Abstract
According to recent findings, variances in autism spectrum disorder (ASD) risk factors might be determined by several factors, including molecular genetic variants. Accumulated evidence has also revealed the important role of biological and chemical pathways in ASD aetiology. In this paper, we assess several reviews with regard to their quality of evidence and provide a brief outline of the presumed mechanisms of the genetic, epigenetic, and environmental risk factors of ASD. We also review some of the critical literature, which supports the basis of each factor in the underlying and specific risk patterns of ASD. Finally, we consider some of the implications of recent research regarding potential molecular targets for future investigations.
1. Introduction
Autistic disorder—or, more broadly, autism spectrum disorders (ASD)—is a lifelong syndrome with a childhood inception, characterised by challenges in social interaction and communication, the presence of stereotype rigidity, and ritualistic/repetitive patterns of behaviour [1]. ASD syndrome has been described by many researchers since it was first observed in 1943 by Kanner, who described 11 cases of children, mostly boys, with severe social and variable language dysfunction [2]. The global prevalence of autism is around one percent, with a male preponderance ratio of four to one [3]. Approximately 50 percent of patients with ASD present with intellectual disabilities (ID), and comorbidity with neurodevelopmental and psychiatric conditions is common [3,4]. These psychiatric and medical conditions may include depression, anxiety, attention deficit hyperactivity disorder (ADHD), sleep illnesses, and gastrointestinal symptoms [5,6,7]. In addition, more than 35 percent of autistic individuals suffer from epilepsy, and epileptic EEG abnormalities can often be found in patients even without the occurrence of seizures [8,9]. However, the main diagnostic principle of autistic disorder is based upon the consensus opinion of expert clinicians, albeit with the consideration of the modified criteria under the new ASD heading [10].
Incontrovertible evidence found in twin studies has proven that genetic factors contribute to susceptibility to this disorder [11,12]. The concordance rate of autism is up to 30 percent in dizygotic twins (DZ), 70–90 percent in monozygotic twins (MZ), and 3–19 percent in siblings in general [13,14,15,16,17]. The higher disorder cooccurrence in MZ twins (identical in their genetic material) compared to DZ twins (sharing about 50 percent of their genetic material like nontwin siblings) seems to support a genetic aetiology [10]. In terms of aetiology, for example, family studies comparing the frequency of autism between the first-degree relatives of affected individuals and the wider population should consider the involvement of genetics in the disease. Another area of evidence that supports the genetic aetiology of ASD can be found in studies on rare genetic syndromes with comorbid autism diagnoses. Findings in these studies have shown that sharing the same in utero environment has a more impactful role than genetics on siblings; however, the mechanism underlying this notion is still unclear [10,14,18]. Moreover, it has been shown that molecular alterations can contribute to ASD aetiology, including altered genetic and epigenetic regulation. Epigenetic mechanisms provide new and critical ways to examine risk estimates for neurodevelopmental disorders (NDDs) beyond genetic risk alone. These mechanisms may include DNA methylation (DNAm), histone modification, and ATP-dependent chromatin remodelling; however, the latter system also modulates the transcriptome and splicing processes, thus impacting transcription initiation and the binding of transcription factors [19,20]. Genomic alterations of genes involved in epigenetics, for example, single-nucleotide polymorphisms (SNPs) or copy number variants (CNVs), can lead to epigenetic dysregulation and, ultimately, ASD (Figure 1).
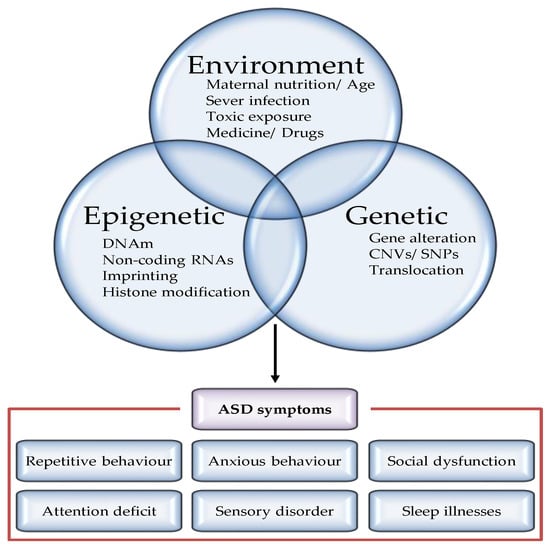
Figure 1.
Factors regulating the pathogenesis of autism spectrum disorders. Although definitive etiology and pathogenesis underlying ASD have not yet been identified, accumulated study has recognized various risk factors, including nature (genes or epigenes) and nurture (environment) factors. Both genetic and epigenetic factors modulate the penetrance of risk genes, resulting in a highly heterogeneous disease phenotype for similar pathogenic variants. Examples of genetic modulators include CNV and mutations. Examples of epigenetic modifiers include methylation, microRNAs (miRNAs), and chromatin remodelling. Furthermore, majority of the environmental factors leading to epigenetic changes on chromatin cause increased ASD risk.
There are over 600 confirmed human genes related to NDD (such as ID and ASD), including DNA methyltransferase 3A (DNMT3A), HECT, UBA, WWE domain-containing E3 ubiquitin-protein ligase 1 (HUWE1), and chromodomain helicase DNA-binding protein 8 (CHD8) [21,22,23]. Emerging evidence suggests that there is a fundamental genetic interference risk in neurodevelopmental and neuropsychiatric disorders [24,25,26]. Many of these genes are essential to the molecular pathways involved in epigenetic regulation, whereas others are encoded to proteins involved in neuronal and synaptic pathways [23,26,27]. However, there is no single underlying cause of ASD or its developmentally related challenges. For a better understanding of the multifactorial aetiologies of ASD, extra work is needed to elaborate on the natural history of its molecular basis and its regulation during significant periods of human development. Apparently, the interaction between genetic and environmental risk factors (GxE) in humans is crucial and is likely facilitated by vital mechanisms such as epigenetics. Thus, the role of genetics, epigenetics, and the environment in ASD risk factors, as well as the collective interaction between them, will require further investigation in order to better classify and diagnose the disorder.
