
Can Immune Suppression and Epigenome Regulation in Placenta Offer Novel Insights into Cancer Immune Evasion and Immunotherapy Resistance?

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
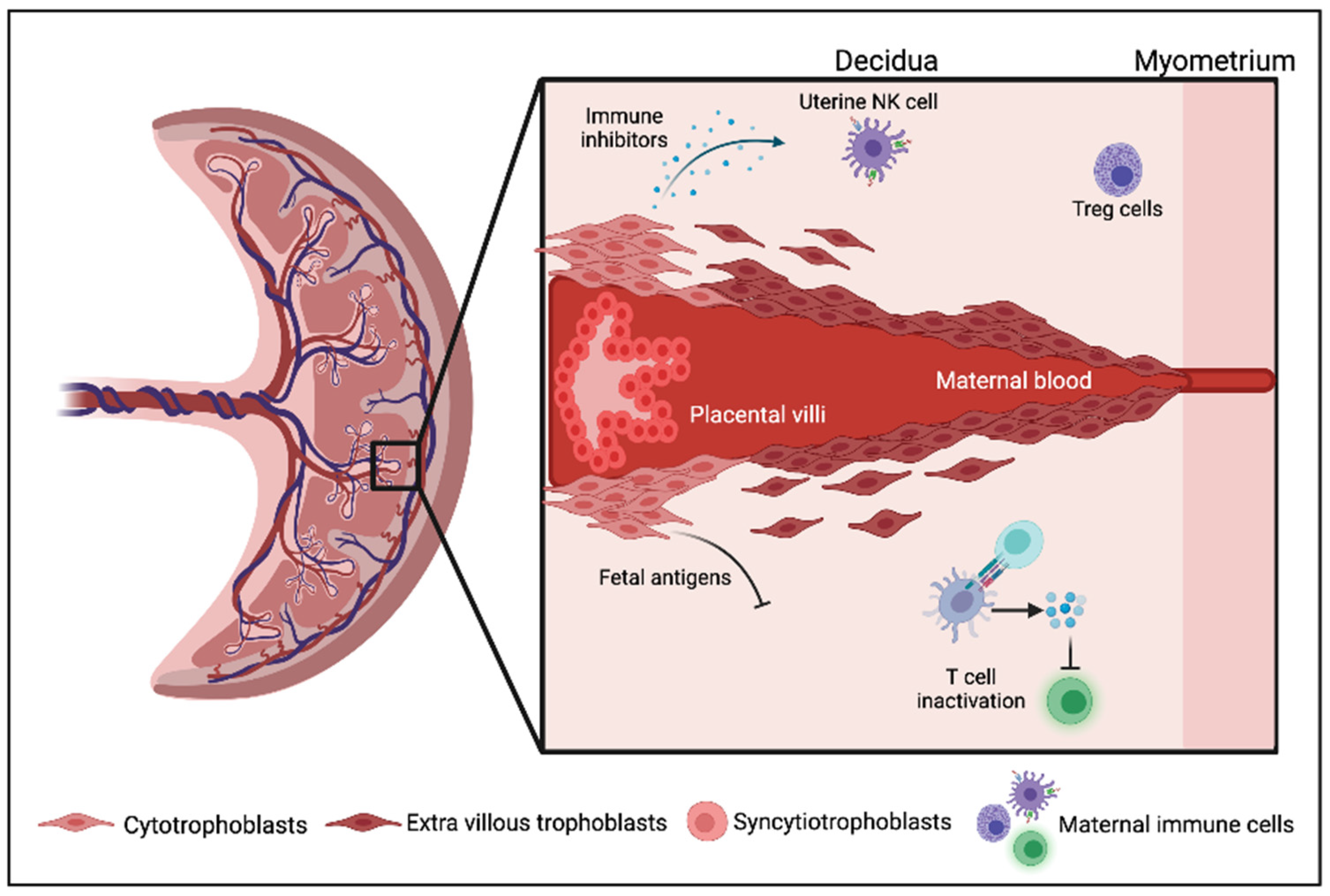
2. Immune Regulation in the Mammalian Placenta and Pregnancy
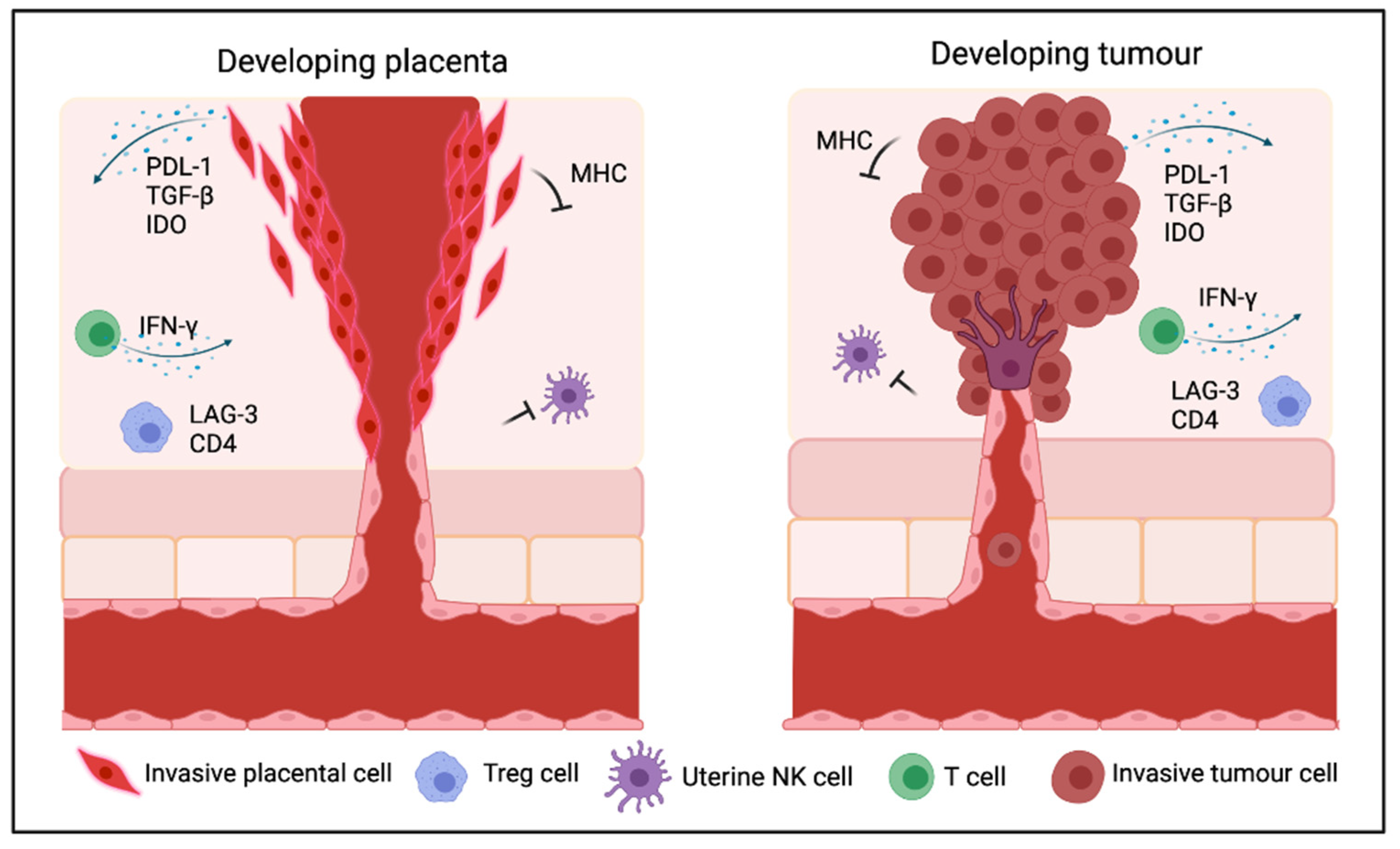
3. Mechanisms Underlying Immunosuppression in Cancer and the Placenta
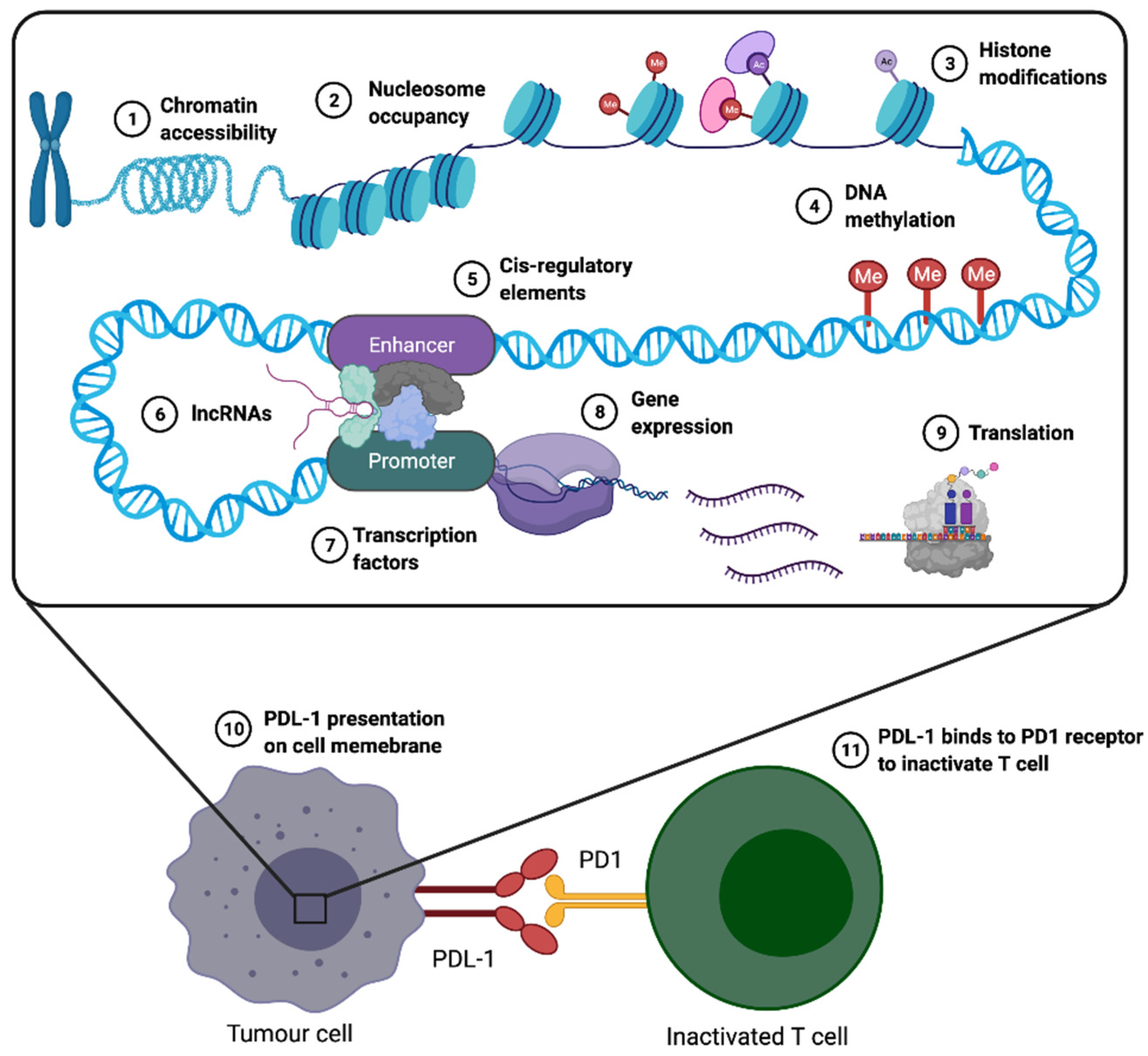
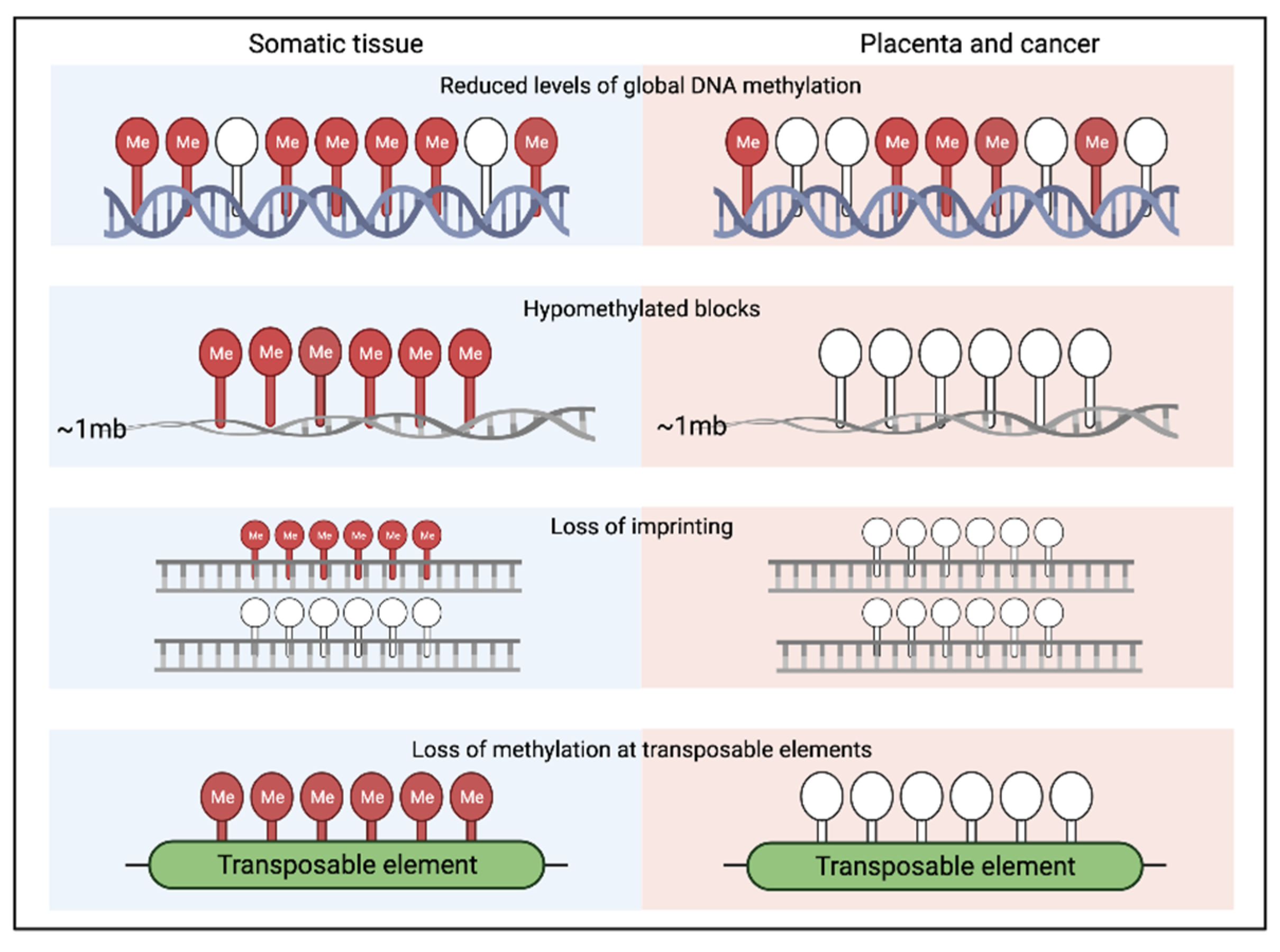
4. Epigenetic Similarities between Early Human Development and Cancer
4.1. Common Epigenetic Mechanisms Frequently Shared between Placenta and Tumours
4.2. Epigenetic Features of Embryonic Stem Cells (ESCs), Compared to Tumours and Cancer Stem Cells (CSCs)
5. Transposable Element (TE) Activation in the Placenta and Cancer; Potential Links to Immune Regulation
6. Human Endogenous Retroviruses (HERVs) and Other Repeat Elements; Potential Roles in Immune Modulation
7. Immune Checkpoint Inhibitors (ICIs)
8. Immunotherapy Resistance in Cancer, a Common Outcome of ICI Therapy
9. Summary and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef]
- Costanzo, V.; Bardelli, A.; Siena, S.; Abrignani, S. Exploring the links between cancer and placenta development. Open Biol. 2018, 8. [Google Scholar] [CrossRef]
- Holtan, S.G.; Creedon, D.J.; Haluska, P.; Markovic, S.N. Cancer and pregnancy: Parallels in growth, invasion, and immune modulation and implications for cancer therapeutic agents. Mayo Clin. Proc. 2009, 84, 985–1000. [Google Scholar] [CrossRef]
- Zhao, H.; Ozen, M.; Wong, R.J.; Stevenson, D.K. Heme oxygenase-1 in pregnancy and cancer: Similarities in cellular invasion, cytoprotection, angiogenesis, and immunomodulation. Front Pharm. 2014, 5, 295. [Google Scholar] [CrossRef]
- Portela, A.; Esteller, M. Epigenetic modifications and human disease. Nat. Biotechnol. 2010, 28, 1057–1068. [Google Scholar] [CrossRef] [PubMed]
- Gan, Q.; Yoshida, T.; McDonald, O.G.; Owens, G.K. Concise review: Epigenetic mechanisms contribute to pluripotency and cell lineage determination of embryonic stem cells. Stem Cells 2007, 25, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Yan, Q. Cancer Epigenetics, Tumor Immunity, and Immunotherapy. Trends Cancer 2020, 6, 580–592. [Google Scholar] [CrossRef]
- Guo, H.; Zhu, P.; Yan, L.; Li, R.; Hu, B.; Lian, Y.; Yan, J.; Ren, X.; Lin, S.; Li, J.; et al. The DNA methylation landscape of human early embryos. Nature 2014, 511, 606–610. [Google Scholar] [CrossRef] [PubMed]
- Mori, M.; Bogdan, A.; Balassa, T.; Csabai, T.; Szekeres-Bartho, J. The decidua-the maternal bed embracing the embryo-maintains the pregnancy. Semin. Immunopathol. 2016, 38, 635–649. [Google Scholar] [CrossRef]
- Knöfler, M.; Haider, S.; Saleh, L.; Pollheimer, J.; Gamage, T.; James, J. Human placenta and trophoblast development: Key molecular mechanisms and model systems. Cell. Mol. Life Sci. 2019, 76, 3479–3496. [Google Scholar] [CrossRef]
- Guleria, I.; Sayegh, M.H. Maternal acceptance of the fetus: True human tolerance. J. Immunol. 2007, 178, 3345–3351. [Google Scholar] [CrossRef]
- Shimizu, J.; Yamazaki, S.; Sakaguchi, S. Induction of tumor immunity by removing CD25+CD4+ T cells: A common basis between tumor immunity and autoimmunity. J. Immunol. 1999, 163, 5211–5218. [Google Scholar]
- Nishikawa, H.; Sakaguchi, S. Regulatory T cells in cancer immunotherapy. Curr. Opin. Immunol. 2014, 27, 1–7. [Google Scholar] [CrossRef]
- Jones, P.A.; Ohtani, H.; Chakravarthy, A.; De Carvalho, D.D. Epigenetic therapy in immune-oncology. Nat. Rev. Cancer 2019, 19, 151–161. [Google Scholar] [CrossRef]
- Li, J.; Byrne, K.T.; Yan, F.; Yamazoe, T.; Chen, Z.; Baslan, T.; Richman, L.P.; Lin, J.H.; Sun, Y.H.; Rech, A.J.; et al. Tumor Cell-Intrinsic Factors Underlie Heterogeneity of Immune Cell Infiltration and Response to Immunotherapy. Immunity 2018, 49, 178–193.e177. [Google Scholar] [CrossRef]
- Hackl, H.; Charoentong, P.; Finotello, F.; Trajanoski, Z. Computational genomics tools for dissecting tumour-immune cell interactions. Nat. Rev. Genet. 2016, 17, 441–458. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hori, S.; Nomura, T.; Sakaguchi, S. Control of regulatory T cell development by the transcription factor Foxp3. Science 2003, 299, 1057–1061. [Google Scholar] [CrossRef]
- Fontenot, J.D.; Gavin, M.A.; Rudensky, A.Y. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat. Immunol. 2003, 4, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Fridman, W.H.; Pagès, F.; Sautès-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, Y.; Nishikawa, H. Roles of regulatory T cells in cancer immunity. Int. Immunol. 2016, 28, 401–409. [Google Scholar] [CrossRef]
- Dwarakanath, B.S.; Farooque, A.; Gupta, S. Targeting regulatory T cells for improving cancer therapy: Challenges and prospects. Cancer Rep. 2018, 1, e21105. [Google Scholar] [CrossRef]
- Jacobs, J.F.; Nierkens, S.; Figdor, C.G.; de Vries, I.J.; Adema, G.J. Regulatory T cells in melanoma: The final hurdle towards effective immunotherapy? Lancet. Oncol. 2012, 13, e32–e42. [Google Scholar] [CrossRef]
- Maeurer, M.J.; Gollin, S.M.; Martin, D.; Swaney, W.; Bryant, J.; Castelli, C.; Robbins, P.; Parmiani, G.; Storkus, W.J.; Lotze, M.T. Tumor escape from immune recognition: Lethal recurrent melanoma in a patient associated with downregulation of the peptide transporter protein TAP-1 and loss of expression of the immunodominant MART-1/Melan-A antigen. J. Clin. Investig. 1996, 98, 1633–1641. [Google Scholar] [CrossRef] [PubMed]
- Polak, M.E.; Borthwick, N.J.; Gabriel, F.G.; Johnson, P.; Higgins, B.; Hurren, J.; McCormick, D.; Jager, M.J.; Cree, I.A. Mechanisms of local immunosuppression in cutaneous melanoma. Br. J. Cancer 2007, 96, 1879–1887. [Google Scholar] [CrossRef]
- Brochez, L.; Chevolet, I.; Kruse, V. The rationale of indoleamine 2,3-dioxygenase inhibition for cancer therapy. Eur. J. Cancer 2017, 76, 167–182. [Google Scholar] [CrossRef]
- Ghiringhelli, F.; Puig, P.E.; Roux, S.; Parcellier, A.; Schmitt, E.; Solary, E.; Kroemer, G.; Martin, F.; Chauffert, B.; Zitvogel, L. Tumor cells convert immature myeloid dendritic cells into TGF-beta-secreting cells inducing CD4+CD25+ regulatory T cell proliferation. J. Exp. Med. 2005, 202, 919–929. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Akhurst, R.J. Differentiation plasticity regulated by TGF-beta family proteins in development and disease. Nat. Cell Biol. 2007, 9, 1000–1004. [Google Scholar] [CrossRef]
