
Emerging Approaches to Profile Accessible Chromatin from Formalin-Fixed Paraffin-Embedded Sections

 , , , , and
, , , , and
Abstract
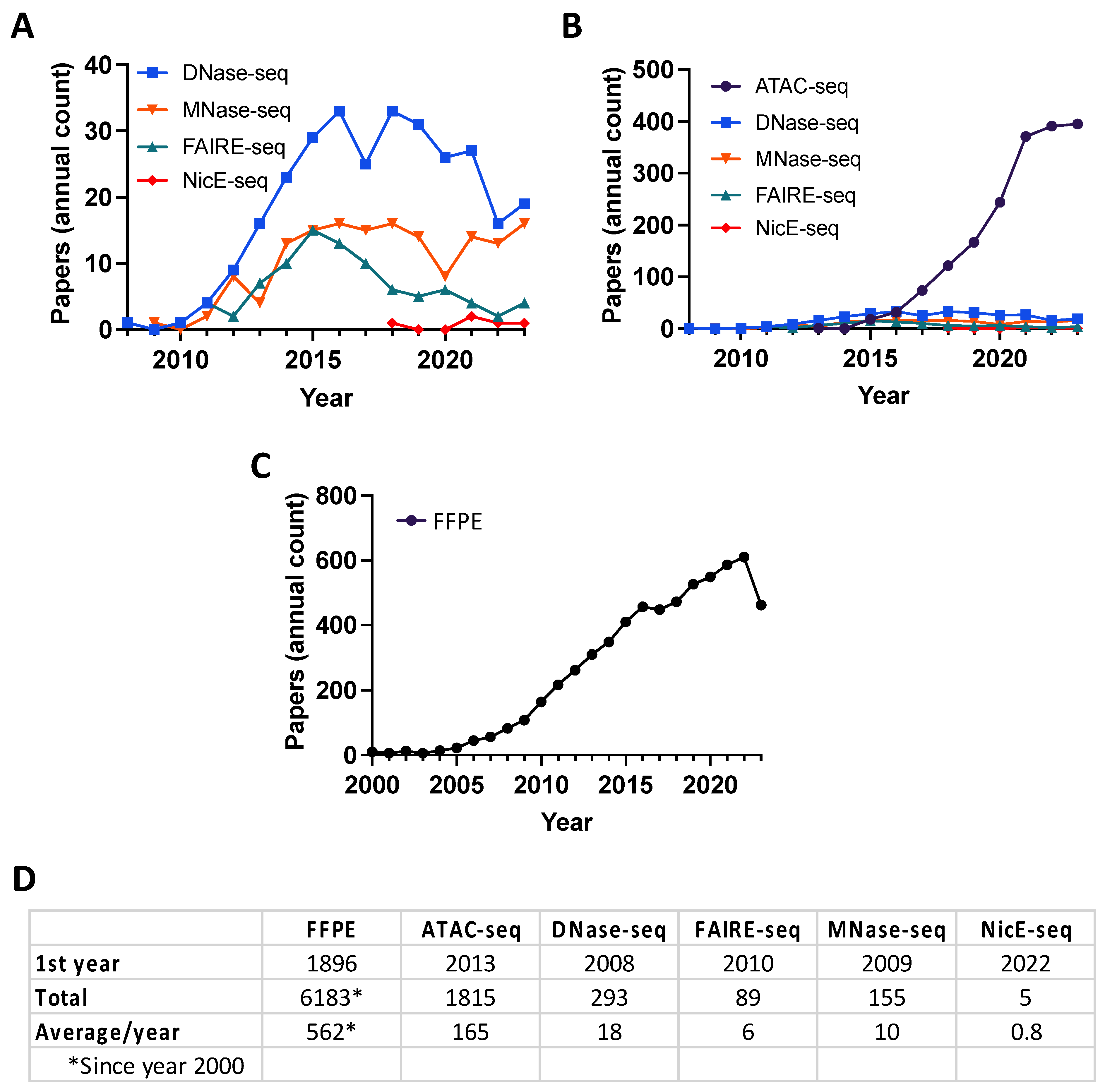
1. Introduction

2. Genome-Wide Profiling of Open Chromatin
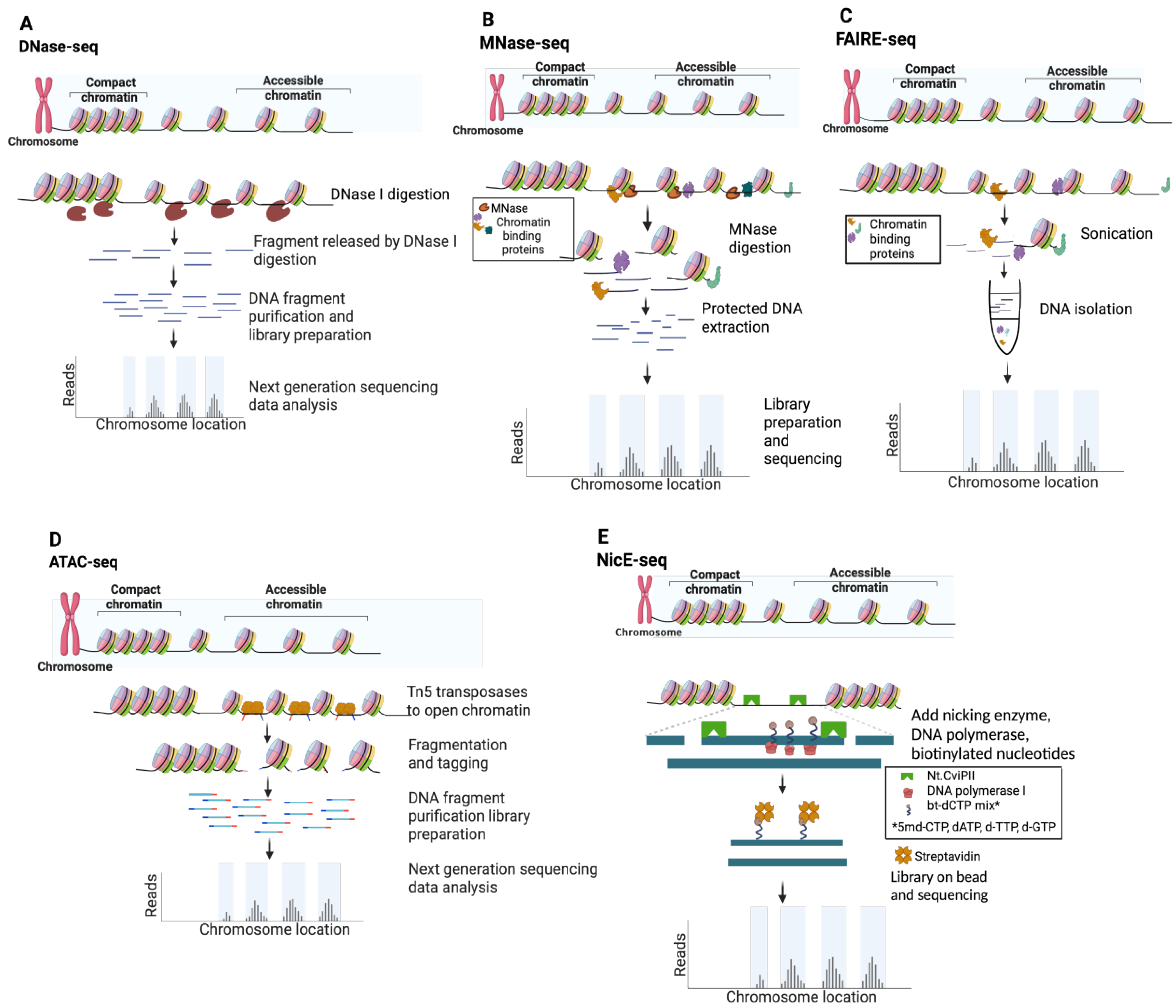
2.1. DNase I Hypersensitivity Mapping Paved the Way for Genome-Wide Open Chromatin Profiling
2.2. Micrococcal Nuclease (MNase) Digestion to Decipher Nucleosome Positioning
2.3. FAIRE-Seq Identifies Accessible Chromatin Regions through Principles of Biochemical Separation and Solubility
2.4. Tn5 Transposon Tagmentation of Accessible Chromatin (ATAC-Seq)
2.5. Nicking Enzyme-Assisted Accessible Chromatin Sequencing (NicE-Seq)
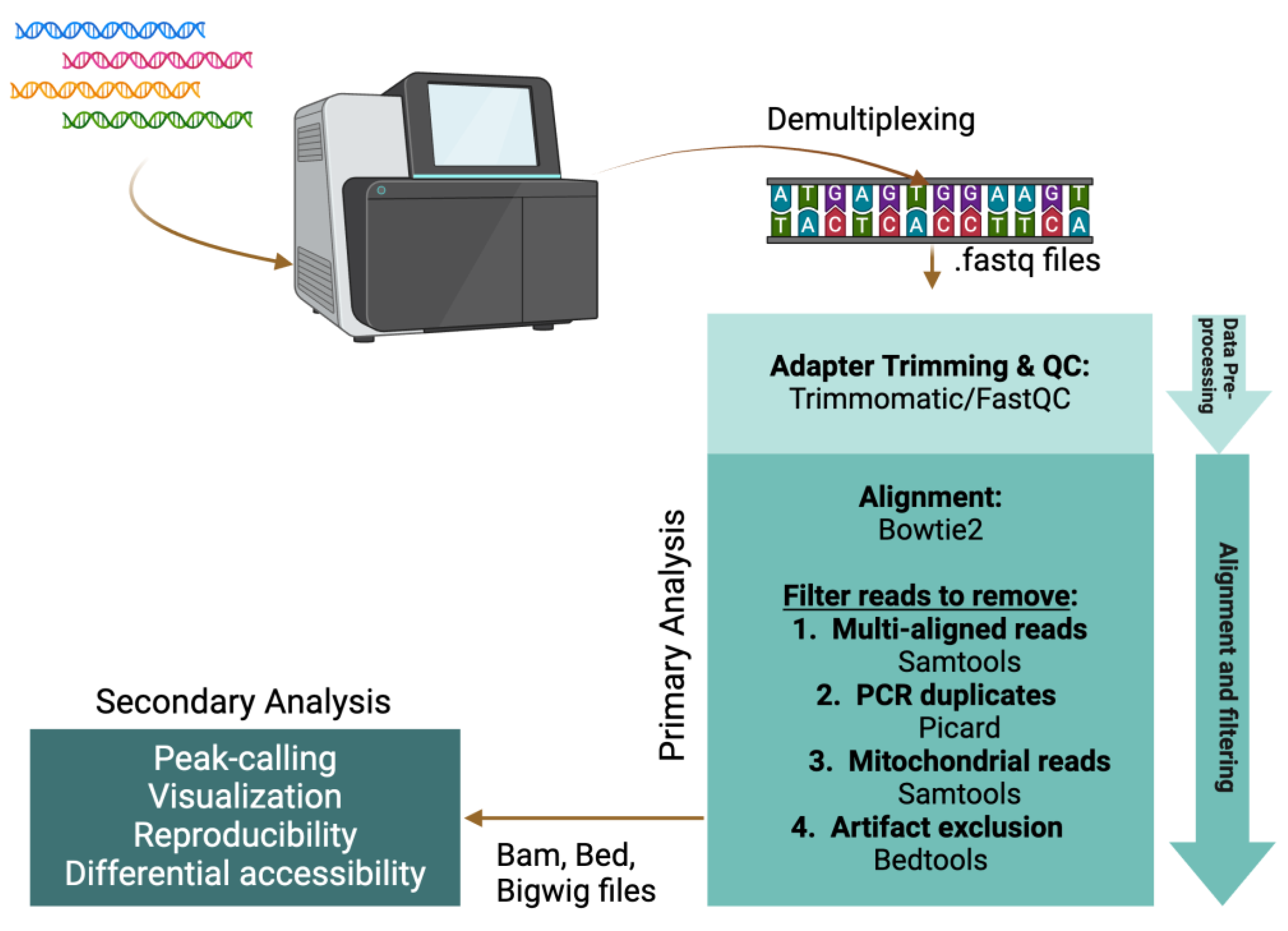
3. Data Analysis
3.1. Read Pre-Processing and Quality Control
3.2. Primary Analysis Pipeline
3.3. Tools for Secondary Analysis
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Berson, A.; Nativio, R.; Berger, S.L.; Bonini, N.M. Epigenetic Regulation in Neurodegenerative Diseases. Trends Neurosci. 2018, 41, 587–598. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Tyler, J.K. Epigenetics and aging. Sci. Adv. 2016, 2, e1600584. [Google Scholar] [CrossRef] [PubMed]
- Rowley, M.J.; Corces, V.G. Organizational principles of 3D genome architecture. Nat. Rev. Genet. 2018, 19, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Wang, X.; Xiao, D.; Liu, S.; Tao, Y. The chromatin-associated RNAs in gene regulation and cancer. Mol. Cancer 2023, 22, 27. [Google Scholar] [CrossRef]
- Bhat, K.P.; Umit Kaniskan, H.; Jin, J.; Gozani, O. Epigenetics and beyond: Targeting writers of protein lysine methylation to treat disease. Nat. Rev. Drug Discov. 2021, 20, 265–286. [Google Scholar] [CrossRef] [PubMed]
- Isbel, L.; Grand, R.S.; Schubeler, D. Generating specificity in genome regulation through transcription factor sensitivity to chromatin. Nat. Rev. Genet. 2022, 23, 728–740. [Google Scholar] [CrossRef] [PubMed]
- Klemm, S.L.; Shipony, Z.; Greenleaf, W.J. Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet. 2019, 20, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Shirvaliloo, M. The landscape of histone modifications in epigenomics since 2020. Epigenomics 2022, 14, 1465–1477. [Google Scholar] [CrossRef] [PubMed]
- Luger, K.; Rechsteiner, T.J.; Flaus, A.J.; Waye, M.M.; Richmond, T.J. Characterization of nucleosome core particles containing histone proteins made in bacteria. J. Mol. Biol. 1997, 272, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Ioshikhes, I.P.; Albert, I.; Zanton, S.J.; Pugh, B.F. Nucleosome positions predicted through comparative genomics. Nat. Genet. 2006, 38, 1210–1215. [Google Scholar] [CrossRef] [PubMed]
- Mavrich, T.N.; Jiang, C.; Ioshikhes, I.P.; Li, X.; Venters, B.J.; Zanton, S.J.; Tomsho, L.P.; Qi, J.; Glaser, R.L.; Schuster, S.C.; et al. Nucleosome organization in the Drosophila genome. Nature 2008, 453, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Schones, D.E.; Cui, K.; Cuddapah, S.; Roh, T.Y.; Barski, A.; Wang, Z.; Wei, G.; Zhao, K. Dynamic regulation of nucleosome positioning in the human genome. Cell 2008, 132, 887–898. [Google Scholar] [CrossRef]