2. Body of the Paper
Currently, ASD is considered to have a strong genetic component, likely resulting from interactions between various genes. Previous and current twin studies have reported an estimate of more than 30 percent heritability [14,28,29]. Due to the use of modern technology, such as next-generation sequencing (NGS), many key points of genetic variability in patients with ASD (as compared to the general population) have emerged. Sequence analysis methods, such as whole-genome sequencing (WGS), have many advantages, and they have been used to discover ASD-relevant mutations in individuals within affected families [30,31]. One study on families of four (parents and two affected siblings) observed similar mutations in 31 percent of siblings compared to ASD patients. Thus, such studies underline the genetic heterogeneity of the disorder even within families [30]. However, for all the advantages of genomic sequencing, genetic variant classification still poses an important challenge. More than 200 ASD-associated genes have been specified in various studies, and risk variants have only been recorded in 25–40 percent of cases [32,33,34,35,36]. Nevertheless, in only one percent of ASD cases can a single genetic mutation or copy number variant be correlated, and, thus, the ASD phenotype is unpredictably impenetrable. According to recent studies, more than one thousand autism genes have been investigated. Using the SFARI gene platform, approximately 212 genes (environment interacting genes) have been studied [37,38]. More recently, studies reported that dysregulated gene expression is associated with inflammatory cytokines and behavioural severity in ASD [39,40]. Overall, various genetic variants and six risk-located loci (1q21.1, 3q29, 7q11.23, 16p11.2, 15q11.2–13, 22q11.2) are recognised as being linked with ASD [41,42,43].
Recent genomic studies have identified many common and rare inherited variants in ASD families [44,45]. In autism, the majority of the genetic alteration tracked by heritability is accounted for by common genetic variation, as quantified by SNP-based heritability which is estimated to be around 50 percent [45]. As with other common neuropsychiatric conditions, common genetic variants contributing to autism have small effect sizes, requiring large population studies for identification. The latest and largest published ASD genome-wide association study (GWAS) reported five genome-wide significant loci in an analysis of 18,381 ASD patients [44]. Larger sample sizes will be necessary to identify further loci, which are expected to be found given the substantial estimate of SNP-based heritability [45,46].
Since 2011, a large number of studies have already been published using WGS in ASD populations, with the identification of rare variants in autism susceptibility genes already described or newly identified [30,47,48,49]. These findings have led to an exponential increase in the number of genes potentially related to autism. However, the increase in de novo events in individuals with ASD is widely described in the literature [50], and several studies highlighted the importance of de novo mutations in a situation of family mutational burden due to the presence of common and rare variants inherited from parents [46,50,51].
Several studies have shown that various ASD genomic risk variants are involved in congregating pathways relevant to the natural basis of ASD. These include cell proliferation and differentiation, neural development, synaptic activity, transcriptional regulators, and chromatin modifiers [41,51,52,53]. Interestingly, many ASD-associated genes are expressed in brain tissues during embryogenesis and formation. These functional groups relate to the neurocognitive phenotype of ASD and can improve our understanding of its molecular mechanisms [25,53]. On the other hand, studies on ASD risk-associated genes initiated by genetic mutation may also help in this attempt at comprehension; their findings suggest that some ASD-related genes can increase the risk factor for other neurological conditions, including schizophrenia, motor impairment, epilepsy, sleep disturbance, ADHD, and ID [25,42,54].
In addition, strong evidence supports the role of epigenetics in the molecular aetiology of ASD, some of which indicates that other genetic syndromes may have direct control over its expression. Studies have proven that they contribute to syndromic ASD in approximately 15 percent of cases [55,56]. Functionally, some epigenetic profiles have been reported to be associated with an increased risk of ASD: for example, histone deacetylases (HDACs); lysine demethylases; proteins containing bromo-, chromo-, or Tudor domains; DNA methyl transferases (DNMTs); histone methyltransferases; acetyltransferases; and chromatin remodelling factors. While the indirect effects of epigenes have also been observed in noncoding RNA (ncRNA), the processing and recruitment of methyl-CpG-binding proteins (MBDs) have also been observed, and these are critical in histone modification and transcription regulation [57,58].
Other lines of evidence implicate other epigenetic markers in the expression of ASD, such as differentially methylated variants (DMVs) at specific CpG sites (CpGs) or differentially methylated regions (DMRs). Studies have identified multiple CpGs in a variety of tissue types, including whole blood [59], post-mortem brain tissue [60,61,62], ectodermal cells [63], and lymphoblastoid cell lines (19), as well as sperm from the fathers of children with ASD [64]. However, in concordance with studies on blood–brain DNA methylation, these high correlations have not been frequently observed at specific sites across tissues, which is likely due to genetic influences [65,66]. Several studies have reported DNAm changes in the promoter region in different tissues taken from ASD samples, which has helped identify candidate genes, including methyl-CpG binding protein 2 (MECP2), reelin (RELN), glutamic acid decarboxylase 65 (GAD65), oxytocin receptor (OXTR), ubiquitin-protein ligase E3A (UBE3A), and SHANK3 [67,68,69,70,71,72]. The effect of changes in DMV at specific promoter CpG regions has been found to be modest (approximately twofold) in targeted genes, with a significant gain of methylation (GOM) overall and in a sex-specific manner. Other studies on DNAm have demonstrated the replication of differentially methylated positions (DMPs), which are hypomethylated in the 3′ untranslated region (3′UTR) of genes in the brain samples of ASD individuals, including chromosome 11 open reading frame 21 (C11orf21), proline-rich transmembrane protein 1 (PRRT1), and tetraspanin 32 (TSPAN32) [61,62]. Other results emphasise the presence of functionally relevant genes, such as phosphatase and tensin homolog (PTEN) [73], AT-rich interaction domain 1B (ARID1B) [74], N-methyl-D-aspartate (NMDA) [75], glutamate ionotropic receptor NMDA type subunit 2B (GRIN2B) [76], neurexin 1 (NRXN1) [64], and PRRT1 [61], which have critical roles in physiological function and other molecular pathways [77].
Previous studies have reported the enrichment of genome-wide results by primarily using quantitative trait loci (QTLs) for the purposes of gene expression; supposing these polymorphisms present some ASD risk through regulatory mechanisms, QTLs offer insights into the functional biology of GWAS variants [78,79,80]. Besides investigating the enrichment of polymorphisms that control epigenetic markers (for example, DNAm), understanding the regulatory effects of the ASD epigenome will clearly answer whether the disease’s genetic risks act, in part, through epigenetic regulation. Moreover, the presence of DNAm, detected using SNP profiling or methylation QTLs (meQTLs), has been reported in both autism-associated GWAS and cases of schizophrenia and bipolar disorder, which may indicate genetic overlap with ASD [78,79,81,82,83].
