

Polycomb Directed Cell Fate Decisions in Development and Cancer

Abstract
1. Introduction
2. PcG Complexes
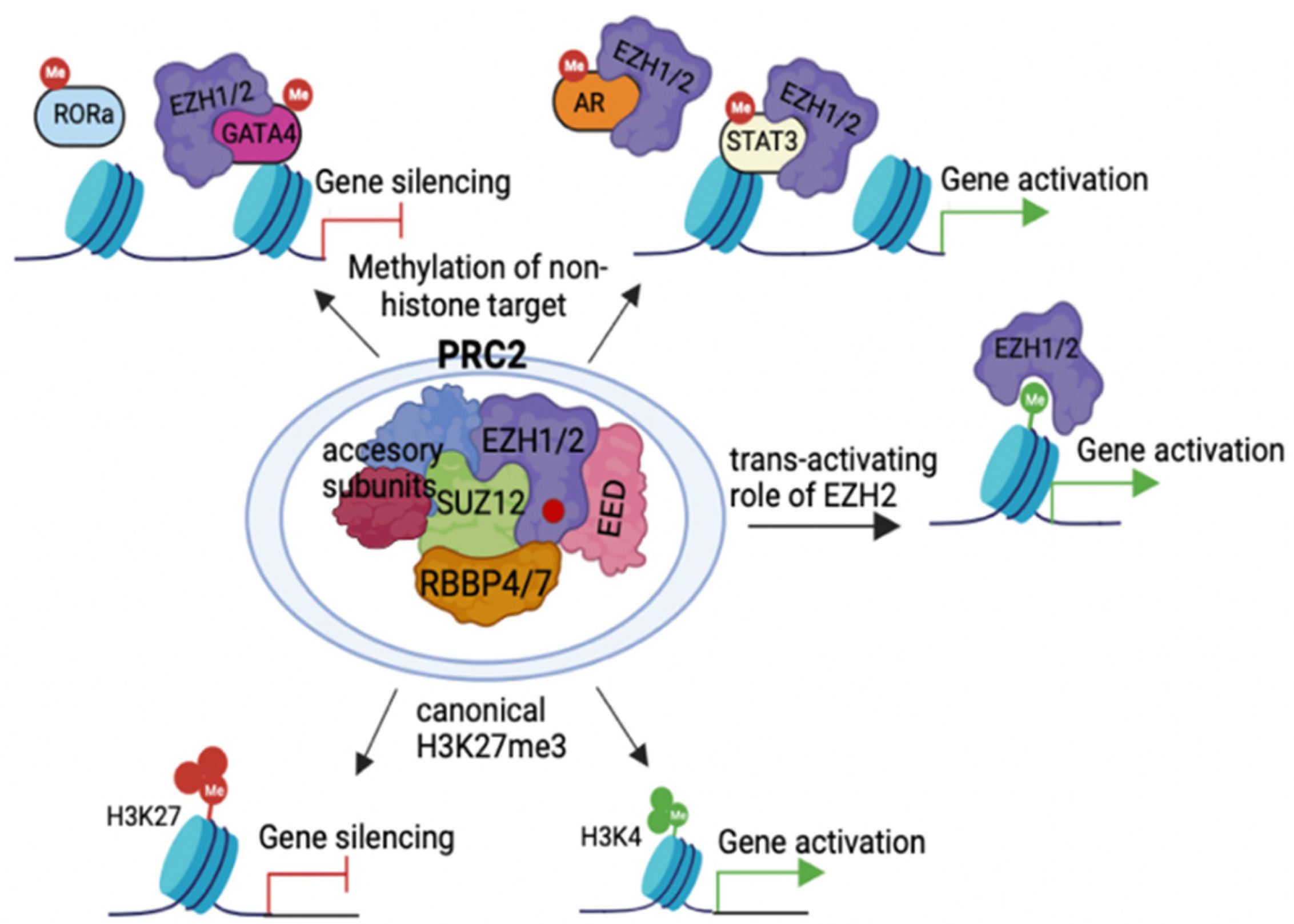
2.1. Action Modes of PRC2 Molecules
2.2. PRC2 Recruitment
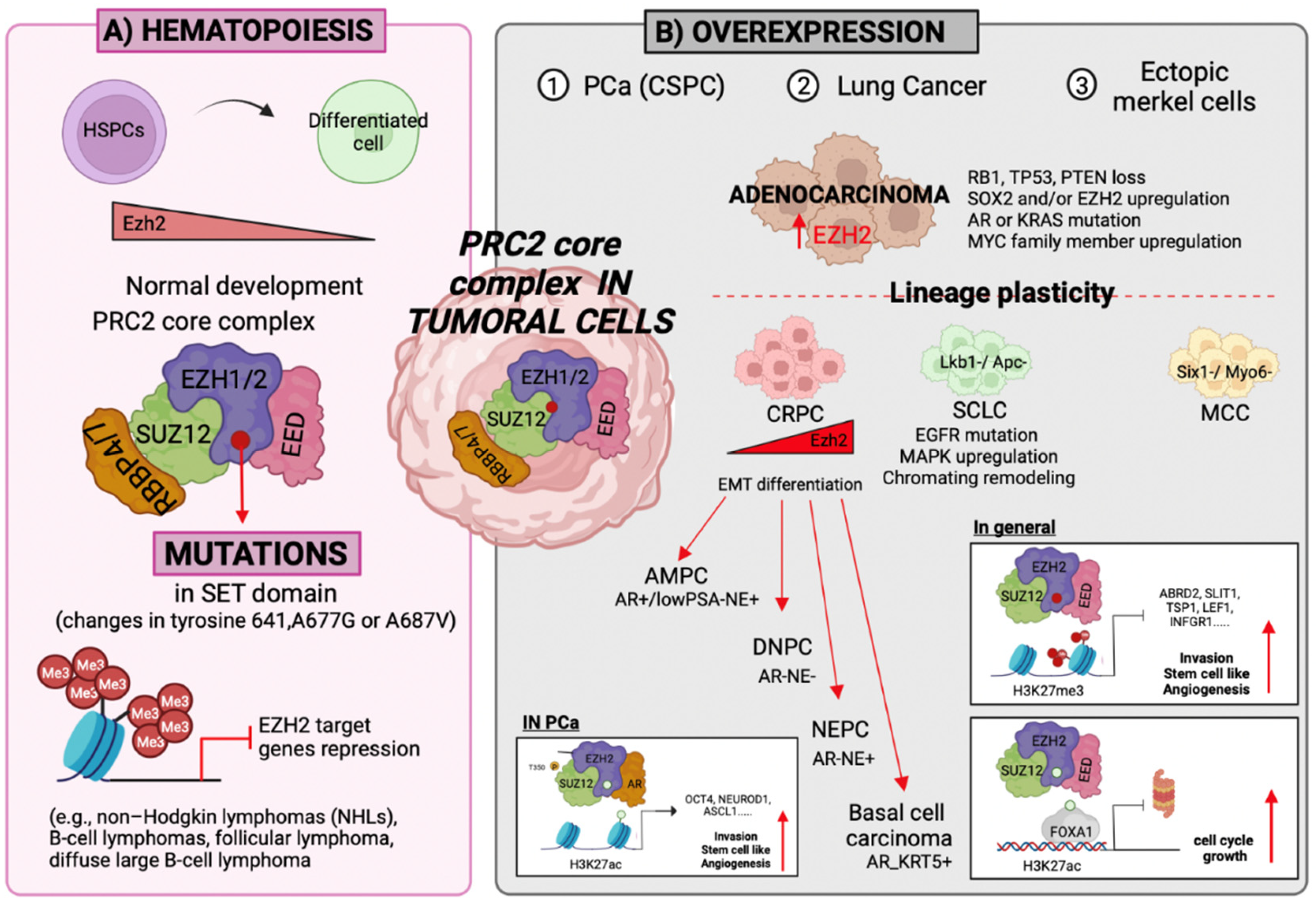
3. PcG Protein Functions and Cell Fate Determination
3.1. Self-Renewal
3.2. Differentiation
3.3. Cell Fate Determination
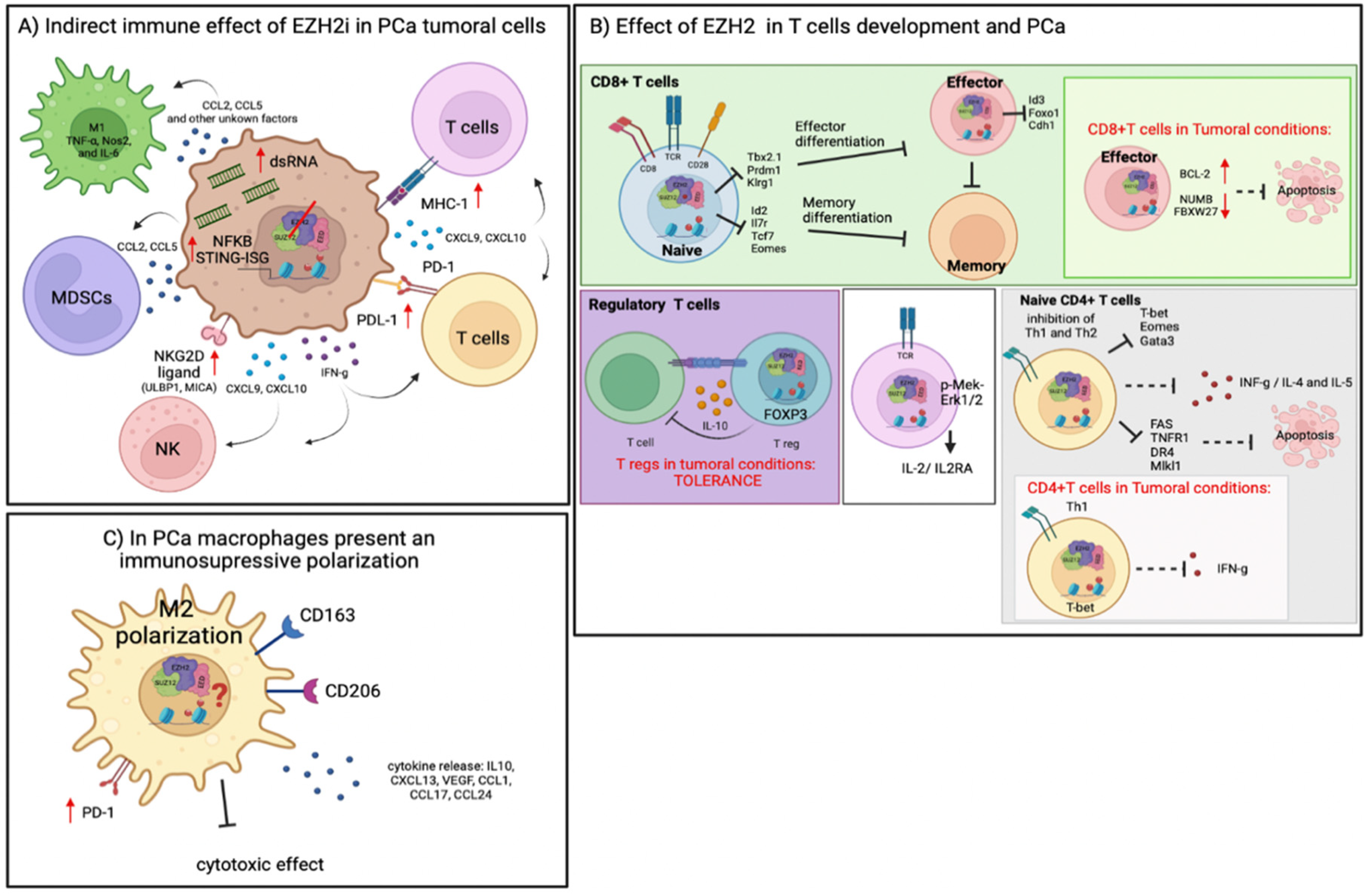
4. Immune Regulation by EZH2 in TME
4.1. Role of EZH2 in T-Cell Differentiation and Cancer
4.2. Role of EZH2 Function in NK Cells Differentiation and Cancer
4.3. Role of EZH2 Reprogramming Tumor Immunosuppressive Cells
5. Role of EZH2 in Tumoral Metabolism
6. PRC2 Therapeutical Options in Cancer
Clinical Trials Using PCR2 Therapy in Cancer
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gibney, E.R.; Nolan, C.M. Epigenetics and gene expression. Heredity 2010, 105, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Mendenhall, E.M.; Bernstein, B.E. Chromatin state maps: New technologies, new insights. Curr. Opin. Genet. Dev. 2008, 18, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Schuebel, K.E. The epigenomic era opens. Nature 2007, 448, 48–49. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chammas, P.; Mocavini, I.; Di Croce, L. Engaging chromatin: PRC2 structure meets function. Br. J. Cancer 2020, 122, 315–328. [Google Scholar] [CrossRef]
- Ehrlich, M.; Gama-Sosa, M.A.; Huang, L.-H.; Midgett, R.M.; Kuo, K.C.; McCune, R.A.; Gehrke, C. Amount and distribution of 5-methylcytosine in human DNA from different types of tissues or cells. Nucleic Acids Res. 1982, 10, 2709–2721. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Vogelstein, B. Hypomethylation distinguishes genes of some human cancers form their normal counterparts. Nature 1983, 301, 89–92. [Google Scholar] [CrossRef]
- Allfrey, V.G.; Mirsky, A.E. Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Biochemistry 1964, 51, 786–794. [Google Scholar] [CrossRef]
- Buschhausen, G.; Wittig, B.; Graessmann, M.; Graessmann, A. Chromatin structure is required to block transcription of the methylated herpers simplex virus thymidine kinase gene. Proc. Natl. Acad. Sci. USA 1987, 84, 1177–1181. [Google Scholar] [CrossRef]
- Keshet, I.; Lieman-Hurwitz, J.; Cedar, H. DNA methylation affects the formation of active chromatin. Cell 1986, 44, 535–543. [Google Scholar] [CrossRef]
- Steenman, M.J.C.; Rainier, S.; Dobry, C.J.; Grundy, P.; Horon, I.L.; Feinberg, A.P. Loss of imprinting of IGF2 is linked to reduced expression and abnormal methylation of H19 in Wilms’ tumour. Nature 1994, 7, 433–439. [Google Scholar] [CrossRef]
- Moulton, T.; Crenshaw, T.; Hao, Y.; Moosikasuwan, J.; Lin, N.; Dembitzer, F.; Hensle, T.; Weiss, T.; McMorrow, L.; Loew, T.; et al. Epigenetic lesions at the H19 locus in Wilms’ tumour patients. Nature 1994, 7, 440–447. [Google Scholar] [CrossRef]
- Moazed, D. Small RNAs in transcriptional gene silencing and genome defence. Nature 2009, 457, 413–420. [Google Scholar] [CrossRef]
- Bartolomei, M.S.; Semel, S.; Tilghman, S.M. Parental imprinting of the mouse H19 gene. Lett. Nat. 1991, 351, 153–155. [Google Scholar] [CrossRef] [PubMed]
- Ranasinghe, R.T.; Challand, M.R.; Ganzinger, K.A.; Lewis, B.W.; Softley, C.; Schmied, W.H.; Horrocks, M.H.; Shivji, N.; Chin, J.W.; Spencer, J.; et al. Detecting RNA base methylations in single cells by in situ hybridization. Nat. Commun. 2018, 9, 655. [Google Scholar] [CrossRef] [PubMed]
- Peterson, C.L.; Herskowitz, I. Characterizationo fthe yeast, SWI1, SWI2, and SWI3 genes, which encode a global activator of transcription. Cell 1992, 68, 573–583. [Google Scholar] [CrossRef]
- Aalfs, J.D.; Narlikar, G.J.; Kingston, R.E. Functional differences between the human ATP-dependent nucleosome remodeling proteins BRG1 and SNF2H. J. Biol. Chem. 2001, 276, 34270–34278. [Google Scholar] [CrossRef]
- Delmas, V.; Stokes, D.G.; Perry, R.P. A mammalian DNA-binding protein that contains a chromodomain and a SNF2/SWI2-like helicase domain. Proc. Natl. Acad. Sci. USA 1993, 90, 2414–2418. [Google Scholar] [CrossRef]
- Shen, X.; Mizuguchi, G.; Hamiche, A.; Wu, C. A chromatin remodelling complex involved in transcription and DNA processing. Lett. Nat. 2000, 406, 541–544. [Google Scholar] [CrossRef]
- Lewis, E.B. A gene complex controlling segmentation in Drosophila. Nature 1978, 276, 565–570. [Google Scholar] [CrossRef]
- Struhl, G. A gene product required for correct initiation of segmental determination in Drosophila. Nature 1981, 293, 36–41. [Google Scholar] [CrossRef]
- Kennison, J.A.; Tamkun, J.W. Dosage-dependent modifiers of Polycomb and Antennapedia mutations in Drosophila. Proc. Natl. Acad. Sci. USA 1988, 85, 8136–8140. [Google Scholar] [CrossRef]
- Schuettengruber, B.; Chourrout, D.; Vervoort, M.; Leblanc, B.; Cavalli, G. Genome regulation by polycomb and trithorax proteins. Cell 2007, 128, 735–745. [Google Scholar] [CrossRef]
- Kassis, J.A.; Brown, J.L. Polycomb group response elements in Drosophila and vertebrates. Adv. Genet. 2013, 81, 83–118. [Google Scholar] [CrossRef]
- Schwartz, Y.B.; Pirrotta, V. Polycomb silencing mechanisms and the management of genomic programmes. Nat. Rev. Genet. 2007, 8, 9–22. [Google Scholar] [CrossRef]
- Bracken, A.P.; Dietrich, N.; Pasini, D.; Hansen, K.H.; Helin, K. Genome-wide mapping of Polycomb target genes unravels their roles in cell fate transitions. Genes Dev. 2006, 20, 1123–1136. [Google Scholar] [CrossRef]
- Brumbaugh, J.; Di Stefano, B.; Sardina, J.L. Editorial: Chromatin Regulation in Cell Fate Decisions. Front. Cell. Dev. Biol. 2021, 9, 734020. [Google Scholar] [CrossRef]
- Dahle, O.; Kumar, A.; Kuehn, M.R. Nodal Signaling Recruits the Histone Demethylase Jmjd3 to counteract Polycomb-mediated repression at target genes. Sci. Signal. 2010, 3, 1–8. [Google Scholar] [CrossRef]
- Das, P.; Taube, J.H. Regulating Methylation at H3K27: A Trick or Treat for Cancer Cell Plasticity. Cancers 2020, 12, 2792. [Google Scholar] [CrossRef]
- Johann, P.D.; Erkek, S.; Zapatka, M.; Kerl, K.; Buchhalter, I.; Hovestadt, V.; Jones, D.T.W.; Sturm, D.; Hermann, C.; Segura Wang, M.; et al. Atypical Teratoid/Rhabdoid Tumors Are Comprised of Three Epigenetic Subgroups with Distinct Enhancer Landscapes. Cancer Cell. 2016, 29, 379–393. [Google Scholar] [CrossRef]
- Oakes, C.C.; Seifert, M.; Assenov, Y.; Gu, L.; Przekopowitz, M.; Ruppert, A.S.; Wang, Q.; Imbusch, C.D.; Serva, A.; Koser, S.D.; et al. DNA methylation dynamics during B cell maturation underlie a continuum of disease phenotypes in chronic lymphocytic leukemia. Nat. Genet. 2016, 48, 253–264. [Google Scholar] [CrossRef]
- Lewis, P.H. PC: Polycomb. Drosophila Infor. Serv. 1949, 21, 69. [Google Scholar]
- Margueron, R.; Reinberg, D. The Polycomb complex PRC2 and its mark in life. Nature 2011, 469, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Pasini, D.; Di Croce, L. Emerging roles for Polycomb proteins in cancer. Curr. Opin. Genet. Dev. 2016, 36, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Schuettengruber, B.; Bourbon, H.M.; Di Croce, L.; Cavalli, G. Genome Regulation by Polycomb and Trithorax: 70 Years and Counting. Cell 2017, 171, 34–57. [Google Scholar] [CrossRef]
- Di Croce, L.; Helin, K. Transcriptional regulation by Polycomb group proteins. Nat. Struct. Mol. Biol. 2013, 20, 1147–1155. [Google Scholar] [CrossRef]
- Entrevan, M.; Schuettengruber, B.; Cavalli, G. Regulation of Genome Architecture and Function by Polycomb Proteins. Trends Cell. Biol. 2016, 26, 511–525. [Google Scholar] [CrossRef]
- Ferrari, K.J.; Scelfo, A.; Jammula, S.; Cuomo, A.; Barozzi, I.; Stutzer, A.; Fischle, W.; Bonaldi, T.; Pasini, D. Polycomb-dependent H3K27me1 and H3K27me2 regulate active transcription and enhancer fidelity. Mol. Cell 2014, 53, 49–62. [Google Scholar] [CrossRef]
- Francis, N.J.; Kingston, R.E.; Woodcock, C.L. Chromatin compaction by a polycomb group protein complex. Science 2004, 306, 1574–1577. [Google Scholar] [CrossRef]
- Eskeland, R.; Leeb, M.; Grimes, G.R.; Kress, C.; Boyle, S.; Sproul, D.; Gilbert, N.; Fan, Y.; Skoultchi, A.I.; Wutz, A.; et al. Ring1B compacts chromatin structure and represses gene expression independent of histone ubiquitination. Mol. Cell. 2010, 38, 452–464. [Google Scholar] [CrossRef]
- Boettiger, A.N.; Bintu, B.; Moffitt, J.R.; Wang, S.; Beliveau, B.J.; Fudenberg, G.; Imakaev, M.; Mirny, L.A.; Wu, C.T.; Zhuang, X. Super-resolution imaging reveals distinct chromatin folding for different epigenetic states. Nature 2016, 529, 418–422. [Google Scholar] [CrossRef]
- Azuara, V.; Perry, P.; Sauer, S.; Spivakov, M.; Jørgensen, H.M.; John, R.M.; Gouti, M.; Casanova, M.; Warnes, G.; Merkenschlager, M.; et al. Chromatin signatures of pluripotent cell lines. Nature 2006, 8, 532–538. [Google Scholar] [CrossRef]
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006, 125, 315–326. [Google Scholar] [CrossRef]
- Voigt, P.; LeRoy, G.; Drury, W.J., 3rd; Zee, B.M.; Son, J.; Beck, D.B.; Young, N.L.; Garcia, B.A.; Reinberg, D. Asymmetrically modified nucleosomes. Cell 2012, 151, 181–193. [Google Scholar] [CrossRef]
- Kinkley, S.; Helmuth, J.; Polansky, J.K.; Dunkel, I.; Gasparoni, G.; Frohler, S.; Chen, W.; Walter, J.; Hamann, A.; Chung, H.R. reChIP-seq reveals widespread bivalency of H3K4me3 and H3K27me3 in CD4(+) memory T cells. Nat. Commun. 2016, 7, 12514. [Google Scholar] [CrossRef]
