
Architecture, Chromatin and Gene Organization of Toxoplasma gondii Subtelomeres

 , ,
, ,  , and
, and
Abstract
1. Introduction
2. Results
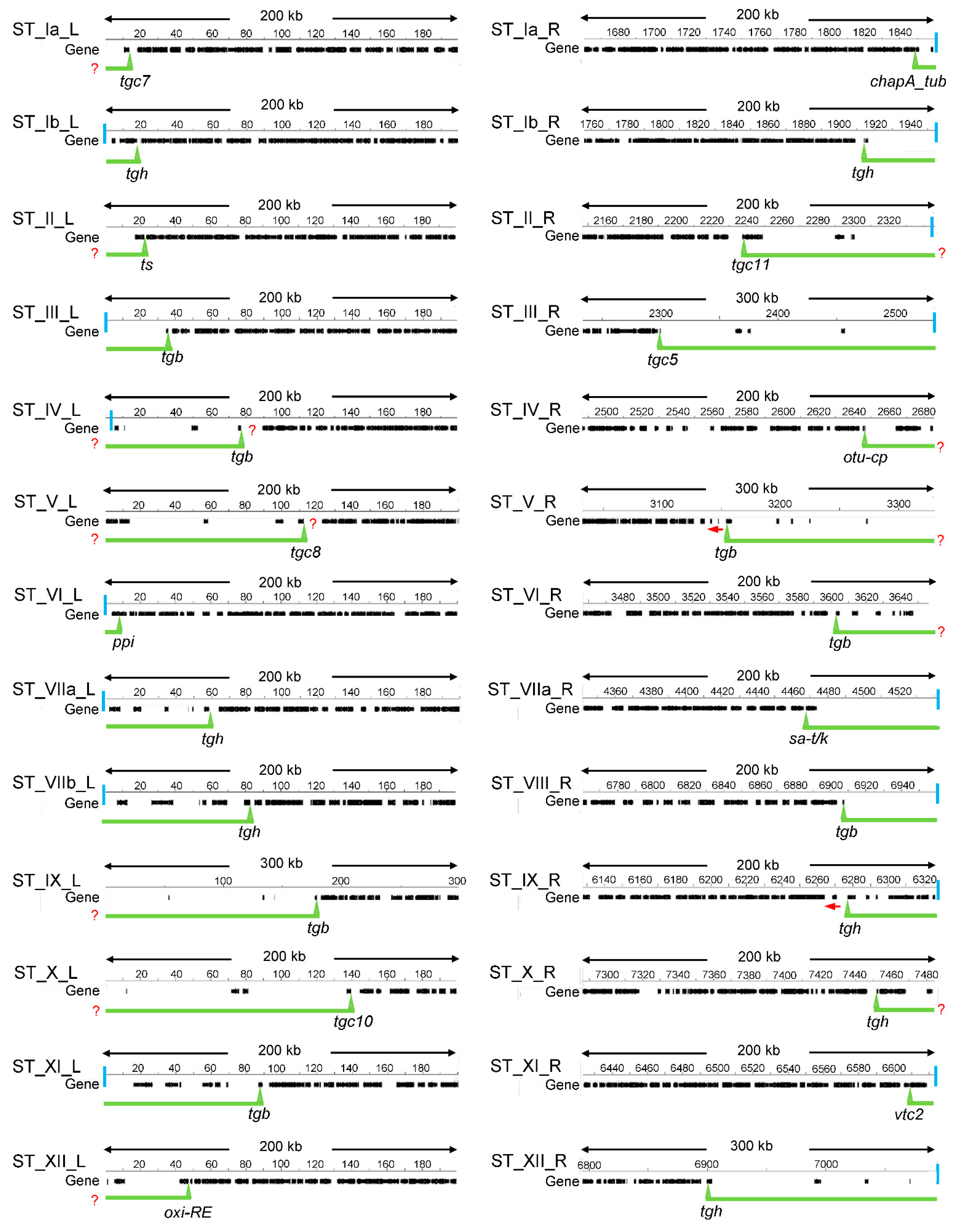
2.1. Defining T. gondii STs
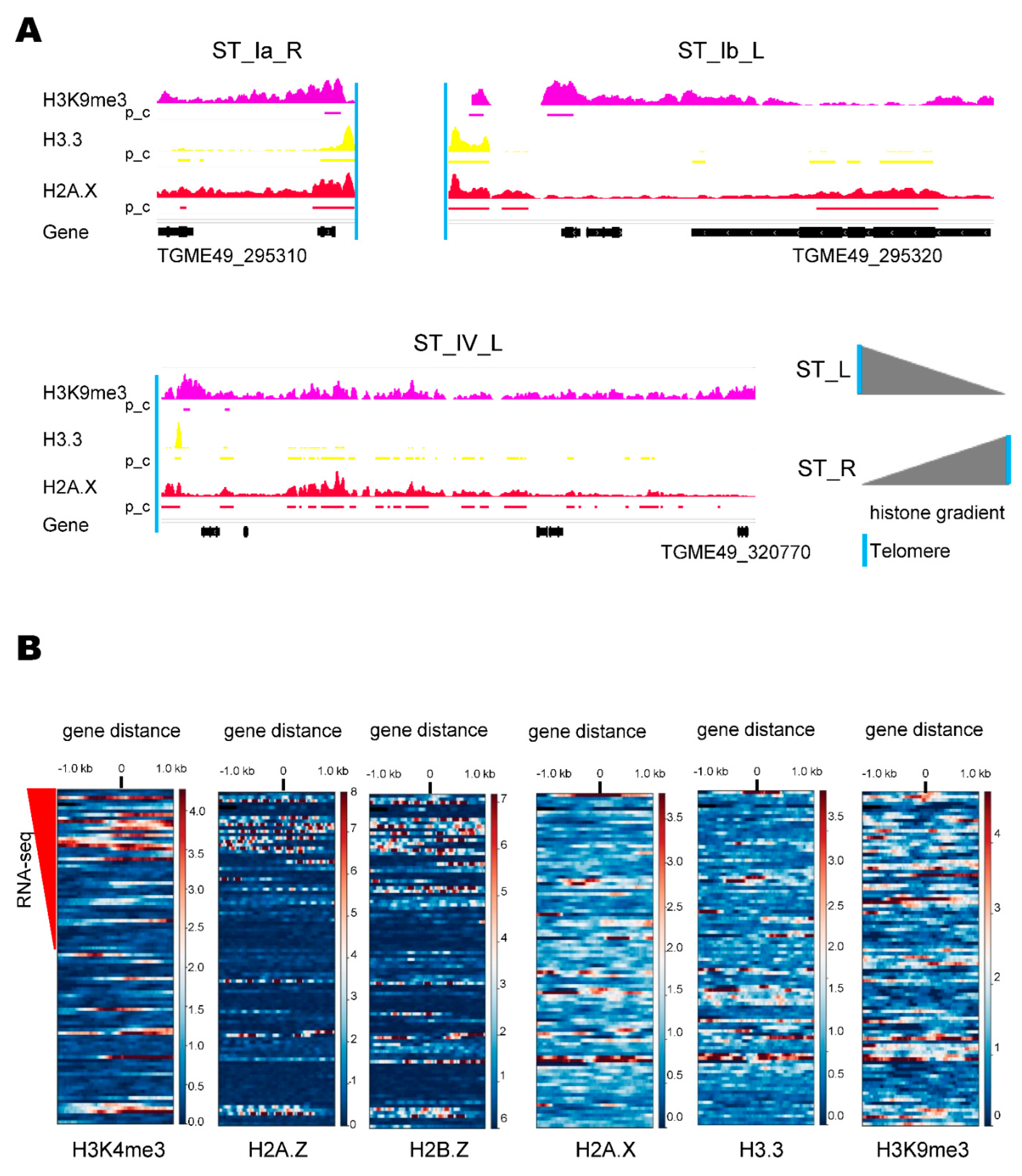
2.2. ST Chromatin Architecture
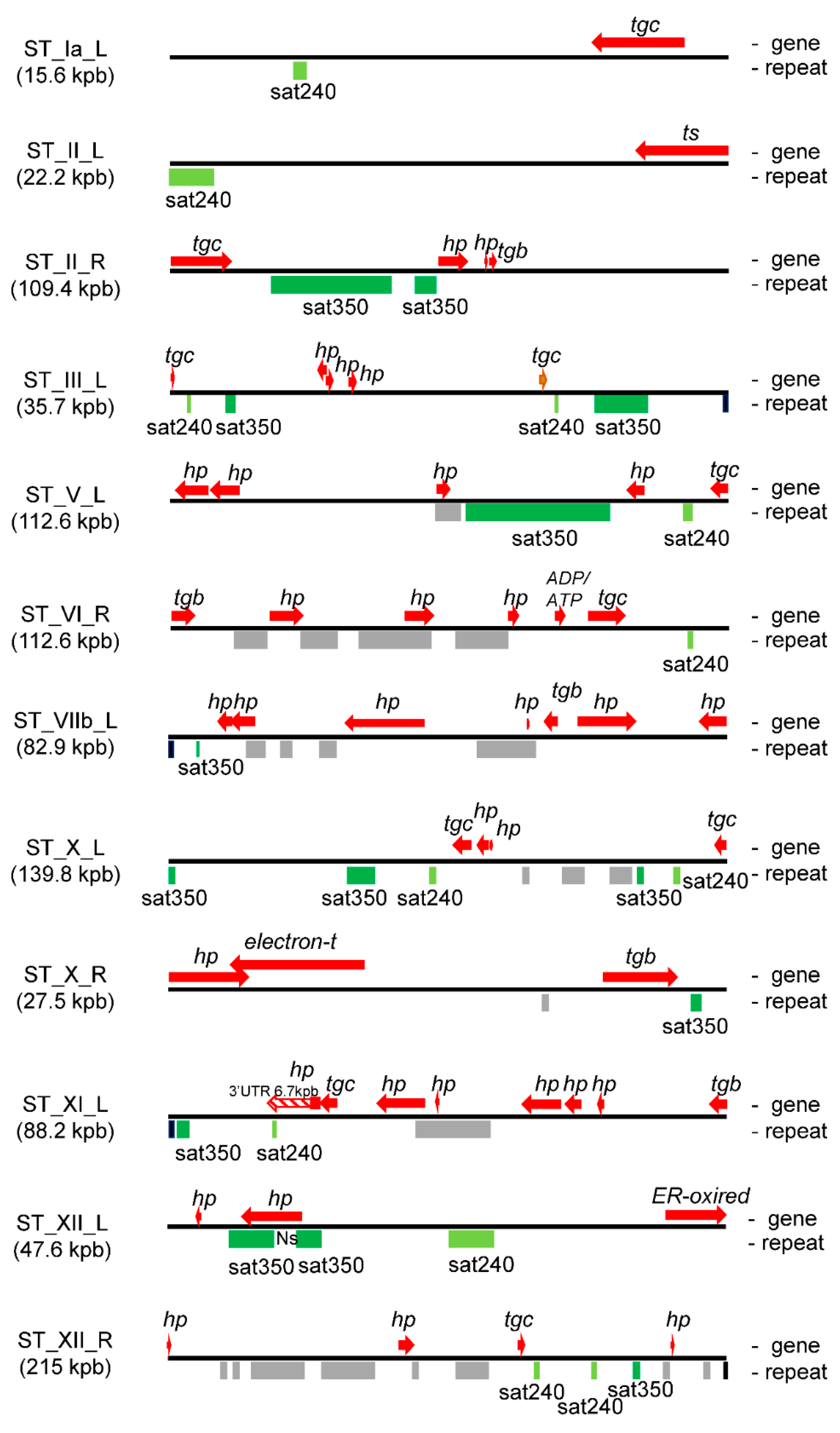
2.3. T. gondii STs Are Enriched in Tandem DNA Repeat Elements
2.4. Satellite DNA and Subtelomeric Genes
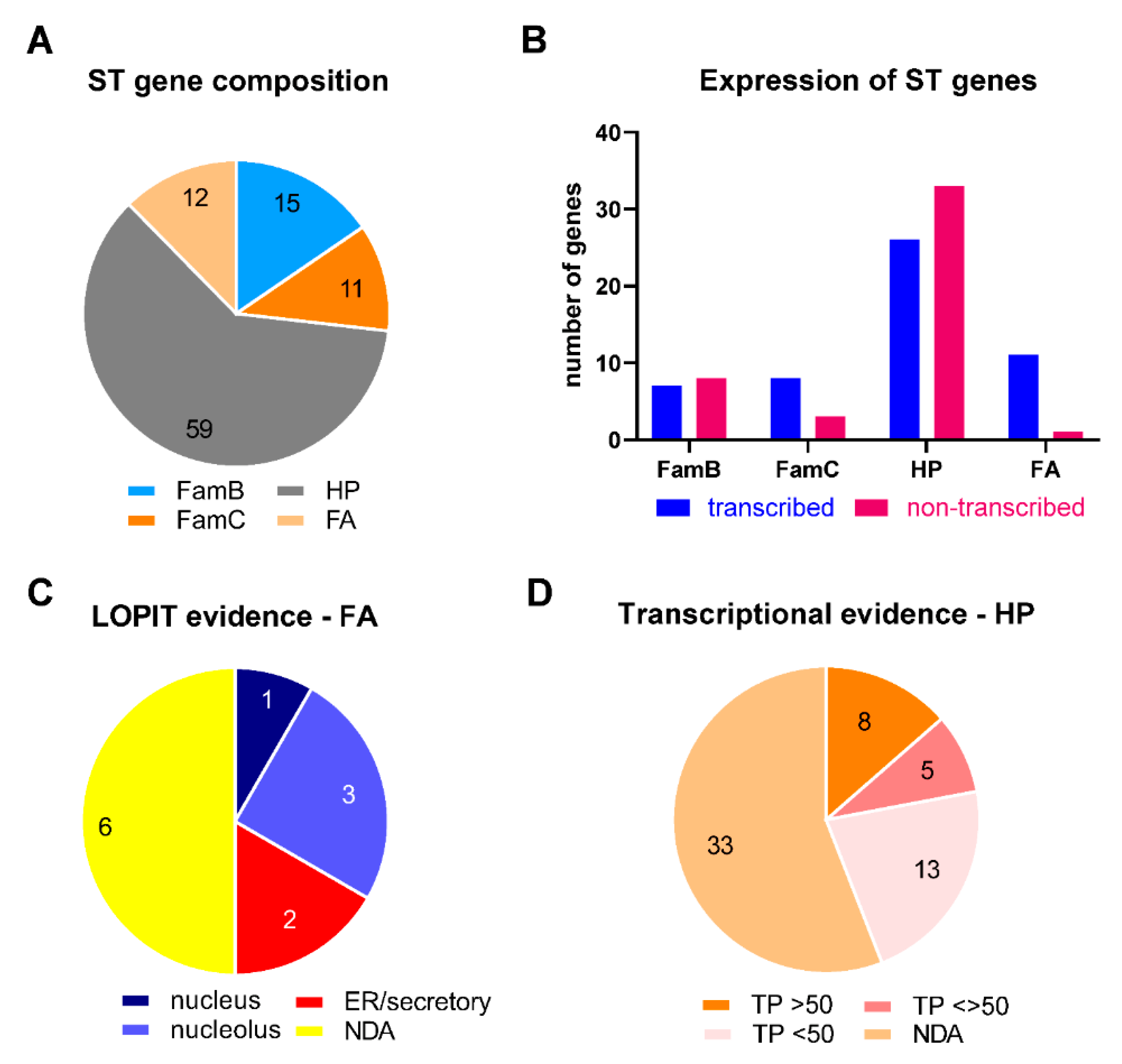
2.5. Features of ST Genes
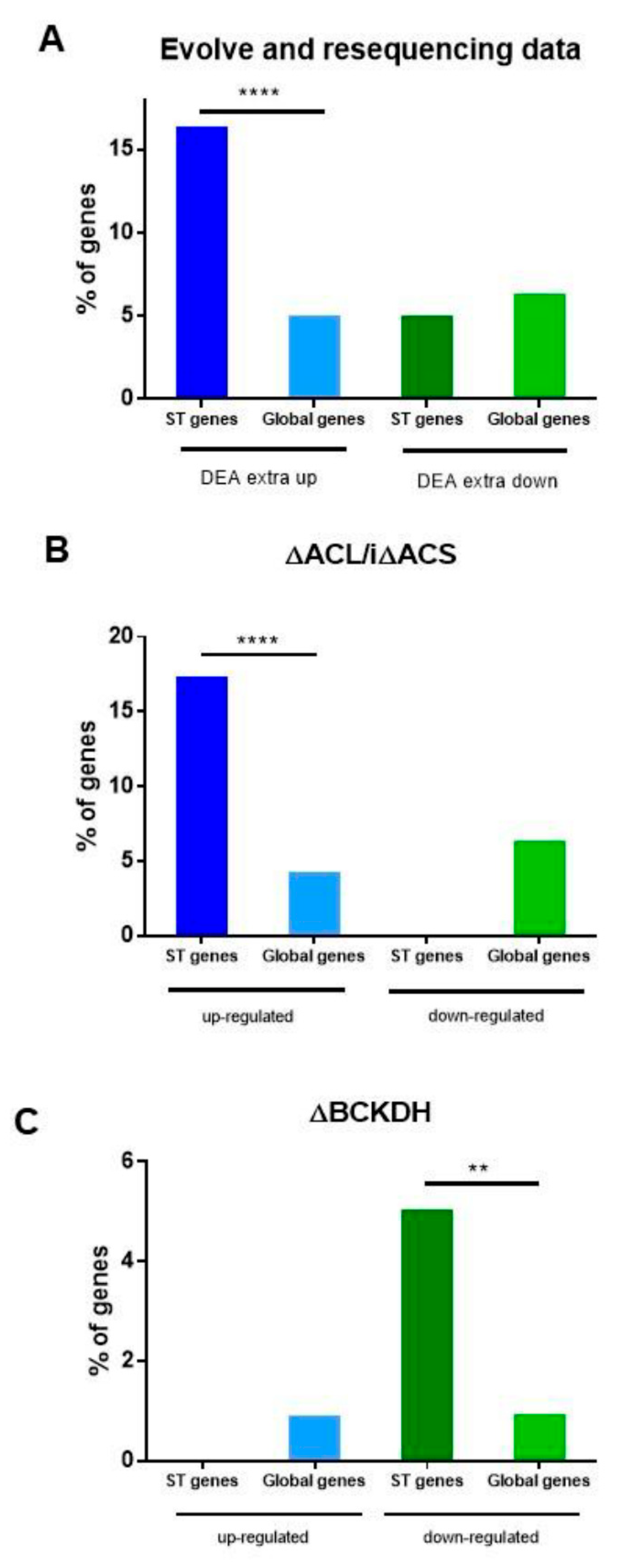
2.6. Role of ST Genes in Adaptability to Different Environmental/Metabolic Situations
3. Discussion
4. Materials and Methods
4.1. Sequence Analysis
4.2. Chromatin Analysis
4.3. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Janssen, A.; Colmenares, S.U.; Karpen, G.H. Heterochromatin: Guardian of the Genome. Annu. Rev. Cell Dev. Biol. 2018, 34, 265–288. [Google Scholar] [CrossRef] [PubMed]
- Pryde, F.E.; Louis, E.J. Limitations of silencing at native yeast telomeres. EMBO J. 1999, 18, 2538–2550. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, L.M.; Freitas-Junior, L.H.; Bottius, E.; Olivo-Marin, J.C.; Scherf, A. A central role for Plasmodium falciparum subtelomeric regions in spatial positioning and telomere length regulation. EMBO J. 2002, 21, 815–824. [Google Scholar] [CrossRef]
- Barry, J.D.; Ginger, M.L.; Burton, P.; McCulloch, R. Why are parasite contingency genes often associated with telomeres? Int. J. Parasitol. 2003, 33, 29–45. [Google Scholar] [CrossRef]
- Loney, E.R.; Inglis, P.W.; Sharp, S.; Pryde, F.E.; Kent, N.A.; Mellor, J.; Louis, E.J. Repressive and non-repressive chromatin at native telomeres in Saccharomyces cerevisiae. Epigenet. Chromatin 2009, 2, 18. [Google Scholar] [CrossRef]
- Hernandez-Rivas, R.; Perez-Toledo, K.; Herrera Solorio, A.M.; Delgadillo, D.M.; Vargas, M. Telomeric heterochromatin in Plasmodium falciparum. J. Biomed. Biotechnol. 2010, 2010, 290501. [Google Scholar] [CrossRef] [PubMed]
- Jezek, M.; Green, E.M. Histone Modifications and the Maintenance of Telomere Integrity. Cells 2019, 8, 199. [Google Scholar] [CrossRef] [PubMed]
- Diotti, R.; Esposito, M.; Shen, C.H. Telomeric and Sub-Telomeric Structure and Implications in Fungal Opportunistic Pathogens. Microorganisms 2021, 9, 1405. [Google Scholar] [CrossRef]
- Yadav, R.K.; Matsuda, A.; Lowe, B.R.; Hiraoka, Y.; Partridge, J.F. Subtelomeric Chromatin in the Fission Yeast S. pombe. Microorganisms 2021, 9, 1977. [Google Scholar] [CrossRef]