Genetic expression levels and epigenetic markers are molecular sensors for many environmental factors, including diet and chemical toxins, over the course of human development [84]. At critical times throughout development, typical cell programming can be dysregulated by environmental exposures, either exogenous or endogenous, leading to unfavourable long-term health outcomes [85]. Accumulated evidence from observational studies has demonstrated the association of endogenous environmental factors with ASD risks, for example, paternal and maternal age [86,87,88]. Combining these factors, along with genetic and epigenetic findings, has helped in identifying several de novo mutations in such disorders, and the majority of cases have been found to be related to paternal age [33,35]. Furthermore, modest increases in ASD and ADHD risk have been found to be associated with maternal prenatal stress [89,90]. However, while OXTR methylation outcomes were predicted in many studies, nevertheless, correlations between maternal stress and autistic traits have not been observed in genetic and methylation changes in the gene [91,92,93]. Prenatal maternal nutrition, particularly with respect to the folate compound, has been identified as reducing the risk of ASD through its critical role in one-carbon metabolism (OCM) and methionine cycles [94,95,96,97]. At the molecular level, the recycling of homocysteine generates amino acids (cysteine and methionine), which are essential in the process of methylation and antioxidative capacity. This biological process accelerates the formation of S-adenosyl methionine (SAM), supporting epigenetic mechanisms that are important for neurobehavioral development and preventing ASD via nutritional supplements [98]. Still, the molecular mechanisms by which these elements reduce ASD risk have not yet been fully illustrated.
Moreover, exogenous exposures, for example, smoking, medication (valproic acid, selective serotonin reuptake inhibitors), alcohol, zinc deficiency, metal ion toxicity, viral infection, and chemical agents (pesticides, metals, bisphenol A), are often associated with adverse foetal neurodevelopmental outcomes [74,99]. However, the alcohol and smoking risk factors are irrelevant with respect to ASD studies, which could be related to differences in exposure conditions between mother and foetus [100]. In addition, zinc is known to be an essential element in foetal growth and development during pregnancy, as well as in the development of children in general [101,102]. Thus, an extended deficiency of zinc in pregnant women might lead to dysfunction in embryonic growth and development, as well as in synaptic systems. Several reports have demonstrated that the absence of zinc prevents the formation of scaffold structures in the ProSAP/Shank family of proteins, which are the key regulator molecules in synapses [103]. A transgenic study of Shank3+/- and Shank-/- deficient mice using a prenatal zinc-deficient autism animal model indicated diverse brain region abnormalities in different models of ASD [104].
Finally, interactions between the genome and epigenome will allow scientists to enhance methylation studies through increased genomic coverage, expanding our knowledge of the role of ASD-specific biomarkers. For instance, changes in specific epigenetic markers (CpG methylation) in noncoding regions, including promoters, enhancers, silencers, insulators, intergenic regions, and ncRNAs, have been observed during neurogenesis [105,106,107,108,109,110]. Most importantly, alternative types of methylation (non-CpG methylation (CpH, where H = A, C, or T) and 5-hydroxymethylcytosine (5-hmC)) have been reported in the pathogenesis of ASD. The nuclear DNA 5-hydroxymethylcytosine, a base molecule involved in the processing of oxidative demethylation, is highly expressed in brain tissue relative to 5-methylcytosine (5mC) and may reveal significant brain-region-specific regulatory epigenetic signals [111]. Therefore, based on various factors, tackling the issue of heterogeneity in ASD-relative risks by studying more homogeneous subsets of individuals will become an evolving method for better explaining these mechanisms.
In summary, the studies cited in this paper should guide the study of ASD by classifying genetic expressions, epigenetic biomarkers, and environmental conditions for the better prediction of individuals with ASD. Ultimately, they have brought to light several important factors that urgently need to be investigated in order to improve both research design and the interpretation of data going forward.
3. Conclusions
Molecular mechanisms can help fill the gap in defining ASD pathogenesis where epigenetic or genetic information alone is inadequate to describe the aetiology of all cases. In terms of effect, genetic and epigenetic profiles are expected to be heterogeneous. Combining genetic, epigenetic, and environmental data is likely to represent a more comprehensive understanding of molecular settings with respect to the aetiology of ASD. The early detection of the molecular basis (genotype, epigenotype) and biological associations with intellectual or ASD-related risk factors will allow health providers to better classify affected individuals, facilitate earlier diagnosis, and improve prognosis. Altogether, improving our molecular understanding of the disorder will also aid in uncovering more homogeneous subgroups of individuals, which will allow for better patient stratification for behavioural and pharmacological therapies.
Author Contributions
Conceptualisation, A.A.K., I.S.A. and D.A.M.; writing—original draft preparation, A.A.K.; writing—review and editing, A.A.K., I.S.A. and D.A.M.; supervision, D.A.M.; funding acquisition, I.S.A., who also contributed for technical support. All authors contributed to the article for reviewing. All authors have read and agreed to the published version of the manuscript.
Funding
This manuscript was supported by the Deanship of Scientific Research at Umm Al-Qura University (UQU) with a grant funded by the Grants and Research Groups Unit (GRGU): Ref (19-Med-1-03-0013).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data pertinent to this study are included herein.
Acknowledgments
The authors wish to thank the Deanship of Scientific Research (DSR) at Umm Al-Qura University for their generous support. We extend our sincere thanks to the Research Department, General Directorate of Health Affairs Makkah Region (GDHAMR) at MOH and to the Medical Genetic Unit, Maternity and Children Hospital (MCH) at Makkah Healthcare Cluster (MHC). Additionally, we would like to thank the Molecular Research and Training Center (MRTC), iGene Center in Jeddah for their assistance.
Conflicts of Interest
The authors declare no conflict of interest regarding the publication of this review article.