- Dahmani, A.; Delisle, J.S. TGF-β in T Cell Biology: Implications for Cancer Immunotherapy. Cancers 2018, 10, 194. [Google Scholar] [CrossRef] [PubMed]
- Jørgensen, N.; Persson, G.; Hviid, T.V.F. The Tolerogenic Function of Regulatory T Cells in Pregnancy and Cancer. Front. Immunol. 2019, 10, 911. [Google Scholar] [CrossRef]
- Huang, Z.; Li, S.; Fan, W.; Ma, Q. Transforming growth factor β1 promotes invasion of human JEG-3 trophoblast cells via TGF-β/Smad3 signaling pathway. Oncotarget 2017, 8, 33560–33570. [Google Scholar] [CrossRef] [PubMed]
- Lafontaine, L.; Chaudhry, P.; Lafleur, M.J.; Van Themsche, C.; Soares, M.J.; Asselin, E. Transforming growth factor Beta regulates proliferation and invasion of rat placental cell lines. Biol. Reprod. 2011, 84, 553–559. [Google Scholar] [CrossRef]
- Xiong, S.; Cheng, J.C.; Klausen, C.; Zhao, J.; Leung, P.C. TGF-β1 stimulates migration of type II endometrial cancer cells by down-regulating PTEN via activation of SMAD and ERK1/2 signaling pathways. Oncotarget 2016, 7, 61262–61272. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Wang, J.; Zhang, F.; Chai, Y.; Brand, D.; Wang, X.; Horwitz, D.A.; Shi, W.; Zheng, S.G. Role of SMAD and non-SMAD signals in the development of Th17 and regulatory T cells. J. Immunol. 2010, 184, 4295–4306. [Google Scholar] [CrossRef]
- Cavalli, G.; Heard, E. Advances in epigenetics link genetics to the environment and disease. Nature 2019, 571, 489–499. [Google Scholar] [CrossRef] [PubMed]
- John, R.M.; Rougeulle, C. Developmental Epigenetics: Phenotype and the Flexible Epigenome. Front. Cell Dev. Biol. 2018, 6, 130. [Google Scholar] [CrossRef]
- Duan, Q.; Zhang, H.; Zheng, J.; Zhang, L. Turning Cold into Hot: Firing up the Tumor Microenvironment. Trends Cancer 2020, 6, 605–618. [Google Scholar] [CrossRef] [PubMed]
- Schadendorf, D.; Hodi, F.S.; Robert, C.; Weber, J.S.; Margolin, K.; Hamid, O.; Patt, D.; Chen, T.T.; Berman, D.M.; Wolchok, J.D. Pooled Analysis of Long-Term Survival Data From Phase II and Phase III Trials of Ipilimumab in Unresectable or Metastatic Melanoma. J. Clin. Oncol. 2015, 33, 1889–1894. [Google Scholar] [CrossRef]
- Sabbatino, F.; Villani, V.; Yearley, J.H.; Deshpande, V.; Cai, L.; Konstantinidis, I.T.; Moon, C.; Nota, S.; Wang, Y.; Al-Sukaini, A.; et al. PD-L1 and HLA Class I Antigen Expression and Clinical Course of the Disease in Intrahepatic Cholangiocarcinoma. Clin. Cancer Res. 2016, 22, 470–478. [Google Scholar] [CrossRef] [PubMed]
- Brandner, J.M.; Haass, N.K. Melanoma’s connections to the tumour microenvironment. Pathology 2013, 45, 443–452. [Google Scholar] [CrossRef]
- Verma, A.; Mathur, R.; Farooque, A.; Kaul, V.; Gupta, S.; Dwarakanath, B.S. T-Regulatory Cells In Tumor Progression And Therapy. Cancer Manage. Res. 2019, 11, 10731–10747. [Google Scholar] [CrossRef] [PubMed]
- Miko, E.; Meggyes, M.; Doba, K.; Barakonyi, A.; Szereday, L. Immune Checkpoint Molecules in Reproductive Immunology. Front. Immunol. 2019, 10, 846. [Google Scholar] [CrossRef] [PubMed]
- Tuncer, Z.S.; Vegh, G.L.; Fulop, V.; Genest, D.R.; Mok, S.C.; Berkowitz, R.S. Expression of epidermal growth factor receptor-related family products in gestational trophoblastic diseases and normal placenta and its relationship with development of postmolar tumor. Gynecol. Oncol. 2000, 77, 389–393. [Google Scholar] [CrossRef]
- Veras, E.; Kurman, R.J.; Wang, T.L.; Shih, I.M. PD-L1 Expression in Human Placentas and Gestational Trophoblastic Diseases. Int. J. Gynecol. Pathol. 2017, 36, 146–153. [Google Scholar] [CrossRef]
- Bellucci, R.; Martin, A.; Bommarito, D.; Wang, K.; Hansen, S.H.; Freeman, G.J.; Ritz, J. Interferon-γ-induced activation of JAK1 and JAK2 suppresses tumor cell susceptibility to NK cells through upregulation of PD-L1 expression. Oncoimmunology 2015, 4, e1008824. [Google Scholar] [CrossRef] [PubMed]
- Messerschmidt, J.L.; Prendergast, G.C.; Messerschmidt, G.L. How Cancers Escape Immune Destruction and Mechanisms of Action for the New Significantly Active Immune Therapies: Helping Nonimmunologists Decipher Recent Advances. Oncologist 2016, 21, 233–243. [Google Scholar] [CrossRef]
- Munn, D.H.; Zhou, M.; Attwood, J.T.; Bondarev, I.; Conway, S.J.; Marshall, B.; Brown, C.; Mellor, A.L. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science 1998, 281, 1191–1193. [Google Scholar] [CrossRef]
- Rezende-Oliveira, K.; Gómez-Hernández, C.; da Silva, M.V.; Helmo, F.R.; Rodrigues, V. Analysis of regulatory T cells and CTLA-4 expression in pregnant women according to seropositivity to Toxoplasma gondii. Parasitology 2020, 147, 810–815. [Google Scholar] [CrossRef]
- Walker, L.S.; Sansom, D.M. Confusing signals: Recent progress in CTLA-4 biology. Trends Immunol. 2015, 36, 63–70. [Google Scholar] [CrossRef]
- Walker, L.S.K. EFIS Lecture: Understanding the CTLA-4 checkpoint in the maintenance of immune homeostasis. Immunol. Lett. 2017, 184, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Dimova, T.; Nagaeva, O.; Stenqvist, A.C.; Hedlund, M.; Kjellberg, L.; Strand, M.; Dehlin, E.; Mincheva-Nilsson, L. Maternal Foxp3 expressing CD4+ CD25+ and CD4+ CD25- regulatory T-cell populations are enriched in human early normal pregnancy decidua: A phenotypic study of paired decidual and peripheral blood samples. Am. J. Reprod. Immunol. 2011, 66 (Suppl. 1), 44–56. [Google Scholar] [CrossRef] [PubMed]
- Arora, S.; Velichinskii, R.; Lesh, R.W.; Ali, U.; Kubiak, M.; Bansal, P.; Borghaei, H.; Edelman, M.J.; Boumber, Y. Existing and Emerging Biomarkers for Immune Checkpoint Immunotherapy in Solid Tumors. Adv. Ther. 2019, 36, 2638–2678. [Google Scholar] [CrossRef]
- Lin, A.; Yan, W.H. Heterogeneity of HLA-G Expression in Cancers: Facing the Challenges. Front. Immunol. 2018, 9, 2164. [Google Scholar] [CrossRef] [PubMed]
- Craenmehr, M.H.C.; Nederlof, I.; Cao, M.; Drabbels, J.J.M.; Spruyt-Gerritse, M.J.; Anholts, J.D.H.; Kapsenberg, H.M.; Stegehuis, J.A.; van der Keur, C.; Fasse, E.; et al. Increased HLA-G Expression in Term Placenta of Women with a History of Recurrent Miscarriage Despite Their Genetic Predisposition to Decreased HLA-G Levels. Int. J. Mol. Sci. 2019, 20, 625. [Google Scholar] [CrossRef]