- Weiner, A.; Lara-Astiaso, D.; Krupalnik, V.; Gafni, O.; David, E.; Winter, D.R.; Hanna, J.H.; Amit, I. Co-ChIP enables genome-wide mapping of histone mark co-occurrence at single-molecule resolution. Nat. Biotechnol. 2016, 34, 953–961. [Google Scholar] [CrossRef]
- Kar, G.; Kim, J.K.; Kolodziejczyk, A.A.; Natarajan, K.N.; Torlai Triglia, E.; Mifsud, B.; Elderkin, S.; Marioni, J.C.; Pombo, A.; Teichmann, S.A. Flipping between Polycomb repressed and active transcriptional states introduces noise in gene expression. Nat. Commun. 2017, 8, 36. [Google Scholar] [CrossRef]
- Brookes, E.; de Santiago, I.; Hebenstreit, D.; Morris, K.J.; Carroll, T.; Xie, S.Q.; Stock, J.K.; Heidemann, M.; Eick, D.; Nozaki, N.; et al. Polycomb associates genome-wide with a specific RNA polymerase II variant, and regulates metabolic genes in ESCs. Cell Stem. Cell 2012, 10, 157–170. [Google Scholar] [CrossRef]
- Shi, Y.; Wang, X.X.; Zhuang, Y.W.; Jiang, Y.; Melcher, K.; Xu, H.E. Structure of the PRC2 complex and application to drug discovery. Acta Pharmacol. Sin. 2017, 38, 963–976. [Google Scholar] [CrossRef]
- Li, Z.; Cao, R.; Wang, M.; Myers, M.P.; Zhang, Y.; Xu, R.M. Structure of a Bmi-1-Ring1B polycomb group ubiquitin ligase complex. J. Biol. Chem. 2006, 281, 20643–20649. [Google Scholar] [CrossRef]
- Boccuni, P.; MacGrogan, D.; Scandura, J.M.; Nimer, S.D. The human L(3)MBT polycomb group protein is a transcriptional repressor and interacts physically and functionally with TEL (ETV6). J. Biol. Chem. 2003, 278, 15412–15420. [Google Scholar] [CrossRef]
- Cao, R.; Zhang, Y. SUZ12 is required for both the histone methyltransferase activity and the silencing function of the EED-EZH2 complex. Mol. Cell. 2004, 15, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Denisenko, O.; Shnyreva, M.; Suzuki, H.; Bomsztyk, K. Point mutations in the WD40 d0main of Eed block its interaction with Ezh2. Mol. Cell. Biol. 1998, 18, 5634–5642. [Google Scholar] [CrossRef] [PubMed]
- Ragazzini, R.; Perez-Palacios, R.; Baymaz, I.H.; Diop, S.; Ancelin, K.; Zielinski, D.; Michaud, A.; Givelet, M.; Borsos, M.; Aflaki, S.; et al. EZHIP constrains Polycomb Repressive Complex 2 activity in germ cells. Nat. Commun. 2019, 10, 3858. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.J.; Moorehead, R.A. Polycomb repressor complex 2 function in breast cancer (Review). Int. J. Oncol. 2020, 57, 1085–1094. [Google Scholar] [CrossRef]
- Copeland, R.A.; Solomon, M.E.; Richon, V.M. Protein methyltransferases as a target class for drug discovery. Nat. Rev. Drug Discov. 2009, 8, 724–732. [Google Scholar] [CrossRef]
- Margueron, R.; Li, G.; Sarma, K.; Blais, A.; Zavadil, J.; Woodcock, C.L.; Dynlacht, B.D.; Reinberg, D. Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol. Cell. 2008, 32, 503–518. [Google Scholar] [CrossRef]
- Wan, L.; Xu, K.; Wei, Y.; Zhang, J.; Han, T.; Fry, C.; Zhang, Z.; Wang, Y.V.; Huang, L.; Yuan, M.; et al. Phosphorylation of EZH2 by AMPK Suppresses PRC2 Methyltransferase Activity and Oncogenic Function. Mol. Cell. 2018, 69, 279–291.e275. [Google Scholar] [CrossRef]
- Chen, S.; Bohrer, L.R.; Rai, A.N.; Pan, Y.; Gan, L.; Zhou, X.; Bagchi, A.; Simon, J.A.; Huang, H. Cyclin-dependent kinases regulate epigenetic gene silencing through phosphorylation of EZH2. Nat. Cell. Biol. 2010, 12, 1108–1114. [Google Scholar] [CrossRef]
- Chen, D.L.; Ju, H.Q.; Lu, Y.X.; Chen, L.Z.; Zeng, Z.L.; Zhang, D.S.; Luo, H.Y.; Wang, F.; Qiu, M.Z.; Wang, D.S.; et al. Long non-coding RNA XIST regulates gastric cancer progression by acting as a molecular sponge of miR-101 to modulate EZH2 expression. J. Exp. Clin. Cancer Res. 2016, 35, 142. [Google Scholar] [CrossRef]
- Wu, L.; Murat, P.; Matak-Vinkovic, D.; Murrell, A.; Balasubramanian, S. Binding interactions between long noncoding RNA HOTAIR and PRC2 proteins. Biochemistry 2013, 52, 9519–9527. [Google Scholar] [CrossRef]
- Yang, C.-C.; LaBaff, A.; Wei, Y.; Nie, L.; Xia, W.; Huo, L.; Yamaguchi, H.; Hsu, Y.-H.; Hsu, J.L.; Liu, D.; et al. Phosphorylation of EZH2 at T416 by CDK2 contributes to the malignancy of triple negative breast cancer. Am. J. Transl. Res. 2015, 7, 1009–1020. [Google Scholar]
- Han, Z.; Xing, X.; Hu, M.; Zhang, Y.; Liu, P.; Chai, J. Structural basis of EZH2 recognition by EED. Structure 2007, 15, 1306–1315. [Google Scholar] [CrossRef][Green Version]
- Pasini, D.; Bracken, A.P.; Hansen, J.B.; Capillo, M.; Helin, K. The polycomb group protein Suz12 is required for embryonic stem cell differentiation. Mol. Cell. Biol. 2007, 27, 3769–3779. [Google Scholar] [CrossRef]
- Chen, S.; Ma, J.; Wu, F.; Xiong, L.J.; Ma, H.; Xu, W.; Lv, R.; Li, X.; Villen, J.; Gygi, S.P.; et al. The histone H3 Lys 27 demethylase JMJD3 regulates gene expression by impacting transcriptional elongation. Genes Dev. 2012, 26, 1364–1375. [Google Scholar] [CrossRef] [PubMed]
- Bracken, A.P.; Helin, K. Polycomb group proteins: Navigators of lineage pathways led astray in cancer. Nat. Rev. Cancer 2009, 9, 773–784. [Google Scholar] [CrossRef]
- He, A.; Shen, X.; Ma, Q.; Cao, J.; von Gise, A.; Zhou, P.; Wang, G.; Marquez, V.E.; Orkin, S.H.; Pu, W.T. PRC2 directly methylates GATA4 and represses its transcriptional activity. Genes Dev. 2012, 26, 37–42. [Google Scholar] [CrossRef]
- Lee, J.M.; Lee, J.S.; Kim, H.; Kim, K.; Park, H.; Kim, J.Y.; Lee, S.H.; Kim, I.S.; Kim, J.; Lee, M.; et al. EZH2 generates a methyl degron that is recognized by the DCAF1/DDB1/CUL4 E3 ubiquitin ligase complex. Mol. Cell 2012, 48, 572–586. [Google Scholar] [CrossRef]
- Gunawan, M.; Venkatesan, N.; Loh, J.T.; Wong, J.F.; Berger, H.; Neo, W.H.; Li, L.Y.; La Win, M.K.; Yau, Y.H.; Guo, T.; et al. The methyltransferase Ezh2 controls cell adhesion and migration through direct methylation of the extranuclear regulatory protein talin. Nat. Immunol. 2015, 16, 505–516. [Google Scholar] [CrossRef]
- Kim, E.; Kim, M.; Woo, D.H.; Shin, Y.; Shin, J.; Chang, N.; Oh, Y.T.; Kim, H.; Rheey, J.; Nakano, I.; et al. Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell 2013, 23, 839–852. [Google Scholar] [CrossRef]
- Xu, K.; Wu, Z.J.; Groner, A.C.; He, H.H.; Cai, C.; Lis, R.T.; Wu, X.; Stack, E.C.; Loda, M.; Liu, T.; et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science 2012, 338, 1465–1469. [Google Scholar] [CrossRef]
- Kim, J.; Lee, Y.; Lu, X.; Song, B.; Fong, K.W.; Cao, Q.; Licht, J.D.; Zhao, J.C.; Yu, J. Polycomb- and Methylation-Independent Roles of EZH2 as a Transcription Activator. Cell Rep. 2018, 25, 2808–2820.e4. [Google Scholar] [CrossRef] [PubMed]
- Parreno, V.; Martinez, A.M.; Cavalli, G. Mechanisms of Polycomb group protein function in cancer. Cell Res. 2022, 32, 231–253. [Google Scholar] [CrossRef] [PubMed]
- Bieluszewski, T.; Xiao, J.; Yang, Y.; Wagner, D. PRC2 activity, recruitment, and silencing: A comparative perspective. Trends Plant Sci. 2021, 26, 1186–1198. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liefke, R.; Jiang, J.; Kurland, J.V.; Tian, W.; Deng, P.; Zhang, W.; He, Q.; Patel, D.J.; Bulyk, M.L.; et al. Polycomb-like proteins link the PRC2 complex to CpG islands. Nature 2017, 549, 287–291. [Google Scholar] [CrossRef]
- Perino, M.; van Mierlo, G.; Karemaker, I.D.; van Genesen, S.; Vermeulen, M.; Marks, H.; van Heeringen, S.J.; Veenstra, G.J.C. MTF2 recruits Polycomb Repressive Complex 2 by helical-shape-selective DNA binding. Nat. Genet. 2018, 50, 1002–1010. [Google Scholar] [CrossRef]
- Cooper, S.; Grijzenhout, A.; Underwood, E.; Ancelin, K.; Zhang, T.; Nesterova, T.B.; Anil-Kirmizitas, B.; Bassett, A.; Kooistra, S.M.; Agger, K.; et al. Jarid2 binds mono-ubiquitylated H2A lysine 119 to mediate crosstalk between Polycomb complexes PRC1 and PRC2. Nat. Commun. 2016, 7, 13661. [Google Scholar] [CrossRef]
- Kalb, R.; Latwiel, S.; Baymaz, H.I.; Jansen, P.W.; Muller, C.W.; Vermeulen, M.; Muller, J. Histone H2A monoubiquitination promotes histone H3 methylation in Polycomb repression. Nat. Struct. Mol. Biol. 2014, 21, 569–571. [Google Scholar] [CrossRef]
- Ringrose, L.; Rehmsmeier, M.; Dura, J.-M.; Paro, R. Genome-Wide prediction of Polycomb/Trithorax response elements in Drosophila melanogaster. Develpmental Cell 2003, 5, 759–771. [Google Scholar] [CrossRef]
- Simon, J.A.; Kingston, R.E. Mechanisms of polycomb gene silencing: Knowns and unknowns. Nat. Rev. Mol. Cell. Biol. 2009, 10, 697–708. [Google Scholar] [CrossRef]
- Riising, E.M.; Comet, I.; Leblanc, B.; Wu, X.; Johansen, J.V.; Helin, K. Gene silencing triggers polycomb repressive complex 2 recruitment to CpG islands genome wide. Mol. Cell 2014, 55, 347–360. [Google Scholar] [CrossRef]
- Laugesen, A.; Hojfeldt, J.W.; Helin, K. Molecular Mechanisms Directing PRC2 Recruitment and H3K27 Methylation. Mol. Cell 2019, 74, 8–18. [Google Scholar] [CrossRef]
- Ku, M.; Koche, R.P.; Rheinbay, E.; Mendenhall, E.M.; Endoh, M.; Mikkelsen, T.S.; Presser, A.; Nusbaum, C.; Xie, X.; Chi, A.S.; et al. Genomewide analysis of PRC1 and PRC2 occupancy identifies two classes of bivalent domains. PLoS Genet. 2008, 4, e1000242. [Google Scholar] [CrossRef]
- Mendenhall, E.M.; Koche, R.P.; Truong, T.; Zhou, V.W.; Issac, B.; Chi, A.S.; Ku, M.; Bernstein, B.E. GC-rich sequence elements recruit PRC2 in mammalian ES cells. PLoS Genet. 2010, 6, e1001244. [Google Scholar] [CrossRef]
- Wu, H.; D’Alessio, A.C.; Ito, S.; Xia, K.; Wang, Z.; Cui, K.; Zhao, K.; Sun, Y.E.; Zhang, Y. Dual functions of Tet1 in transcriptional regulation in mouse embryonic stem cells. Nature 2011, 473, 389–393. [Google Scholar] [CrossRef]