References
- Lai, M.C.; Lombardo, M.V.; Ruigrok, A.N.; Chakrabarti, B.; Auyeung, B.; Szatmari, P.; Happé, F.; Baron-Cohen, S. Quantifying and exploring camouflaging in men and women with autism. Autism 2017, 21, 690–702. [Google Scholar] [CrossRef] [PubMed]
- Kanner, L. Autistic disturbances of affective contact. Nerv. Child. 1943, 2, 217–250. [Google Scholar]
- Anagnostou, E.; Zwaigenbaum, L.; Szatmari, P.; Fombonne, E.; Fernandez, B.A.; Woodbury-Smith, M.; Brian, J.; Bryson, S.; Smith, I.M.; Drmic, I.; et al. Autism spectrum disorder: Advances in evidence-based practice. Cmaj 2014, 186, 509–519. [Google Scholar] [CrossRef]
- Baio, J.; Wiggins, L.; Christensen, D.L.; Maenner, M.J.; Daniels, J.; Warren, Z.; Kurzius-Spencer, M.; Zahorodny, W.; Robinson Rosenberg, C.; White, T.; et al. Prevalence of autism spectrum disorder among children aged 8 years autism and developmental disabilities monitoring network, 11 sites, United States, 2014. MMWR Surveill. Summ. 2018, 67, 1. [Google Scholar] [CrossRef]
- Valicenti-McDermott, M.; Mcvicar, K.; Rapin, I.; Wershil, B.K.; Cohen, H.; Shinnar, S. Frequency of gastrointestinal symptoms in children with autistic spectrum disorders and association with family history of autoimmune disease. J. Dev. Behav. Pediatr. 2006, 27, S128–S136. [Google Scholar] [CrossRef] [PubMed]
- Richdale, A.L.; Schreck, K.A. Sleep problems in autism spectrum disorders: Prevalence, nature, & possible biopsychosocial aetiologies. Sleep Med. Rev. 2009, 13, 403–411. [Google Scholar] [PubMed]
- White, S.W.; Oswald, D.; Ollendick, T.; Scahill, L. Anxiety in children and adolescents with autism spectrum disorders. Clin. Psychol. Rev. 2009, 29, 216–229. [Google Scholar] [CrossRef] [PubMed]
- Yasuhara, A. Correlation between EEG abnormalities and symptoms of autism spectrum disorder (ASD). Brain Dev. 2010, 32, 791–798. [Google Scholar] [CrossRef]
- Canitano, R. Epilepsy in autism spectrum disorders. Eur. Child Adolesc. Psychiatry 2007, 16, 61–66. [Google Scholar] [CrossRef]
- Geschwind, D.H. Genetics of autism spectrum disorders. Trends Cogn. Sci. 2011, 15, 409–416. [Google Scholar] [CrossRef]
- Folstein, S.; Rutter, M. Infantile autism: A genetic study of 21 twin pairs. J. Child Psychol. Psychiatry 1977, 18, 297–321. [Google Scholar] [CrossRef] [PubMed]
- Folstein, S.; Rutter, M. Genetic influences and infantile autism. Nature 1977, 265, 726–728. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, R.E.; Law, J.K.; Yenokyan, G.; McGready, J.; Kaufmann, W.E.; Law, P.A. Characteristics and concordance of autism spectrum disorders among 277 twin pairs. Arch. Pediatr. Adolesc. Med. 2009, 163, 907–914. [Google Scholar] [CrossRef]
- Hallmayer, J.; Cleveland, S.; Torres, A.; Phillips, J.; Cohen, B.; Torigoe, T.; Miller, J.; Fedele, A.; Collins, J.; Smith, K.; et al. Genetic heritability and shared environmental factors among twin pairs with autism. Arch. Gen. Psychiatry 2011, 68, 1095–1102. [Google Scholar] [CrossRef]
- Ronald, A.; Hoekstra, R. Progress in understanding the causes of autism spectrum disorders and autistic traits: Twin studies from 1977 to the present day. Behav. Genet. Psychopathol. 2014, 2, 33–65. [Google Scholar]
- Ozonoff, S.; Young, G.S.; Carter, A.; Messinger, D.; Yirmiya, N.; Zwaigenbaum, L.; Bryson, S.; Carver, L.J.; Constantino, J.N.; Dobkins, K.; et al. Recurrence risk for autism spectrum disorders: A Baby Siblings Research Consortium study. Pediatrics 2011, 128, e488–e495. [Google Scholar] [CrossRef]
- Constantino, J.N.; Todorov, A.; Hilton, C.; Law, P.; Zhang, Y.; Molloy, E.; Fitzgerald, R.; Geschwind, D. Autism recurrence in half siblings: Strong support for genetic mechanisms of transmission in ASD. Mol. psychiatry 2013, 18, 137–138. [Google Scholar] [CrossRef]
- Brian, J.; Bryson, S.E.; Garon, N.; Roberts, W.; Smith, I.M.; Szatmari, P. Clinical assessment of autism in high-risk 18-month-olds. Autism 2008, 12, 433–456. [Google Scholar] [CrossRef]
- Nguyen, A.; Rauch, T.A.; Pfeifer, G.P.; Hu, V.W. Global methylation profiling of lymphoblastoid cell lines reveals epigenetic contributions to autism spectrum disorders and a novel autism candidate gene, RORA, whose protein product is reduced in autistic brain. FASEB J. 2010, 24, 3036–3051. [Google Scholar] [CrossRef]
- Tseng, C.-E.J.; McDougle, C.J.; Hooker, J.M.; Zürcher, N.R. Epigenetics of Autism Spectrum Disorder: Histone Deacetylases. Biol. Psychiatry 2022, 91, 922–933. [Google Scholar] [CrossRef]
- Van Bokhoven, H. Genetic and epigenetic networks in intellectual disabilities. Ann. Rev. Gen. 2011, 45, 81–104. [Google Scholar] [CrossRef] [PubMed]
- Turinsky, A.L.; Turner, B.; Borja, R.C.; Gleeson, J.A.; Heath, M.; Pu, S.; Switzer, T.; Dong, D.; Gong, Y.; On, T.; et al. DAnCER: Disease-annotated chromatin epigenetics resource. Nucleic Acids Res. 2011, 39, D889–D894. [Google Scholar] [CrossRef] [PubMed][Green Version]
- McCarthy, S.E.; Gillis, J.; Kramer, M.; Lihm, J.; Yoon, S.; Berstein, Y.; Mistry, M.; Pavlidis, P.; Solomon, R.; Ghiban, E.; et al. De novo mutations in schizophrenia implicate chromatin remodeling and support a genetic overlap with autism and intellectual disability. Mol. Psychiatry 2014, 19, 652–658. [Google Scholar] [CrossRef]