- Goldman-Wohl, D.S.; Ariel, I.; Greenfield, C.; Hanoch, J.; Yagel, S. HLA-G expression in extravillous trophoblasts is an intrinsic property of cell differentiation: A lesson learned from ectopic pregnancies. Mol. Hum. Reprod. 2000, 6, 535–540. [Google Scholar] [CrossRef]
- Xiao, Q.; Nobre, A.; Piñeiro, P.; Berciano-Guerrero, M.; Alba, E.; Cobo, M.; Lauschke, V.M.; Barragán, I. Genetic and Epigenetic Biomarkers of Immune Checkpoint Blockade Response. J. Clin. Med. 2020, 9, 286. [Google Scholar] [CrossRef]
- Whitehurst, A.W. Cause and consequence of cancer/testis antigen activation in cancer. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 251–272. [Google Scholar] [CrossRef]
- Simpson, A.J.; Caballero, O.L.; Jungbluth, A.; Chen, Y.T.; Old, L.J. Cancer/testis antigens, gametogenesis and cancer. Nat. Rev. Cancer 2005, 5, 615–625. [Google Scholar] [CrossRef]
- Azizi, F.; Ghafouri-Fard, S. Outer Dense Fiber Proteins: Bridging between Male Infertility and Cancer. Arch. Iran. Med. 2017, 20, 320–325. [Google Scholar]
- Knuth, A.; Wölfel, T.; Klehmann, E.; Boon, T.; Meyer zum Büschenfelde, K.H. Cytolytic T-cell clones against an autologous human melanoma: Specificity study and definition of three antigens by immunoselection. Proc. Natl. Acad. Sci. USA 1989, 86, 2804–2808. [Google Scholar] [CrossRef] [PubMed]
- Faramarzi, S.; Ghafouri-Fard, S. Melanoma: A prototype of cancer-testis antigen-expressing malignancies. Immunotherapy 2017, 9, 1103–1113. [Google Scholar] [CrossRef]
- Gedye, C.; Quirk, J.; Browning, J.; Svobodová, S.; John, T.; Sluka, P.; Dunbar, P.R.; Corbeil, D.; Cebon, J.; Davis, I.D. Cancer/testis antigens can be immunological targets in clonogenic CD133+ melanoma cells. Cancer Immunol. Immunother. 2009, 58, 1635–1646. [Google Scholar] [CrossRef]
- Souri, Z.; Wierenga, A.P.A.; Mulder, A.; Jochemsen, A.G.; Jager, M.J. HLA Expression in Uveal Melanoma: An Indicator of Malignancy and a Modifiable Immunological Target. Cancers 2019, 11, 1132. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.; Kim, H.S.; Kim, J.Y.; Sun, J.M.; Ahn, J.S.; Ahn, M.J.; Park, K.; Esteller, M.; Lee, S.H.; Choi, J.K. DNA methylation loss promotes immune evasion of tumours with high mutation and copy number load. Nat. Commun. 2019, 10, 4278. [Google Scholar] [CrossRef]
- Hoevenaar, W.H.M.; Janssen, A.; Quirindongo, A.I.; Ma, H.; Klaasen, S.J.; Teixeira, A.; van Gerwen, B.; Lansu, N.; Morsink, F.H.M.; Offerhaus, G.J.A.; et al. Degree and site of chromosomal instability define its oncogenic potential. Nat. Commun. 2020, 11, 1501. [Google Scholar] [CrossRef]
- Li, J.; Duran, M.A.; Dhanota, N.; Chatila, W.K.; Bettigole, S.E.; Kwon, J.; Sriram, R.K.; Humphries, M.P.; Salto-Tellez, M.; James, J.A.; et al. Metastasis and immune evasion from extracellular cGAMP hydrolysis. Cancer Discov. 2020. [Google Scholar] [CrossRef]
- Wouters, J.; Vizoso, M.; Martinez-Cardus, A.; Carmona, F.J.; Govaere, O.; Laguna, T.; Joseph, J.; Dynoodt, P.; Aura, C.; Foth, M.; et al. Comprehensive DNA methylation study identifies novel progression-related and prognostic markers for cutaneous melanoma. BMC Med. 2017, 15, 101. [Google Scholar] [CrossRef]
- Chatterjee, A.; Rodger, E.J.; Eccles, M.R. Epigenetic drivers of tumourigenesis and cancer metastasis. Semin. Cancer Biol. 2018, 51, 149–159. [Google Scholar] [CrossRef]
- Wu, H.; Zhao, M.; Tan, L.; Lu, Q. The key culprit in the pathogenesis of systemic lupus erythematosus: Aberrant DNA methylation. Autoimmun. Rev. 2016, 15, 684–689. [Google Scholar] [CrossRef]
- Li, E.; Zhang, Y. DNA methylation in mammals. Cold Spring Harb. Perspect. Biol. 2014, 6, a019133. [Google Scholar] [CrossRef]
- Deaton, A.M.; Bird, A. CpG islands and the regulation of transcription. Genes Dev. 2011, 25, 1010–1022. [Google Scholar] [CrossRef]
- Esteller, M. The necessity of a human epigenome project. Carcinogenesis 2006, 27, 1121–1125. [Google Scholar] [CrossRef]
- Baylin, S.B. DNA methylation and gene silencing in cancer. Nat. Clin. Pr. Oncol. 2005, 2 (Suppl. 1), S4–S11. [Google Scholar] [CrossRef]
- Zhou, W.; Liang, G.; Molloy, P.L.; Jones, P.A. DNA methylation enables transposable element-driven genome expansion. Proc. Natl. Acad. Sci. USA 2020, 117, 19359–19366. [Google Scholar] [CrossRef]
- Smith, J.; Sen, S.; Weeks, R.J.; Eccles, M.R.; Chatterjee, A. Promoter DNA Hypermethylation and Paradoxical Gene Activation. Trends Cancer 2020, 6, 392–406. [Google Scholar] [CrossRef]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef]
- Vidal, E.; Sayols, S.; Moran, S.; Guillaumet-Adkins, A.; Schroeder, M.P.; Royo, R.; Orozco, M.; Gut, M.; Gut, I.; Lopez-Bigas, N.; et al. A DNA methylation map of human cancer at single base-pair resolution. Oncogene 2017, 36, 5648–5657. [Google Scholar] [CrossRef]
- Chatterjee, A.; Macaulay, E.C.; Rodger, E.J.; Stockwell, P.A.; Parry, M.F.; Roberts, H.E.; Slatter, T.L.; Hung, N.A.; Devenish, C.J.; Morison, I.M. Placental Hypomethylation Is More Pronounced in Genomic Loci Devoid of Retroelements. G3 2016, 6, 1911–1921. [Google Scholar] [CrossRef]
- Jiang, C.; Pugh, B.F. Nucleosome positioning and gene regulation: Advances through genomics. Nat. Rev. Genet. 2009, 10, 161–172. [Google Scholar] [CrossRef]
- Novakovic, B.; Saffery, R. Placental pseudo-malignancy from a DNA methylation perspective: Unanswered questions and future directions. Front. Genet. 2013, 4, 285. [Google Scholar] [CrossRef][Green Version]
- Novakovic, B.; Saffery, R. DNA methylation profiling highlights the unique nature of the human placental epigenome. Epigenomics 2010, 2, 627–638. [Google Scholar] [CrossRef]
- Macaulay, E.C.; Chatterjee, A.; Cheng, X.; Baguley, B.C.; Eccles, M.R.; Morison, I.M. The Genes of Life and Death: A Potential Role for Placental-Specific Genes in Cancer: Active retrotransposons in the placenta encode unique functional genes that may also be used by cancer cells to promote malignancy. BioEssays News Rev. Mol. Cell. Dev. Biol. 2017, 39. [Google Scholar] [CrossRef]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in cancer. Carcinogenesis 2010, 31, 27–36. [Google Scholar] [CrossRef]