- Hojfeldt, J.W.; Hedehus, L.; Laugesen, A.; Tatar, T.; Wiehle, L.; Helin, K. Non-core Subunits of the PRC2 Complex Are Collectively Required for Its Target-Site Specificity. Mol. Cell 2019, 76, 423–436.e3. [Google Scholar] [CrossRef]
- Farcas, A.M.; Blackledge, N.P.; Sudbery, I.; Long, H.K.; McGouran, J.F.; Rose, N.R.; Lee, S.; Sims, D.; Cerase, A.; Sheahan, T.W.; et al. KDM2B links the Polycomb Repressive Complex 1 (PRC1) to recognition of CpG islands. eLife 2012, 1, e00205. [Google Scholar] [CrossRef]
- Singh, N.; Ramnarine, V.R.; Song, J.H.; Pandey, R.; Padi, S.K.R.; Nouri, M.; Olive, V.; Kobelev, M.; Okumura, K.; McCarthy, D.; et al. The long noncoding RNA H19 regulates tumor plasticity in neuroendocrine prostate cancer. Nat. Commun. 2021, 12, 7349. [Google Scholar] [CrossRef]
- Morey, L.; Helin, K. Polycomb group protein-mediated repression of transcription. Trends Biochem. Sci. 2010, 35, 323–332. [Google Scholar] [CrossRef]
- Yuan, S.; Norgard, R.J.; Stanger, B.Z. Cellular Plasticity in Cancer. Cancer Discov. 2019, 9, 837–851. [Google Scholar] [CrossRef]
- O’Carroll, D.; Erhardt, S.; Pagani, M.; Barton, S.C.; Surani, M.A.; Jenuwein, T. The polycomb-group gene Ezh2 is required for early mouse development. Mol. Cell. Biol. 2001, 21, 4330–4336. [Google Scholar] [CrossRef]
- Pasini, D.; Bracken, A.P.; Jensen, M.R.; Lazzerini Denchi, E.; Helin, K. Suz12 is essential for mouse development and for EZH2 histone methyltransferase activity. EMBO J. 2004, 23, 4061–4071. [Google Scholar] [CrossRef] [PubMed]
- Faust, C.; Schumacher, A.; Holdener, B.; Magnuson, T. The eed mutation disrupts anterior mesoderm production in mice. Development 1995, 121, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Kleer, C.G.; Cao, Q.; Varambally, S.; Shen, R.; Ota, I.; Tomlins, S.A.; Ghosh, D.; Sewalt, R.G.; Otte, A.P.; Hayes, D.F.; et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc. Natl. Acad. Sci. USA 2003, 100, 11606–11611. [Google Scholar] [CrossRef] [PubMed]
- Endoh, M.; Endo, T.A.; Endoh, T.; Fujimura, Y.; Ohara, O.; Toyoda, T.; Otte, A.P.; Okano, M.; Brockdorff, N.; Vidal, M.; et al. Polycomb group proteins Ring1A/B are functionally linked to the core transcriptional regulatory circuitry to maintain ES cell identity. Development 2008, 135, 1513–1524. [Google Scholar] [CrossRef]
- Chamberlain, S.J.; Yee, D.; Magnuson, T. Polycomb repressive complex 2 is dispensable for maintenance of embryonic stem cell pluripotency. Stem Cells 2008, 26, 1496–1505. [Google Scholar] [CrossRef]
- Leeb, M.; Wutz, A. Ring1B is crucial for the regulation of developmental control genes and PRC1 proteins but not X inactivation in embryonic cells. J. Cell. Biol. 2007, 178, 219–229. [Google Scholar] [CrossRef]
- Ezhkova, E.; Lien, W.H.; Stokes, N.; Pasolli, H.A.; Silva, J.M.; Fuchs, E. EZH1 and EZH2 cogovern histone H3K27 trimethylation and are essential for hair follicle homeostasis and wound repair. Genes Dev. 2011, 25, 485–498. [Google Scholar] [CrossRef]
- Shen, X.; Liu, Y.; Hsu, Y.J.; Fujiwara, Y.; Kim, J.; Mao, X.; Yuan, G.C.; Orkin, S.H. EZH1 mediates methylation on histone H3 lysine 27 and complements EZH2 in maintaining stem cell identity and executing pluripotency. Mol. Cell 2008, 32, 491–502. [Google Scholar] [CrossRef]
- Kloet, S.L.; Makowski, M.M.; Baymaz, H.I.; van Voorthuijsen, L.; Karemaker, I.D.; Santanach, A.; Jansen, P.; Di Croce, L.; Vermeulen, M. The dynamic interactome and genomic targets of Polycomb complexes during stem-cell differentiation. Nat. Struct. Mol. Biol. 2016, 23, 682–690. [Google Scholar] [CrossRef]
- Hauri, S.; Comoglio, F.; Seimiya, M.; Gerstung, M.; Glatter, T.; Hansen, K.; Aebersold, R.; Paro, R.; Gstaiger, M.; Beisel, C. A High-Density Map for Navigating the Human Polycomb Complexome. Cell. Rep. 2016, 17, 583–595. [Google Scholar] [CrossRef]
- Pomerantz, M.M.; Qiu, X.; Zhu, Y.; Takeda, D.Y.; Pan, W.; Baca, S.C.; Gusev, A.; Korthauer, K.D.; Severson, T.M.; Ha, G.; et al. Prostate cancer reactivates developmental epigenomic programs during metastatic progression. Nat. Genet. 2020, 52, 790–799. [Google Scholar] [CrossRef]
- Su, I.H.; Basavaraj, A.; Krutchinsky, A.N.; Hobert, O.; Ullrich, A.; Chait, B.T.; Tarakhovsky, A. Ezh2 controls B cell development through histone H3 methylation and Igh rearrangement. Nat. Immunol. 2003, 4, 124–131. [Google Scholar] [CrossRef]
- Beguelin, W.; Popovic, R.; Teater, M.; Jiang, Y.; Bunting, K.L.; Rosen, M.; Shen, H.; Yang, S.N.; Wang, L.; Ezponda, T.; et al. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell 2013, 23, 677–692. [Google Scholar] [CrossRef]
- Beguelin, W.; Teater, M.; Gearhart, M.D.; Calvo Fernandez, M.T.; Goldstein, R.L.; Cardenas, M.G.; Hatzi, K.; Rosen, M.; Shen, H.; Corcoran, C.M.; et al. EZH2 and BCL6 Cooperate to Assemble CBX8-BCOR Complex to Repress Bivalent Promoters, Mediate Germinal Center Formation and Lymphomagenesis. Cancer Cell 2016, 30, 197–213. [Google Scholar] [CrossRef]
- Caganova, M.; Carrisi, C.; Varano, G.; Mainoldi, F.; Zanardi, F.; Germain, P.L.; George, L.; Alberghini, F.; Ferrarini, L.; Talukder, A.K.; et al. Germinal center dysregulation by histone methyltransferase EZH2 promotes lymphomagenesis. J. Clin. Investig. 2013, 123, 5009–5022. [Google Scholar] [CrossRef]
- Morin, R.D.; Johnson, N.A.; Severson, T.M.; Mungall, A.J.; An, J.; Goya, R.; Paul, J.E.; Boyle, M.; Woolcock, B.W.; Kuchenbauer, F.; et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat. Genet. 2010, 42, 181–185. [Google Scholar] [CrossRef]
- Okosun, J.; Bodor, C.; Wang, J.; Araf, S.; Yang, C.Y.; Pan, C.; Boller, S.; Cittaro, D.; Bozek, M.; Iqbal, S.; et al. Integrated genomic analysis identifies recurrent mutations and evolution patterns driving the initiation and progression of follicular lymphoma. Nat. Genet. 2014, 46, 176–181. [Google Scholar] [CrossRef]
- Sneeringer, C.J.; Scott, M.P.; Kuntz, K.W.; Knutson, S.K.; Pollock, R.M.; Richon, V.M.; Copeland, R.A. Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B-cell lymphomas. Proc. Natl. Acad. Sci. USA 2010, 107, 20980–20985. [Google Scholar] [CrossRef]
- McCabe, M.T.; Ott, H.M.; Ganji, G.; Korenchuk, S.; Thompson, C.; Van Aller, G.S.; Liu, Y.; Graves, A.P.; Della Pietra, A., 3rd; Diaz, E.; et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 2012, 492, 108–112. [Google Scholar] [CrossRef]
- Hatzi, K.; Melnick, A. Breaking bad in the germinal center: How deregulation of BCL6 contributes to lymphomagenesis. Trends Mol. Med. 2014, 20, 343–352. [Google Scholar] [CrossRef]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.Y.; Jones, D.T.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.A.; Tonjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Hou, Y.; Yi, X.; Sun, S.; Guo, J.; He, X.; Li, T.; Cai, J.; Wang, Z. EZH2 activates CHK1 signaling to promote ovarian cancer chemoresistance by maintaining the properties of cancer stem cells. Theranostics 2021, 11, 1795–1813. [Google Scholar] [CrossRef] [PubMed]
- Comet, I.; Riising, E.M.; Leblanc, B.; Helin, K. Maintaining cell identity: PRC2-mediated regulation of transcription and cancer. Nat. Rev. Cancer 2016, 16, 803–810. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Roberts, C.W. Targeting EZH2 in cancer. Nat. Med. 2016, 22, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Souroullas, G.P.; Jeck, W.R.; Parker, J.S.; Simon, J.M.; Liu, J.Y.; Paulk, J.; Xiong, J.; Clark, K.S.; Fedoriw, Y.; Qi, J.; et al. An oncogenic Ezh2 mutation induces tumors through global redistribution of histone 3 lysine 27 trimethylation. Nat. Med. 2016, 22, 632–640. [Google Scholar] [CrossRef]
- Kuzmichev, A.; Margueron, R.; Vaquero, A.; Preissner, T.S.; Scher, M.; Kirmizis, A.; Ouyang, X.; Brockdorff, N.; Abate-Shen, C.; Farnham, P.; et al. Composition and histone substrates of polycomb repressive group complexes change during cellular differentiation. Proc. Natl. Acad. Sci. USA 2005, 102, 1859–1864. [Google Scholar] [CrossRef]
- Cebria, F.; Kobayashi, C.; Umesono, Y.; Nakazawa, M.; Mineta, K.; Ikeo, K.; Gojobori, T.; Itoh, M.; Taira, M.; Sanchez Alvarado, A.; et al. FGFR-related gene nou-darake restricts brain tissues to the head region of planarians. Nature 2002, 419, 620–624. [Google Scholar] [CrossRef]
- Zhao, J.C.; Yu, J.; Runkle, C.; Wu, L.; Hu, M.; Wu, D.; Liu, J.S.; Wang, Q.; Qin, Z.S.; Yu, J. Cooperation between Polycomb and androgen receptor during oncogenic transformation. Genome Res. 2012, 22, 322–331. [Google Scholar] [CrossRef]
- Kadoch, C.; Hargreaves, D.C.; Hodges, C.; Elias, L.; Ho, L.; Ranish, J.; Crabtree, G.R. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat. Genet. 2013, 45, 592–601. [Google Scholar] [CrossRef]
- Kim, K.H.; Kim, W.; Howard, T.P.; Vazquez, F.; Tsherniak, A.; Wu, J.N.; Wang, W.; Haswell, J.R.; Walensky, L.D.; Hahn, W.C.; et al. SWI/SNF-mutant cancers depend on catalytic and non-catalytic activity of EZH2. Nat. Med. 2015, 21, 1491–1496. [Google Scholar] [CrossRef]
- Van der Vaart, A.; Godfrey, M.; Portegijs, V.; Van Den Heuvel, S. Dose-dependent functions of SWI/SN BAF in permitimg and inhibiting cell proliferation in vivo. Sci. Adv. 2020, 6, 1–14. [Google Scholar] [CrossRef]
- Wilson, B.G.; Wang, X.; Shen, X.; McKenna, E.S.; Lemieux, M.E.; Cho, Y.J.; Koellhoffer, E.C.; Pomeroy, S.L.; Orkin, S.H.; Roberts, C.W. Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell 2010, 18, 316–328. [Google Scholar] [CrossRef]