- Carroll, L.S.; Owen, M.J. Genetic overlap between autism, schizophrenia and bipolar disorder. Genome Med. 2009, 1, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Cukier, H.N.; Dueker, N.D.; Slifer, S.H.; Lee, J.M.; Whitehead, P.L.; Lalanne, E.; Leyva, N.; Konidari, I.; Gentry, R.C.; Hulme, W.F.; et al. Exome sequencing of extended families with autism reveals genes shared across neurodevelopmental and neuropsychiatric disorders. Mol. Autism 2014, 5, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Network, T. Pathway Analysis Subgroup of the Psychiatric Genomics Consortium. Psychiatric genome-wide association study analyses implicate neuronal, immune and histone pathways. Nat. Neurosci. 2015, 18, 199. [Google Scholar]
- Kiser, D.P.; Rivero, O.; Lesch, K.P. Annual Research Review: The (epi) genetics of neurodevelopmental disorders in the era of whole-genome sequencing-unveiling the dark matter. J. Child Psychol. Psychiatry 2015, 56, 278–295. [Google Scholar] [CrossRef]
- Freitag, C.M. The genetics of autistic disorders and its clinical relevance: A review of the literature. Mol. Psychiatry 2007, 12, 2–22. [Google Scholar] [CrossRef]
- Smalley, S.L.; Asarnow, R.F.; Spence, M.A. Autism and genetics: A decade of research. Arch. Gen. Psychiatry 1988, 45, 953–961. [Google Scholar] [CrossRef]
- Yuen, R.K.; Thiruvahindrapuram, B.; Merico, D.; Walker, S.; Tammimies, K.; Hoang, N.; Chrysler, C.; Nalpathamkalam, T.; Pellecchia, G.; Liu, Y.; et al. Whole-genome sequencing of quartet families with autism spectrum disorder. Nat. Med. 2015, 21, 185–191. [Google Scholar] [CrossRef]
- Gratten, J.; Wray, N.R.; Keller, M.C.; Visscher, P.M. Large-scale genomics unveils the genetic architecture of psychiatric disorders. Nat. Neuroscie. 2014, 17, 782–790. [Google Scholar] [CrossRef] [PubMed]
- Betancur, C. Etiological heterogeneity in autism spectrum disorders: More than 100 genetic and genomic disorders and still counting. Brain Res. 2011, 1380, 42–77. [Google Scholar] [CrossRef] [PubMed]
- Iossifov, I.; OG_Âroak, B.J.; Sanders, S.J.; Ronemus, M.; Krumm, N.; Levy, D.; Stessman, H.A.; Witherspoon, K.T.; Vives, L.; Patterson, K.E.; et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 2014, 515, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Iossifov, I.; Ronemus, M.; Levy, D.; Wang, Z.; Hakker, I.; Rosenbaum, J.; Yamrom, B.; Lee, Y.; Narzisi, G.; Leotta, A.; et al. De novo gene disruptions in children on the autistic spectrum. Neuron 2012, 74, 285–299. [Google Scholar] [CrossRef]
- Sanders, S.J.; Murtha, M.T.; Gupta, A.R.; Murdoch, J.D.; Raubeson, M.J.; Willsey, A.J.; Willsey, J.A.; Ercan-Sencicek, A.G.; DiLullo, N.M.; Parikshak, N.N.; et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 2012, 485, 237–241. [Google Scholar] [CrossRef]
- Banerjee-Basu, S.; Packer, A. SFARI Gene: An evolving database for the autism research community. Dis. Model. Mech. 2010, 3, 133–135. [Google Scholar] [CrossRef]
- Xiong, J.; Chen, S.; Pang, N.; Deng, X.; Yang, L.; He, F.; Wu, L.; Chen, C.; Yin, F.; Peng, J. Neurological diseases with autism spectrum disorder: Role of ASD risk genes. Front. Neurosci. 2019, 13, 349. [Google Scholar] [CrossRef]
- Santos, J.Ë.X.; Rasga, C.; Marques, A.R.; Martiniano, H.F.; Asif, M.; Vilela, J.; Oliveira, G.; Sousa, L.; Nunes, A.; Astrid, M.; et al. A role for gene-environment interactions in Autism Spectrum Disorder is suggested by variants in genes regulating exposure to environmental factors. bioRxiv 2019, 16, 520544. [Google Scholar]
- Hughes, H.K.; Onore, C.E.; Careaga, M.; Rogers, S.J.; Ashwood, P. Increased Monocyte Production of IL-6 after Toll-like Receptor Activation in Children with Autism Spectrum Disorder (ASD) Is Associated with Repetitive and Restricted Behaviors. Brain Sci. 2022, 12, 220. [Google Scholar] [CrossRef]
- Hughes, H.K.; Rowland, M.E.; Onore, C.E.; Rogers, S.; Ciernia, A.V.; Ashwood, P. Dysregulated gene expression associated with inflammatory and translation pathways in activated monocytes from children with autism spectrum disorder. Transl. Psychiatry 2022, 12, 1–9. [Google Scholar] [CrossRef]
- Sanders, S.J.; He, X.; Willsey, A.J.; Ercan-Sencicek, A.G.; Samocha, K.E.; Cicek, A.E.; Murtha, M.t.; Bal, V.H.; Bishop, S.L.; Dong, S.; et al. Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron 2015, 87, 1215–1233. [Google Scholar] [CrossRef] [PubMed]
- Jeste, S.S.; Geschwind, D.H. Disentangling the heterogeneity of autism spectrum disorder through genetic findings. Nat. Rev. Neurol. 2014, 10, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Marshall, C.R.; Noor, A.; Vincent, J.B.; Lionel, A.C.; Feuk, L.; Skaug, J.; Shago, M.; Moessner, R.; Pinto, D.; Ren, Y.; et al. Structural variation of chromosomes in autism spectrum disorder. Am. J. Hum. Genet. 2008, 82, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Toma, C. Genetic Variation across Phenotypic Severity of Autism. Trends Genet. 2020, 36, 228–231. [Google Scholar] [CrossRef] [PubMed]
- Gaugler, T.; Klei, L.; Sanders, S.J.; Bodea, C.A.; Goldberg, A.P.; Lee, A.B.; Mahajan, M.; Manaa, D.; Pawitan, Y.; Reichert, J.; et al. Most genetic risk for autism resides with common variation. Nat. Genet. 2014, 46, 881–885. [Google Scholar] [CrossRef] [PubMed]
- Choi, L.; An, J.Y. Genetic architecture of autism spectrum disorder: Lessons from large-scale genomic studies. Neurosci. & Biobehav. Revi. 2021, 128, 244–257. [Google Scholar]
- Doan, R.N.; Lim, E.T.; De Rubeis, S.; Betancur, C.; Cutler, D.J.; Chiocchetti, A.G.; Overman, L.M.; Soucy, A.; Goetze, S.; Autism Sequencing Consortium; et al. Recessive gene disruptions in autism spectrum disorder. Nat. Genet. 2019, 51, 1092–1098. [Google Scholar] [CrossRef]