- Ng, H.K.; Novakovic, B.; Hiendleder, S.; Craig, J.M.; Roberts, C.T.; Saffery, R. Distinct patterns of gene-specific methylation in mammalian placentas: Implications for placental evolution and function. Placenta 2010, 31, 259–268. [Google Scholar] [CrossRef]
- Toyota, M.; Ahuja, N.; Ohe-Toyota, M.; Herman, J.G.; Baylin, S.B.; Issa, J.P. CpG island methylator phenotype in colorectal cancer. Proc. Natl. Acad. Sci. USA 1999, 96, 8681–8686. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, D.I.; Blair, J.D.; Lott, P.; Yu, H.O.; Hong, D.; Crary, F.; Ashwood, P.; Walker, C.; Korf, I.; Robinson, W.P.; et al. The human placenta methylome. Proc. Natl. Acad. Sci. USA 2013, 110, 6037–6042. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.M.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef]
- Chatterjee, A.; Stockwell, P.A.; Ahn, A.; Rodger, E.J.; Leichter, A.L.; Eccles, M.R. Genome-wide methylation sequencing of paired primary and metastatic cell lines identifies common DNA methylation changes and a role for EBF3 as a candidate epigenetic driver of melanoma metastasis. Oncotarget 2017, 8, 6085–6101. [Google Scholar] [CrossRef] [PubMed]
- Nordor, A.V.; Nehar-Belaid, D.; Richon, S.; Klatzmann, D.; Bellet, D.; Dangles-Marie, V.; Fournier, T.; Aryee, M.J. The early pregnancy placenta foreshadows DNA methylation alterations of solid tumors. Epigenetics 2017, 12, 793–803. [Google Scholar] [CrossRef]
- Rahat, B.; Mahajan, A.; Bagga, R.; Hamid, A.; Kaur, J. Epigenetic modifications at DMRs of placental genes are subjected to variations in normal gestation, pathological conditions and folate supplementation. Sci. Rep. 2017, 7, 40774. [Google Scholar] [CrossRef]
- Smith, Z.D.; Chan, M.M.; Humm, K.C.; Karnik, R.; Mekhoubad, S.; Regev, A.; Eggan, K.; Meissner, A. DNA methylation dynamics of the human preimplantation embryo. Nature 2014, 511, 611–615. [Google Scholar] [CrossRef]
- Hadjimichael, C.; Chanoumidou, K.; Papadopoulou, N.; Arampatzi, P.; Papamatheakis, J.; Kretsovali, A. Common stemness regulators of embryonic and cancer stem cells. World J. Stem Cells 2015, 7, 1150–1184. [Google Scholar] [CrossRef]
- Glumac, P.M.; LeBeau, A.M. The role of CD133 in cancer: A concise review. Clin. Transl. Med. 2018, 7, 18. [Google Scholar] [CrossRef]
- Yi, J.M.; Tsai, H.C.; Glöckner, S.C.; Lin, S.; Ohm, J.E.; Easwaran, H.; James, C.D.; Costello, J.F.; Riggins, G.; Eberhart, C.G.; et al. Abnormal DNA methylation of CD133 in colorectal and glioblastoma tumors. Cancer Res. 2008, 68, 8094–8103. [Google Scholar] [CrossRef]
- Wang, H.; Gong, P.; Li, J.; Fu, Y.; Zhou, Z.; Liu, L. Role of CD133 in human embryonic stem cell proliferation and teratoma formation. Stem Cell Res. Ther. 2020, 11, 208. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Mathews, L.A.; Cabarcas, S.M.; Zhang, X.; Yang, A.; Zhang, Y.; Young, M.R.; Klarmann, K.D.; Keller, J.R.; Farrar, W.L. Epigenetic regulation of SOX9 by the NF-κB signaling pathway in pancreatic cancer stem cells. Stem Cells 2013, 31, 1454–1466. [Google Scholar] [CrossRef]
- Costa, Y.; Ding, J.; Theunissen, T.W.; Faiola, F.; Hore, T.A.; Shliaha, P.V.; Fidalgo, M.; Saunders, A.; Lawrence, M.; Dietmann, S.; et al. NANOG-dependent function of TET1 and TET2 in establishment of pluripotency. Nature 2013, 495, 370–374. [Google Scholar] [CrossRef] [PubMed]
- Suelves, M.; Carrió, E.; Núñez-Álvarez, Y.; Peinado, M.A. DNA methylation dynamics in cellular commitment and differentiation. Brief. Funct. Genom. 2016, 15, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Aponte, P.M.; Caicedo, A. Stemness in Cancer: Stem Cells, Cancer Stem Cells, and Their Microenvironment. Stem Cells Int. 2017, 2017, 5619472. [Google Scholar] [CrossRef] [PubMed]
- Valk-Lingbeek, M.E.; Bruggeman, S.W.; van Lohuizen, M. Stem cells and cancer; the polycomb connection. Cell 2004, 118, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Gal-Yam, E.N.; Egger, G.; Iniguez, L.; Holster, H.; Einarsson, S.; Zhang, X.; Lin, J.C.; Liang, G.; Jones, P.A.; Tanay, A. Frequent switching of Polycomb repressive marks and DNA hypermethylation in the PC3 prostate cancer cell line. Proc. Natl. Acad. Sci. USA 2008, 105, 12979–12984. [Google Scholar] [CrossRef]
- French, R.; Pauklin, S. Epigenetic regulation of cancer stem cell formation and maintenance. Int. J. Cancer 2020. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Li, Y.; Kulik, M.; Tiedemann, R.L.; Robertson, K.D.; Dalton, S.; Zhao, S. Nucleosome positioning changes during human embryonic stem cell differentiation. Epigenetics 2016, 11, 426–437. [Google Scholar] [CrossRef]
- Ye, Y.; Chen, X.; Zhang, W. Mammalian SWI/SNF Chromatin Remodeling Complexes in Embryonic Stem Cells: Regulating the Balance Between Pluripotency and Differentiation. Front. Cell Dev. Biol. 2020, 8, 626383. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Hore, T.A.; Reik, W. Reprogramming the methylome: Erasing memory and creating diversity. Cell Stem Cell 2014, 14, 710–719. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.S.; Shah, N.M.; Du, A.Y.; Dailey, Z.Z.; Pehrsson, E.C.; Godoy, P.M.; Zhang, D.; Li, D.; Xing, X.; Kim, S.; et al. Transposable elements drive widespread expression of oncogenes in human cancers. Nat. Genet. 2019, 51, 611–617. [Google Scholar] [CrossRef]
- Anwar, S.L.; Wulaningsih, W.; Lehmann, U. Transposable Elements in Human Cancer: Causes and Consequences of Deregulation. Int. J. Mol. Sci. 2017, 18, 974. [Google Scholar] [CrossRef]
- Chuong, E.B.; Rumi, M.A.; Soares, M.J.; Baker, J.C. Endogenous retroviruses function as species-specific enhancer elements in the placenta. Nat. Genet. 2013, 45, 325–329. [Google Scholar] [CrossRef] [PubMed]
- Slotkin, R.K.; Martienssen, R. Transposable elements and the epigenetic regulation of the genome. Nat. Rev. Genet. 2007, 8, 272–285. [Google Scholar] [CrossRef]
- Sugimoto, J.; Schust, D.J. Review: Human endogenous retroviruses and the placenta. Reprod. Sci. 2009, 16, 1023–1033. [Google Scholar] [CrossRef]
- Fedoroff, N.V. Presidential address. Transposable elements, epigenetics, and genome evolution. Science 2012, 338, 758–767. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-López, M.; García-Pérez, J.L. DNA transposons: Nature and applications in genomics. Curr. Genom. 2010, 11, 115–128. [Google Scholar] [CrossRef]