- Hu, X.; Liu, R.; Hou, J.; Peng, W.; Wan, S.; Xu, M.; Li, Y.; Zhang, G.; Zhai, X.; Liang, P.; et al. SMARCE1 promotes neuroblastoma tumorigenesis through assisting MYCN-mediated transcriptional activation. Oncogene 2022, 1–12. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Aloia, L.; Di Stefano, B.; Di Croce, L. Polycomb complexes in stem cells and embryonic development. Development 2013, 140, 2525–2534. [Google Scholar] [CrossRef]
- Cohen, I.; Zhao, D.; Bar, C.; Valdes, V.J.; Dauber-Decker, K.L.; Nguyen, M.B.; Nakayama, M.; Rendl, M.; Bickmore, W.A.; Koseki, H.; et al. PRC1 Fine-tunes Gene Repression and Activation to Safeguard Skin Development and Stem Cell Specification. Cell Stem. Cell 2018, 22, 726–739.e7. [Google Scholar] [CrossRef]
- Koppens, M.; van Lohuizen, M. Context-dependent actions of Polycomb repressors in cancer. Oncogene 2016, 35, 1341–1352. [Google Scholar] [CrossRef]
- Serresi, M.; Gargiulo, G.; Proost, N.; Siteur, B.; Cesaroni, M.; Koppens, M.; Xie, H.; Sutherland, K.D.; Hulsman, D.; Citterio, E.; et al. Polycomb Repressive Complex 2 Is a Barrier to KRAS-Driven Inflammation and Epithelial-Mesenchymal Transition in Non-Small-Cell Lung Cancer. Cancer Cell 2016, 29, 17–31. [Google Scholar] [CrossRef]
- Jacobs, J.J.L.; Kieboom, K.; Marino, S.; DePinho, R.A.; Van Lohuizen, M. The oncogene and Polycomb-group gene bmi-1 regulated cell proliferation and senescence through the ink4a locus. Nature 1999, 397, 164–168. [Google Scholar] [CrossRef]
- Bracken, A.P.; Kleine-Kohlbrecher, D.; Dietrich, N.; Pasini, D.; Gargiulo, G.; Beekman, C.; Theilgaard-Monch, K.; Minucci, S.; Porse, B.T.; Marine, J.C.; et al. The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes Dev. 2007, 21, 525–530. [Google Scholar] [CrossRef]
- Jones, P.A.; Issa, J.P.; Baylin, S. Targeting the cancer epigenome for therapy. Nat. Rev. Genet. 2016, 17, 630–641. [Google Scholar] [CrossRef] [PubMed]
- Scelfo, A.; Piunti, A.; Pasini, D. The controversial role of the Polycomb group proteins in transcription and cancer: How much do we not understand Polycomb proteins? FEBS J. 2015, 282, 1703–1722. [Google Scholar] [CrossRef] [PubMed]
- Gazdar, A.F.; Bunn, P.A.; Minna, J.D. Small-cell lung cancer: What we know, what we need to know and the path forward. Nat. Rev. Cancer 2017, 17, 725–737. [Google Scholar] [CrossRef] [PubMed]
- Nadal, R.; Schweizer, M.; Kryvenko, O.N.; Epstein, J.I.; Eisenberger, M.A. Small cell carcinoma of the prostate. Nat. Rev. Urol. 2014, 11, 213–219. [Google Scholar] [CrossRef]
- Zhang, H.; Fillmore Brainson, C.; Koyama, S.; Redig, A.J.; Chen, T.; Li, S.; Gupta, M.; Garcia-de-Alba, C.; Paschini, M.; Herter-Sprie, G.S.; et al. Lkb1 inactivation drives lung cancer lineage switching governed by Polycomb Repressive Complex 2. Nat. Commun. 2017, 8, 14922. [Google Scholar] [CrossRef]
- Yang, D.; Jones, M.G.; Naranjo, S.; Rideout, W.M., 3rd; Min, K.H.J.; Ho, R.; Wu, W.; Replogle, J.M.; Page, J.L.; Quinn, J.J.; et al. Lineage tracing reveals the phylodynamics, plasticity, and paths of tumor evolution. Cell 2022, 185, 1905–1923.e25. [Google Scholar] [CrossRef]
- Mukhopadhyay, A.; Berrett, K.C.; Kc, U.; Clair, P.M.; Pop, S.M.; Carr, S.R.; Witt, B.L.; Oliver, T.G. Sox2 cooperates with Lkb1 loss in a mouse model of squamous cell lung cancer. Cell Rep. 2014, 8, 40–49. [Google Scholar] [CrossRef]
- Ferone, G.; Song, J.Y.; Sutherland, K.D.; Bhaskaran, R.; Monkhorst, K.; Lambooij, J.P.; Proost, N.; Gargiulo, G.; Berns, A. SOX2 Is the Determining Oncogenic Switch in Promoting Lung Squamous Cell Carcinoma from Different Cells of Origin. Cancer Cell. 2016, 30, 519–532. [Google Scholar] [CrossRef]
- Harms, K.L.; Chubb, H.; Zhao, L.; Fullen, D.R.; Bichakjian, C.K.; Johnson, T.M.; Carskadon, S.; Palanisamy, N.; Harms, P.W. Increased expression of EZH2 in Merkel cell carcinoma is associated with disease progression and poorer prognosis. Hum. Pathol. 2017, 67, 78–84. [Google Scholar] [CrossRef]
- Gartin, A.K.; Frost, T.C.; Cushman, C.H.; Leeper, B.A.; Gokhale, P.C.; DeCaprio, J.A. Merkel Cell Carcinoma Sensitivity to EZH2 Inhibition Is Mediated by SIX1 Derepression. J. Investig. Dermatol. 2022. [Google Scholar] [CrossRef]
- Kamminga, L.M.; Bystrykh, L.V.; de Boer, A.; Houwer, S.; Douma, J.; Weersing, E.; Dontje, B.; de Haan, G. The Polycomb group gene Ezh2 prevents hematopoietic stem cell exhaustion. Blood 2006, 107, 2170–2179. [Google Scholar] [CrossRef]
- Xie, M.; Lu, C.; Wang, J.; McLellan, M.D.; Johnson, K.J.; Wendl, M.C.; McMichael, J.F.; Schmidt, H.K.; Yellapantula, V.; Miller, C.A.; et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med. 2014, 20, 1472–1478. [Google Scholar] [CrossRef]
- Lessard, J.; Schumacher, A.; Thorsteinsdottir, U.; Van Lohuizen, M.; Magnuson, T.; Sauvageau, G. Functional antagonism of the Polycomb-Group genes eed and Bmi1 in hemopoietic cell proliferation. Genes Dev. 1999, 13, 2691–2703. [Google Scholar] [CrossRef]
- Mochizuki-Kashio, M.; Mishima, Y.; Miyagi, S.; Negishi, M.; Saraya, A.; Konuma, T.; Shinga, J.; Koseki, H.; Iwama, A. Dependency on the polycomb gene Ezh2 distinguishes fetal from adult hematopoietic stem cells. Blood 2011, 118, 6553–6561. [Google Scholar] [CrossRef]
- Hidalgo, I.; Herrera-Merchan, A.; Ligos, J.M.; Carramolino, L.; Nunez, J.; Martinez, F.; Dominguez, O.; Torres, M.; Gonzalez, S. Ezh1 is required for hematopoietic stem cell maintenance and prevents senescence-like cell cycle arrest. Cell Stem. Cell 2012, 11, 649–662. [Google Scholar] [CrossRef]
- Chen, H.; Gu, X.; Su, I.H.; Bottino, R.; Contreras, J.L.; Tarakhovsky, A.; Kim, S.K. Polycomb protein Ezh2 regulates pancreatic beta-cell Ink4a/Arf expression and regeneration in diabetes mellitus. Genes Dev. 2009, 23, 975–985. [Google Scholar] [CrossRef]
- Juan, A.H.; Derfoul, A.; Feng, X.; Ryall, J.G.; Dell’Orso, S.; Pasut, A.; Zare, H.; Simone, J.M.; Rudnicki, M.A.; Sartorelli, V. Polycomb EZH2 controls self-renewal and safeguards the transcriptional identity of skeletal muscle stem cells. Genes Dev. 2011, 25, 789–794. [Google Scholar] [CrossRef]
- Chiacchiera, F.; Rossi, A.; Jammula, S.; Zanotti, M.; Pasini, D. PRC2 preserves intestinal progenitors and restricts secretory lineage commitment. EMBO J. 2016, 35, 2301–2314. [Google Scholar] [CrossRef]
- Di Foggia, V.; Zhang, X.; Licastro, D.; Gerli, M.F.; Phadke, R.; Muntoni, F.; Mourikis, P.; Tajbakhsh, S.; Ellis, M.; Greaves, L.C.; et al. Bmi1 enhances skeletal muscle regeneration through MT1-mediated oxidative stress protection in a mouse model of dystrophinopathy. J. Exp. Med. 2014, 211, 2617–2633. [Google Scholar] [CrossRef]
- Jacobs, J.J.L.; Scheijen, B.; Voncken, J.-W.; Kieboom, K.; Berns, A.; Van Lohuizen, M. Bmi-1 collaborates with c-Myc in tumorigenesis by inhibiting c-Myc-induced apoptosis via INK4a/ARF. Genes Dev. 1999, 13, 2678–2690. [Google Scholar] [CrossRef]
- Gil, J.; Peters, G. Regulation of the INK4b-ARF-INK4a tumour suppressor locus: All for one or one for all. Nat. Rev. Mol. Cell Biol. 2006, 7, 667–677. [Google Scholar] [CrossRef]
- Voorhoeve, P.M.; Agami, R. The tumor-suppressive functions of the human INK4A locus. Cancer Cell 2003, 4, 311–319. [Google Scholar] [CrossRef]
- Shields, C.E.; Potlapalli, S.; Cuya-Smith, S.M.; Chappell, S.K.; Chen, D.; Martinez, D.; Pogoriler, J.; Rathi, K.S.; Patel, S.A.; Oristian, K.M.; et al. Epigenetic regulator BMI1 promotes alveolar rhabdomyosarcoma proliferation and constitutes a novel therapeutic target. Mol. Oncol. 2021, 15, 2156–2171. [Google Scholar] [CrossRef]
- Shields, C.E.; Schnepp, R.W.; Haynes, K.A. Differential Epigenetic Effects of BMI Inhibitor PTC-028 on Fusion-Positive Rhabdomyosarcoma Cell Lines from Distinct Metastatic Sites. Regen. Eng. Transl. Med. 2022, 1–10. [Google Scholar] [CrossRef]
- Chaffer, C.L.; Marjanovic, N.D.; Lee, T.; Bell, G.; Kleer, C.G.; Reinhardt, F.; D’Alessio, A.C.; Young, R.A.; Weinberg, R.A. Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell 2013, 154, 61–74. [Google Scholar] [CrossRef]
- Mallen-St Clair, J.; Soydaner-Azeloglu, R.; Lee, K.E.; Taylor, L.; Livanos, A.; Pylayeva-Gupta, Y.; Miller, G.; Margueron, R.; Reinberg, D.; Bar-Sagi, D. EZH2 couples pancreatic regeneration to neoplastic progression. Genes Dev. 2012, 26, 439–444. [Google Scholar] [CrossRef]
- Lang, S.H.; Frame, F.M.; Collins, A.T. Prostate cancer stem cells. J. Pathol. 2009, 217, 299–306. [Google Scholar] [CrossRef]
- Cao, W.; Ribeiro Rde, O.; Liu, D.; Saintigny, P.; Xia, R.; Xue, Y.; Lin, R.; Mao, L.; Ren, H. EZH2 promotes malignant behaviors via cell cycle dysregulation and its mRNA level associates with prognosis of patient with non-small cell lung cancer. PLoS ONE 2012, 7, e52984. [Google Scholar] [CrossRef]
- Liu, C.; Kelnar, K.; Liu, B.; Chen, X.; Calhoun-Davis, T.; Li, H.; Patrawala, L.; Yan, H.; Jeter, C.; Honorio, S.; et al. The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nat. Med. 2011, 17, 211–215. [Google Scholar] [CrossRef]
- Bai, Y.; Zhang, Z.; Cheng, L.; Wang, R.; Chen, X.; Kong, Y.; Feng, F.; Ahamd, N.; Li, L.; Liu, X. Inhibition of enhancer of zeste homolog 2 (EZH2) overcomes enzalutamide resistance in castration-resistant prostate cancer. J. Biol. Chem. 2019, 294, 9911–9923. [Google Scholar] [CrossRef]
- Patel, G.K.; Chugh, N.; Tripathi, M. Neuroendocrine Differentiation of Prostate Cancer-An Intriguing Example of Tumor Evolution at Play. Cancers 2019, 11, 1405. [Google Scholar] [CrossRef] [PubMed]