- Kraushaar, D.C.; Jin, W.; Maunakea, A.; Abraham, B.; Ha, M.; Zhao, K. Genome-wide incorporation dynamics reveal distinct categories of turnover for the histone variant H3.3. Genome Biol. 2013, 14, R121. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wei, T.; Panchenko, A.R. Histone variant H2A.Z modulates nucleosome dynamics to promote DNA accessibility. Nat. Commun. 2023, 14, 769. [Google Scholar] [CrossRef] [PubMed]
- Weiner, A.; Hsieh, T.H.; Appleboim, A.; Chen, H.V.; Rahat, A.; Amit, I.; Rando, O.J.; Friedman, N. High-resolution chromatin dynamics during a yeast stress response. Mol. Cell. 2015, 58, 371–386. [Google Scholar] [CrossRef] [PubMed]
- Consortium, E.P. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Consortium, E.P.; Moore, J.E.; Purcaro, M.J.; Pratt, H.E.; Epstein, C.B.; Shoresh, N.; Adrian, J.; Kawli, T.; Davis, C.A.; Dobin, A.; et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 2020, 583, 699–710. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.J.; Statham, A.; Stirzaker, C.; Molloy, P.L.; Frommer, M. DNA methylation: Bisulphite modification and analysis. Nat. Protoc. 2006, 1, 2353–2364. [Google Scholar] [CrossRef] [PubMed]
- Vaisvila, R.; Ponnaluri, V.K.C.; Sun, Z.; Langhorst, B.W.; Saleh, L.; Guan, S.; Dai, N.; Campbell, M.A.; Sexton, B.S.; Marks, K.; et al. Enzymatic methyl sequencing detects DNA methylation at single-base resolution from picograms of DNA. Genome Res. 2021, 31, 1280–1289. [Google Scholar] [CrossRef] [PubMed]
- Kaya-Okur, H.S.; Wu, S.J.; Codomo, C.A.; Pledger, E.S.; Bryson, T.D.; Henikoff, J.G.; Ahmad, K.; Henikoff, S. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat. Commun. 2019, 10, 1930. [Google Scholar] [CrossRef] [PubMed]
- Skene, P.J.; Henikoff, S. An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. eLife 2017, 6, e21856. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Mortazavi, A. Integrating ChIP-seq with other functional genomics data. Brief. Funct. Genom. 2018, 17, 104–115. [Google Scholar] [CrossRef]
- Shimizu, M.; Roth, S.Y.; Szent-Gyorgyi, C.; Simpson, R.T. Nucleosomes are positioned with base pair precision adjacent to the alpha 2 operator in Saccharomyces cerevisiae. EMBO J. 1991, 10, 3033–3041. [Google Scholar] [CrossRef] [PubMed]
- Weintraub, H.; Groudine, M. Chromosomal subunits in active genes have an altered conformation. Science 1976, 193, 848–856. [Google Scholar] [CrossRef]
- Boyle, A.P.; Davis, S.; Shulha, H.P.; Meltzer, P.; Margulies, E.H.; Weng, Z.; Furey, T.S.; Crawford, G.E. High-resolution mapping and characterization of open chromatin across the genome. Cell 2008, 132, 311–322. [Google Scholar] [CrossRef]
- Simpson, R.T. Chromatin structure and analysis of mechanisms of activators and repressors. Methods 1998, 15, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Vierstra, J.; Stamatoyannopoulos, J.A. Genomic footprinting. Nat. Methods 2016, 13, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Crawford, G.E.; Holt, I.E.; Whittle, J.; Webb, B.D.; Tai, D.; Davis, S.; Margulies, E.H.; Chen, Y.; Bernat, J.A.; Ginsburg, D.; et al. Genome-wide mapping of DNase hypersensitive sites using massively parallel signature sequencing (MPSS). Genome Res. 2006, 16, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Albert, I.; Mavrich, T.N.; Tomsho, L.P.; Qi, J.; Zanton, S.J.; Schuster, S.C.; Pugh, B.F. Translational and rotational settings of H2A.Z nucleosomes across the Saccharomyces cerevisiae genome. Nature 2007, 446, 572–576. [Google Scholar] [CrossRef] [PubMed]