- Saleh, A.; Macia, A.; Muotri, A.R. Transposable Elements, Inflammation, and Neurological Disease. Front. Neurol. 2019, 10, 894. [Google Scholar] [CrossRef]
- Ishak, C.A.; De Carvalho, D.D.; De Carvalho, D.D. Reactivation of Endogenous Retroelements in Cancer Development and Therapy. Annu. Rev. Cancer Biol. 2020, 4, 159–176. [Google Scholar] [CrossRef]
- Thompson, P.J.; Macfarlan, T.S.; Lorincz, M.C. Long Terminal Repeats: From Parasitic Elements to Building Blocks of the Transcriptional Regulatory Repertoire. Mol. Cell 2016, 62, 766–776. [Google Scholar] [CrossRef]
- Percharde, M.; Sultana, T.; Ramalho-Santos, M. What Doesn’t Kill You Makes You Stronger: Transposons as Dual Players in Chromatin Regulation and Genomic Variation. BioEssays News Rev. Mol. Cell. Dev. Biol. 2020, 42, e1900232. [Google Scholar] [CrossRef]
- Kunarso, G.; Chia, N.Y.; Jeyakani, J.; Hwang, C.; Lu, X.; Chan, Y.S.; Ng, H.H.; Bourque, G. Transposable elements have rewired the core regulatory network of human embryonic stem cells. Nat. Genet. 2010, 42, 631–634. [Google Scholar] [CrossRef]
- Chuong, E.B.; Elde, N.C.; Feschotte, C. Regulatory evolution of innate immunity through co-option of endogenous retroviruses. Science 2016, 351, 1083–1087. [Google Scholar] [CrossRef]
- Macaulay, E.C.; Weeks, R.J.; Andrews, S.; Morison, I.M. Hypomethylation of functional retrotransposon-derived genes in the human placenta. Mamm. Genome 2011, 22, 722–735. [Google Scholar] [CrossRef] [PubMed]
- Roulois, D.; Loo Yau, H.; Singhania, R.; Wang, Y.; Danesh, A.; Shen, S.Y.; Han, H.; Liang, G.; Jones, P.A.; Pugh, T.J.; et al. DNA-Demethylating Agents Target Colorectal Cancer Cells by Inducing Viral Mimicry by Endogenous Transcripts. Cell 2015, 162, 961–973. [Google Scholar] [CrossRef]
- Kong, Y.; Rose, C.M.; Cass, A.A.; Williams, A.G.; Darwish, M.; Lianoglou, S.; Haverty, P.M.; Tong, A.J.; Blanchette, C.; Albert, M.L.; et al. Transposable element expression in tumors is associated with immune infiltration and increased antigenicity. Nat. Commun. 2019, 10, 5228. [Google Scholar] [CrossRef] [PubMed]
- Ye, M.; Goudot, C.; Hoyler, T.; Lemoine, B.; Amigorena, S.; Zueva, E. Specific subfamilies of transposable elements contribute to different domains of T lymphocyte enhancers. Proc. Natl. Acad. Sci. USA 2020, 117, 7905–7916. [Google Scholar] [CrossRef]
- Lynch, V.J.; Leclerc, R.D.; May, G.; Wagner, G.P. Transposon-mediated rewiring of gene regulatory networks contributed to the evolution of pregnancy in mammals. Nat. Genet. 2011, 43, 1154–1159. [Google Scholar] [CrossRef]
- Lynch-Sutherland, C.F.; Chatterjee, A.; Stockwell, P.A.; Eccles, M.R.; Macaulay, E.C. Reawakening the Developmental Origins of Cancer Through Transposable Elements. Front. Oncol. 2020, 10, 468. [Google Scholar] [CrossRef] [PubMed]
- Ferretti, C.; Bruni, L.; Dangles-Marie, V.; Pecking, A.P.; Bellet, D. Molecular circuits shared by placental and cancer cells, and their implications in the proliferative, invasive and migratory capacities of trophoblasts. Hum. Reprod. Update 2007, 13, 121–141. [Google Scholar] [CrossRef]
- Beaman, K.D.; Dambaeva, S.; Katara, G.K.; Kulshrestha, A.; Gilman-Sachs, A. The immune response in pregnancy and in cancer is active and supportive of placental and tumor cell growth not their destruction. Gynecol. Oncol. 2017, 145, 476–480. [Google Scholar] [CrossRef]
- Gama-Sosa, M.A.; Slagel, V.A.; Trewyn, R.W.; Oxenhandler, R.; Kuo, K.C.; Gehrke, C.W.; Ehrlich, M. The 5-methylcytosine content of DNA from human tumors. Nucleic Acids Res. 1983, 11, 6883–6894. [Google Scholar] [CrossRef] [PubMed]
- Fuke, C.; Shimabukuro, M.; Petronis, A.; Sugimoto, J.; Oda, T.; Miura, K.; Miyazaki, T.; Ogura, C.; Okazaki, Y.; Jinno, Y. Age related changes in 5-methylcytosine content in human peripheral leukocytes and placentas: An HPLC-based study. Ann. Hum. Genet. 2004, 68, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Denner, J. Expression and function of endogenous retroviruses in the placenta. APMIS Acta Pathol. Microbiol. Immunol. Scand. 2016, 124, 31–43. [Google Scholar] [CrossRef]
- Gonzalez-Cao, M.; Iduma, P.; Karachaliou, N.; Santarpia, M.; Blanco, J.; Rosell, R. Human endogenous retroviruses and cancer. Cancer Biol. Med. 2016, 13, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Chuong, E.B.; Elde, N.C.; Feschotte, C. Regulatory activities of transposable elements: From conflicts to benefits. Nat. Rev. Genet. 2017, 18, 71–86. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.C.; Beckermann, K.E.; Bortone, D.S.; De Cubas, A.A.; Bixby, L.M.; Lee, S.J.; Panda, A.; Ganesan, S.; Bhanot, G.; Wallen, E.M.; et al. Endogenous retroviral signatures predict immunotherapy response in clear cell renal cell carcinoma. J. Clin. Investig. 2018, 128, 4804–4820. [Google Scholar] [CrossRef]
- Lin, D.Y.; Huang, C.C.; Hsieh, Y.T.; Lin, H.C.; Pao, P.C.; Tsou, J.H.; Lai, C.Y.; Hung, L.Y.; Wang, J.M.; Chang, W.C.; et al. Analysis of the interaction between Zinc finger protein 179 (Znf179) and promyelocytic leukemia zinc finger (Plzf). J. Biomed. Sci. 2013, 20, 98. [Google Scholar] [CrossRef]
- Li, X.; Xiang, Y.; Li, F.; Yin, C.; Li, B.; Ke, X. WNT/β-Catenin Signaling Pathway Regulating T Cell-Inflammation in the Tumor Microenvironment. Front. Immunol. 2019, 10, 2293. [Google Scholar] [CrossRef]
- Trujillo, J.A.; Sweis, R.F.; Bao, R.; Luke, J.J. T Cell-Inflamed versus Non-T Cell-Inflamed Tumors: A Conceptual Framework for Cancer Immunotherapy Drug Development and Combination Therapy Selection. Cancer Immunol. Res. 2018, 6, 990–1000. [Google Scholar] [CrossRef]
- Kovacs, D.; Migliano, E.; Muscardin, L.; Silipo, V.; Catricalà, C.; Picardo, M.; Bellei, B. The role of Wnt/β-catenin signaling pathway in melanoma epithelial-to-mesenchymal-like switching: Evidences from patients-derived cell lines. Oncotarget 2016, 7, 43295–43314. [Google Scholar] [CrossRef]
- Yang, L.; Li, A.; Lei, Q.; Zhang, Y. Tumor-intrinsic signaling pathways: Key roles in the regulation of the immunosuppressive tumor microenvironment. J. Hematol. Oncol. 2019, 12, 125. [Google Scholar] [CrossRef] [PubMed]
