
Global m6A RNA Methylation in SARS-CoV-2 Positive Nasopharyngeal Samples in a Mexican Population: A First Approximation Study

 , , , and
, , , and 
Abstract
:1. Introduction
2. Results
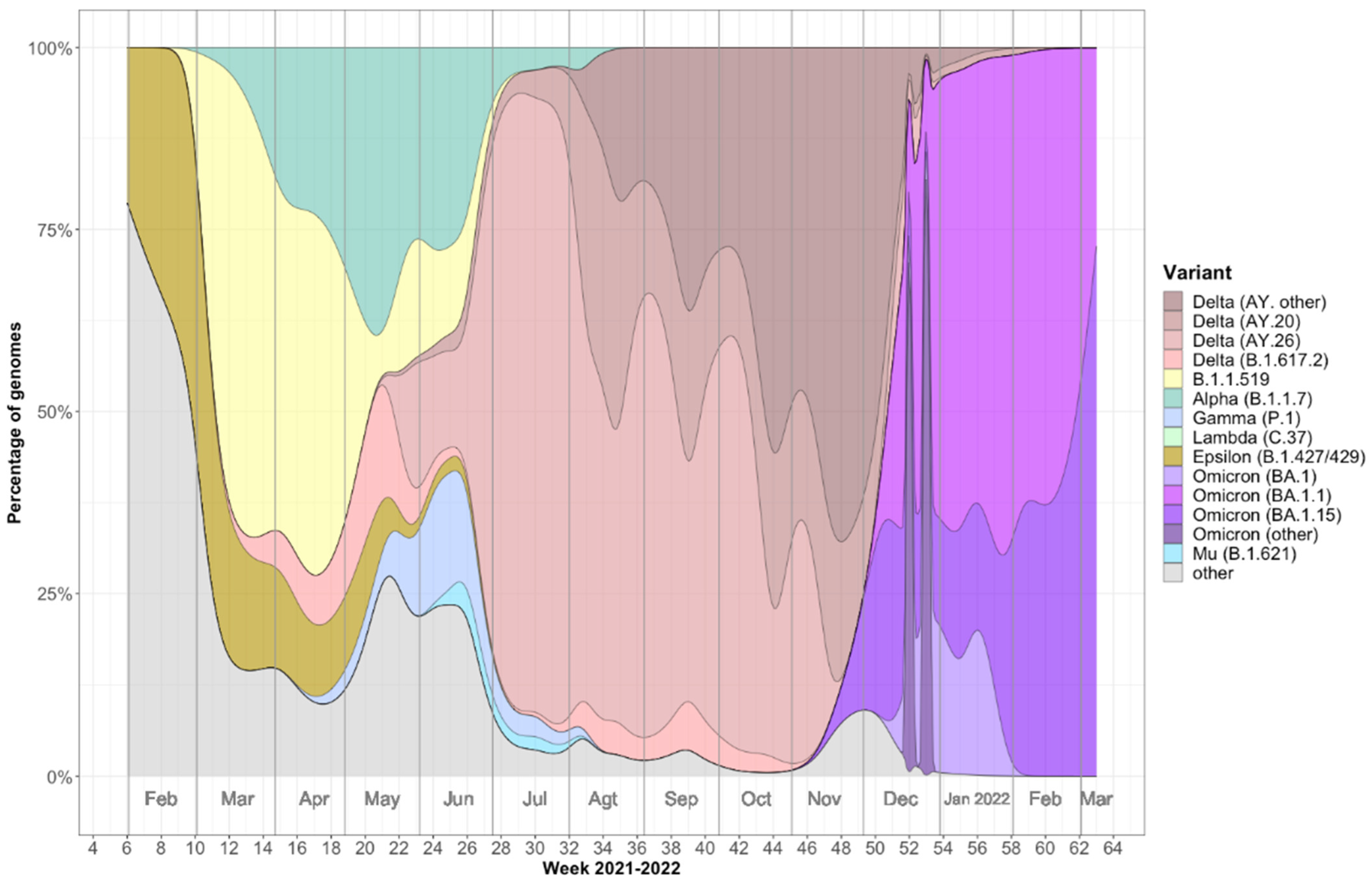
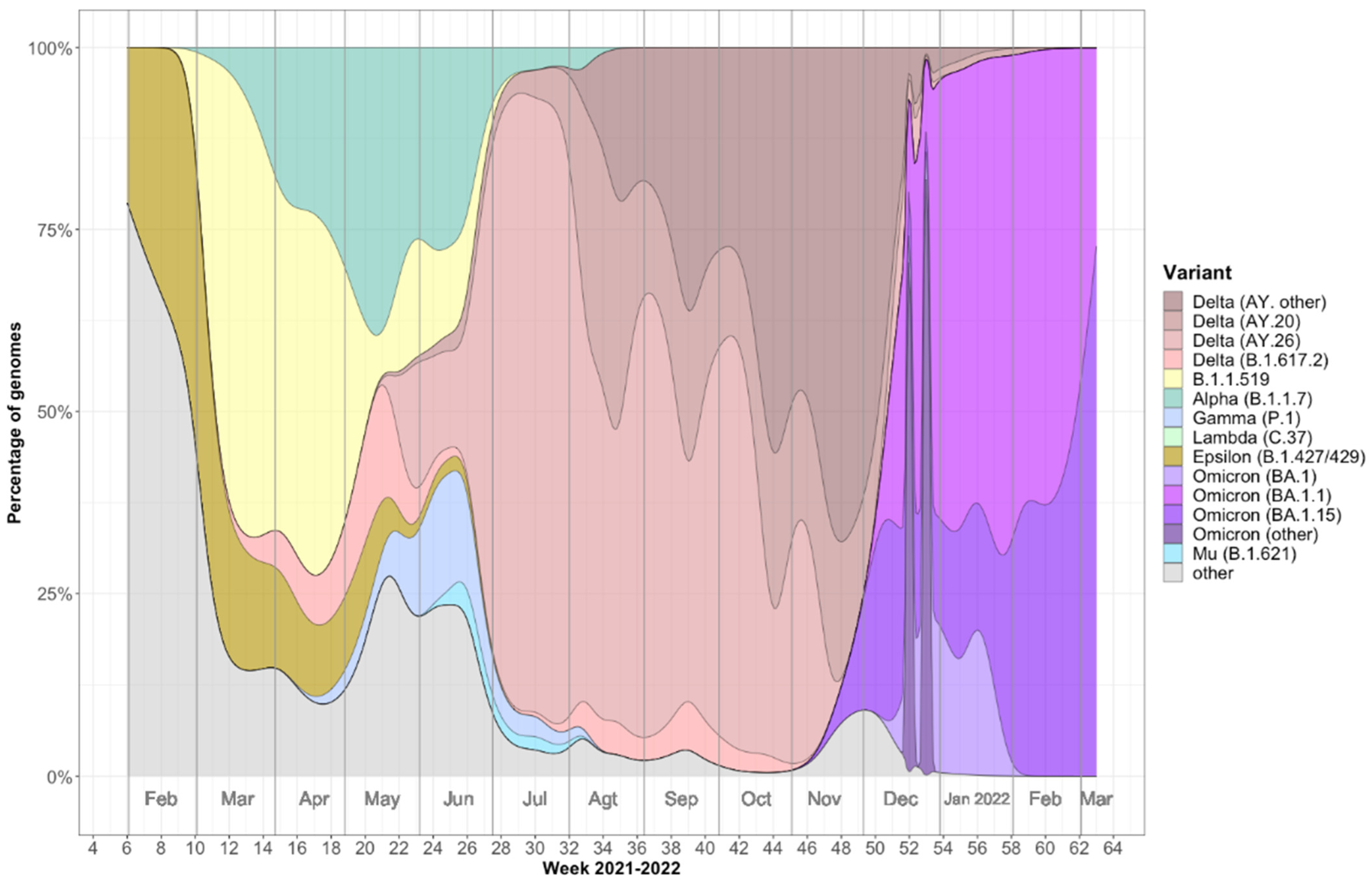
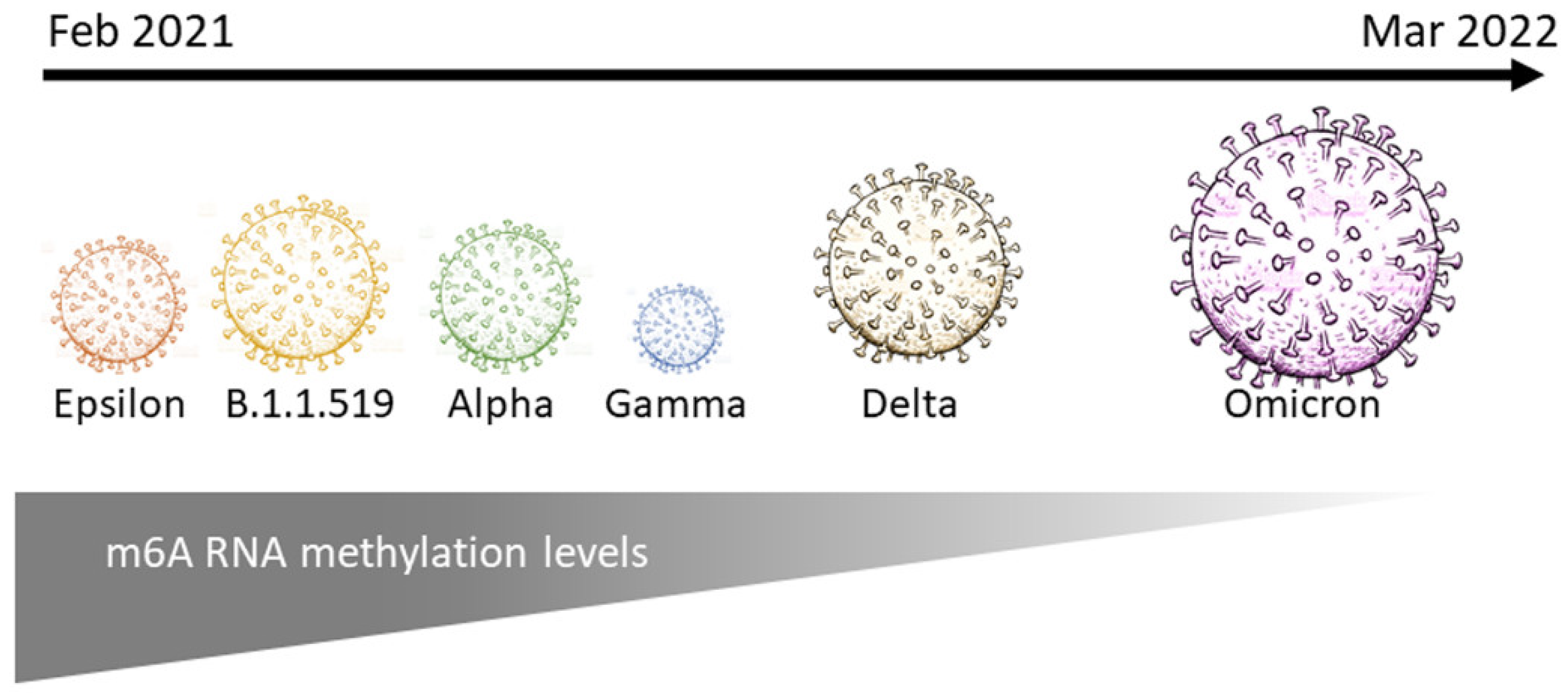
2.1. Viral Variants Detected in Mazatlán, Mexico during the COVID-19 Pandemic (February 2021–March 2022)
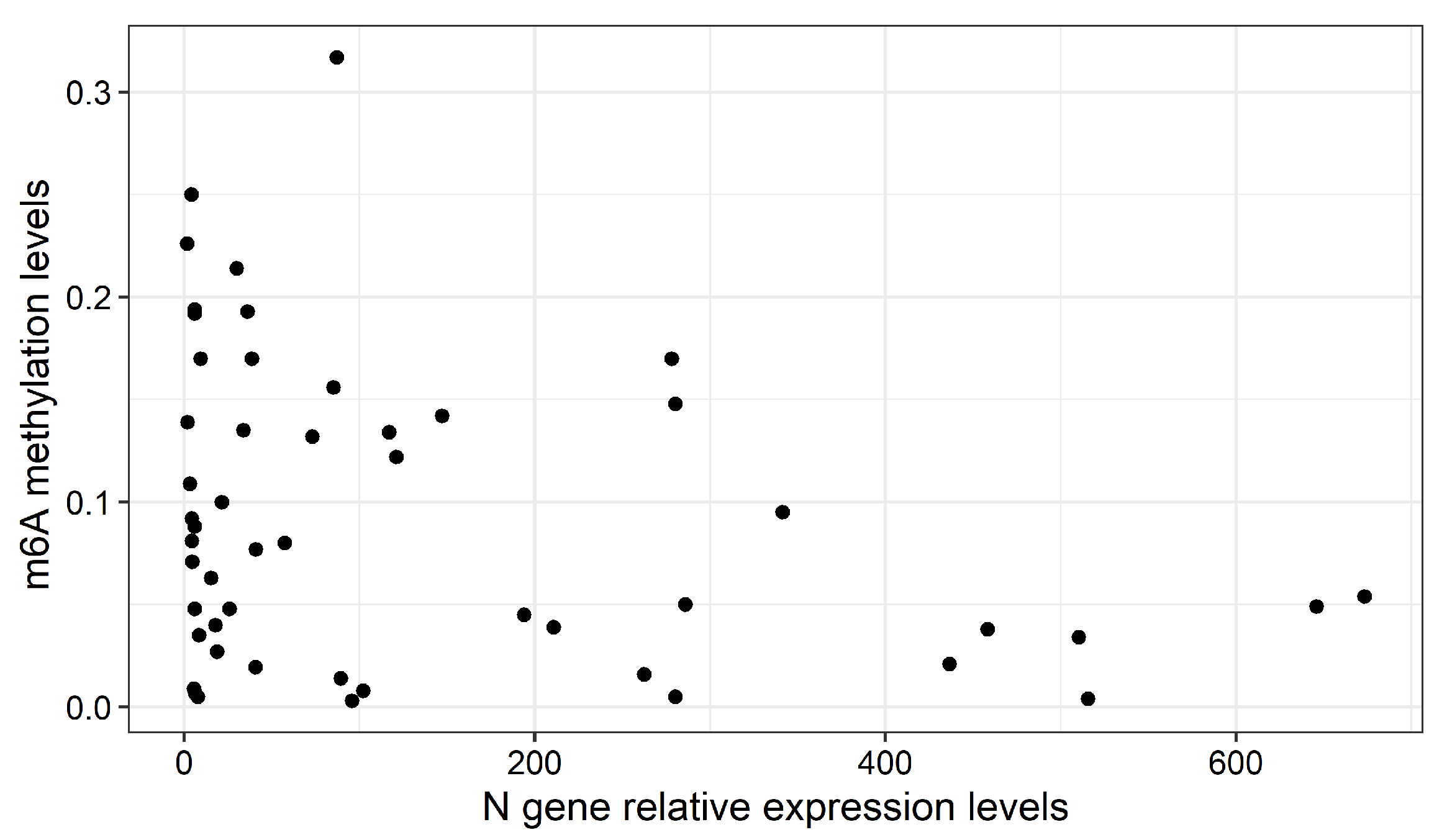
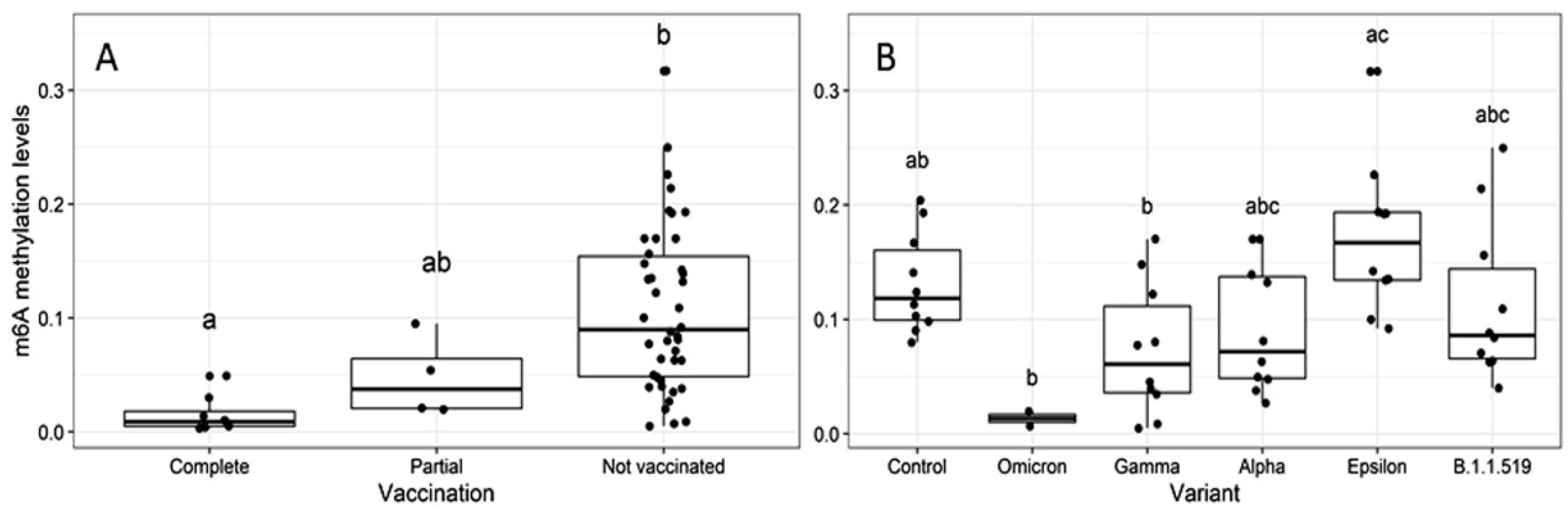
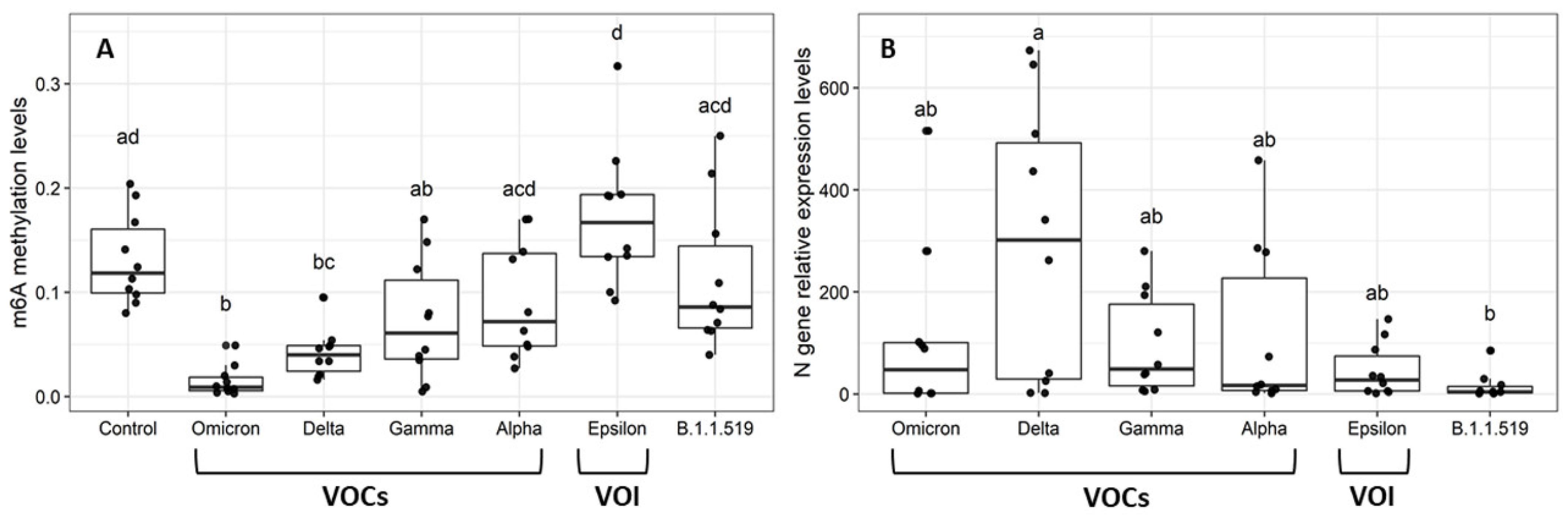
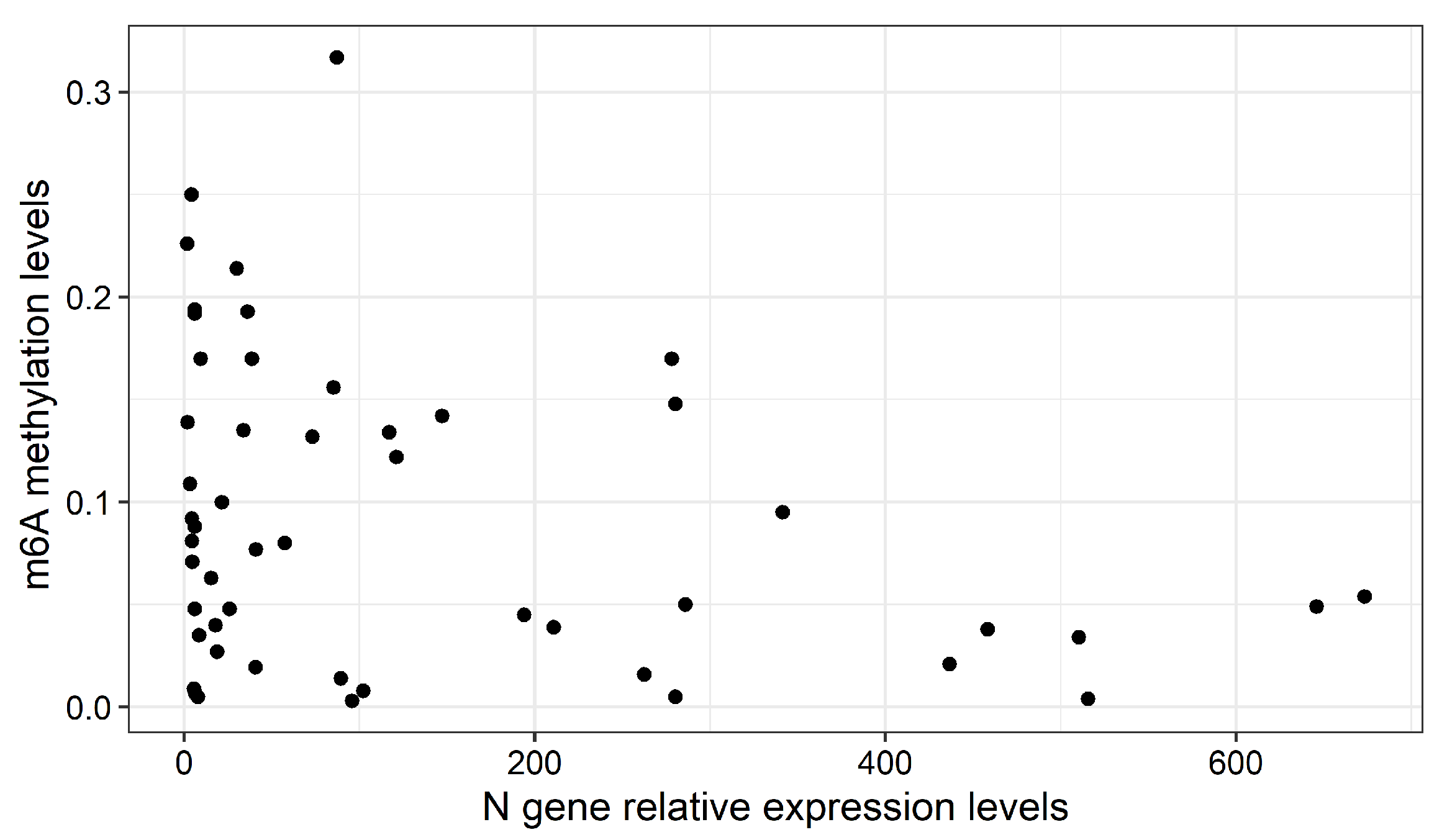
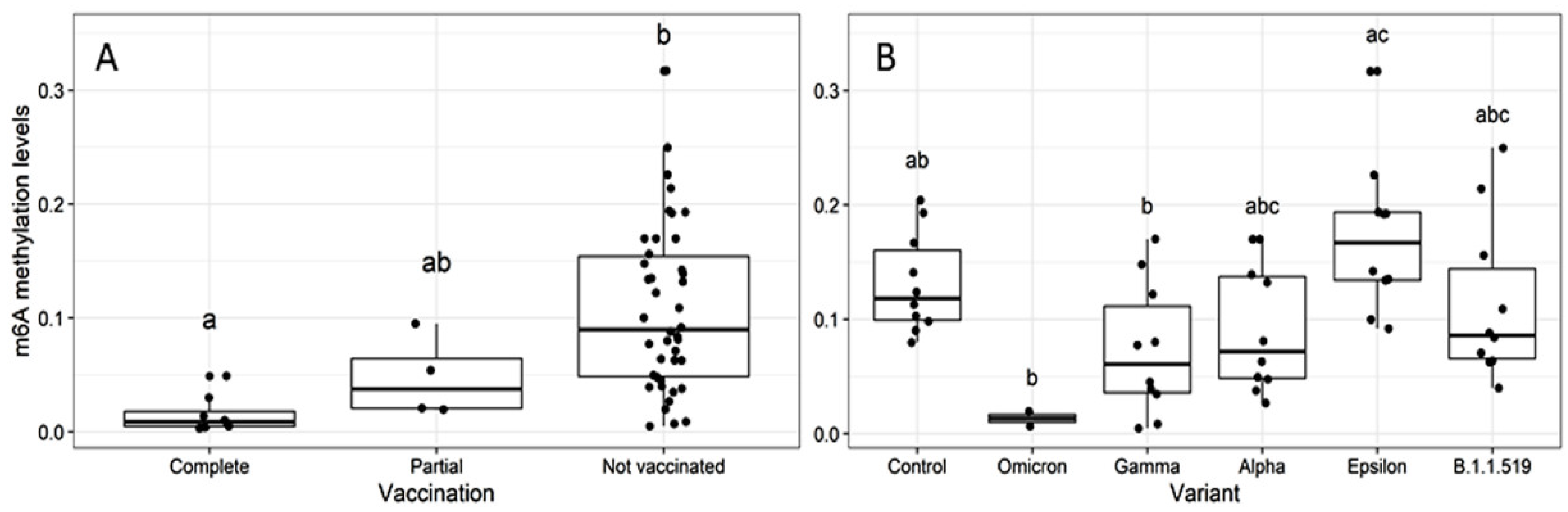
2.2. Global m6A Levels and Nucleocapsid (N) Gene Expression in Nasopharyngeal Samples
3. Discussion
4. Materials and Methods
4.1. Samples
4.2. Identification and Sequencing of SARS-CoV-2 Positive Samples
4.3. Global RNA Methylation Assay