- Tsompana, M.; Buck, M.J. Chromatin accessibility: A window into the genome. Epigenetics Chromatin 2014, 7, 33. [Google Scholar] [CrossRef] [PubMed]
- Klein, D.C.; Hainer, S.J. Genomic methods in profiling DNA accessibility and factor localization. Chromosome Res. 2020, 28, 69–85. [Google Scholar] [CrossRef] [PubMed]
- Mansisidor, A.R.; Risca, V.I. Chromatin accessibility: Methods, mechanisms, and biological insights. Nucleus 2022, 13, 236–276. [Google Scholar] [CrossRef] [PubMed]
- Waldron, L.; Simpson, P.; Parmigiani, G.; Huttenhower, C. Report on emerging technologies for translational bioinformatics: A symposium on gene expression profiling for archival tissues. BMC Cancer 2012, 12, 124. [Google Scholar] [CrossRef] [PubMed]
- Kokkat, T.J.; Patel, M.S.; McGarvey, D.; LiVolsi, V.A.; Baloch, Z.W. Archived formalin-fixed paraffin-embedded (FFPE) blocks: A valuable underexploited resource for extraction of DNA, RNA, and protein. Biopreserv. Biobank. 2013, 11, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Blum, F. Notiz über die Anwendung des Formaldehyds (Formol) als Härtungs-und Konservierungsmittel. Anat. Anz. 1894, 9, 229–231. [Google Scholar]
- Donczo, B.; Guttman, A. Biomedical analysis of formalin-fixed, paraffin-embedded tissue samples: The Holy Grail for molecular diagnostics. J. Pharm. Biomed. Anal. 2018, 155, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Steiert, T.A.; Parra, G.; Gut, M.; Arnold, N.; Trotta, J.R.; Tonda, R.; Moussy, A.; Gerber, Z.; Abuja, P.M.; Zatloukal, K.; et al. A critical spotlight on the paradigms of FFPE-DNA sequencing. Nucleic Acids Res. 2023, 51, 7143–7162. [Google Scholar] [CrossRef] [PubMed]
- Guyard, A.; Boyez, A.; Pujals, A.; Robe, C.; Tran Van Nhieu, J.; Allory, Y.; Moroch, J.; Georges, O.; Fournet, J.C.; Zafrani, E.S.; et al. DNA degrades during storage in formalin-fixed and paraffin-embedded tissue blocks. Virchows Arch. 2017, 471, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Nicieza, R.G.; Huergo, J.; Connolly, B.A.; Sanchez, J. Purification, characterization, and role of nucleases and serine proteases in Streptomyces differentiation. Analogies with the biochemical processes described in late steps of eukaryotic apoptosis. J. Biol. Chem. 1999, 274, 20366–20375. [Google Scholar] [CrossRef] [PubMed]
- Koohy, H.; Down, T.A.; Hubbard, T.J. Chromatin accessibility data sets show bias due to sequence specificity of the DNase I enzyme. PLoS ONE 2013, 8, e69853. [Google Scholar] [CrossRef]
- Nordstrom, K.J.V.; Schmidt, F.; Gasparoni, N.; Salhab, A.; Gasparoni, G.; Kattler, K.; Muller, F.; Ebert, P.; Costa, I.G.; DEEP Consortium; et al. Unique and assay specific features of NOMe-, ATAC- and DNase I-seq data. Nucleic Acids Res. 2019, 47, 10580–10596. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.; Tang, Q.; Wan, M.; Cui, K.; Zhang, Y.; Ren, G.; Ni, B.; Sklar, J.; Przytycka, T.M.; Childs, R.; et al. Genome-wide detection of DNase I hypersensitive sites in single cells and FFPE tissue samples. Nature 2015, 528, 142–146. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.; Ding, Y.; Song, J.; Zhao, K. Genome-wide mapping of DNase I hypersensitive sites in rare cell populations using single-cell DNase sequencing. Nat. Protoc. 2017, 12, 2342–2354. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, G.; Millan-Zambrano, G.; Medina, D.A.; Jordan-Pla, A.; Perez-Ortin, J.E.; Penate, X.; Chavez, S. Subtracting the sequence bias from partially digested MNase-seq data reveals a general contribution of TFIIS to nucleosome positioning. Epigenetics Chromatin 2017, 10, 58. [Google Scholar] [CrossRef] [PubMed]