- Percharde, M.; Lin, C.J.; Yin, Y.; Guan, J.; Peixoto, G.A.; Bulut-Karslioglu, A.; Biechele, S.; Huang, B.; Shen, X.; Ramalho-Santos, M. A LINE1-Nucleolin Partnership Regulates Early Development and ESC Identity. Cell 2018, 174, 391–405.e319. [Google Scholar] [CrossRef]
- Blaise, S.; de Parseval, N.; Bénit, L.; Heidmann, T. Genomewide screening for fusogenic human endogenous retrovirus envelopes identifies syncytin 2, a gene conserved on primate evolution. Proc. Natl. Acad. Sci. USA 2003, 100, 13013–13018. [Google Scholar] [CrossRef]
- Zhang, Y.; Cheng, T.C.; Huang, G.; Lu, Q.; Surleac, M.D.; Mandell, J.D.; Pontarotti, P.; Petrescu, A.J.; Xu, A.; Xiong, Y.; et al. Transposon molecular domestication and the evolution of the RAG recombinase. Nature 2019, 569, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Chiappinelli, K.B.; Strissel, P.L.; Desrichard, A.; Li, H.; Henke, C.; Akman, B.; Hein, A.; Rote, N.S.; Cope, L.M.; Snyder, A.; et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell 2015, 162, 974–986. [Google Scholar] [CrossRef] [PubMed]
- Hur, S. Double-Stranded RNA Sensors and Modulators in Innate Immunity. Annu. Rev. Immunol. 2019, 37, 349–375. [Google Scholar] [CrossRef]
- Rodić, N.; Sharma, R.; Sharma, R.; Zampella, J.; Dai, L.; Taylor, M.S.; Hruban, R.H.; Iacobuzio-Donahue, C.A.; Maitra, A.; Torbenson, M.S.; et al. Long interspersed element-1 protein expression is a hallmark of many human cancers. Am. J. Pathol. 2014, 184, 1280–1286. [Google Scholar] [CrossRef] [PubMed]
- Rooney, M.S.; Shukla, S.A.; Wu, C.J.; Getz, G.; Hacohen, N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015, 160, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Disis, M.L. Mechanism of action of immunotherapy. Semin. Oncol. 2014, 41 (Suppl. 5), S3–S13. [Google Scholar] [CrossRef] [PubMed]
- Passarelli, A.; Mannavola, F.; Stucci, L.S.; Tucci, M.; Silvestris, F. Immune system and melanoma biology: A balance between immunosurveillance and immune escape. Oncotarget 2017, 8, 106132–106142. [Google Scholar] [CrossRef]
- Koppolu, V.; Rekha Vasigala, V.K. Checkpoint immunotherapy by nivolumab for treatment of metastatic melanoma. J. Cancer Res. Ther. 2018, 14, 1167–1175. [Google Scholar] [CrossRef]
- Zhao, Y.; Yang, W.; Huang, Y.; Cui, R.; Li, X.; Li, B. Evolving Roles for Targeting CTLA-4 in Cancer Immunotherapy. Cell. Physiol. Biochem. 2018, 47, 721–734. [Google Scholar] [CrossRef]
- Boasso, A.; Herbeuval, J.P.; Hardy, A.W.; Winkler, C.; Shearer, G.M. Regulation of indoleamine 2,3-dioxygenase and tryptophanyl-tRNA-synthetase by CTLA-4-Fc in human CD4+ T cells. Blood 2005, 105, 1574–1581. [Google Scholar] [CrossRef] [PubMed]
- Grohmann, U.; Orabona, C.; Fallarino, F.; Vacca, C.; Calcinaro, F.; Falorni, A.; Candeloro, P.; Belladonna, M.L.; Bianchi, R.; Fioretti, M.C.; et al. CTLA-4-Ig regulates tryptophan catabolism in vivo. Nat. Immunol. 2002, 3, 1097–1101. [Google Scholar] [CrossRef]
- Parry, R.V.; Chemnitz, J.M.; Frauwirth, K.A.; Lanfranco, A.R.; Braunstein, I.; Kobayashi, S.V.; Linsley, P.S.; Thompson, C.B.; Riley, J.L. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol. Cell. Biol. 2005, 25, 9543–9553. [Google Scholar] [CrossRef]
- Kubsch, S.; Graulich, E.; Knop, J.; Steinbrink, K. Suppressor activity of anergic T cells induced by IL-10-treated human dendritic cells: Association with IL-2- and CTLA-4-dependent G1 arrest of the cell cycle regulated by p27Kip1. Eur. J. Immunol. 2003, 33, 1988–1997. [Google Scholar] [CrossRef] [PubMed]
- Olsson, C.; Riesbeck, K.; Dohlsten, M.; Michaëlsson, E. CTLA-4 ligation suppresses CD28-induced NF-kappaB and AP-1 activity in mouse T cell blasts. J. Biol. Chem. 1999, 274, 14400–14405. [Google Scholar] [CrossRef]
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front. Oncol. 2018, 8, 86. [Google Scholar] [CrossRef]
- Yang, J.C.; Hughes, M.; Kammula, U.; Royal, R.; Sherry, R.M.; Topalian, S.L.; Suri, K.B.; Levy, C.; Allen, T.; Mavroukakis, S.; et al. Ipilimumab (anti-CTLA4 antibody) causes regression of metastatic renal cell cancer associated with enteritis and hypophysitis. J. Immunother. 2007, 30, 825–830. [Google Scholar] [CrossRef] [PubMed]
- Marconcini, R.; Spagnolo, F.; Stucci, L.S.; Ribero, S.; Marra, E.; Rosa, F.; Picasso, V.; Di Guardo, L.; Cimminiello, C.; Cavalieri, S.; et al. Current status and perspectives in immunotherapy for metastatic melanoma. Oncotarget 2018, 9, 12452–12470. [Google Scholar] [CrossRef]
- Sharpe, A.H.; Pauken, K.E. The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 2018, 18, 153–167. [Google Scholar] [CrossRef]
- Chatterjee, A.; Rodger, E.J.; Ahn, A.; Stockwell, P.A.; Parry, M.; Motwani, J.; Gallagher, S.J.; Shklovskaya, E.; Tiffen, J.; Eccles, M.R.; et al. Marked Global DNA Hypomethylation Is Associated with Constitutive PD-L1 Expression in Melanoma. iScience 2018, 4, 312–325. [Google Scholar] [CrossRef] [PubMed]
- Chikuma, S.; Terawaki, S.; Hayashi, T.; Nabeshima, R.; Yoshida, T.; Shibayama, S.; Okazaki, T.; Honjo, T. PD-1-mediated suppression of IL-2 production induces CD8+ T cell anergy in vivo. J. Immunol. 2009, 182, 6682–6689. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Ni, Z.; Liu, X.; Feng, S.; Dong, X.; Shi, X.; Zhai, J.; Mai, S.; Jiang, J.; Wang, Z.; et al. Beyond T Cells: Understanding the Role of PD-1/PD-L1 in Tumor-Associated Macrophages. J. Immunol. Res. 2019, 2019, 1919082. [Google Scholar] [CrossRef]
- Maleki Vareki, S.; Garrigos, C.; Duran, I. Biomarkers of response to PD-1/PD-L1 inhibition. Crit. Rev. Oncol. Hematol. 2017, 116, 116–124. [Google Scholar] [CrossRef]
- Chen, J.; Jiang, C.C.; Jin, L.; Zhang, X.D. Regulation of PD-L1: A novel role of pro-survival signalling in cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2016, 27, 409–416. [Google Scholar] [CrossRef]
- Nguyen, N.; Bellile, E.; Thomas, D.; McHugh, J.; Rozek, L.; Virani, S.; Peterson, L.; Carey, T.E.; Walline, H.; Moyer, J.; et al. Tumor infiltrating lymphocytes and survival in patients with head and neck squamous cell carcinoma. Head Neck 2016, 38, 1074–1084. [Google Scholar] [CrossRef]