- Ruzzo, E.K.; Pérez-Cano, L.; Jung, J.-Y.; Wang, L.-K.; Kashef-Haghighi, D.; Hartl, C.; Singh, C.; Xu, J.; Hoekstra, J.N.; Leventhal, O.; et al. Inherited and de novo genetic risk for autism impacts shared networks. Cell 2019, 178, 850–866. [Google Scholar] [CrossRef]
- Satterstrom, F.K.; Kosmicki, J.A.; Wang, J.; Breen, M.S.; De Rubeis, S.; An, J.Y.; Peng, M.; Collins, R.; Grove, J.; Klei, L.; et al. Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell 2020, 180, 568–584. [Google Scholar] [CrossRef]
- Chaste, P.; Roeder, K.; Devlin, B. The Yin and Yang of autism genetics: How rare de novo and common variations affect liability. Ann. Rev. Genom. Hum. Genet. 2017, 18, 167–187. [Google Scholar] [CrossRef]
- Pinto, D.; Delaby, E.; Merico, D.; Barbosa, M.; Merikangas, A.; Klei, L.; Thiruvahindrapuram, B.; Xu, X.; Ziman, R.; Wang, Z.; et al. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am. J. Hum. Genet. 2014, 94, 677–694. [Google Scholar] [CrossRef] [PubMed]
- Lasalle, J.M. Autism genes keep turning up chromatin. OA Autism 2013, 1, 14. [Google Scholar] [CrossRef] [PubMed]
- Elhawary, N.A.; Tayeb, M.T.; Sindi, I.A.; Qutub, N.; Rashad, M.; Mufti, A.; Arab, A.H.; Khogeer, A.A.; Elhawary, E.N.; Dannoun, A.; et al. Genetic biomarkers predict susceptibility to autism spectrum disorder through interactive models of inheritance in a Saudi community. Cogent Biol. 2019, 5, 1606555. [Google Scholar] [CrossRef]
- Li, J.; Cai, T.; Jiang, Y.; Chen, H.; He, X.; Chen, C.; Li, X.; Shao, Q.; Ran, X.; Li, Z.; et al. Genes with de novo mutations are shared by four neuropsychiatric disorders discovered from NPdenovo database. Mol. Psychiatry 2016, 21, 290–297. [Google Scholar] [CrossRef]
- Harris, S.W.; Hessl, D.; Goodlin-Jones, B.; Ferranti, J.; Bacalman, S.; Barbato, I.; Tassone, F.; Hagerman, P.J.; Herman, K.; Hagerman, R.J.; et al. Autism profiles of males with fragile X syndrome. Am. J. Ment. Retard. 2008, 113, 427–438. [Google Scholar] [CrossRef]
- Zafeiriou, D.I.; Ververi, A.; Dafoulis, V.; Kalyva, E.; Vargiami, E. Autism spectrum disorders: The quest for genetic syndromes. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2013, 162, 327–366. [Google Scholar] [CrossRef]
- Parikshak, N.N.; Swarup, V.; Belgard, T.G.; Irimia, M.; Ramaswami, G.; Gandal, M.J.; Hartl, C.; Leppa, V.; Ubieta, L.T.; Huang, J.; et al. Genome-wide changes in lncRNA, splicing, and regional gene expression patterns in autism. Nature 2016, 540, 423–427. [Google Scholar] [CrossRef]
- Ziats, M.N.; Rennert, O.M. Aberrant expression of long noncoding RNAs in autistic brain. J. Mol. Neurosci. 2013, 49, 589–593. [Google Scholar] [CrossRef]
- Wong, C.C.Y.; Meaburn, E.L.; Ronald, A.; Price, T.S.; Jeffries, A.R.; Schalkwyk, L.C.; Plomin, R.; Mill, J. Methylomic analysis of monozygotic twins discordant for autism spectrum disorder and related behavioural traits. Mol. Psychiatry 2014, 19, 495–503. [Google Scholar] [CrossRef]
- Ginsberg, M.R.; Rubin, R.A.; Falcone, T.; Ting, A.H.; Natowicz, M.R. Brain transcriptional and epigenetic associations with autism. PLoS ONE 2012, 7, e44736. [Google Scholar] [CrossRef]
- Ladd-Acosta, C.; Hansen, K.D.; Briem, E.; Fallin, M.D.; Kaufmann, W.E.; Feinberg, A.P. Common DNA methylation alterations in multiple brain regions in autism. Mol. Psychiatry 2014, 19, 862–871. [Google Scholar] [CrossRef] [PubMed]
- Nardone, S.; Sharan Sams, D.; Reuveni, E.; Getselter, D.; Oron, O.; Karpuj, M.; Elliott, E. DNA methylation analysis of the autistic brain reveals multiple dysregulated biological pathways. Transl. Psychiatry 2014, 4, e433. [Google Scholar] [CrossRef] [PubMed]
- Berko, E.R.; Suzuki, M.; Beren, F.; Lemetre, C.; Alaimo, C.M.; Calder, R.B.; Ballaban-Gil, K.; Gounder, B.; Kampf, K.; Kirschen, J.; et al. Mosaic epigenetic dysregulation of ectodermal cells in autism spectrum disorder. PLoS Genet. 2014, 10, e1004402. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, J.I.; Bakulski, K.M.; Jaffe, A.E.; Tryggvadottir, R.; Brown, S.C.; Goldman, L.R.; Croen, L.A.; Hertz-Picciotto, I.; Newschaffer, C.J.; Fallin, M.D.; et al. Paternal sperm DNA methylation associated with early signs of autism risk in an autism-enriched cohort. Int. J. Epidemiol. 2015, 44, 1199–1210. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.N.; Volta, M.; Pidsley, R.; Lunnon, K.; Dixit, A.; Lovestone, S.; Coarfa, C.; Harris, R.A.; Milosavljevic, A.; Troakes, C.; et al. Functional annotation of the human brain methylome identifies tissue-specific epigenetic variation across brain and blood. Genome Biol. 2012, 13, 1–14. [Google Scholar] [CrossRef]
- Hannon, E.; Lunnon, K.; Schalkwyk, L.; Mill, J. Interindividual methylomic variation across blood, cortex, and cerebellum: Implications for epigenetic studies of neurological and neuropsychiatric phenotypes. Epigenetics 2015, 10, 1024–1032. [Google Scholar] [CrossRef]
- Elagoz Yuksel, M.; Yuceturk, B.; Karatas, O.F.; Ozen, M.; Dogangun, B. The altered promoter methylation of oxytocin receptor gene in autism. J. Neurogenet. 2016, 30, 280–284. [Google Scholar] [CrossRef]
- Gregory, S.G.; Connelly, J.J.; Towers, A.J.; Johnson, J.; Biscocho, D.; Markunas, C.A.; Lintas, C.; Abramson, R.K.; Wright, H.H.; Ellis, P.; et al. Genomic and epigenetic evidence for oxytocin receptor deficiency in autism. BMC Med. 2009, 7, 62. [Google Scholar] [CrossRef]