- Mieczkowski, J.; Cook, A.; Bowman, S.K.; Mueller, B.; Alver, B.H.; Kundu, S.; Deaton, A.M.; Urban, J.A.; Larschan, E.; Park, P.J.; et al. MNase titration reveals differences between nucleosome occupancy and chromatin accessibility. Nat. Commun. 2016, 7, 11485. [Google Scholar] [CrossRef] [PubMed]
- Nagy, P.L.; Price, D.H. Formaldehyde-assisted isolation of regulatory elements. Wiley Interdiscip. Rev. Syst. Biol. Med. 2009, 1, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Buenrostro, J.D.; Wu, B.; Chang, H.Y.; Greenleaf, W.J. ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr. Protoc. Mol. Biol. 2015, 109, 21.29.1–21.29.9. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Schulz, M.H.; Look, T.; Begemann, M.; Zenke, M.; Costa, I.G. Identification of transcription factor binding sites using ATAC-seq. Genome Biol. 2019, 20, 45. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.H.; Zhu, Z.; Dunigan, D.D.; Van Etten, J.L.; Xu, S.Y. Cloning of Nt.CviQII nicking endonuclease and its cognate methyltransferase: M.CviQII methylates AG sequences. Protein Expr. Purif. 2006, 49, 138–150. [Google Scholar] [CrossRef] [PubMed]
- Esteve, P.O.; Vishnu, U.S.; Chin, H.G.; Pradhan, S. Visualization and Sequencing of Accessible Chromatin Reveals Cell Cycle and Post-HDAC inhibitor Treatment Dynamics. J. Mol. Biol. 2020, 432, 5304–5321. [Google Scholar] [CrossRef] [PubMed]
- Vishnu, U.S.; Esteve, P.O.; Chin, H.G.; Pradhan, S. One-pot universal NicE-seq: All enzymatic downstream processing of 4% formaldehyde crosslinked cells for chromatin accessibility genomics. Epigenetics Chromatin 2021, 14, 53. [Google Scholar] [CrossRef] [PubMed]
- Axel, R. Cleavage of DNA in nuclei and chromatin with staphylococcal nuclease. Biochemistry 1975, 14, 2921–2925. [Google Scholar] [CrossRef] [PubMed]
- Martins, A.L.; Walavalkar, N.M.; Anderson, W.D.; Zang, C.; Guertin, M.J. Universal correction of enzymatic sequence bias reveals molecular signatures of protein/DNA interactions. Nucleic Acids Res. 2018, 46, e9. [Google Scholar] [CrossRef] [PubMed]
- Henikoff, J.G.; Belsky, J.A.; Krassovsky, K.; MacAlpine, D.M.; Henikoff, S. Epigenome characterization at single base-pair resolution. Proc. Natl. Acad. Sci. USA 2011, 108, 18318–18323. [Google Scholar] [CrossRef] [PubMed]
- Chereji, R.V.; Bryson, T.D.; Henikoff, S. Quantitative MNase-seq accurately maps nucleosome occupancy levels. Genome Biol. 2019, 20, 198. [Google Scholar] [CrossRef]
- Nagy, P.L.; Cleary, M.L.; Brown, P.O.; Lieb, J.D. Genomewide demarcation of RNA polymerase II transcription units revealed by physical fractionation of chromatin. Proc. Natl. Acad. Sci. USA 2003, 100, 6364–6369. [Google Scholar] [CrossRef] [PubMed]
- Giresi, P.G.; Kim, J.; McDaniell, R.M.; Iyer, V.R.; Lieb, J.D. FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome Res. 2007, 17, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Simon, J.M.; Giresi, P.G.; Davis, I.J.; Lieb, J.D. Using formaldehyde-assisted isolation of regulatory elements (FAIRE) to isolate active regulatory DNA. Nat. Protoc. 2012, 7, 256–267. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Ji, Z.; Fang, W.; Ji, H. Global prediction of chromatin accessibility using small-cell-number and single-cell RNA-seq. Nucleic Acids Res. 2019, 47, e121. [Google Scholar] [CrossRef] [PubMed]
