
The EpiDiverse Plant Epigenome-Wide Association Studies (EWAS) Pipeline

 , , , , ,
, , , , ,  and
and
Abstract
1. Introduction
2. Results and Discussion
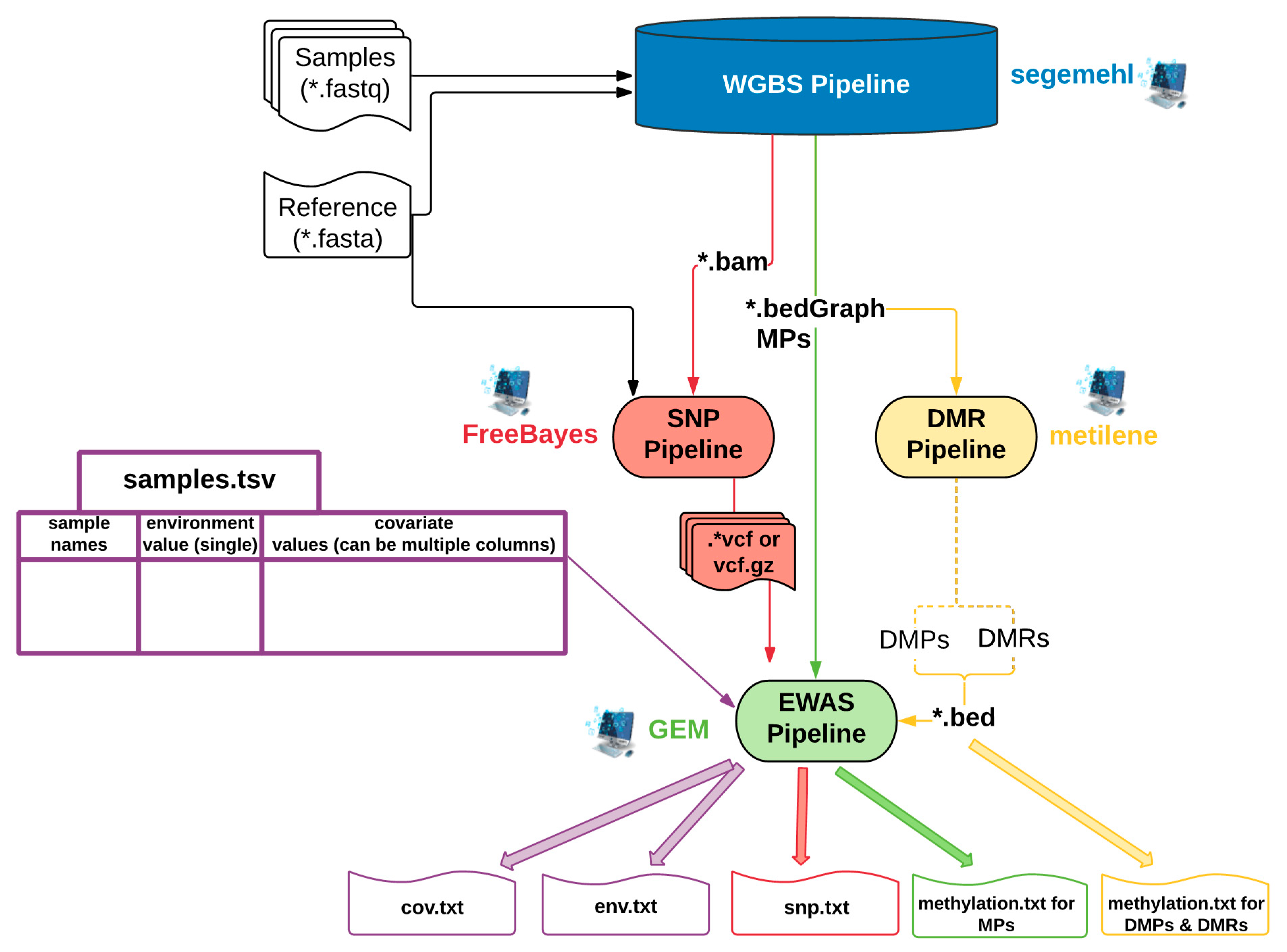