- Bunn, P.A.; Minna, J.D.; Augustyn, A.; Gazdar, A.F.; Ouadah, Y.; Krasnow, M.A.; Berns, A.; Brambilla, E.; Rekhtman, N.; Massion, P.P.; et al. Small Cell Lung Cancer: Can Recent Advances in Biology and Molecular Biology Be Translated into Improved Outcomes? J. Thorac. Oncol. 2016, 11, 453–474. [Google Scholar] [CrossRef] [PubMed]
- Collinson, A.; Collier, A.J.; Morgan, N.P.; Sienerth, A.R.; Chandra, T.; Andrews, S.; Rugg-Gunn, P.J. Deletion of the Polycomb-Group Protein EZH2 Leads to Compromised Self-Renewal and Differentiation Defects in Human Embryonic Stem Cells. Cell Rep. 2016, 17, 2700–2714. [Google Scholar] [CrossRef] [PubMed]
- Boyer, L.A.; Plath, K.; Zeitlinger, J.; Brambrink, T.; Medeiros, L.A.; Lee, T.I.; Levine, S.S.; Wernig, M.; Tajonar, A.; Ray, M.K.; et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 2006, 441, 349–353. [Google Scholar] [CrossRef]
- Lee, T.I.; Jenner, R.G.; Boyer, L.A.; Guenther, M.G.; Levine, S.S.; Kumar, R.M.; Chevalier, B.; Johnstone, S.E.; Cole, M.F.; Isono, K.; et al. Control of developmental regulators by Polycomb in human embryonic stem cells. Cell 2006, 125, 301–313. [Google Scholar] [CrossRef]
- Klauke, K.; Radulovic, V.; Broekhuis, M.; Weersing, E.; Zwart, E.; Olthof, S.; Ritsema, M.; Bruggeman, S.; Wu, X.; Helin, K.; et al. Polycomb Cbx family members mediate the balance between haematopoietic stem cell self-renewal and differentiation. Nat. Cell. Biol. 2013, 15, 353–362. [Google Scholar] [CrossRef]
- Plass, C.; Pfister, S.M.; Lindroth, A.M.; Bogatyrova, O.; Claus, R.; Lichter, P. Mutations in regulators of the epigenome and their connections to global chromatin patterns in cancer. Nat. Rev. Genet. 2013, 14, 765–780. [Google Scholar] [CrossRef]
- Majewski, I.J.; Ritchie, M.E.; Phipson, B.; Corbin, J.; Pakusch, M.; Ebert, A.; Busslinger, M.; Koseki, H.; Hu, Y.; Smyth, G.K.; et al. Opposing roles of polycomb repressive complexes in hematopoietic stem and progenitor cells. Blood 2010, 116, 731–739. [Google Scholar] [CrossRef]
- Lee, H.G.; Kahn, T.G.; Simcox, A.; Schwartz, Y.B.; Pirrotta, V. Genome-wide activities of Polycomb complexes control pervasive transcription. Genome Res. 2015, 25, 1170–1181. [Google Scholar] [CrossRef]
- Xiao, J.; Jin, R.; Yu, X.; Shen, M.; Wagner, J.D.; Pai, A.; Song, C.; Zhuang, M.; Klasfeld, S.; He, C.; et al. Cis and trans determinants of epigenetic silencing by Polycomb repressive complex 2 in Arabidopsis. Nat. Genet. 2017, 49, 1546–1552. [Google Scholar] [CrossRef]
- Villa, R.; Pasini, D.; Gutierrez, A.; Morey, L.; Occhionorelli, M.; Vire, E.; Nomdedeu, J.F.; Jenuwein, T.; Pelicci, P.G.; Minucci, S.; et al. Role of the polycomb repressive complex 2 in acute promyelocytic leukemia. Cancer Cell 2007, 11, 513–525. [Google Scholar] [CrossRef]
- Boukarabila, H.; Saurin, A.J.; Batsche, E.; Mossadegh, N.; van Lohuizen, M.; Otte, A.P.; Pradel, J.; Muchardt, C.; Sieweke, M.; Duprez, E. The PRC1 Polycomb group complex interacts with PLZF/RARA to mediate leukemic transformation. Genes Dev. 2009, 23, 1195–1206. [Google Scholar] [CrossRef]
- Neff, T.; Sinha, A.U.; Kluk, M.J.; Zhu, N.; Khattab, M.H.; Stein, L.; Xie, H.; Orkin, S.H.; Armstrong, S.A. Polycomb repressive complex 2 is required for MLL-AF9 leukemia. Proc. Natl. Acad. Sci. USA 2012, 109, 5028–5033. [Google Scholar] [CrossRef]
- Tanaka, S.; Miyagi, S.; Sashida, G.; Chiba, T.; Yuan, J.; Mochizuki-Kashio, M.; Suzuki, Y.; Sugano, S.; Nakaseko, C.; Yokote, K.; et al. Ezh2 augments leukemogenicity by reinforcing differentiation blockage in acute myeloid leukemia. Blood 2012, 120, 1107–1117. [Google Scholar] [CrossRef]
- Kinkel, S.A.; Galeev, R.; Flensburg, C.; Keniry, A.; Breslin, K.; Gilan, O.; Lee, S.; Liu, J.; Chen, K.; Gearing, L.J.; et al. Jarid2 regulates hematopoietic stem cell function by acting with polycomb repressive complex 2. Blood 2015, 125, 1890–1900. [Google Scholar] [CrossRef]
- Davies, A.; Nouruzi, S.; Ganguli, D.; Namekawa, T.; Thaper, D.; Linder, S.; Karaoglanoglu, F.; Omur, M.E.; Kim, S.; Kobelev, M.; et al. An androgen receptor switch underlies lineage infidelity in treatment-resistant prostate cancer. Nat. Cell. Biol. 2021, 23, 1023–1034. [Google Scholar] [CrossRef]
- Ezhkova, E.; Pasolli, H.A.; Parker, J.S.; Stokes, N.; Su, I.-H.; Hannon, G.; Tarakhovsky, A.; Fuchs, E. Ezh2 Orchestrates Gene Expression for the Stepwise Differentiation of Tissue-Specific Stem Cells. Cell 2009, 136, 1122–1135. [Google Scholar] [CrossRef]
- Flora, P.; Li, M.Y.; Galbo, P.M., Jr.; Astorkia, M.; Zheng, D.; Ezhkova, E. Polycomb repressive complex 2 in adult hair follicle stem cells is dispensable for hair regeneration. PLoS Genet. 2021, 17, e1009948. [Google Scholar] [CrossRef]
- Linder, S.; Hoogstraat, M.; Stelloo, S.; Schuurman, K.; de Barros, H.; Alkemade, M.; Sanders, J.; Kim, Y.; Bekers, E.; de Jong, J.; et al. Drug-induced epigenomic plasticity reprograms circadian rhythm regulation to drive prostate cancer towards androgen-independence. Cancer Discov 2022, 12, 2074–2097. [Google Scholar] [CrossRef] [PubMed]
- Labrecque, M.P.; Coleman, I.M.; Brown, L.G.; True, L.D.; Kollath, L.; Lakely, B.; Nguyen, H.M.; Yang, Y.C.; da Costa, R.M.G.; Kaipainen, A.; et al. Molecular profiling stratifies diverse phenotypes of treatment-refractory metastatic castration-resistant prostate cancer. J. Clin. Investig. 2019, 129, 4492–4505. [Google Scholar] [CrossRef]
- Kleb, B.; Estecio, M.R.; Zhang, J.; Tzelepi, V.; Chung, W.; Jelinek, J.; Navone, N.M.; Tahir, S.; Marquez, V.E.; Issa, J.P.; et al. Differentially methylated genes and androgen receptor re-expression in small cell prostate carcinomas. Epigenetics 2016, 11, 184–193. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.T.; Yao, Y.H.; Li, B.G.; Tang, Y.; Chang, J.W.; Zhang, J. Neuroendocrine Prostate Cancer (NEPC) progressing from conventional prostatic adenocarcinoma: Factors associated with time to development of NEPC and survival from NEPC diagnosis-a systematic review and pooled analysis. J. Clin. Oncol. 2014, 32, 3383–3390. [Google Scholar] [CrossRef] [PubMed]
- Dardenne, E.; Beltran, H.; Benelli, M.; Gayvert, K.; Berger, A.; Puca, L.; Cyrta, J.; Sboner, A.; Noorzad, Z.; MacDonald, T.; et al. N-Myc Induces an EZH2-Mediated Transcriptional Program Driving Neuroendocrine Prostate Cancer. Cancer Cell 2016, 30, 563–577. [Google Scholar] [CrossRef] [PubMed]
- Beltran, H.; Rickman, D.S.; Park, K.; Chae, S.S.; Sboner, A.; MacDonald, T.Y.; Wang, Y.; Sheikh, K.L.; Terry, S.; Tagawa, S.T.; et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov. 2011, 1, 487–495. [Google Scholar] [CrossRef]
- Mosquera, J.M.; Beltran, H.; Park, K.; MacDonald, T.Y.; Robinson, B.D.; Tagawa, S.T.; Perner, S.; Bismar, T.A.; Erbersdobler, A.; Dhir, R.; et al. Concurrent AURKA and MYCN gene amplifications are harbingers of lethal treatment-related neuroendocrine prostate cancer. Neoplasia 2013, 15, 1–10. [Google Scholar] [CrossRef]
- Berger, A.; Brady, N.J.; Bareja, R.; Robinson, B.; Conteduca, V.; Augello, M.A.; Puca, L.; Ahmed, A.; Dardenne, E.; Lu, X.; et al. N-Myc-mediated epigenetic reprogramming drives lineage plasticity in advanced prostate cancer. J. Clin. Investig. 2019, 129, 3924–3940. [Google Scholar] [CrossRef]
- Qiu, X.; Boufaied, N.; Hallal, T.; Feit, A.; de Polo, A.; Luoma, A.M.; Alahmadi, W.; Larocque, J.; Zadra, G.; Xie, Y.; et al. MYC drives aggressive prostate cancer by disrupting transcriptional pause release at androgen receptor targets. Nat. Commun. 2022, 13, 2559. [Google Scholar] [CrossRef]
- Peifer, M.; Fernandez-Cuesta, L.; Sos, M.L.; George, J.; Seidel, D.; Kasper, L.H.; Plenker, D.; Leenders, F.; Sun, R.; Zander, T.; et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat. Genet. 2012, 44, 1104–1110. [Google Scholar] [CrossRef]
- Ku, S.Y.; Rosario, S.; Wang, Y.; Mu, P.; Seshadri, M.; Goodrich, Z.W.; Goodrich, M.M.; Labbe, D.P.; Gomez, E.C.; Wang, J.; et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 2017, 355, 78–83. [Google Scholar] [CrossRef]
- Kim, K.B.; Dunn, C.T.; Park, K.S. Recent progress in mapping the emerging landscape of the small-cell lung cancer genome. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef]
- Baca, S.C.; Takeda, D.Y.; Seo, J.H.; Hwang, J.; Ku, S.Y.; Arafeh, R.; Arnoff, T.; Agarwal, S.; Bell, C.; O’Connor, E.; et al. Reprogramming of the FOXA1 cistrome in treatment-emergent neuroendocrine prostate cancer. Nat. Commun. 2021, 12, 1979. [Google Scholar] [CrossRef]
- Borromeo, M.D.; Savage, T.K.; Kollipara, R.K.; He, M.; Augustyn, A.; Osborne, J.K.; Girard, L.; Minna, J.D.; Gazdar, A.F.; Cobb, M.H.; et al. ASCL1 and NEUROD1 Reveal Heterogeneity in Pulmonary Neuroendocrine Tumors and Regulate Distinct Genetic Programs. Cell. Rep. 2016, 16, 1259–1272. [Google Scholar] [CrossRef]
- Nouruzi, S.; Ganguli, D.; Tabrizian, N.; Kobelev, M.; Sivak, O.; Namekawa, T.; Thaper, D.; Baca, S.C.; Freedman, M.L.; Aguda, A.; et al. ASCL1 activates neuronal stem cell-like lineage programming through remodeling of the chromatin landscape in prostate cancer. Nat. Commun. 2022, 13, 2282. [Google Scholar] [CrossRef]
- Loh, C.H.; van Genesen, S.; Perino, M.; Bark, M.R.; Veenstra, G.J.C. Loss of PRC2 subunits primes lineage choice during exit of pluripotency. Nat. Commun. 2021, 12, 6985. [Google Scholar] [CrossRef]