- Steglich, B.; Stralfors, A.; Khorosjutina, O.; Persson, J.; Smialowska, A.; Javerzat, J.P.; Ekwall, K. The Fun30 chromatin remodeler Fft3 controls nuclear organization and chromatin structure of insulators and subtelomeres in fission yeast. PLoS Genet. 2015, 11, e1005101. [Google Scholar] [CrossRef]
- Tashiro, S.; Nishihara, Y.; Kugou, K.; Ohta, K.; Kanoh, J. Subtelomeres constitute a safeguard for gene expression and chromosome homeostasis. Nucleic Acids Res. 2017, 45, 10333–10349. [Google Scholar] [CrossRef] [PubMed]
- Ottaviani, A.; Gilson, E.; Magdinier, F. Telomeric position effect: From the yeast paradigm to human pathologies? Biochimie 2008, 90, 93–107. [Google Scholar] [CrossRef] [PubMed]
- Wyse, B.A.; Oshidari, R.; Jeffery, D.C.; Yankulov, K.Y. Parasite epigenetics and immune evasion: Lessons from budding yeast. Epigenet. Chromatin 2013, 6, 40. [Google Scholar] [CrossRef]
- Azzalin, C.M.; Lingner, J. Telomere functions grounding on TERRA firma. Trends Cell Biol. 2015, 25, 29–36. [Google Scholar] [CrossRef]
- Brown, C.A.; Murray, A.W.; Verstrepen, K.J. Rapid expansion and functional divergence of subtelomeric gene families in yeasts. Curr. Biol. CB 2010, 20, 895–903. [Google Scholar] [CrossRef]
- Teunissen, A.W.; Steensma, H.Y. Review: The dominant flocculation genes of Saccharomyces cerevisiae constitute a new subtelomeric gene family. Yeast 1995, 11, 1001–1013. [Google Scholar] [CrossRef]
- Hasin, Y.; Olender, T.; Khen, M.; Gonzaga-Jauregui, C.; Kim, P.M.; Urban, A.E.; Snyder, M.; Gerstein, M.B.; Lancet, D.; Korbel, J.O. High-resolution copy-number variation map reflects human olfactory receptor diversity and evolution. PLoS Genet. 2008, 4, e1000249. [Google Scholar] [CrossRef]
- Tembo, D.; Montgomery, J. Var gene expression and human Plasmodium pathogenesis. Future Microbiol. 2010, 5, 801–815. [Google Scholar] [CrossRef]
- Cross, G.A.; Kim, H.S.; Wickstead, B. Capturing the variant surface glycoprotein repertoire (the VSGnome) of Trypanosoma brucei Lister 427. Mol. Biochem. Parasitol. 2014, 195, 59–73. [Google Scholar] [CrossRef]
- Cestari, I.; Stuart, K. Transcriptional Regulation of Telomeric Expression Sites and Antigenic Variation in Trypanosomes. Curr. Genom. 2018, 19, 119–132. [Google Scholar] [CrossRef]
- Ramirez, J.L. An Evolutionary View of Trypanosoma Cruzi Telomeres. Front. Cell. Infect. Microbiol. 2019, 9, 439. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Leveau, A.; Zhao, Q.; Feng, Q.; Lu, H.; Miao, J.; Xue, Z.; Martin, A.C.; Wegel, E.; Wang, J.; et al. Subtelomeric assembly of a multi-gene pathway for antimicrobial defense compounds in cereals. Nat. Commun. 2021, 12, 2563. [Google Scholar] [CrossRef] [PubMed]