- Jiang, Y.H.; Sahoo, T.; Michaelis, R.C.; Bercovich, D.; Bressler, J.; Kashork, C.D.; Liu, Q.; Shaffer, L.G.; Schroer, R.J.; Stockton, D.W.; et al. A mixed epigenetic/genetic model for oligogenic inheritance of autism with a limited role for UBE3A. Am. J. Med. Genet. Part A 2004, 131, 1–10. [Google Scholar] [CrossRef]
- Nagarajan, R.; Hogart, A.; Gwye, Y.; Martin, M.R.; Lasalle, J.M. Reduced MeCP2 expression is frequent in autism frontal cortex and correlates with aberrant MECP2 promoter methylation. Epigenetics 2006, 1, 172–182. [Google Scholar] [CrossRef]
- Zhu, L.; Wang, X.; Li, X.L.; Towers, A.; Cao, X.; Wang, P.; Bowman, R.; Yang, H.; Goldstein, J.; Li, Y.J.; et al. Epigenetic dysregulation of SHANK3 in brain tissues from individuals with autism spectrum disorders. Hum. Mol. Genet. 2014, 23, 1563–1578. [Google Scholar] [CrossRef] [PubMed]
- Zhubi, A.; Chen, Y.; Dong, E.; Cook, E.H.; Guidotti, A.; Grayson, D.R. Increased binding of MeCP2 to the GAD1 and RELN promoters may be mediated by an enrichment of 5-hmC in autism spectrum disorder (ASD) cerebellum. Transl. Psychiatry 2014, 4, e349. [Google Scholar] [CrossRef] [PubMed]
- Berry, K.P.; Lu, Q.R. Chromatin modification and epigenetic control in functional nerve regeneration. Semin. Cell Dev. Biol. 2020, 97, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.H.; Choi, J.; Lee, W.J.; Do, J.T. Genetic and Epigenetic Etiology Underlying Autism Spectrum Disorder. J. Clin. Med. 2020, 9, 966. [Google Scholar] [CrossRef]
- Liu, S.; Zhou, L.; Yuan, H.; Vieira, M.; Sanz-Clemente, A.; Badger, J.D.; Lu, W.; Traynelis, S.F.; Roche, K.W. A rare variant identified within the GluN2B C-terminus in a patient with autism affects NMDA receptor surface expression and spine density. J. Neurosci. 2017, 37, 4093–4102. [Google Scholar] [CrossRef]
- Liu, S.; Zhou, L.; Yuan, H.; Vieira, M.; Sanz-Clemente, A.; Badger, J.D.; Lu, W.; Traynelis, S.F.; Roche, K.W. Genetics and epigenetics of autism spectrum disorder-current evidence in the field. J. Appl. Genet. 2019, 60, 37–47. [Google Scholar]
- Wang, W.; Corominas, R.; Lin, G.N. De novo mutations from whole exome sequencing in neurodevelopmental and psychiatric disorders: From discovery to application. Front. Genet. 2019, 10, 258. [Google Scholar] [CrossRef]
- Gamazon, E.R.; Badner, J.A.; Cheng, L.; Zhang, C.; Zhang, D.; Cox, N.J.; Gershon, E.S.; Kelsoe, J.R.; Greenwood, T.A.; Nievergelt, C.M.; et al. Enrichment of cis-regulatory gene expression SNPs and methylation quantitative trait loci among bipolar disorder susceptibility variants. Mol. Psychiatry 2013, 18, 340–346. [Google Scholar] [CrossRef]
- Van Eijk, K.R.; De Jong, S.; Strengman, E.; Buizer-Voskamp, J.E.; Kahn, R.Ñ.S.; Boks, M.P.; Horvath, S.; Ophoff, R.O. Identification of schizophrenia-associated loci by combining DNA methylation and gene expression data from whole blood. Eur. J. Hum. Genet. 2015, 23, 1106–1110. [Google Scholar] [CrossRef]
- Davis, L.K.; Gamazon, E.R.; Kistner-Griffin, E.; Badner, J.A.; Liu, C.; Cook, E.H.; Sutcliffe, J.S.; Cox, N.J. Loci nominally associated with autism from genome-wide analysis show enrichment of brain expression quantitative trait loci but not lymphoblastoid cell line expression quantitative trait loci. Mol. Autism 2012, 3, 1–11. [Google Scholar] [CrossRef]
- Hannon, E.; Spiers, H.; Viana, J.; Pidsley, R.; Burrage, J.; Murphy, T.M.; Troakes, C.; Turecki, G.; O’Donovan, M.C.; Schalkwyk, L.C.; et al. Methylation QTLs in the developing brain and their enrichment in schizophrenia risk loci. Nat. Neurosci. 2016, 19, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Cross-Disorder Group of the Psychiatric Genomics Consortium. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat. Genet. 2013, 45, 984. [Google Scholar] [CrossRef] [PubMed]
- Aspra, Q.; Cabrera-Mendoza, B.; Morales-Mar_Õn, M.E.; M_Érquez, C.; Chicalote, C.; Ballesteros, A.; Aguilar, M.; Castro, X.; Gómez-Cotero, A.; Balboa-Verduzco, A.; et al. Epigenome-Wide Analysis Reveals DNA Methylation Alteration in ZFP57 and Its Target RASGFR2 in a Mexican Population Cohort with Autism. Children 2022, 9, 462. [Google Scholar] [CrossRef] [PubMed]
- Vellingiri, B.; Mahalaxmi, I.; Raj, N.; Narayanasamy, A.; Gopalakrishnan, A.V. New insights into Epigenetics as an Influencer: An associative study between maternal prenatal factors in Autism Spectrum Disorder (ASD). Neurol. Perspect. 2022, 2, 78–86. [Google Scholar]
- El Hajj, N.; Schneider, E.; Lehnen, H.; Haaf, T. Epigenetics and life-long consequences of an adverse nutritional and diabetic intrauterine environment. Reproduction 2014, 148, R111–R120. [Google Scholar] [CrossRef] [PubMed]
- Sandin, S.; Lichtenstein, P.; Kuja-Halkola, R.; Larsson, H.; Hultman, C.M.; Reichenberg, A. The familial risk of autism. JAMA 2014, 311, 1770–1777. [Google Scholar] [CrossRef]
- Sandin, S.; Schendel, D.; Magnusson, P.; Hultman, C.; Sur_Ñn, P.; Susser, E.; Grønborg, T.; Gissler, M.; Gunnes, N.; Gross, R.; et al. Autism risk associated with parental age and with increasing difference in age between the parents. Mol. Psychiatry 2016, 21, 693–700. [Google Scholar] [CrossRef]
- Frans, E.M.; Lichtenstein, P.; Hultman, C.M.; Kuja-Halkola, R. Age at fatherhood: Heritability and associations with psychiatric disorders. Psychol. Med. 2016, 46, 2981–2988. [Google Scholar] [CrossRef]
- Ronald, A.; Pennell, C.E.; Whitehouse, A.J. Prenatal maternal stress associated with ADHD and autistic traits in early childhood. Front. Psychol. 2011, 1, 223. [Google Scholar] [CrossRef]