- Jiang, L.; Su, X.; Zhang, T.; Yin, X.; Zhang, M.; Fu, H.; Han, H.; Sun, Y.; Dong, L.; Qian, J.; et al. PD-L1 expression and its relationship with oncogenic drivers in non-small cell lung cancer (NSCLC). Oncotarget 2017, 8, 26845–26857. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Lian, Z.; Wang, S.; Xing, L.; Yu, J. Interactions between EGFR and PD-1/PD-L1 pathway: Implications for treatment of NSCLC. Cancer Lett. 2018, 418, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.T.; Lee, S.H.; Heo, Y.S. Molecular Interactions of Antibody Drugs Targeting PD-1, PD-L1, and CTLA-4 in Immuno-Oncology. Molecules 2019, 24, 1190. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Cornel, A.M.; Mimpen, I.L.; Nierkens, S. MHC Class I Downregulation in Cancer: Underlying Mechanisms and Potential Targets for Cancer Immunotherapy. Cancers 2020, 12, 1760. [Google Scholar] [CrossRef]
- Anfossi, N.; André, P.; Guia, S.; Falk, C.S.; Roetynck, S.; Stewart, C.A.; Breso, V.; Frassati, C.; Reviron, D.; Middleton, D.; et al. Human NK cell education by inhibitory receptors for MHC class I. Immunity 2006, 25, 331–342. [Google Scholar] [CrossRef]
- Lee, J.C.; Lee, K.M.; Kim, D.W.; Heo, D.S. Elevated TGF-beta1 secretion and down-modulation of NKG2D underlies impaired NK cytotoxicity in cancer patients. J. Immunol. 2004, 172, 7335–7340. [Google Scholar] [CrossRef]
- Spel, L.; Boelens, J.J.; van der Steen, D.M.; Blokland, N.J.; van Noesel, M.M.; Molenaar, J.J.; Heemskerk, M.H.; Boes, M.; Nierkens, S. Natural killer cells facilitate PRAME-specific T-cell reactivity against neuroblastoma. Oncotarget 2015, 6, 35770–35781. [Google Scholar] [CrossRef]
- Jonges, L.E.; Giezeman-Smits, K.M.; van Vlierberghe, R.L.; Ensink, N.G.; Hagenaars, M.; Joly, E.; Eggermont, A.M.; van de Velde, C.J.; Fleuren, G.J.; Kuppen, P.J. NK cells modulate MHC class I expression on tumor cells and their susceptibility to lysis. Immunobiology 2000, 202, 326–338. [Google Scholar] [CrossRef]
- Long, L.; Zhang, X.; Chen, F.; Pan, Q.; Phiphatwatchara, P.; Zeng, Y.; Chen, H. The promising immune checkpoint LAG-3: From tumor microenvironment to cancer immunotherapy. Genes Cancer 2018, 9, 176–189. [Google Scholar] [CrossRef]
- He, Y.; Cao, J.; Zhao, C.; Li, X.; Zhou, C.; Hirsch, F.R. TIM-3, a promising target for cancer immunotherapy. OncoTargets Ther. 2018, 11, 7005–7009. [Google Scholar] [CrossRef] [PubMed]
- Huard, B.; Gaulard, P.; Faure, F.; Hercend, T.; Triebel, F. Cellular expression and tissue distribution of the human LAG-3-encoded protein, an MHC class II ligand. Immunogenetics 1994, 39, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Maruhashi, T.; Sugiura, D.; Okazaki, I.M.; Okazaki, T. LAG-3: From molecular functions to clinical applications. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Baixeras, E.; Huard, B.; Miossec, C.; Jitsukawa, S.; Martin, M.; Hercend, T.; Auffray, C.; Triebel, F.; Piatier-Tonneau, D. Characterization of the lymphocyte activation gene 3-encoded protein. A new ligand for human leukocyte antigen class II antigens. J. Exp. Med. 1992, 176, 327–337. [Google Scholar] [CrossRef]
- Huang, C.T.; Workman, C.J.; Flies, D.; Pan, X.; Marson, A.L.; Zhou, G.; Hipkiss, E.L.; Ravi, S.; Kowalski, J.; Levitsky, H.I.; et al. Role of LAG-3 in regulatory T cells. Immunity 2004, 21, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Andrews, L.P.; Marciscano, A.E.; Drake, C.G.; Vignali, D.A. LAG3 (CD223) as a cancer immunotherapy target. Immunol. Rev. 2017, 276, 80–96. [Google Scholar] [CrossRef]
- Zhang, Y.H.; Sun, H.X. Immune checkpoint molecules in pregnancy: Focus on regulatory T cells. Eur. J. Immunol. 2020, 50, 160–169. [Google Scholar] [CrossRef]
- Camisaschi, C.; Casati, C.; Rini, F.; Perego, M.; De Filippo, A.; Triebel, F.; Parmiani, G.; Belli, F.; Rivoltini, L.; Castelli, C. LAG-3 expression defines a subset of CD4(+)CD25(high)Foxp3(+) regulatory T cells that are expanded at tumor sites. J. Immunol. 2010, 184, 6545–6551. [Google Scholar] [CrossRef]
- Kouo, T.; Huang, L.; Pucsek, A.B.; Cao, M.; Solt, S.; Armstrong, T.; Jaffee, E. Galectin-3 Shapes Antitumor Immune Responses by Suppressing CD8+ T Cells via LAG-3 and Inhibiting Expansion of Plasmacytoid Dendritic Cells. Cancer Immunol. Res. 2015, 3, 412–423. [Google Scholar] [CrossRef]
- Wang, J.; Sanmamed, M.F.; Datar, I.; Su, T.T.; Ji, L.; Sun, J.; Chen, L.; Chen, Y.; Zhu, G.; Yin, W.; et al. Fibrinogen-like Protein 1 Is a Major Immune Inhibitory Ligand of LAG-3. Cell 2019, 176, 334–347.e312. [Google Scholar] [CrossRef]
- Woo, S.R.; Turnis, M.E.; Goldberg, M.V.; Bankoti, J.; Selby, M.; Nirschl, C.J.; Bettini, M.L.; Gravano, D.M.; Vogel, P.; Liu, C.L.; et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012, 72, 917–927. [Google Scholar] [CrossRef]
- Matsuzaki, J.; Gnjatic, S.; Mhawech-Fauceglia, P.; Beck, A.; Miller, A.; Tsuji, T.; Eppolito, C.; Qian, F.; Lele, S.; Shrikant, P.; et al. Tumor-infiltrating NY-ESO-1-specific CD8+ T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 7875–7880. [Google Scholar] [CrossRef] [PubMed]
- Guerrouahen, B.S.; Maccalli, C.; Cugno, C.; Rutella, S.; Akporiaye, E.T. Reverting Immune Suppression to Enhance Cancer Immunotherapy. Front. Oncol. 2019, 9, 1554. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Anderson, A.C.; Schubart, A.; Xiong, H.; Imitola, J.; Khoury, S.J.; Zheng, X.X.; Strom, T.B.; Kuchroo, V.K. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat. Immunol. 2005, 6, 1245–1252. [Google Scholar] [CrossRef] [PubMed]
- Das, M.; Zhu, C.; Kuchroo, V.K. Tim-3 and its role in regulating anti-tumor immunity. Immunol. Rev. 2017, 276, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Chiba, S.; Baghdadi, M.; Akiba, H.; Yoshiyama, H.; Kinoshita, I.; Dosaka-Akita, H.; Fujioka, Y.; Ohba, Y.; Gorman, J.V.; Colgan, J.D.; et al. Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat. Immunol. 2012, 13, 832–842. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Liu, X.; Ma, H.; Zhang, H.; Song, X.; Gao, L.; Liang, X.; Ma, C. Tim-3 fosters HCC development by enhancing TGF-β-mediated alternative activation of macrophages. Gut 2015, 64, 1593–1604. [Google Scholar] [CrossRef]
- Cai, C.; Xu, Y.F.; Wu, Z.J.; Dong, Q.; Li, M.Y.; Olson, J.C.; Rabinowitz, Y.M.; Wang, L.H.; Sun, Y. Tim-3 expression represents dysfunctional tumor infiltrating T cells in renal cell carcinoma. World J. Urol. 2016, 34, 561–567. [Google Scholar] [CrossRef]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef]