- Gottschling, D.E.; Aparicio, O.M.; Billington, B.L.; Zakian, V.A. Position effect at S. cerevisiae telomeres: Reversible repression of Pol II transcription. Cell 1990, 63, 751–762. [Google Scholar] [CrossRef]
- Duffy, M.F.; Selvarajah, S.A.; Josling, G.A.; Petter, M. The role of chromatin in Plasmodium gene expression. Cell. Microbiol. 2012, 14, 819–828. [Google Scholar] [CrossRef]
- Tenter, A.M.; Heckeroth, A.R.; Weiss, L.M. Toxoplasma gondii: From animals to humans. Int. J. Parasitol. 2000, 30, 1217–1258. [Google Scholar] [CrossRef]
- Berna, L.; Marquez, P.; Cabrera, A.; Greif, G.; Francia, M.E.; Robello, C. Reevaluation of the Toxoplasma gondii and Neospora caninum genomes reveals misassembly, karyotype differences, and chromosomal rearrangements. Genome Res. 2021, 31, 823–833. [Google Scholar] [CrossRef]
- Xia, J.; Venkat, A.; Bainbridge, R.E.; Reese, M.L.; Le Roch, K.G.; Ay, F.; Boyle, J.P. Third-generation sequencing revises the molecular karyotype for Toxoplasma gondii and identifies emerging copy number variants in sexual recombinants. Genome Res. 2021, 31, 834–851. [Google Scholar] [CrossRef]
- Dalmasso, M.C.; Carmona, S.J.; Angel, S.O.; Aguero, F. Characterization of Toxoplasma gondii subtelomeric-like regions: Identification of a long-range compositional bias that is also associated with gene-poor regions. BMC Genom. 2014, 15, 21. [Google Scholar] [CrossRef][Green Version]
- Gissot, M.; Walker, R.; Delhaye, S.; Huot, L.; Hot, D.; Tomavo, S. Toxoplasma gondii chromodomain protein 1 binds to heterochromatin and colocalises with centromeres and telomeres at the nuclear periphery. PLoS ONE 2012, 7, e32671. [Google Scholar] [CrossRef]
- Braun, L.; Cannella, D.; Ortet, P.; Barakat, M.; Sautel, C.F.; Kieffer, S.; Garin, J.; Bastien, O.; Voinnet, O.; Hakimi, M.A. A complex small RNA repertoire is generated by a plant/fungal-like machinery and effected by a metazoan-like Argonaute in the single-cell human parasite Toxoplasma gondii. PLoS Pathog. 2010, 6, e1000920. [Google Scholar] [CrossRef]
- Nardelli, S.C.; Silmon de Monerri, N.C.; Vanagas, L.; Wang, X.; Tampaki, Z.; Sullivan, W.J., Jr.; Angel, S.O.; Kim, K. Genome-wide localization of histone variants in Toxoplasma gondii implicates variant exchange in stage-specific gene expression. BMC Genom. 2022, 23, 128. [Google Scholar] [CrossRef]
- Reid, A.J.; Vermont, S.J.; Cotton, J.A.; Harris, D.; Hill-Cawthorne, G.A.; Konen-Waisman, S.; Latham, S.M.; Mourier, T.; Norton, R.; Quail, M.A.; et al. Comparative genomics of the apicomplexan parasites Toxoplasma gondii and Neospora caninum: Coccidia differing in host range and transmission strategy. PLoS Pathog. 2012, 8, e1002567. [Google Scholar] [CrossRef]