2.1. EpiDiverse EWAS Pipeline Workflow
2.1.1. Input Types for the EWAS Pipeline
2.1.2. Available Models
2.1.3. NA Filtering and Imputation with Methylation and SNP Datasets
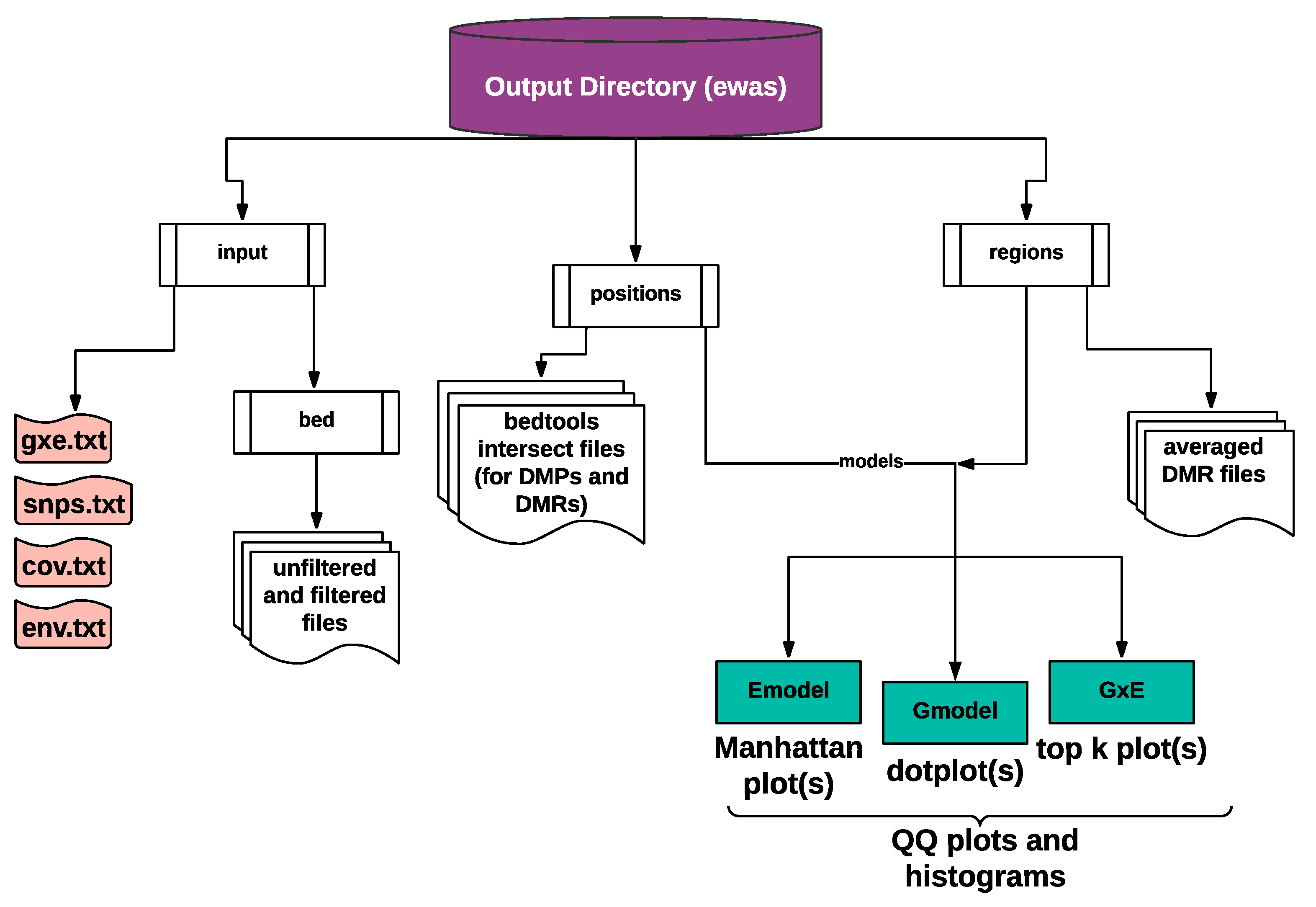