- Giresi, P.G.; Lieb, J.D. Isolation of active regulatory elements from eukaryotic chromatin using FAIRE (Formaldehyde Assisted Isolation of Regulatory Elements). Methods 2009, 48, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Gaulton, K.J.; Nammo, T.; Pasquali, L.; Simon, J.M.; Giresi, P.G.; Fogarty, M.P.; Panhuis, T.M.; Mieczkowski, P.; Secchi, A.; Bosco, D.; et al. A map of open chromatin in human pancreatic islets. Nat. Genet. 2010, 42, 255–259. [Google Scholar] [CrossRef]
- Buchert, E.M.; Fogarty, E.A.; Uyehara, C.M.; McKay, D.J.; Buttitta, L.A. A tissue dissociation method for ATAC-seq and CUT&RUN in Drosophila pupal tissues. Fly 2023, 17, 2209481. [Google Scholar] [CrossRef] [PubMed]
- Berg, D.E. Julian Davies and the discovery of kanamycin resistance transposon Tn5. J. Antibiot. 2017, 70, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Reznikoff, W.S. Tn5 as a model for understanding DNA transposition. Mol. Microbiol. 2003, 47, 1199–1206. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Jin, K.; Bai, Y.; Fu, H.; Liu, L.; Liu, B. Tn5 Transposase Applied in Genomics Research. Int. J. Mol. Sci. 2020, 21, 8329. [Google Scholar] [CrossRef] [PubMed]
- Buenrostro, J.D.; Giresi, P.G.; Zaba, L.C.; Chang, H.Y.; Greenleaf, W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 2013, 10, 1213–1218. [Google Scholar] [CrossRef] [PubMed]
- Wolpe, J.B.; Martins, A.L.; Guertin, M.J. Correction of transposase sequence bias in ATAC-seq data with rule ensemble modeling. NAR Genom. Bioinform. 2023, 5, lqad054. [Google Scholar] [CrossRef]
- Schmidt, F.; Gasparoni, N.; Gasparoni, G.; Gianmoena, K.; Cadenas, C.; Polansky, J.K.; Ebert, P.; Nordstrom, K.; Barann, M.; Sinha, A.; et al. Combining transcription factor binding affinities with open-chromatin data for accurate gene expression prediction. Nucleic Acids Res. 2017, 45, 54–66. [Google Scholar] [CrossRef]
- Corces, M.R.; Trevino, A.E.; Hamilton, E.G.; Greenside, P.G.; Sinnott-Armstrong, N.A.; Vesuna, S.; Satpathy, A.T.; Rubin, A.J.; Montine, K.S.; Wu, B.; et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 2017, 14, 959–962. [Google Scholar] [CrossRef]
- Grandi, F.C.; Modi, H.; Kampman, L.; Corces, M.R. Chromatin accessibility profiling by ATAC-seq. Nat. Protoc. 2022, 17, 1518–1552. [Google Scholar] [CrossRef]
- Henikoff, S.; Henikoff, J.G.; Ahmad, K.; Paranal, R.M.; Janssens, D.H.; Russell, Z.R.; Szulzewsky, F.; Kugel, S.; Holland, E.C. Epigenomic analysis of formalin-fixed paraffin-embedded samples by CUT&Tag. Nat. Commun. 2023, 14, 5930. [Google Scholar] [CrossRef] [PubMed]
- Yadav, R.P.; Polavarapu, V.K.; Xing, P.; Chen, X. FFPE-ATAC: A Highly Sensitive Method for Profiling Chromatin Accessibility in Formalin-Fixed Paraffin-Embedded Samples. Curr. Protoc. 2022, 2, e535. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Polavarapu, V.K.; Xing, P.; Zhao, M.; Mathot, L.; Zhao, L.; Rosen, G.; Swartling, F.J.; Sjoblom, T.; Chen, X. Profiling chromatin accessibility in formalin-fixed paraffin-embedded samples. Genome Res. 2022, 32, 150–161. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Polavarapu, V.K.; Yadav, R.P.; Xing, P.; Chen, X. A Highly Sensitive Method to Efficiently Profile the Histone Modifications of FFPE Samples. Bio Protoc. 2022, 12, e4418. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Xing, P.; Polavarapu, V.K.; Zhao, M.; Valero-Martinez, B.; Dang, Y.; Maturi, N.; Mathot, L.; Neves, I.; Yildirim, I.; et al. FACT-seq: Profiling histone modifications in formalin-fixed paraffin-embedded samples with low cell numbers. Nucleic Acids Res. 2021, 49, e125. [Google Scholar] [CrossRef] [PubMed]