- Lorenzi, H.; Khan, A.; Behnke, M.S.; Namasivayam, S.; Swapna, L.S.; Hadjithomas, M.; Karamycheva, S.; Pinney, D.; Brunk, B.P.; Ajioka, J.W.; et al. Local admixture of amplified and diversified secreted pathogenesis determinants shapes mosaic Toxoplasma gondii genomes. Nat. Commun. 2016, 7, 10147. [Google Scholar] [CrossRef]
- Primo, V.A., Jr.; Rezvani, Y.; Farrell, A.; Murphy, C.Q.; Lou, J.; Vajdi, A.; Marth, G.T.; Zarringhalam, K.; Gubbels, M.J. The Extracellular Milieu of Toxoplasma’s Lytic Cycle Drives Lab Adaptation, Primarily by Transcriptional Reprogramming. mSystems 2021, 6, e0119621. [Google Scholar] [CrossRef] [PubMed]
- Kloehn, J.; Oppenheim, R.D.; Siddiqui, G.; De Bock, P.J.; Kumar Dogga, S.; Coute, Y.; Hakimi, M.A.; Creek, D.J.; Soldati-Favre, D. Multi-omics analysis delineates the distinct functions of sub-cellular acetyl-CoA pools in Toxoplasma gondii. BMC Biol. 2020, 18, 67. [Google Scholar] [CrossRef] [PubMed]
- Radke, J.R.; Behnke, M.S.; Mackey, A.J.; Radke, J.B.; Roos, D.S.; White, M.W. The transcriptome of Toxoplasma gondii. BMC Biol. 2005, 3, 26. [Google Scholar] [CrossRef]
- Gissot, M.; Kelly, K.A.; Ajioka, J.W.; Greally, J.M.; Kim, K. Epigenomic modifications predict active promoters and gene structure in Toxoplasma gondii. PLoS Pathog. 2007, 3, e77. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Ramos, M.A. Satellite DNA: An Evolving Topic. Genes 2017, 8, 230. [Google Scholar] [CrossRef]
- Homan, W.L.; Vercammen, M.; De Braekeleer, J.; Verschueren, H. Identification of a 200- to 300-fold repetitive 529 bp DNA fragment in Toxoplasma gondii, and its use for diagnostic and quantitative PCR. Int. J. Parasitol. 2000, 30, 69–75. [Google Scholar] [CrossRef]
- Walsh, D.; Katris, N.J.; Sheiner, L.; Botte, C.Y. Toxoplasma metabolic flexibility in different growth conditions. Trends Parasitol. 2022, 38, 775–790. [Google Scholar] [CrossRef]
- Moraes Barros, R.R.; Marini, M.M.; Antonio, C.R.; Cortez, D.R.; Miyake, A.M.; Lima, F.M.; Ruiz, J.C.; Bartholomeu, D.C.; Chiurillo, M.A.; Ramirez, J.L.; et al. Anatomy and evolution of telomeric and subtelomeric regions in the human protozoan parasite Trypanosoma cruzi. BMC Genom. 2012, 13, 229. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, J.; Rice, J.C.; Strahl, B.D.; Allis, C.D.; Grewal, S.I. Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science 2001, 292, 110–113. [Google Scholar] [CrossRef] [PubMed]