- Rijlaarsdam, J.; Pappa, I.; Walton, E.; Bakermans-Kranenburg, M.J.; Mileva-Seitz, V.R.; Rippe, R.C.; Roza, S.J.; Jaddoe, V.W.V.; Verhulst, F.C.; Felix, J.F.; et al. An epigenome-wide association meta-analysis of prenatal maternal stress in neonates: A model approach for replication. Epigenetics 2016, 11, 140–149. [Google Scholar] [CrossRef]
- Chen, X.; Nishitani, S.; Haroon, E.; Smith, A.K.; Rilling, J.K. OXTR methylation modulates exogenous oxytocin effects on human brain activity during social interaction. Genes Brain Behav. 2020, 19, e12555. [Google Scholar] [CrossRef] [PubMed]
- Ebner, N.C.; Lin, T.; Muradoglu, M.; Weir, D.H.; Plasencia, G.M.; Lillard, T.S.; Pournajafi-Nazarloo, H.; Cohen, R.A.; Carter, C.S.; Connelly, J.J. Associations between oxytocin receptor gene (OXTR) methylation, plasma oxytocin, and attachment across adulthood. Int. J. Psychophysiol. 2019, 136, 22–32. [Google Scholar] [PubMed]
- Siu, M.T.; Weksberg, R. Epigenetics of Autism Spectrum Disorder. In Neuroepigenomics in Aging and Disease; Delgado-Morales, R., Ed.; Springer International Publishing: Cham, Switzerland, 2017; pp. 63–90. [Google Scholar]
- Schaevitz, L.R.; Berger-Sweeney, J.E. Gene_Environment Interactions and Epigenetic Pathways in Autism: The Importance of One-Carbon Metabolism. ILAR J. 2012, 53, 322–340. [Google Scholar] [CrossRef]
- Schmidt, R.J. Maternal folic acid supplements associated with reduced autism risk in the child. BMJ Evid.-Based Med. 2013, 18, e53. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, R.J.; Tancredi, D.J.; Ozonoff, S.; Hansen, R.L.; Hartiala, J.; Allayee, H.; Schmidt, L.C.; Tassone, F.; Hertz-Picciotto, I. Maternal periconceptional folic acid intake and risk of autism spectrum disorders and developmental delay in the CHARGE (CHildhood Autism Risks from Genetics and Environment) case-control study. Am. J. Clin. Nutr. 2012, 96, 80–89. [Google Scholar] [CrossRef]
- Suren, P.; Roth, C.; Bresnahan, M.; Haugen, M.; Hornig, M.; Hirtz, D.; Lie, K.K.; Lipkin, W.I.; Magnus, P.; Reichborn-Kjennerud, T.; et al. Association between maternal use of folic acid supplements and risk of autism spectrum disorders in children. JAMA 2013, 309, 570–577. [Google Scholar] [CrossRef]
- Ornoy, A.; Weinstein-Fudim, L.; Tfilin, M.; Ergaz, Z.; Yanai, J.; Szyf, M.; Turgeman, G. S-adenosyl methionine prevents ASD like behaviors triggered by early postnatal valproic acid exposure in very young mice. Neurotoxicol. Teratol. 2019, 71, 64–74. [Google Scholar] [CrossRef]
- Dufour-Rainfray, D.; Vourc’h, P.; Tourlet, S.; Guilloteau, D.; Chalon, S.; Andres, C.R. Fetal exposure to teratogens: Evidence of genes involved in autism. Neurosci. Biobehav. Rev. 2011, 35, 1254–1265. [Google Scholar] [CrossRef]
- Lyall, K.; Schmidt, R.J.; Hertz-Picciotto, I. Maternal lifestyle and environmental risk factors for autism spectrum disorders. Int. J. Epidemiol. 2014, 43, 443–464. [Google Scholar] [CrossRef]
- Simmer, K.; Thompson, R.P.H. Zinc in the fetus and newborn. Acta P_Îdiatrica 1985, 74, 158–163. [Google Scholar] [CrossRef]
- Fabris, N.; Mocchegiani, E. Zinc, human diseases and aging. Aging Clin. Exp. Res. 1995, 7, 77–93. [Google Scholar] [CrossRef] [PubMed]
- Grabrucker, S.; Jannetti, L.; Eckert, M.; Gaub, S.; Chhabra, R.; Pfaender, S.; Mangus, K.; Reddy, P.P.; Rankovic, V.; Schmeisser, M.J.; et al. Zinc deficiency dysregulates the synaptic ProSAP/Shank scaffold and might contribute to autism spectrum disorders. Brain 2014, 137, 137–152. [Google Scholar] [CrossRef] [PubMed]
- Schoen, M.; Asoglu, H.; Bauer, H.F.; M__ller, H.P.; Abaei, A.; Sauer, A.K.; Zhang, R.; Song, T.; Bockmann, J.; Kassubek, J.; et al. Shank3 transgenic and prenatal zinc-deficient autism mouse models show convergent and individual alterations of brain structures in MRI. Front. Neural Circuits 2019, 13, 6. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Mukamel, E.A.; Nery, J.R.; Urich, M.; Puddifoot, C.A.; Johnson, N.D.; Jacinta Lucero, J.; Huang, Y.; Dwork, A.J.; Schultz, M.D.; et al. Global epigenomic reconfiguration during mammalian brain development. Science 2013, 341, 1237905. [Google Scholar] [CrossRef]
- Guo, J.U.; Szulwach, K.E.; Su, Y.; Li, Y.; Yao, B.; Xu, Z.; Shin, J.H.; Xie, B.; Gao, Y.; Ming, G.; et al. Genome-wide antagonism between 5-hydroxymethylcytosine and DNA methylation in the adult mouse brain. Front. Biol. 2014, 9, 66–74. [Google Scholar] [CrossRef]
- An, J.Y.; Lin, K.; Zhu, L.; Werling, D.M.; Dong, S.; Brand, H.; Wang, H.Z.; Zhao, X.; Schwartz, G.B.; Collins, R.L.; et al. Genome-wide de novo risk score implicates promoter variation in autism spectrum disorder. Science 2018, 362, eaat6576. [Google Scholar] [CrossRef]
- Padhi, E.M.; Hayeck, T.J.; Mannion, B.; Chatterjee, S.; Byrska-Bishop, M.; Musunuri, R.; Giuseppe, N.; Avinash, A.; Zhang, C.; Riana, D.H.; et al. De Novo Mutation in an Enhancer of EBF3 in simplex autism. bioRxiv 2020, 1–25. [Google Scholar]
- Turner, T.N.; Eichler, E.E. The Role of De Novo Noncoding Regulatory Mutations in Neurodevelopmental Disorders. Trends Neurosci. 2019, 42, 115–127. [Google Scholar] [CrossRef]
- Ning, Z.; Williams, J.M.; Kumari, R.; Baranov, P.V.; Moore, T. Opposite Expression Patterns of Spry3 and p75NTR in Cerebellar Vermis Suggest a Male-Specific Mechanism of Autism Pathogenesis. Front. Psychiatry 2019, 10, article 416. [Google Scholar] [CrossRef]
- Kriaucionis, S.; Heintz, N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 2009, 324, 929–930. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).