- Park, Y.J.; Kuen, D.S.; Chung, Y. Future prospects of immune checkpoint blockade in cancer: From response prediction to overcoming resistance. Exp. Mol. Med. 2018, 50, 109. [Google Scholar] [CrossRef]
- Bai, R.; Chen, N.; Li, L.; Du, N.; Bai, L.; Lv, Z.; Tian, H.; Cui, J. Mechanisms of Cancer Resistance to Immunotherapy. Front. Oncol. 2020, 10, 1290. [Google Scholar] [CrossRef] [PubMed]
- Ventola, C.L. Cancer Immunotherapy, Part 3: Challenges and Future Trends. P&T Peer-Rev. J. Formul. Manage. 2017, 42, 514–521. [Google Scholar]
- Emran, A.A.; Chatterjee, A.; Rodger, E.J.; Tiffen, J.C.; Gallagher, S.J.; Eccles, M.R.; Hersey, P. Targeting DNA Methylation and EZH2 Activity to Overcome Melanoma Resistance to Immunotherapy. Trends Immunol. 2019, 40, 328–344. [Google Scholar] [CrossRef] [PubMed]
- Jessurun, C.A.C.; Vos, J.A.M.; Limpens, J.; Luiten, R.M. Biomarkers for Response of Melanoma Patients to Immune Checkpoint Inhibitors: A Systematic Review. Front. Oncol. 2017, 7, 233. [Google Scholar] [CrossRef]
- Qian, F.F.; Han, B.H. Mechanisms of resistance to immune checkpoint inhibitors and strategies to reverse drug resistance in lung cancer. Chin. Med. J. 2020, 133, 2444–2455. [Google Scholar] [CrossRef]
- Rotte, A. Combination of CTLA-4 and PD-1 blockers for treatment of cancer. J. Exp. Clin. Cancer Res. CR 2019, 38, 255. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Chehrazi-Raffle, A.; Reddi, S.; Salgia, R. Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: A comprehensive review of registration trials and future considerations. J. Immunother. Cancer 2018, 6, 8. [Google Scholar] [CrossRef] [PubMed]
- Vaddepally, R.K.; Kharel, P.; Pandey, R.; Garje, R.; Chandra, A.B. Review of Indications of FDA-Approved Immune Checkpoint Inhibitors per NCCN Guidelines with the Level of Evidence. Cancers 2020, 12, 738. [Google Scholar] [CrossRef]
- Jackson, C.M.; Choi, J.; Lim, M. Mechanisms of immunotherapy resistance: Lessons from glioblastoma. Nat. Immunol. 2019, 20, 1100–1109. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef]
- Lastwika, K.J.; Wilson, W., 3rd; Li, Q.K.; Norris, J.; Xu, H.; Ghazarian, S.R.; Kitagawa, H.; Kawabata, S.; Taube, J.M.; Yao, S.; et al. Control of PD-L1 Expression by Oncogenic Activation of the AKT-mTOR Pathway in Non-Small Cell Lung Cancer. Cancer Res. 2016, 76, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Parsa, A.T.; Waldron, J.S.; Panner, A.; Crane, C.A.; Parney, I.F.; Barry, J.J.; Cachola, K.E.; Murray, J.C.; Tihan, T.; Jensen, M.C.; et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat. Med. 2007, 13, 84–88. [Google Scholar] [CrossRef]
- Akbay, E.A.; Koyama, S.; Carretero, J.; Altabef, A.; Tchaicha, J.H.; Christensen, C.L.; Mikse, O.R.; Cherniack, A.D.; Beauchamp, E.M.; Pugh, T.J.; et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov. 2013, 3, 1355–1363. [Google Scholar] [CrossRef]
- Casey, S.C.; Tong, L.; Li, Y.; Do, R.; Walz, S.; Fitzgerald, K.N.; Gouw, A.M.; Baylot, V.; Gütgemann, I.; Eilers, M.; et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science 2016, 352, 227–231. [Google Scholar] [CrossRef]
- Dorand, R.D.; Nthale, J.; Myers, J.T.; Barkauskas, D.S.; Avril, S.; Chirieleison, S.M.; Pareek, T.K.; Abbott, D.W.; Stearns, D.S.; Letterio, J.J.; et al. Cdk5 disruption attenuates tumor PD-L1 expression and promotes antitumor immunity. Science 2016, 353, 399–403. [Google Scholar] [CrossRef]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2017, 168, 542. [Google Scholar] [CrossRef]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Van Rooij, N.; van Buuren, M.M.; Philips, D.; Velds, A.; Toebes, M.; Heemskerk, B.; van Dijk, L.J.; Behjati, S.; Hilkmann, H.; El Atmioui, D.; et al. Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2013, 31, e439–e442. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Goodman, A.; Walavalkar, V.; Barkauskas, D.A.; Sharabi, A.; Kurzrock, R. Hyperprogressors after Immunotherapy: Analysis of Genomic Alterations Associated with Accelerated Growth Rate. Clin. Cancer Res. 2017, 23, 4242–4250. [Google Scholar] [CrossRef]
- Amaral, T.; Garbe, C. Acquired resistance mechanisms to immunotherapy. Ann. Transl. Med. 2016, 4, 547. [Google Scholar] [CrossRef]
- Nagarsheth, N.; Peng, D.; Kryczek, I.; Wu, K.; Li, W.; Zhao, E.; Zhao, L.; Wei, S.; Frankel, T.; Vatan, L.; et al. PRC2 Epigenetically Silences Th1-Type Chemokines to Suppress Effector T-Cell Trafficking in Colon Cancer. Cancer Res. 2016, 76, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Thommen, D.S.; Schreiner, J.; Müller, P.; Herzig, P.; Roller, A.; Belousov, A.; Umana, P.; Pisa, P.; Klein, C.; Bacac, M.; et al. Progression of Lung Cancer Is Associated with Increased Dysfunction of T Cells Defined by Coexpression of Multiple Inhibitory Receptors. Cancer Immunol. Res. 2015, 3, 1344–1355. [Google Scholar] [CrossRef]
- You, J.S.; Jones, P.A. Cancer genetics and epigenetics: Two sides of the same coin? Cancer Cell 2012, 22, 9–20. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hossain, S.M.; Lynch-Sutherland, C.F.; Chatterjee, A.; Macaulay, E.C.; Eccles, M.R. Can Immune Suppression and Epigenome Regulation in Placenta Offer Novel Insights into Cancer Immune Evasion and Immunotherapy Resistance? Epigenomes 2021, 5, 16. https://doi.org/10.3390/epigenomes5030016
Hossain SM, Lynch-Sutherland CF, Chatterjee A, Macaulay EC, Eccles MR. Can Immune Suppression and Epigenome Regulation in Placenta Offer Novel Insights into Cancer Immune Evasion and Immunotherapy Resistance? Epigenomes. 2021; 5(3):16. https://doi.org/10.3390/epigenomes5030016
Chicago/Turabian StyleHossain, Sultana Mehbuba, Chiemi F. Lynch-Sutherland, Aniruddha Chatterjee, Erin C. Macaulay, and Michael R. Eccles. 2021. "Can Immune Suppression and Epigenome Regulation in Placenta Offer Novel Insights into Cancer Immune Evasion and Immunotherapy Resistance?" Epigenomes 5, no. 3: 16. https://doi.org/10.3390/epigenomes5030016
APA StyleHossain, S. M., Lynch-Sutherland, C. F., Chatterjee, A., Macaulay, E. C., & Eccles, M. R. (2021). Can Immune Suppression and Epigenome Regulation in Placenta Offer Novel Insights into Cancer Immune Evasion and Immunotherapy Resistance? Epigenomes, 5(3), 16. https://doi.org/10.3390/epigenomes5030016