- Mah, L.J.; El-Osta, A.; Karagiannis, T.C. gammaH2AX: A sensitive molecular marker of DNA damage and repair. Leukemia 2010, 24, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.; Kim, K.; Chang, D.Y.; Kang, H.B.; Shin, E.C.; Kwon, J.; Choi, J.K. Genome-wide reorganization of histone H2AX toward particular fragile sites on cell activation. Nucleic Acids Res. 2014, 42, 1016–1025. [Google Scholar] [CrossRef]
- Fraschka, S.A.; Henderson, R.W.; Bartfai, R. H3.3 demarcates GC-rich coding and subtelomeric regions and serves as potential memory mark for virulence gene expression in Plasmodium falciparum. Sci. Rep. 2016, 6, 31965. [Google Scholar] [CrossRef]
- Santenard, A.; Ziegler-Birling, C.; Koch, M.; Tora, L.; Bannister, A.J.; Torres-Padilla, M.E. Heterochromatin formation in the mouse embryo requires critical residues of the histone variant H3.3. Nat. Cell Biol. 2010, 12, 853–862. [Google Scholar] [CrossRef]
- Udugama, M.; Vinod, B.; Chan, F.L.; Hii, L.; Garvie, A.; Collas, P.; Kalitsis, P.; Steer, D.; Das, P.P.; Tripathi, P.; et al. Histone H3.3 phosphorylation promotes heterochromatin formation by inhibiting H3K9/K36 histone demethylase. Nucleic Acids Res. 2022, 8, 4500–4514. [Google Scholar] [CrossRef]
- Clemente, M.; de Miguel, N.; Lia, V.V.; Matrajt, M.; Angel, S.O. Structure analysis of two Toxoplasma gondii and Neospora caninum satellite DNA families and evolution of their common monomeric sequence. J. Mol. Evol. 2004, 58, 557–567. [Google Scholar] [CrossRef]
- Feliciello, I.; Pezer, Z.; Sermek, A.; Bruvo Madaric, B.; Ljubic, S.; Ugarkovic, D. Satellite DNA-Mediated Gene Expression Regulation: Physiological and Evolutionary Implication. Prog. Mol. Subcell. Biol. 2021, 60, 145–167. [Google Scholar] [CrossRef]
- Lukito, Y.; Chujo, T.; Hale, T.K.; Mace, W.; Johnson, L.J.; Scott, B. Regulation of subtelomeric fungal secondary metabolite genes by H3K4me3 regulators CclA and KdmB. Mol. Microbiol. 2019, 112, 837–853. [Google Scholar] [CrossRef]
- Etchegaray, J.P.; Mostoslavsky, R. Interplay between Metabolism and Epigenetics: A Nuclear Adaptation to Environmental Changes. Mol. Cell 2016, 62, 695–711. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Tu, B.P. Acetyl-CoA and the regulation of metabolism: Mechanisms and consequences. Curr. Opin. Cell Biol. 2015, 33, 125–131. [Google Scholar] [CrossRef]
- Noe, L.; Kucherov, G. YASS: Enhancing the sensitivity of DNA similarity search. Nucleic Acids Res. 2005, 33, W540–W543. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdottir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef]
- Feng, J.; Liu, T.; Qin, B.; Zhang, Y.; Liu, X.S. Identifying ChIP-seq enrichment using MACS. Nat. Protoc. 2012, 7, 1728–1740. [Google Scholar] [CrossRef]