- Zingg, D.; Debbache, J.; Schaefer, S.M.; Tuncer, E.; Frommel, S.C.; Cheng, P.; Arenas-Ramirez, N.; Haeusel, J.; Zhang, Y.; Bonalli, M.; et al. The epigenetic modifier EZH2 controls melanoma growth and metastasis through silencing of distinct tumour suppressors. Nat. Commun. 2015, 6, 6051. [Google Scholar] [CrossRef]
- Serrano-Gomez, S.J.; Maziveyi, M.; Alahari, S.K. Regulation of epithelial-mesenchymal transition through epigenetic and post-translational modifications. Mol. Cancer 2016, 15, 18. [Google Scholar] [CrossRef]
- Nose, A.; Takeichi, M. A novel Cadherin cell adhesion molecule: Its expression patterns associated with implantation and organogenesis of mouse embryos. J. Cell Biol. 1986, 103, 2649–2658. [Google Scholar] [CrossRef]
- Xu, F.; Li, X.; Wu, L.; Zhang, Q.; Yang, R.; Yang, Y.; Zhang, Z.; He, Q.; Chang, C. Overexpression of the EZH2, RING1 and BMI1 genes is common in myelodysplastic syndromes: Relation to adverse epigenetic alteration and poor prognostic scoring. Ann. Hematol. 2011, 90, 643–653. [Google Scholar] [CrossRef]
- Tong, Z.T.; Cai, M.Y.; Wang, X.G.; Kong, L.L.; Mai, S.J.; Liu, Y.H.; Zhang, H.B.; Liao, Y.J.; Zheng, F.; Zhu, W.; et al. EZH2 supports nasopharyngeal carcinoma cell aggressiveness by forming a co-repressor complex with HDAC1/HDAC2 and Snail to inhibit E-cadherin. Oncogene 2012, 31, 583–594. [Google Scholar] [CrossRef]
- Zhang, Q.; Dong, P.; Liu, X.; Sakuragi, N.; Guo, S.W. Enhancer of Zeste homolog 2 (EZH2) induces epithelial-mesenchymal transition in endometriosis. Sci. Rep. 2017, 7, 6804. [Google Scholar] [CrossRef]
- Cao, Q.; Yu, J.; Dhanasekaran, S.M.; Kim, J.H.; Mani, R.S.; Tomlins, S.A.; Mehra, R.; Laxman, B.; Cao, X.; Yu, J.; et al. Repression of E-cadherin by the polycomb group protein EZH2 in cancer. Oncogene 2008, 27, 7274–7284. [Google Scholar] [CrossRef]
- Byles, V.; Zhu, L.; Lovaas, J.D.; Chmilewski, L.K.; Wang, J.; Faller, D.V.; Dai, Y. SIRT1 induces EMT by cooperating with EMT transcription factors and enhances prostate cancer cell migration and metastasis. Oncogene 2012, 31, 4619–4629. [Google Scholar] [CrossRef]
- Peng, D.; Kryczek, I.; Nagarsheth, N.; Zhao, L.; Wei, S.; Wang, W.; Sun, Y.; Zhao, E.; Vatan, L.; Szeliga, W.; et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 2015, 527, 249–253. [Google Scholar] [CrossRef]
- Gao, J.; Shi, L.Z.; Zhao, H.; Chen, J.; Xiong, L.; He, Q.; Chen, T.; Roszik, J.; Bernatchez, C.; Woodman, S.E.; et al. Loss of IFN-gamma Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell 2016, 167, 397–404.e9. [Google Scholar] [CrossRef]
- Sade-Feldman, M.; Jiao, Y.J.; Chen, J.H.; Rooney, M.S.; Barzily-Rokni, M.; Eliane, J.P.; Bjorgaard, S.L.; Hammond, M.R.; Vitzthum, H.; Blackmon, S.M.; et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat. Commun. 2017, 8, 1136. [Google Scholar] [CrossRef]
- Burr, M.L.; Sparbier, C.E.; Chan, K.L.; Chan, Y.C.; Kersbergen, A.; Lam, E.Y.N.; Azidis-Yates, E.; Vassiliadis, D.; Bell, C.C.; Gilan, O.; et al. An Evolutionarily Conserved Function of Polycomb Silences the MHC Class I Antigen Presentation Pathway and Enables Immune Evasion in Cancer. Cancer Cell 2019, 36, 385–401.e8. [Google Scholar] [CrossRef]
- Canadas, I.; Thummalapalli, R.; Kim, J.W.; Kitajima, S.; Jenkins, R.W.; Christensen, C.L.; Campisi, M.; Kuang, Y.; Zhang, Y.; Gjini, E.; et al. Tumor innate immunity primed by specific interferon-stimulated endogenous retroviruses. Nat. Med. 2018, 24, 1143–1150. [Google Scholar] [CrossRef]
- Morel, K.L.; Sheahan, A.V.; Burkhart, D.L.; Baca, S.C.; Boufaied, N.; Liu, Y.; Qiu, X.; Canadas, I.; Roehle, K.; Heckler, M.; et al. EZH2 inhibition activates a dsRNA-STING-interferon stress axis that potentiates response to PD-1 checkpoint blockade in prostate cancer. Nat. Cancer 2021, 2, 444–456. [Google Scholar] [CrossRef]
- Pech, M.F.; Fong, L.E.; Villalta, J.E.; Chan, L.J.; Kharbanda, S.; O’Brien, J.J.; McAllister, F.E.; Firestone, A.J.; Jan, C.H.; Settleman, J. Systematic identification of cancer cell vulnerabilities to natural killer cell-mediated immune surveillance. Elife 2019, 8, e47362. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Colaprico, A.; Silva, T.C.; Chen, J.; An, H.; Ban, Y.; Huang, H.; Wang, L.; James, J.L.; Balko, J.M.; et al. Multi-omics analysis identifies therapeutic vulnerabilities in triple-negative breast cancer subtypes. Nat. Commun. 2021, 12, 6276. [Google Scholar] [CrossRef]
- Mahadevan, N.R.; Knelson, E.H.; Wolff, J.O.; Vajdi, A.; Saigi, M.; Campisi, M.; Hong, D.; Thai, T.C.; Piel, B.; Han, S.; et al. Intrinsic Immunogenicity of Small Cell Lung Carcinoma Revealed by Its Cellular Plasticity. Cancer Discov. 2021, 11, 1952–1969. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Wang, Y.; Yang, M.; Wang, Z.; Wang, Y.; Chaurasia, S.; Wu, Z.; Zhang, M.; Yadav, G.G.; Rathod, S.; et al. LincRNA-immunity landscape analysis identifies EPIC1 as a regulator of tumor immune evasion and immunotherapy resistance. Sci. Adv. 2021, 7, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Xiao, G.; Jin, L.L.; Liu, C.Q.; Wang, Y.C.; Meng, Y.M.; Zhou, Z.G.; Chen, J.; Yu, X.J.; Zhang, Y.J.; Xu, J.; et al. EZH2 negatively regulates PD-L1 expression in hepatocellular carcinoma. J. Immunother. Cancer 2019, 7, 300. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Bado, I.L.; Hu, J.; Wan, Y.W.; Wu, L.; Wang, H.; Gao, Y.; Jeong, H.H.; Xu, Z.; Hao, X.; et al. The bone microenvironment invigorates metastatic seeds for further dissemination. Cell 2021, 184, 2471–2486.e20. [Google Scholar] [CrossRef]
- Samsonov, R.; Shtam, T.; Burdakov, V.; Glotov, A.; Tsyrlina, E.; Berstein, L.; Nosov, A.; Evtushenko, V.; Filatov, M.; Malek, A. Lectin-induced agglutination method of urinary exosomes isolation followed by mi-RNA analysis: Application for prostate cancer diagnostic. Prostate 2016, 76, 68–79. [Google Scholar] [CrossRef]
- Tiffen, J.C.; Gallagher, S.J.; Tseng, H.Y.; Filipp, F.V.; Fazekas de St. Groth, B.; Hersey, P. EZH2 as a mediator of treatment resistance in melanoma. Pigment Cell Melanoma Res. 2016, 29, 500–507. [Google Scholar] [CrossRef]
- Zingg, D.; Arenas-Ramirez, N.; Sahin, D.; Rosalia, R.A.; Antunes, A.T.; Haeusel, J.; Sommer, L.; Boyman, O. The Histone Methyltransferase Ezh2 Controls Mechanisms of Adaptive Resistance to Tumor Immunotherapy. Cell Rep. 2017, 20, 854–867. [Google Scholar] [CrossRef]
- Nagarsheth, N.; Peng, D.; Kryczek, I.; Wu, K.; Li, W.; Zhao, E.; Zhao, L.; Wei, S.; Frankel, T.; Vatan, L.; et al. PRC2 Epigenetically Silences Th1-Type Chemokines to Suppress Effector T-Cell Trafficking in Colon Cancer. Cancer Res. 2016, 76, 275–282. [Google Scholar] [CrossRef]
- Karantanos, T.; Chistofides, A.; Barhdan, K.; Li, L.; Boussiotis, V.A. Regulation of T Cell Differentiation and Function by EZH2. Front. Immunol. 2016, 7, 172. [Google Scholar] [CrossRef]
- Yi, S.; Sun, J.; Qiu, L.; Fu, W.; Wang, A.; Liu, X.; Yang, Y.; Kadin, M.E.; Tu, P.; Wang, Y. Dual Role of EZH2 in Cutaneous Anaplastic Large Cell Lymphoma: Promoting Tumor Cell Survival and Regulating Tumor Microenvironment. J. Investig. Dermatol. 2018, 138, 1126–1136. [Google Scholar] [CrossRef]
- Zhao, J.; Li, H.; Zhao, S.; Wang, E.; Zhu, J.; Feng, D.; Zhu, Y.; Dou, W.; Fan, Q.; Hu, J.; et al. Epigenetic silencing of miR-144/451a cluster contributes to HCC progression via paracrine HGF/MIF-mediated TAM remodeling. Mol. Cancer 2021, 20, 46. [Google Scholar] [CrossRef]
- Qi, B.; Yang, C.; Zhu, Z.; Chen, H. EZH2-Inhibited MicroRNA-454-3p Promotes M2 Macrophage Polarization in Glioma. Front. Cell Dev. Biol. 2020, 8, 574940. [Google Scholar] [CrossRef]
- Yin, H.; Wang, Y.; Wu, Y.; Zhang, X.; Zhang, X.; Liu, J.; Wang, T.; Fan, J.; Sun, J.; Yang, A.; et al. EZH2-mediated Epigenetic Silencing of miR-29/miR-30 targets LOXL4 and contributes to Tumorigenesis, Metastasis, and Immune Microenvironment Remodeling in Breast Cancer. Theranostics 2020, 10, 8494–8512. [Google Scholar] [CrossRef]
- Zheng, Y.; Wang, Z.; Wei, S.; Liu, Z.; Chen, G. Epigenetic silencing of chemokine CCL2 represses macrophage infiltration to potentiate tumor development in small cell lung cancer. Cancer Lett. 2021, 499, 148–163. [Google Scholar] [CrossRef]
- Xia, L.; Zhu, X.; Zhang, L.; Xu, Y.; Chen, G.; Luo, J. EZH2 enhances expression of CCL5 to promote recruitment of macrophages and invasion in lung cancer. Biotechnol. Appl. Biochem. 2020, 67, 1011–1019. [Google Scholar] [CrossRef]
- Watanabe, K.; Jose, P.J.; Rankin, S.M. Eotaxin-2 generation is differentially regulated by lypopolysaccaride and IL-4 in monocytes and macrophages. J. Immunol. 2002, 168, 1911–1918. [Google Scholar] [CrossRef]
- Yang, X.P.; Jiang, K.; Hirahara, K.; Vahedi, G.; Afzali, B.; Sciume, G.; Bonelli, M.; Sun, H.W.; Jankovic, D.; Kanno, Y.; et al. EZH2 is crucial for both differentiation of regulatory T cells and T effector cell expansion. Sci. Rep. 2015, 5, 10643. [Google Scholar] [CrossRef]
- Qiu, J.; Sharma, S.; Rollins, R.A.; Paul, T.A. The complex role of EZH2 in the tumor microenvironment: Opportunities and challenges for immunotherapy combinations. Future Med. Chem. 2020, 15, 1415–1430. [Google Scholar] [CrossRef]
- Poli, V.; Fagnocchi, L.; Zippo, A. Tumorigenic Cell Reprogramming and Cancer Plasticity: Interplay between Signaling, Microenvironment, and Epigenetics. Stem Cells Int. 2018, 2018, 4598195. [Google Scholar] [CrossRef]
- Chou, R.-H.; Yu, Y.-L.; Hung, M.-C. The roles of EZH2 in cell lineage commitment. Am. J. Transl. Res. 2011, 3, 243–250. [Google Scholar]