4.4. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mariano, G.; Farthing, R.J.; Lale-Farjat, S.L.M.; Bergeron, J.R.C. Structural characterization of SARS-CoV-2: Where we are, and where we need to be. Front. Mol. Biosci. 2020, 7, 605236. [Google Scholar] [CrossRef] [PubMed]
- Bai, Z.; Cao, Y.; Liu, W.; Li, J. The SARS-CoV-2 nucleocapsid protein and its role in viral structure, biological functions, and a potential target for drug or vaccine mitigation. Viruses 2021, 13, 1115. [Google Scholar] [CrossRef] [PubMed]
- Le Bert, N.; Tan, A.T.; Kunasegaran, K.; Tham, C.Y.L.; Hafezi, M.; Chia, A.; Chng, M.H.Y.; Lin, M.; Tan, N.; Linster, M.; et al. SARS-CoV-2-specific T cell immunity in cases of COVID-19 and SARS, and uninfected controls. Nature 2020, 584, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, Q.; Guo, D. Emerging coronaviruses: Genome structure, replication, and pathogenesis. J. Med. Virol. 2020, 92, 418–423. [Google Scholar] [CrossRef]
- Davies, N.G.; Abbott, S.; Barnard, R.C.; Jarvis, C.I.; Kucharski, A.J.; Munday, J.D.; Pearson, C.A.B.; Russell, T.W.; Tully, D.C.; Washburne, A.D.; et al. Estimated transmissibility and impact of SARS-CoV-2 lineage B.1.1.7 in England. Science 2021, 372, eabg3055. [Google Scholar] [CrossRef]
- Bager, P.; Wohlfahrt, J.; Fonager, J.; Rasmussen, M.; Albertsen, M.; Michaelsen, T.Y.; Møller, C.H.; Ethelberg, S.; Legarth, R.; Button, M.; et al. Risk of hospitalisation associated with infection with SARS-CoV-2 lineage B.1.1.7 in Denmark: An observational cohort study. Lancet Infect. Dis. 2021, 21, 1507–1517. [Google Scholar] [CrossRef]
- Desrosiers, R.; Friderici, K.; Rottman, F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc. Natl. Acad. Sci. USA 1974, 71, 3971–3975. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Wong, C.M. The emerging roles of N6-methyladenosine (m6A) deregulation in liver carcinogenesis. Mol. Cancer 2020, 19, 44. [Google Scholar] [CrossRef] [Green Version]
- Manners, O.; Baquero-Perez, B.; Whitehouse, A. m6A: Widespread regulatory control in virus replication. Biochim. Biophys. Acta Gene Regul. Mech. 2019, 1862, 370–381. [Google Scholar] [CrossRef]
- Fu, Y.; Dominissini, D.; Rechavi, G.; He, C. Gene expression regulation mediated through reversible m6A RNA methylation. Nat. Rev. Genet. 2014, 15, 293–306. [Google Scholar] [CrossRef]
- Ke, S.; Pandya-Jones, A.; Saito, Y.; Fak, J.J.; Vågbø, C.B.; Geula, S.; Hanna, J.H.; Black, D.L.; Darnell, J.E., Jr.; Darnell, R.B. m6A mRNA modifications are deposited in nascent pre-mRNA and are not required for splicing but do specify cytoplasmic turnover. Genes Dev. 2017, 31, 990–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaccara, S.; Ries, J.R.; Jaffrey, R.S. Reading, writing and erasing mRNA methylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 608–624. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Hui, H.; Bray, B.; Gonzalez, G.M.; Zeller, M.; Anderson, K.G.; Knight, R.; Smith, D.; Wang, Y.; Carlin, A.F.; et al. METTL3 regulates viral m6A RNA modification and host cell innate immune responses during SARS-CoV-2 infection. Cell Rep. 2021, 35, 109091. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Xu, Y.P.; Li, K.; Ye, Q.; Zhou, H.Y.; Sun, H.; Li, X.; Yu, L.; Deng, Y.Q.; Li, R.T.; et al. The m6A methylome of SARS-CoV-2 in host cells. Cell Res. 2021, 31, 404–414. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Hao, H.; Ma, L.; Zhang, Y.; Hu, X.; Chen, Z.; Liu, D.; Yuan, J.; Hu, Z.; Guan, W. Methyltransferase-like 3 modulates severe acute respiratory syndrome coronavirus-2 RNA N6-methyladenosine modification and replication. mBio 2021, 12, e01067-21. [Google Scholar] [CrossRef]