- Ramirez, F.; Ryan, D.P.; Gruning, B.; Bhardwaj, V.; Kilpert, F.; Richter, A.S.; Heyne, S.; Dundar, F.; Manke, T. deepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016, 44, W160–W165. [Google Scholar] [CrossRef]
- Croken, M.M.; Qiu, W.; White, M.W.; Kim, K. Gene Set Enrichment Analysis (GSEA) of Toxoplasma gondii expression datasets links cell cycle progression and the bradyzoite developmental program. BMC Genom. 2014, 15, 515. [Google Scholar] [CrossRef]
- Perea-Resa, C.; Blower, M.D. Centromere Biology: Transcription Goes on Stage. Mol. Cell. Biol. 2018, 38, e00263-18. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ST | Length (kpb) | Nr of Genes | CG% 1 | Telomere Sequences | Satellite DNA |
|---|---|---|---|---|---|
| ST_Ia_L | 15.6 | 1 | 50.1 | no | yes |
| ST_Ia_R | 10.5 | 2 | 49.0 | yes | no |
| ST_Ib_L | 18.4 | 3 | 52.0 | yes | no |
| ST_Ib_R | 40 | 2 | 53.3 | yes | yes |
| ST_II_L | 22.2 | 1 | 49.7 | no | no |
| ST_II_R | 109.4 | 4 | 53.3 | yes * | yes |
| ST_III_L | 35.7 | 1 | 53.3 | yes | no |
| ST_III_R | 232.4 | 5 | 52.5 | yes | yes |
| ST_IV_L | 77 | 5 | 54.8 | yes ** | yes |
| ST_IV_R | 40.2 | 4 | 50.8 | no | yes |
| ST_V_L | 112.6 | 5 | 54.3 | no | yes |
| ST_V_R | 177.9 | 5 | 54.4 | no | yes |
| ST_VI_L | 8.1 | 1 | 50.1 | yes | no |
| ST_VI_R | 53.7 | 7 | 52.7 | no | yes |
| ST_VIIa_L | 58.4 | 6 | 52.6 | yes | yes |
| ST_VIIa_R | 76.3 | 1 | 53.7 | yes | no |
| ST_VIIb_L + | 82.9 | 7 | 53.4 | yes | yes |
| ST_VIII_R + | 53.1 | 1 | 54.9 | yes | yes |
| ST_IX_L | 179.1 | 4 | 53.9 | no | yes |
| ST_IX_R | 49.6 | 8 | 52.2 | yes | no |
| ST_X_L | 139.8 | 5 | 52.3 | no | yes |
| ST_X_R | 27.5 | 3 | 50.1 | no | yes |
| ST_XI_L | 88.2 | 7 | 51.4 | yes | yes |
| ST_XI_R | 13.2 | 1 | 53.1 | yes | no |
| ST_XII_L | 47.6 | 3 | 51.6 | no | yes |
| ST_XII_R | 215 | 4 | 52.3 | yes | yes |
| average | 76.3 | 3.7 | 52.37 | - | - |
| sum | 1985.4 | 96 | - | 16 (yes) | 18 (yes) |
| Me49 Chr ++ | 54,997.637 | 8920 | - | - | - |
| Gene | geneID (TGME49) | Ch | Protein Length (aa) | TM | SP Score | Phenotype 1 | Transcription 1,§ Me49/RH | Proteomic 1 | LOPIT 1 |
|---|---|---|---|---|---|---|---|---|---|
| T. gondii FA 2: | |||||||||
| chapA-tub | _295310 | I a | 189 | 0 | <0.01 | −4.78 | 85.7/85.6 | yes | nucleolus |
| ts | _220840 | II | 571 | 0 | <0.01 | 1.76 | 20.7/7.66 | Oocyst ^ | NDA |
| otu-cp familiy | _237894 | IV | 219 | 0 | <0.01 | −4.37 | 39.9/38.8 | NDA | NDA |
| enoyl-CoA | _317705 | IV | 371 | 0 | <0.01 | −0.45 | 93/92.3 | yes | nucleolus |
