2A.Z incorporation within gene bodies. Nucleic Acids Res. 2017, 45, 12715–12722. [Google Scholar] [CrossRef] [PubMed]
- Lewis, T.S.; Sokolova, V.; Jung, H.; Ng, H.; Tan, D. Structural basis of chromatin regulation by histone variant H2A.Z. Nucleic Acids Res. 2021, 49, 11379–11391. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Dai, L.; Li, C.; Shi, L.; Huang, Y.; Guo, Z.; Wu, F.; Zhu, P.; Zhou, Z. Structural basis of nucleosome dynamics modulation by histone variants H2A.B and H2A.Z.2.2. EMBO J. 2021, 40, e105907. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.D.; Banaszynski, L.A.; Noh, K.M.; Lewis, P.W.; Elsaesser, S.J.; Stadler, S.; Dewell, S.; Law, M.; Guo, X.; Li, X.; et al. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell 2010, 140, 678–691. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Zang, C.; Wei, G.; Cui, K.; Peng, W.; Zhao, K.; Felsenfeld, G. H3.3/H2A.Z double variant-containing nucleosomes mark ‘nucleosome-free regions’ of active promoters and other regulatory regions. Nat. Genet. 2009, 41, 941–945. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, B.E.; Ahmad, K. Transcriptional activation triggers deposition and removal of the histone variant H3.3. Genes Dev. 2005, 19, 804–814. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Felsenfeld, G. Nucleosome stability mediated by histone variants H3.3 and H2A.Z. Genes Dev. 2007, 21, 1519–1529. [Google Scholar] [CrossRef] [PubMed]
- Tachiwana, H.; Osakabe, A.; Shiga, T.; Miya, Y.; Kimura, H.; Kagawa, W.; Kurumizaka, H. Structures of human nucleosomes containing major histone H3 variants. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 578–583. [Google Scholar] [CrossRef]
- Chen, P.; Zhao, J.; Wang, Y.; Wang, M.; Long, H.; Liang, D.; Huang, L.; Wen, Z.; Li, W.; Li, X.; et al. H3.3 actively marks enhancers and primes gene transcription via opening higher-ordered chromatin. Genes Dev. 2013, 27, 2109–2124. [Google Scholar] [CrossRef]
- Thakar, A.; Gupta, P.; Ishibashi, T.; Finn, R.; Silva-Moreno, B.; Uchiyama, S.; Fukui, K.; Tomschik, M.; Ausio, J.; Zlatanova, J. H2A.Z and H3.3 histone variants affect nucleosome structure: Biochemical and biophysical studies. Biochemistry 2009, 48, 10852–10857. [Google Scholar] [CrossRef] [PubMed]
- Horikoshi, N.; Arimura, Y.; Taguchi, H.; Kurumizaka, H. Crystal structures of heterotypic nucleosomes containing histones H2A.Z and H2A. Open Biol. 2016, 6, 160127. [Google Scholar] [CrossRef] [PubMed]
- Sokolova, V.; Sarkar, S.; Tan, D. Histone variants and chromatin structure, update of advances. Comput. Struct. Biotechnol. J. 2023, 21, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Dyer, P.N.; Edayathumangalam, R.S.; White, C.L.; Bao, Y.; Chakravarthy, S.; Muthurajan, U.M.; Luger, K. Reconstitution of nucleosome core particles from recombinant histones and DNA. Methods Enzymol. 2004, 375, 23–44. [Google Scholar] [PubMed]
- Zheng, S.Q.; Palovcak, E.; Armache, J.P.; Verba, K.A.; Cheng, Y.; Agard, D.A. MotionCor2: Anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 2017, 14, 331–332. [Google Scholar] [CrossRef]
- Rohou, A.; Grigorieff, N. CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 2015, 192, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Zivanov, J.; Nakane, T.; Forsberg, B.O.; Kimanius, D.; Hagen, W.J.; Lindahl, E.; Scheres, S.H. New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife 2018, 7, e42166. [Google Scholar] [CrossRef] [PubMed]
- Meng, E.C.; Goddard, T.D.; Pettersen, E.F.; Couch, G.S.; Pearson, Z.J.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Tools for structure building and analysis. Protein Sci. 2023, 32, e4792. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef]
- Adams, P.D.; Afonine, P.V.; Bunkóczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef]
- Schalch, T.; Duda, S.; Sargent, D.F.; Richmond, T.J. X-ray structure of a tetranucleosome and its implications for the chromatin fibre. Nature 2005, 436, 138–141. [Google Scholar] [CrossRef] [PubMed]
- Luger, K.; Mader, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Siddaway, R.; Milos, S.; Coyaud, É.; Yun, H.Y.; Morcos, S.M.; Pajovic, S.; Campos, E.I.; Raught, B.; Hawkins, C. The in vivo Interaction Landscape of Histones H3.1 and H3.3. Mol. Cell Proteom. 2022, 21, 100411. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Mizuguchi, G.; Hamiche, A.; Wu, C. A chromatin remodelling complex involved in transcription and DNA processing. Nature 2000, 406, 541–544. [Google Scholar] [CrossRef] [PubMed]
- Papamichos-Chronakis, M.; Peterson, C.L. The Ino80 chromatin-remodeling enzyme regulates replisome function and stability. Nat. Struct. Mol. Biol. 2008, 15, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Sokolova, V.; Lee, G.; Mullins, A.; Mody, P.; Watanabe, S.; Tan, D. DNA-translocation-independent role of INO80 remodeler in DNA damage repairs. J. Biol. Chem. 2023, 299, 105245. [Google Scholar] [CrossRef]
- OWillhoft; Bythell-Douglas, R.; McCormack, E.A.; Wigley, D.B. Synergy and antagonism in regulation of recombinant human INO80 chromatin remodeling complex. Nucleic Acids Res. 2016, 44, 8179–8188. [Google Scholar]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jung, H.; Sokolova, V.; Lee, G.; Stevens, V.R.; Tan, D. Structural and Biochemical Characterization of the Nucleosome Containing Variants H3.3 and H2A.Z. Epigenomes 2024, 8, 21. https://doi.org/10.3390/epigenomes8020021
Jung H, Sokolova V, Lee G, Stevens VR, Tan D. Structural and Biochemical Characterization of the Nucleosome Containing Variants H3.3 and H2A.Z. Epigenomes. 2024; 8(2):21. https://doi.org/10.3390/epigenomes8020021
Chicago/Turabian StyleJung, Harry, Vladyslava Sokolova, Gahyun Lee, Victoria Rose Stevens, and Dongyan Tan. 2024. "Structural and Biochemical Characterization of the Nucleosome Containing Variants H3.3 and H2A.Z" Epigenomes 8, no. 2: 21. https://doi.org/10.3390/epigenomes8020021
APA StyleJung, H., Sokolova, V., Lee, G., Stevens, V. R., & Tan, D. (2024). Structural and Biochemical Characterization of the Nucleosome Containing Variants H3.3 and H2A.Z. Epigenomes, 8(2), 21. https://doi.org/10.3390/epigenomes8020021