- Burgess, H.M.; Depledge, D.P.; Thompson, L.; Srinivas, K.P.; Grande, R.C.; Vink, E.I.; Abebe, J.S.; Blackaby, W.P.; Hendrick, A.; Albertella, M.R.; et al. Targeting the m6A RNA modification pathway blocks SARS-CoV-2 and HCoV-OC43 replication. Genes Dev. 2021, 35, 1005–1019. [Google Scholar] [CrossRef]
- Meng, Y.; Zhang, Q.; Wang, K.; Zhang, X.; Yang, R.; Bi, K.; Chen, W.; Diao, H. RBM15-mediated N6-methyladenosine modification affects COVID-19 severity by regulating the expression of multitarget genes. Cell Death Dis. 2021, 12, 732. [Google Scholar] [CrossRef]
- An, S.; Xie, Z.; Liao, Y.; Jiang, J.; Dong, W.; Yin, F.; Li, W.X.; Ye, L.; Lin, J.; Liang, H. Systematic analysis of clinical relevance and molecular characterization of m6A in COVID-19 patients. Genes Dis. 2022, in press. [Google Scholar] [CrossRef]
- Liu, C.; Yang, Z.; Li, R.; Wu, Y.; Chi, M.; Gao, S.; Sun, X.; Meng, X.; Wang, B. Potential roles of N6-methyladenosine (m6A) in immune cells. J. Transl. Med. 2021, 19, 251. [Google Scholar] [CrossRef]
- Lu, M.; Xue, M.; Wang, H.T.; Kairis, E.L.; Ahmad, S.; Wei, J.; Zhang, Z.; Liu, Q.; Zhang, Y.; Gao, Y.; et al. Nonsegmented negative-sense RNA viruses utilize N6-methyladenosine (m6A) as a common strategy to evade host innate immunity. J. Virol. 2021, 95, e01939-20. [Google Scholar] [CrossRef]
- Brocard, M.; Ruggieri, A.; Locker, N. m6A RNA methylation, a new hallmark in virus-host interactions. J. Gen. Virol. 2017, 98, 2207–2214. [Google Scholar] [CrossRef] [PubMed]
- Statement on Omicron Sublineage BA.2. Available online: https://www.who.int/news/item/22-02-2022-statement-on-omicron-sublineage-ba.2 (accessed on 29 March 2022).