| eIF3-D | _317720 | IV | 609 | 0 | <0.01 | −4.87 | 90.8/92.7 | yes | nucleus |
| otu-cp | _237900 | IV | 486 | 0 | <0.01 | NDA | 39.9/38.8 | yes | NDA |
| ppi | _238000 | VI | 283 | 0 | <0.01 | −6.27 | 83.2/87.1 | yes | nucleolus |
| adp/atp | _237700 | VI | 61 | 0 | <0.01 | −0.76 | NDA | NDA | NDA |
| sa-t/k | _282230 | VII a | 634 | 0 | <0.01 | 0.94 | 10.2/6.1 | Oocyst ^ | NDA |
| electron-t | _207250 | X | 450 | 0 | <0.01 | 0.73 | 33.4/16.6 | Oocyst ^ | NDA |
| vtc2 | _298630 | XI | 1308 | 2 | <0.01 | −3.3 | 31.9/35.4 | yes | ER/Golgi/PM |
| ER-oxired | _300380 | XII | 587 | 1 | 0.02 | 0.82 | 63.7/74.8 | yes | ER |
| FamB and FamC multigene families: | |||||||||
| TgB1 | _200480 | VIII | 167 | 1 | 0.19/ + | 1.23 | NDA | NDA | NDA |
| TgB2 | _244920 | VI | 162 | 1 | 0.39 | −0.82 | NDA | NDA | NDA |
| TgB3 | _264225 | VIIb | 208 | 2 Φ | 0.72 | 2.03 | NDA | NDA | NDA |
| TgB4 | _274290 | IX | 136 | 1 | <0.01 | 0.9 | 91.8/87.7 | NDA | NDA |
| TgB5 | _276230 | III | 163 | 1 | 0.87/ + | 2.38 | 69.9/85.4 | NDA | NDA |
| TgB6 | _279460 | IX | 149 | 1 | 0.82/ + | 0.43 | NDA | NDA | NDA |
| TgB7 | _283450 | V | 265 | 1 | 0.35 | 1.72 | 67.0/25.3 | NDA | NDA |
| TgB8 | _295300 | Ia | 158 | 1 | 0.87/ + | 0.42 | 60.7/24.7 | NDA | NDA |
| TgB9 | _298090 | II | 172 | 1 | 0.91/ + | −0.24 | NDA | NDA | NDA |
| TgB10 | _299570 | IX | 150 | 1 | 0.85/ + | −1.12 | 50.9/31.0 | NDA | NDA |
| TgB11 | _301890 | X | 165 | 1 | 0.012 | NDA | 91.0/82/2 | NDA | NDA |
| TgB12 | _307045 | XI | 258 | 1 | 0.17/ + | 1.30 | 63.7/37.9 | NDA | NDA |
| TgB13 | _307460 | V | 212 | 1 | <0.01 | 1.16 | NDA | NDA | NDA |
| TgB14 | _307480 | V | 177 | 1 | <0.01 | 0.28 | NDA | NDA | NDA |
| TgB15 | _320770 | IV | 182 | 1 | 0.89/ + | −0.05 | NDA | NDA | NDA |
| HhB3 | _264225 | NA | 264 | 2 Φ | 0.064 | NDA | NDA | NDA | NDA |
| HhB6 | _279460 | NA | 152 | 1 | 0.84/ + | NDA | NDA | NDA | NDA |
| HhB7 | _283450 | NA | 126 | 1 | <0.01 | NDA | NDA | NDA | NDA |
| HhB8 | _295300 | NA | 158 | 1 | 0.92/ + | NDA | NDA | NDA | NDA |
| HhB9 | _298090 | NA | 137 | 0 | 0.92/ + | NDA | NDA | NDA | NDA |
| HhB10 | _299570 | NA | 107 | 1 | 0.91/ + | NDA | NDA | NDA | NDA |
| HhB16 | _454170 | NA | 183 | 1 | 0.79/ + | NDA | NDA | NDA | NDA |
| NcB13 | CEL64573 * | Ib a | 163 | 1 | 0.71 | NDA | NDA | NDA | NDA |
| NcB17 | _060290 | XII | 147 | 1 | <0.01 | NDA | NDA | NDA | NDA |
| NcB18 | CEL66112 | NDA | 199 | 1 | 0.30 | NDA | NDA | NDA | NDA |
| NcB19 | CEL64819 * | III b | 173 | 1 | <0.01 | NDA | NDA | NDA | NDA |
| TgC1 | _321170 | III | 232 | 2 Φ | 0.13 | 0.52 | 44.7/61.2 | NDA | NDA |
| TgC2 | _200700 | X | 139 | 1 Φ | 0.05 | NDA | NDA | NDA | NDA |