- Huang, J.; Zhang, J.; Guo, Z.; Li, C.; Tan, Z.; Wang, J.; Yang, J.; Xue, L. Easy or Not-The Advances of EZH2 in Regulating T Cell Development, Differentiation, and Activation in Antitumor Immunity. Front. Immunol. 2021, 12, 741302. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Quiros, J.; Mahuron, K.; Pai, C.C.; Ranzani, V.; Young, A.; Silveria, S.; Harwin, T.; Abnousian, A.; Pagani, M.; et al. Targeting EZH2 Reprograms Intratumoral Regulatory T Cells to Enhance Cancer Immunity. Cell. Rep. 2018, 23, 3262–3274. [Google Scholar] [CrossRef] [PubMed]
- Kakaradov, B.; Arsenio, J.; Widjaja, C.E.; He, Z.; Aigner, S.; Metz, P.J.; Yu, B.; Wehrens, E.J.; Lopez, J.; Kim, S.H.; et al. Early transcriptional and epigenetic regulation of CD8(+) T cell differentiation revealed by single-cell RNA sequencing. Nat. Immunol. 2017, 18, 422–432. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Wang, J.; Kato, K.; Xie, F.; Varambally, S.; Mineishi, S.; Kuick, R.; Mochizuki, K.; Liu, Y.; Nieves, E.; et al. Inhibition of histone methylation arrests ongoing graft-versus-host disease in mice by selectively inducing apoptosis of alloreactive effector T cells. Blood 2012, 119, 1274–1282. [Google Scholar] [CrossRef]
- Zhao, E.; Maj, T.; Kryczek, I.; Li, W.; Wu, K.; Zhao, L.; Wei, S.; Crespo, J.; Wan, S.; Vatan, L.; et al. Cancer mediates effector T cell dysfunction by targeting microRNAs and EZH2 via glycolysis restriction. Nat. Immunol. 2016, 17, 95–103. [Google Scholar] [CrossRef]
- Zhang, Y.; Kinkel, S.; Maksimovic, J.; Bandala-Sanchez, E.; Tanzer, M.C.; Naselli, G.; Zhang, J.G.; Zhan, Y.; Lew, A.M.; Silke, J.; et al. The polycomb repressive complex 2 governs life and death of peripheral T cells. Blood 2014, 124, 737–749. [Google Scholar] [CrossRef]
- He, S.; Liu, Y.; Meng, L.; Sun, H.; Wang, Y.; Ji, Y.; Purushe, J.; Chen, P.; Li, C.; Madzo, J.; et al. Ezh2 phosphorylation state determines its capacity to maintain CD8(+) T memory precursors for antitumor immunity. Nat. Commun. 2017, 8, 2125. [Google Scholar] [CrossRef]
- Zhang, J.; Ding, L.; Holmfeldt, L.; Wu, G.; Heatley, S.L.; Payne-Turner, D.; Easton, J.; Chen, X.; Wang, J.; Rusch, M.; et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 2012, 481, 157–163. [Google Scholar] [CrossRef]
- Broux, M.; Prieto, C.; Demeyer, S.; Bempt, M.V.; Alberti-Servera, L.; Lodewijckx, I.; Vandepoel, R.; Mentens, N.; Gielen, O.; Jacobs, K.; et al. Suz12 inactivation cooperates with JAK3 mutant signaling in the development of T-cell acute lymphoblastic leukemia. Blood 2019, 134, 1323–1336. [Google Scholar] [CrossRef]
- Chiang, S.C.; Theorell, J.; Entesarian, M.; Meeths, M.; Mastafa, M.; Al-Herz, W.; Frisk, P.; Gilmour, K.C.; Ifversen, M.; Langenskiold, C.; et al. Comparison of primary human cytotoxic T-cell and natural killer cell responses reveal similar molecular requirements for lytic granule exocytosis but differences in cytokine production. Blood 2013, 121, 1345–1356. [Google Scholar] [CrossRef]
- Nagel, S.; Venturini, L.; Marquez, V.E.; Meyer, C.; Kaufmann, M.; Scherr, M.; MacLeod, R.A.F.; Drexler, H.G. Polycomb repressor complex 2 regulates HOXA9 and HOXA10, activating ID2 in NK/T-cell lines. Mol. Cancer 2010, 9, 151. [Google Scholar] [CrossRef]
- Yin, J.; Leavenworth, J.W.; Li, Y.; Luo, Q.; Xie, H.; Liu, X.; Huang, S.; Yan, H.; Fu, Z.; Zhang, L.Y.; et al. Ezh2 regulates differentiation and function of natural killer cells through histone methyltransferase activity. Proc. Natl. Acad. Sci. USA 2015, 112, 15988–15993. [Google Scholar] [CrossRef] [PubMed]
- Bugide, S.; Green, M.R.; Wajapeyee, N. Inhibition of Enhancer of zeste homolog 2 (EZH2) induces natural killer cell-mediated eradication of hepatocellular carcinoma cells. Proc. Natl. Acad. Sci. USA 2018, 115, E3509–E3518. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, S.; Granger, V.; Rak, M.; Hu, Q.; Attwood, K.; Aquila, L.; Krishnan, N.; Osiecki, R.; Azabdaftari, G.; Guru, K.; et al. Inhibition of EZH2 induces NK cell-mediated differentiation and death in muscle-invasive bladder cancer. Cell Death Differ. 2019, 26, 2100–2114. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Wang, Z.; Zhou, J.; Huang, J.; Zhou, L.; Luo, J.; Wan, Y.Y.; Long, H.; Zhu, B. EZH2 Inhibitor GSK126 Suppresses Antitumor Immunity by Driving Production of Myeloid-Derived Suppressor Cells. Cancer Res. 2019, 79, 2009–2020. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Tao, T.; Chen, M.; Jiang, R.; Guan, H.; Huang, Y.; Su, H.; Hu, Q.; Han, X.; Xiao, J. Involvement of EZH2 in aerobic glycolysis of prostate cancer through miR-181b/HK2 axis. Oncol. Rep. 2017, 37, 1430–1436. [Google Scholar] [CrossRef]
- Pang, B.; Rong, X.-R.; Tian, J.-X.; Gao, T.-H.; Gu, G.-Y.; Zhang, R.; Fu, Y.-B.; Pang, Q.; Li, X.-G.; Liu, Q. EZH2 promotes metabolic reprogramming in glioblastomas through epigenetic repression of EAF2-HIF1α signaling. Oncotarget 2016, 7, 45134–45143. [Google Scholar] [CrossRef]
- El Maaty, A.M.; Terzic, J.; Keime, C.; Rovito, D.; Lutzing, R.; Yanushko, D.; Parisotto, M.; Grelet, E.; Namer, I.J.; Lindner, V.; et al. Hypoxia-mediated stabilization of HIF1A in prostatic intraepithelial neoplasia promotes cell plasticity and malignant progression. Sci. Adv. 2022, 8, 1–14. [Google Scholar] [CrossRef]
- Zhang, T.; Gong, Y.; Meng, H.; Li, C.; Xue, L. Symphony of epigenetic and metabolic regulation-interaction between the histone methyltransferase EZH2 and metabolism of tumor. Clin. Epigenet. 2020, 12, 72. [Google Scholar] [CrossRef]
- Schiliro, C.; Firestein, B.L. Mechanisms of Metabolic Reprogramming in Cancer Cells Supporting Enhanced Growth and Proliferation. Cells 2021, 10, 1056. [Google Scholar] [CrossRef]
- Ahmad, F.; Patrick, S.; Sheikh, T.; Sharma, V.; Pathak, P.; Malgulwar, P.B.; Kumar, A.; Joshi, S.D.; Sarkar, C.; Sen, E. Telomerase reverse transcriptase (TERT)—Enhancer of zeste homolog 2 (EZH2) network regulates lipid metabolism and DNA damage responses in glioblastoma. J. Neurochem. 2017, 143, 671–683. [Google Scholar] [CrossRef]
- Munir, R.; Lisec, J.; Swinnen, J.V.; Zaidi, N. Lipid metabolism in cancer cells under metabolic stress. Br. J. Cancer 2019, 120, 1090–1098. [Google Scholar] [CrossRef]
- Cheng, S.C.; Quintin, J.; Cramer, R.A.; Shepardson, K.M.; Saeed, S.; Kumar, V.; Giamarellos-Bourboulis, E.J.; Martens, J.H.; Rao, N.A.; Aghajanirefah, A.; et al. mTOR- and HIF-1alpha-mediated aerobic glycolysis as metabolic basis for trained immunity. Science 2014, 345, 1250684. [Google Scholar] [CrossRef]
- Krawczyk, C.M.; Holowka, T.; Sun, J.; Blagih, J.; Amiel, E.; DeBerardinis, R.J.; Cross, J.R.; Jung, E.; Thompson, C.B.; Jones, R.G.; et al. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 2010, 115, 4742–4749. [Google Scholar] [CrossRef]
- Quintin, J.; Saeed, S.; Martens, J.H.A.; Giamarellos-Bourboulis, E.J.; Ifrim, D.C.; Logie, C.; Jacobs, L.; Jansen, T.; Kullberg, B.J.; Wijmenga, C.; et al. Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe 2012, 12, 223–232. [Google Scholar] [CrossRef]
- Shen, Y.; Kapfhamer, D.; Minnella, A.M.; Kim, J.E.; Won, S.J.; Chen, Y.; Huang, Y.; Low, L.H.; Massa, S.M.; Swanson, R.A. Bioenergetic state regulates innate inflammatory responses through the transcriptional co-repressor CtBP. Nat. Commun. 2017, 8, 624. [Google Scholar] [CrossRef]
- Wang, M.; Guo, Y.; Wang, M.; Zhou, T.; Xue, Y.; Du, G.; Wei, X.; Wang, J.; Qi, L.; Zhang, H.; et al. The Glial Cell-Derived Neurotrophic Factor (GDNF)-responsive Phosphoprotein Landscape Identifies Raptor Phosphorylation Required for Spermatogonial Progenitor Cell Proliferation. Mol. Cell Proteom. 2017, 16, 982–997. [Google Scholar] [CrossRef]
- Yan, C.; Koda, S.; Wu, J.; Zhang, B.B.; Yu, Q.; Netea, M.G.; Tang, R.X.; Zheng, K.Y. Roles of Trained Immunity in the Pathogenesis of Cholangiopathies: A Therapeutic Target. Hepatology 2020, 72, 1838–1850. [Google Scholar] [CrossRef]
- Hardbower, D.M.; Asim, M.; Luis, P.B.; Singh, K.; Barry, D.P.; Yang, C.; Steeves, M.A.; Cleveland, J.L.; Schneider, C.; Piazuelo, M.B.; et al. Ornithine decarboxylase regulates M1 macrophage activation and mucosal inflammation via histone modifications. Proc. Natl. Acad. Sci. USA 2017, 114, E751–E760. [Google Scholar] [CrossRef]
- Bian, Y.; Li, W.; Kremer, D.M.; Sajjakulnukit, P.; Li, S.; Crespo, J.; Nwosu, Z.C.; Zhang, L.; Czerwonka, A.; Pawlowska, A.; et al. Cancer SLC43A2 alters T cell methionine metabolism and histone methylation. Nature 2020, 585, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, S.; Santosa, E.K.; Lau, C.M.; Violante, S.; Giovanelli, P.; Kim, H.; Cross, J.R.; Li, M.O.; Sun, J.C. Lactate dehydrogenase A-dependent aerobic glycolysis promotes natural killer cell anti-viral and anti-tumor function. Cell Rep. 2021, 35, 109210. [Google Scholar] [CrossRef] [PubMed]
- Duan, R.; Du, W.; Guo, W. EZH2: A novel target for cancer treatment. J. Hematol. Oncol. 2020, 13, 104. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.C.; Zeng, G.; Yu, J.; Schiltz, G.E. Small Molecule Approaches for Targeting the Polycomb Repressive Complex 2 (PRC2) in Cancer. J. Med. Chem. 2020, 63, 15344–15370. [Google Scholar] [CrossRef] [PubMed]
- Eich, M.L.; Athar, M.; Ferguson, J.E., 3rd; Varambally, S. EZH2-Targeted Therapies in Cancer: Hype or a Reality. Cancer Res. 2020, 80, 5449–5458. [Google Scholar] [CrossRef] [PubMed]
- Makita, S.; Tobinai, K. Targeting EZH2 with tazemetostat. Lancet Oncol. 2018, 19, 586–587. [Google Scholar] [CrossRef]