- Da Silva, S.; de Lima, S.C.; da Silva, R.C.; Kohl, A.; Pena, L. Viral load in COVID-19 patients: Implications for prognosis and vaccine efficacy in the context of emerging SARS-CoV-2 variants. Front. Med. 2022, 8, 836826. [Google Scholar] [CrossRef] [PubMed]
- McCallum, M.; Czudnochowski, N.; Rosen, L.E.; Zepeda, S.K.; Bowen, J.E.; Walls, A.C.; Hauser, K.; Joshi, A.; Stewart, C.; Dillen, J.R.; et al. Structural basis of SARS-CoV-2 Omicron immune evasion and receptor engagement. Science 2022, 375, 864–868. [Google Scholar] [CrossRef] [PubMed]
- Campos, J.H.C.; Maricato, J.T.; Braconi, C.T.; Antoneli, F.; Janini, L.M.R.; Briones, M.R.S. Direct RNA sequencing reveals SARS-CoV-2 m6A sites and possible differential drach motif methylation among variants. Viruses 2021, 13, 2108. [Google Scholar] [CrossRef]
- Qiu, X.; Hua, X.; Li, Q.; Zhou, Q.; Chen, J. m6A regulator-mediated methylation modification patterns and characteristics of immunity in blood leukocytes of COVID-19 patients. Front. Immunol. 2021, 12, 774776. [Google Scholar] [CrossRef]
- Gensous, N.; Franceschi, C.; Blomberg, B.B.; Pirazzini, C.; Ravaioli, F.; Gentilini, D.; Di Blasio, A.M.; Garagnani, P.; Frasca, D.; Bacalini, M.G. Responders and non-responders to influenza vaccination: A DNA methylation approach on blood cells. Exp. Gerontol. 2018, 105, 94–100. [Google Scholar] [CrossRef]
- Zimmermann, M.T.; Oberg, A.L.; Grill, D.E.; Ovsyannikova, I.G.; Haralambieva, I.H.; Kennedy, R.B.; Poland, G.A. System-wide associations between DNA methylation, gene expression, and humoral immune response to influenza vaccination. PLoS ONE 2016, 11, e0152034. [Google Scholar] [CrossRef]
- Luo, C.H.; Morris, C.P.; Sachithanandham, J.; Amadi, A.; Gaston, D.C.; Li, M.; Swanson, N.J.; Schwartz, M.; Klein, E.Y.; Pekosz, A.; et al. Infection with the SARS-CoV-2 Delta variant is associated with higher recovery of infectious virus compared to the Alpha variant in both unvaccinated and vaccinated individuals. Clin. Infect. Dis. 2021, 18, ciab986. [Google Scholar] [CrossRef]
- Ong, S.; Chiew, C.J.; Ang, L.W.; Mak, T.M.; Cui, L.; Toh, M.; Lim, Y.D.; Lee, P.H.; Lee, T.H.; Chia, P.Y.; et al. Clinical and virological features of SARS-CoV-2 variants of concern: A retrospective cohort study comparing B.1.1.7 (Alpha), B.1.315 (Beta), and B.1.617.2 (Delta). Clin. Infect. Dis. 2021, ciab721. [Google Scholar] [CrossRef]
- Kissler, S.M.; Fauver, J.R.; Mack, C.; Tai, C.G.; Breban, M.I.; Watkins, A.E.; Samant, R.M.; Anderson, D.J.; Metti, J.; Khullar, G.; et al. Viral dynamics of SARS-CoV-2 variants in vaccinated and unvaccinated persons. N. Engl. J. Med. 2021, 385, 2489–2491. [Google Scholar] [CrossRef]
- Duarte, C.M.; Ketcheson, D.I.; Eguíluz, V.M.; Agustí, S.; Fernández-Gracia, J.; Jamil, T.; Laiolo, E.; Gojobori, T.; Alam, I. Rapid evolution of SARS-CoV-2 challenges human defenses. Sci. Rep. 2022, 12, 6457. [Google Scholar] [CrossRef] [PubMed]
- Rao, X.; Huang, X.; Zhou, Z.; Lin, X. An improvement of the 2ˆ(-delta delta CT) method for quantitative real-time polymerase chain reaction data analysis. Biostat. Bioinform. Biomath. 2013, 3, 71–85. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variant | Average Age ± SD | Sex Distribution (n = 70) | Vaccination Status (n = 64) | |
|---|---|---|---|---|
| Males | Females | |||
| Control | 46 ± 7 | 5 | 5 | Not vaccinated (n = 10) |
| Omicron | 42 ± 8 | 5 | 5 | Complete (n = 8) Not vaccinated (n = 2) |
| Delta | 39 ± 10 | 5 | 5 | Partial (n = 4) |
| Gamma | 44 ± 7 | 5 | 5 | Not vaccinated (n = 10) |
| Alpha | 49 ± 8 | 5 | 5 | Not vaccinated (n = 10) |
| Epsilon | 45 ± 10 | 5 | 5 | Not vaccinated (n = 10) |
| B.1.1.519 | 47 ± 11 | 5 | 5 | Not vaccinated (n = 10) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Batista-Roche, J.L.; Gómez-Gil, B.; Lund, G.; Berlanga-Robles, C.A.; García-Gasca, A. Global m6A RNA Methylation in SARS-CoV-2 Positive Nasopharyngeal Samples in a Mexican Population: A First Approximation Study. Epigenomes 2022, 6, 16. https://doi.org/10.3390/epigenomes6030016
Batista-Roche JL, Gómez-Gil B, Lund G, Berlanga-Robles CA, García-Gasca A. Global m6A RNA Methylation in SARS-CoV-2 Positive Nasopharyngeal Samples in a Mexican Population: A First Approximation Study. Epigenomes. 2022; 6(3):16. https://doi.org/10.3390/epigenomes6030016
Chicago/Turabian StyleBatista-Roche, Jorge Luis, Bruno Gómez-Gil, Gertrud Lund, César Alejandro Berlanga-Robles, and Alejandra García-Gasca. 2022. "Global m6A RNA Methylation in SARS-CoV-2 Positive Nasopharyngeal Samples in a Mexican Population: A First Approximation Study" Epigenomes 6, no. 3: 16. https://doi.org/10.3390/epigenomes6030016
APA StyleBatista-Roche, J. L., Gómez-Gil, B., Lund, G., Berlanga-Robles, C. A., & García-Gasca, A. (2022). Global m6A RNA Methylation in SARS-CoV-2 Positive Nasopharyngeal Samples in a Mexican Population: A First Approximation Study. Epigenomes, 6(3), 16. https://doi.org/10.3390/epigenomes6030016