- Amatori, S.; Fanelli, M. The Current State of Chromatin Immunoprecipitation (ChIP) from FFPE Tissues. Int. J. Mol. Sci. 2022, 23, 1103. [Google Scholar] [CrossRef] [PubMed]
- Oba, U.; Kohashi, K.; Sangatsuda, Y.; Oda, Y.; Sonoda, K.H.; Ohga, S.; Yoshimoto, K.; Arai, Y.; Yachida, S.; Shibata, T.; et al. An efficient procedure for the recovery of DNA from formalin-fixed paraffin-embedded tissue sections. Biol. Methods Protoc. 2022, 7, bpac014. [Google Scholar] [CrossRef] [PubMed]
- Henikoff, S.; Henikoff, J.G.; Paranal, R.M.; Greene, J.E.; Zheng, Y.; Russell, Z.R.; Szulzewsky, F.; Kugel, S.; Holland, E.C.; Ahmad, K. Direct measurement of RNA Polymerase II hypertranscription in cancer FFPE samples. bioRxiv 2024. [Google Scholar] [CrossRef] [PubMed]
- Ponnaluri, V.K.C.; Zhang, G.; Esteve, P.O.; Spracklin, G.; Sian, S.; Xu, S.Y.; Benoukraf, T.; Pradhan, S. NicE-seq: High resolution open chromatin profiling. Genome Biol. 2017, 18, 122. [Google Scholar] [CrossRef]
- Chin, H.G.; Sun, Z.; Vishnu, U.S.; Hao, P.; Cejas, P.; Spracklin, G.; Esteve, P.O.; Xu, S.Y.; Long, H.W.; Pradhan, S. Universal NicE-seq for high-resolution accessible chromatin profiling for formaldehyde-fixed and FFPE tissues. Clin. Epigenetics 2020, 12, 143. [Google Scholar] [CrossRef] [PubMed]
- Pranzatelli, T.J.F.; Michael, D.G.; Chiorini, J.A. ATAC2GRN: Optimized ATAC-seq and DNase1-seq pipelines for rapid and accurate genome regulatory network inference. BMC Genom. 2018, 19, 563. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.P.; Sheffield, N.C. Analytical Approaches for ATAC-seq Data Analysis. Curr. Protoc. Hum. Genet. 2020, 106, e101. [Google Scholar] [CrossRef]
- Yan, F.; Powell, D.R.; Curtis, D.J.; Wong, N.C. From reads to insight: A hitchhiker’s guide to ATAC-seq data analysis. Genome Biol. 2020, 21, 22. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing, S. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R. BEDTools: The Swiss-Army Tool for Genome Feature Analysis. Curr. Protoc. Bioinform. 2014, 47, 11.12.1–11.12.34. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Amemiya, H.M.; Kundaje, A.; Boyle, A.P. The ENCODE Blacklist: Identification of Problematic Regions of the Genome. Sci. Rep. 2019, 9, 9354. [Google Scholar] [CrossRef] [PubMed]
- Liu, T. Use model-based Analysis of ChIP-Seq (MACS) to analyze short reads generated by sequencing protein-DNA interactions in embryonic stem cells. Methods Mol. Biol. 2014, 1150, 81–95. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, F.; Ryan, D.P.; Gruning, B.; Bhardwaj, V.; Kilpert, F.; Richter, A.S.; Heyne, S.; Dundar, F.; Manke, T. deepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016, 44, W160–W165. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdottir, H.; Turner, D.; Mesirov, J.P. igv.js: An embeddable JavaScript implementation of the Integrative Genomics Viewer (IGV). Bioinformatics 2023, 39, btac830. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Talebi, A.; Thiery, J.P.; Kerachian, M.A. Fusion transcript discovery using RNA sequencing in formalin-fixed paraffin-embedded specimen. Crit. Rev. Oncol. Hematol. 2021, 160, 103303. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, A.; Oana, Y.; Ohi, Y.; Maeda, Y.; Omori, M.; Takada, Y.; Ikeda, T.; Sotome, K.; Maeda, H.; Yanagisawa, T.; et al. Using the DNA Integrity Number (DIN) to analyze DNA quality in specimens collected from liquid-based cytology after fine needle aspiration of breast tumors and lesions. Acta Cytol. 2024, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Siegel, E.M.; Berglund, A.E.; Riggs, B.M.; Eschrich, S.A.; Putney, R.M.; Ajidahun, A.O.; Coppola, D.; Shibata, D. Expanding epigenomics to archived FFPE tissues: An evaluation of DNA repair methodologies. Cancer Epidemiol. Biomark. Prev. 2014, 23, 2622–2631. [Google Scholar] [CrossRef] [PubMed]
- Cejas, P.; Li, L.; O’Neill, N.K.; Duarte, M.; Rao, P.; Bowden, M.; Zhou, C.W.; Mendiola, M.; Burgos, E.; Feliu, J.; et al. Chromatin immunoprecipitation from fixed clinical tissues reveals tumor-specific enhancer profiles. Nat. Med. 2016, 22, 685–691. [Google Scholar] [CrossRef] [PubMed]
- Kashima, Y.; Reteng, P.; Haga, Y.; Yamagishi, J.; Suzuki, Y. Single-cell analytical technologies: Uncovering the mechanisms behind variations in immune responses. FEBS J. 2024, 291, 819–831. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Bartosovic, M.; Ma, S.; Zhang, D.; Kukanja, P.; Xiao, Y.; Su, G.; Liu, Y.; Qin, X.; Rosoklija, G.B.; et al. Spatial profiling of chromatin accessibility in mouse and human tissues. Nature 2022, 609, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Janesick, A.; Shelansky, R.; Gottscho, A.D.; Wagner, F.; Williams, S.R.; Rouault, M.; Beliakoff, G.; Morrison, C.A.; Oliveira, M.F.; Sicherman, J.T.; et al. High resolution mapping of the tumor microenvironment using integrated single-cell, spatial and in situ analysis. Nat. Commun. 2023, 14, 8353. [Google Scholar] [CrossRef] [PubMed]
- Kumar, T.; Nee, K.; Wei, R.; He, S.; Nguyen, Q.H.; Bai, S.; Blake, K.; Pein, M.; Gong, Y.; Sei, E.; et al. A spatially resolved single-cell genomic atlas of the adult human breast. Nature 2023, 620, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Bartosovic, M.; Kukanja, P.; Zhang, D.; Liu, Y.; Su, G.; Enninful, A.; Bai, Z.; Castelo-Branco, G.; Fan, R. Spatial-CUT&Tag: Spatially resolved chromatin modification profiling at the cellular level. Science 2022, 375, 681–686. [Google Scholar] [CrossRef] [PubMed]
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [PubMed]
- Willroider, M.; Roeber, S.; Horn, A.K.E.; Arzberger, T.; Scheifele, M.; Respondek, G.; Sabri, O.; Barthel, H.; Patt, M.; Mishchenko, O.; et al. Superiority of Formalin-Fixed Paraffin-Embedded Brain Tissue for in vitro Assessment of Progressive Supranuclear Palsy Tau Pathology With [18F]PI-2620. Front. Neurol. 2021, 12, 684523. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DNase-Seq | MNase-Seq | FAIRE-Seq | ATAC-Seq | NicE-Seq | |
|---|---|---|---|---|---|
| Type of input cells/tissue | Fresh/formaldehyde cross-linked/FFPE (Formalin-Fixed Paraffin-Embedded) | Fresh/formaldehyde cross-linked | Formaldehyde cross-linked | Fresh/formaldehyde cross-linked (less efficient in fixed) | Formaldehyde cross-linked/FFPE |