- Yap, T.A.; Winter, J.N.; Giulino-Roth, L.; Longley, J.; Lopez, J.; Michot, J.M.; Leonard, J.P.; Ribrag, V.; McCabe, M.T.; Creasy, C.L.; et al. Phase I Study of the Novel Enhancer of Zeste Homolog 2 (EZH2) Inhibitor GSK2816126 in Patients with Advanced Hematologic and Solid Tumors. Clin. Cancer Res. 2019, 25, 7331–7339. [Google Scholar] [CrossRef]
- Oruetxebarria, I.; Venturini, F.; Kekarainen, T.; Houweling, A.; Zuijderduijn, L.M.; Mohd-Sarip, A.; Vries, R.G.; Hoeben, R.C.; Verrijzer, C.P. P16INK4a is required for hSNF5 chromatin remodeler-induced cellular senescence in malignant rhabdoid tumor cells. J. Biol. Chem. 2004, 279, 3807–3816. [Google Scholar] [CrossRef]
- Knutson, S.K.; Warholic, N.M.; Wigle, T.J.; Klaus, C.R.; Allain, C.J.; Raimondi, A.; Porter Scott, M.; Chesworth, R.; Moyer, M.P.; Copeland, R.A.; et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc. Natl. Acad. Sci. USA 2013, 110, 7922–7927. [Google Scholar] [CrossRef]
- Kurmasheva, R.T.; Sammons, M.; Favours, E.; Wu, J.; Kurmashev, D.; Cosmopoulos, K.; Keilhack, H.; Klaus, C.R.; Houghton, P.J.; Smith, M.A. Initial testing (stage 1) of tazemetostat (EPZ-6438), a novel EZH2 inhibitor, by the Pediatric Preclinical Testing Program. Pediatr. Blood Cancer 2017, 64, e26218. [Google Scholar] [CrossRef]
- Morishima, S.; Ishitsuka, K.; Izutsu, K.; Kusumoto, S.; Makiyama, J.; Utsunomiya, A.; Nosaka, K.; Ishida, T.; Imaizumi, Y.; Yamauchi, N.; et al. First-in-Human Study of the EZH1/2 Dual Inhibitor Valemetostat in Relapsed or Refractory Non-Hodgkin Lymphoma (NHL)—Updated Results Focusing on Adult T-Cell Leukemia-Lymphoma (ATL). Blood 2019, 134, 4025. [Google Scholar] [CrossRef]
- Schmid, P.; Rugo, H.S.; Adams, S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Henschel, V.; Molinero, L.; Chui, S.Y.; et al. Atezolizumab plus nab-paclitaxel as first-line treatment for unresectable, locally advanced or metastatic triple-negative breast cancer (IMpassion130): Updated efficacy results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2020, 21, 44–59. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef]
- Samstein, R.M.; Lee, C.H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet 2019, 51, 202–206. [Google Scholar] [CrossRef]
- Goswami, S.; Apostolou, I.; Zhang, J.; Skepner, J.; Anandhan, S.; Zhang, X.; Xiong, L.; Trojer, P.; Aparicio, A.; Subudhi, S.K.; et al. Modulation of EZH2 expression in T cells improves efficacy of anti-CTLA-4 therapy. J. Clin. Investig. 2018, 128, 3813–3818. [Google Scholar] [CrossRef]
- Cromm, P.M.; Crews, C.M. Targeted Protein Degradation: From Chemical Biology to Drug Discovery. Cell Chem. Biol. 2017, 24, 1181–1190. [Google Scholar] [CrossRef]
- Pettersson, M.; Crews, C.M. PROteolysis TArgeting Chimeras (PROTACs)—Past, present and future. Drug Discov. Today Technol. 2019, 31, 15–27. [Google Scholar] [CrossRef]
- Nalawansha, D.A.; Crews, C.M. PROTACs: An Emerging Therapeutic Modality in Precision Medicine. Cell Chem. Biol. 2020, 27, 998–1014. [Google Scholar] [CrossRef]
- Gao, S.B.; Xu, B.; Ding, L.H.; Zheng, Q.L.; Zhang, L.; Zheng, Q.F.; Li, S.H.; Feng, Z.J.; Wei, J.; Yin, Z.Y.; et al. The functional and mechanistic relatedness of EZH2 and menin in hepatocellular carcinoma. J. Hepatol. 2014, 61, 832–839. [Google Scholar] [CrossRef]
- Chang, J.W.; Gwak, S.Y.; Shim, G.A.; Liu, L.; Lim, Y.C.; Kim, J.M.; Jung, M.G.; Koo, B.S. EZH2 is associated with poor prognosis in head-and-neck squamous cell carcinoma via regulating the epithelial-to-mesenchymal transition and chemosensitivity. Oral. Oncol. 2016, 52, 66–74. [Google Scholar] [CrossRef]
- Tang, F.; Tie, Y.; Wei, Y.Q.; Tu, C.Q.; Wei, X.W. Targeted and immuno-based therapies in sarcoma: Mechanisms and advances in clinical trials. Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188606. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mammals | Drosophila | Arabidopsis | Characteristic Domain | Activity | |
|---|---|---|---|---|---|
| PRC1 | RING1A/RING1B Bim-1 | dRing/Sce | AtRING1A/AtRING1B | RING finger domain [49] | E3 ubiquitin ligase activity for H2A |
| PCGF1-6 | Psc | AtBMI1A/ AtBMI1B/ AtBMI1C | RING finger domain | Co-factors for H2A monoubiquitination | |
| CBX2/4/6/7/8 | Pc | EMF1 LHP1(TFL2)VRN1 VAL1/2/3 | Chromodomain | Recognizes and binds to H3K27me3 | |
| PHC1/ PHC2/ PCH3 | Ph | UNKNOWN | Sterile Alpha Motif (SAM) domain and Zinc finger domain | Mediates monoubiquitinaiton of histone H2A [50] | |
| PRC2 | EZH1/2 | E(z) | CLF/ SWN/ MEA | SET domain | H3K27 methyltransferase [51] |
| SUZ12 | Su(z)12 | EMF2/ VRN2/ FIS2 | Zinc finger | Mediates core PRC2 and accessory components’ interaction [51] | |
| EED | Esc | FIE | WD-40 repeat domain | Stabilizes and enhances E(z) [52] | |
| RBAP48/46 | P55/Nurf55 | MSI1-5 | WD-40 repeat domain | Binds to histones and Su(z)12 | |
| EZHIP | tissue-specific cofactor of PRC2 | Limits PRC2-mediated H3K27me3 deposition [53] | |||
| PRC2.1 | PCL1/2/3 | Pcl | PHD finger, TUDOR | Promotes PRC2 recruitment to CpG islands that lack H3K27m3 mark | |
| EPOP or PALI1/2 | |||||
| PRC2.2 | JARID2 | Zinc finger, ARID domain, JmjC and JmjN | Promotes the PRC2 recruitment to chromatin that has PcG-dependent modifications | ||
| AEBP2 | Zinc finger |
| Subgroup | Compound | Clinical Trial | Phase | Clinical Trial Identifier | Status |
|---|---|---|---|---|---|
| EZH2 inhibitors | Tazemetostat | Patients with relapsed, refractory follicular lymphoma | III | NCT04224493 | Recruiting |
| In combination with pembrolizumab for patients with locally advanced or metastatic urothelial carcinoma | I/II | NCT03854474 | Recruiting | ||
| Patients with moderate and severe hepatic impairment with advanced malignancies | I | NCT04241835 | Recruiting | ||
| Patients with refractory B-cell non-Hodgkin’s lymphoma with EZH2 gene mutation | II | NCT03456726 | Active | ||
| Patients with recurrent ovarian or endometrial cancer | II | NCT03348631 | Suspended | ||
| Patients with B-cell lymphoma or advance solid tumors | I | NCT03010982 | Completed | ||
| Patients with mCRPC (+abiraterone/prednisone or enzalutamide) | II | NCT02875548 | Ongoing | ||
| Patients with advanced epithelioid sarcoma in combination with doxorubicin | III | NCT04204841 | Recruiting | ||
| Prelapsed or refractory INI-1 negative tumors or synovial sarcoma, rhabdoid tumors, malignant rhabdoid tumors of ovary | I | NCT02601937 | Recruiting | ||
| In combination with Atezolizumab and Obinutuzumab in relapsed/refractory follicular Lymphoma and diffuse Large B-cell Lymphoma | I | NTC02220842 | Completed | ||
| In combination with doxorubicin and HCI for advanced soft-tissue sarcoma or epitheloid sarcoma | III | NCT04204941 | Recruiting | ||
| CPI-1205 | ProSTAT: Patients with mCRPC in combination with abiraterone/prednisone (ARPI) | II | NCT03480646 | Recruiting | |
| ORIO-E: Patients with advanced solid tumors in combination with ipilimumab | I/II | NCT03525795 | Recruiting | ||
| Patients with B-cell lymphomas | I | NCT02395601 | Completed | ||
| Hepatic impairment advanced malignant solid tumor | I | NCT04241835 | Recruiting | ||
| CPI-0209 | Patients with advanced solid tumors in combination with irinotecam | I/II | NCT04104776 | Recruiting | |
| Valemetostat | Patients with acute myelogenous leukemia or acute lymphocytic leukaemia | I/II | NCT03110354 | Completed | |
| Patients with recurrent SCLC in combination with irinotecam | I/II | NCT03879798 | Recruiting | ||
| Patients with non-Hodgkin lymphoma (NHL) | I | NCT02732275 | Active | ||
| Patients with relapsed/refractory adult T-cell leukaemia or lymphoma | II | NCT04102150 | Active | ||
| Participants with hepatic impairments (single-dose) | I | NCT04276662 | Completed | ||
| PF-06821497 | Patients with follicular lymphoma, CRPC and relapsed/refractory small cell lung cancer (SCLC) | I | NCT03460977 | Recruiting | |
| SHR2554 | Relapsed or refractory mature lymphoid neoplams | I | NCT03741712 | Recruiting | |
| SHR2554 | In combination with SHR3680 (inhibits androgen-mediated translocation of AR) in CRPC | I/II | NCT03460977 | Recruiting | |
| EPZ-6438+ARPI | CELLO-1: Patients with mCRPC who have not received chemotherapy | I/II | NCT04179864 | Recruiting | |
| Dual EZH2 inhibitors | DS-3201b | Patients with hepatic impairment (single dose DS-3201b) | I | NCT04276662 | Recruiting |
| Patients with adult T-cell leukemia/lymphoma | II | NCT04102150 | Recruiting | ||
| Patients with SCLC with irinotecam | I/II | NCT3879798 | Recruiting | ||
| Patients with lymphomas | I | NCT02732275 | Recruiting | ||
| EED inhibitors | MAK683 | Patients with advanced malignancies (diffuse large B cell lymphoma) | I/II | NCT02900561 | Recruiting |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
German, B.; Ellis, L. Polycomb Directed Cell Fate Decisions in Development and Cancer. Epigenomes 2022, 6, 28. https://doi.org/10.3390/epigenomes6030028
German B, Ellis L. Polycomb Directed Cell Fate Decisions in Development and Cancer. Epigenomes. 2022; 6(3):28. https://doi.org/10.3390/epigenomes6030028
Chicago/Turabian StyleGerman, Beatriz, and Leigh Ellis. 2022. "Polycomb Directed Cell Fate Decisions in Development and Cancer" Epigenomes 6, no. 3: 28. https://doi.org/10.3390/epigenomes6030028
APA StyleGerman, B., & Ellis, L. (2022). Polycomb Directed Cell Fate Decisions in Development and Cancer. Epigenomes, 6(3), 28. https://doi.org/10.3390/epigenomes6030028