| TgC3 | _307260 | XI | 206 | 1 | 0.14 + | 0.96 | 75.2/64.8 | NDA | NDA |
| TgC4 | _200130 | XII | 238 | 1 | 0.04 + | NDA | NDA | NDA | NDA |
| TgC5 | _298960 | III | 282 | 1 Φ | <0.01 | 0.06 | 53.9/40.5 | NDA | NDA |
| TgC6 | _200590 | X | 201 | 1 | 0.01 + | 2.09 | NDA | NDA | NDA |
| TgC7 | _287280 | Ia | 225 | 1 | 0.06 + | NDA | 53.0/36.9 | NDA | NDA |
| TgC8 | _220080 | V | 306 | 1 | <0.01 | 0.44 | 23.7/22.4 | Membrane | NDA |
| TgC10 | _228780 | X | 286 | 2 | <0.01 | 1.49 | 15.2/23.9 | NDA | NDA |
| TgC11 | _298060 | II | 305 | 3 Φ | <0.01 | 1.22 | 14,5/32.6 | NDA | NDA |
| TgC12 | _300990 | IV | 252 | 1 | <0.01 | 0.04 | 18.4/16.0 | NDA | NDA |
| TgC13 | _292610 | V | 265 | 1 | 0.06 + | NDA | 53.0/36.9 | NDA | NDA |
| TgC14 | _329400 | NA | 225 | 1 | 0.06 + | NDA | 53.0/36.9 | NDA | NDA |
| HhC6a | _321170 | NA | 216 | 1 | 0.02 | NDA | NDA | NDA | NDA |
| HhC6b | _200590 | NA | 193 | 1 | 0.06 + | NDA | NDA | NDA | NDA |
| HhC8 | _220080 | NA | 180 | 1c | 0.004 | NDA | NDA | NDA | NDA |
| HhC11 | _298060 | NA | 330 | 1 | <0.01 | NDA | NDA | NDA | NDA |
| NcC15 | CEL68745 * | IX | 313 | 2 Φ | 0.01 | NDA | NDA | NDA | NDA |
| NcC16 | _069620 | NA | 242 | 1 | 0.03 | NDA | NDA | NDA | NDA |
| NcC17 | CEL66113 * | VIIa c | 146 | 1 | 0.005 | NDA | NDA | NDA | NDA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Contreras, S.M.; Zambrano Siri, R.T.; Rivera, E.M.; Cristaldi, C.; Kamenetzky, L.; Kim, K.; Clemente, M.; Ocampo, J.; Vanagas, L.; Angel, S.O. Architecture, Chromatin and Gene Organization of Toxoplasma gondii Subtelomeres. Epigenomes 2022, 6, 29. https://doi.org/10.3390/epigenomes6030029
Contreras SM, Zambrano Siri RT, Rivera EM, Cristaldi C, Kamenetzky L, Kim K, Clemente M, Ocampo J, Vanagas L, Angel SO. Architecture, Chromatin and Gene Organization of Toxoplasma gondii Subtelomeres. Epigenomes. 2022; 6(3):29. https://doi.org/10.3390/epigenomes6030029
Chicago/Turabian StyleContreras, Susana M., Romina T. Zambrano Siri, Elías M. Rivera, Constanza Cristaldi, Laura Kamenetzky, Kami Kim, Marina Clemente, Josefina Ocampo, Laura Vanagas, and Sergio O. Angel. 2022. "Architecture, Chromatin and Gene Organization of Toxoplasma gondii Subtelomeres" Epigenomes 6, no. 3: 29. https://doi.org/10.3390/epigenomes6030029
APA StyleContreras, S. M., Zambrano Siri, R. T., Rivera, E. M., Cristaldi, C., Kamenetzky, L., Kim, K., Clemente, M., Ocampo, J., Vanagas, L., & Angel, S. O. (2022). Architecture, Chromatin and Gene Organization of Toxoplasma gondii Subtelomeres. Epigenomes, 6(3), 29. https://doi.org/10.3390/epigenomes6030029