| Application to FFPE (PubMed) | 1 | 0 | 1 | 2 | 2 |
| Number of input cells | 106–107 | 103–107 | 103–107 | 1 cell—5 × 104 | 25 cells—105 |
| Fragment size (i.e., resolution) | ~200 bp | ~200 bp | ~300 bp | ~100–200 bp | ~300 bp |
| Key features | DNase I (endonuclease) cuts unprotected DNA | MNase (endo-exonuclease) digests unprotected DNA | Sonicate unprotected DNA in cross-linked material | Tn5 transposase tagments open region with DNA adapters | Nt-CviPII nickase cuts/labels CCD sites in unprotected DNA |
| Sequencing type | Single/paired end | Single/paired end | Single/paired end | Single/paired end | Single/paired end |
| Target region | NDR | Linker DNA between Nucleosomes | NDR | NDR | NDR |
| Sequencing depth (human genome; ~3 billion bp) | 20–50 million mapped reads | 150–200 million mapped reads | 20–50 million mapped reads | 25–30 million mapped reads (non-mitochondrial) | 20–30 million mapped reads |
| Cleavage bias | Yes | Yes | No | Yes | Yes |
| Advantages / disadvantages | No prior knowledge of the sequence or binding protein is required / time consuming, requires laborious enzyme titrations and calibrations, requires high sequencing depth | Nucleosome positioning can be inferred / requires laborious enzyme titrations and calibrations, requires high sequencing depth, indirect profiling of open regions | No enzymes optimization or titration required / low signal-to-noise ratio, relatively complex computational data analysis and interpretation, results are highly fixation-dependent | Simple, fast, and sensitive approach; high signal-to-noise ratio / High mitochondrial DNA counts (unless nuclei isolated), requires two independent tagmentation events in opposite orientation, Tn5 sequence bias and promoter-enrichment bias | Simple enzymatic approach, <5% mitochondrial DNA counts, optimal in fixed or FFPE samples, can be used in clinical settings, efficiently profiles promoters and enhancers / AT-rich sequences may be underrepresented |
| References | [26,28] | [12,13,14,44,45] | [46] | [47,48] | [49,50,51] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sunitha Kumary, V.U.N.; Venters, B.J.; Raman, K.; Sen, S.; Estève, P.-O.; Cowles, M.W.; Keogh, M.-C.; Pradhan, S. Emerging Approaches to Profile Accessible Chromatin from Formalin-Fixed Paraffin-Embedded Sections. Epigenomes 2024, 8, 20. https://doi.org/10.3390/epigenomes8020020
Sunitha Kumary VUN, Venters BJ, Raman K, Sen S, Estève P-O, Cowles MW, Keogh M-C, Pradhan S. Emerging Approaches to Profile Accessible Chromatin from Formalin-Fixed Paraffin-Embedded Sections. Epigenomes. 2024; 8(2):20. https://doi.org/10.3390/epigenomes8020020
Chicago/Turabian StyleSunitha Kumary, Vishnu Udayakumaran Nair, Bryan J. Venters, Karthikeyan Raman, Sagnik Sen, Pierre-Olivier Estève, Martis W. Cowles, Michael-Christopher Keogh, and Sriharsa Pradhan. 2024. "Emerging Approaches to Profile Accessible Chromatin from Formalin-Fixed Paraffin-Embedded Sections" Epigenomes 8, no. 2: 20. https://doi.org/10.3390/epigenomes8020020
APA StyleSunitha Kumary, V. U. N., Venters, B. J., Raman, K., Sen, S., Estève, P.-O., Cowles, M. W., Keogh, M.-C., & Pradhan, S. (2024). Emerging Approaches to Profile Accessible Chromatin from Formalin-Fixed Paraffin-Embedded Sections. Epigenomes, 8(2), 20. https://doi.org/10.3390/epigenomes8020020