2.1.4. Text and Graphical Outputs
2.2. Evaluation of the EpiDiverse EWAS Pipeline
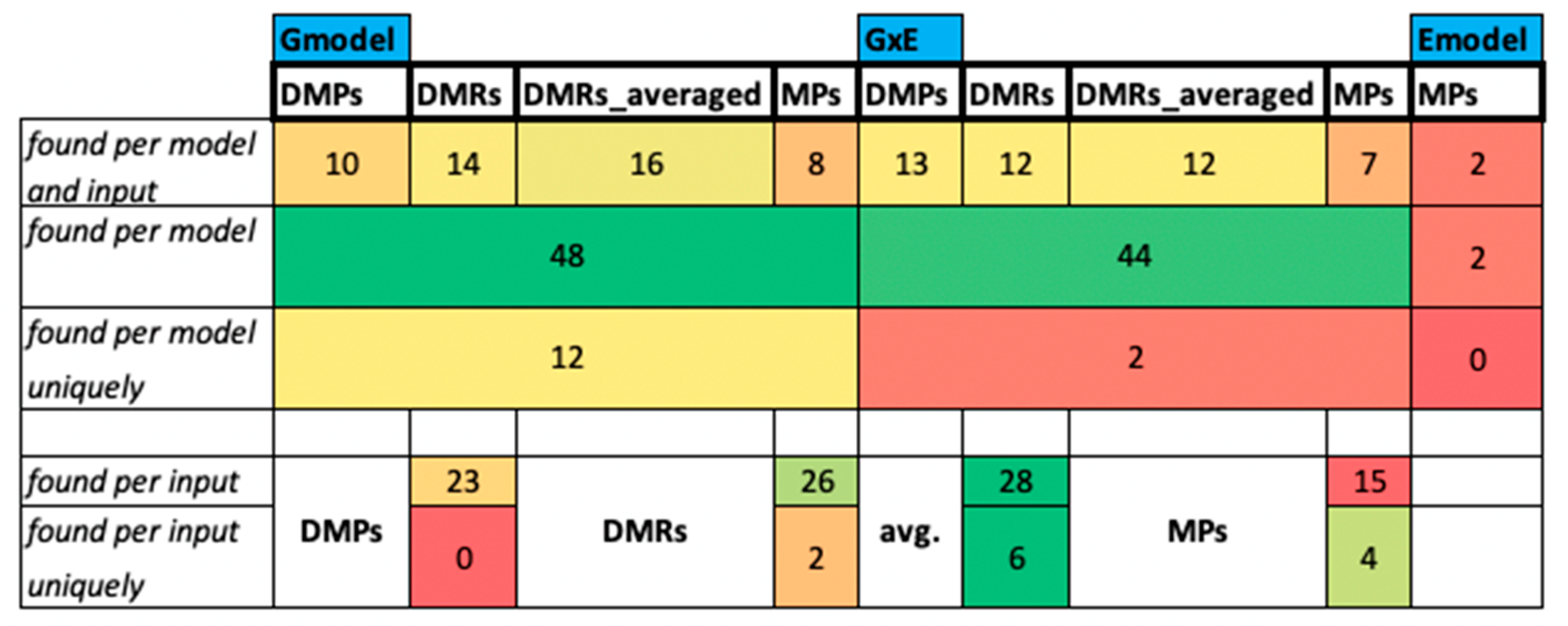
2.2.1. Analysis of Q. lobata Dataset
2.2.2. Analysis of P. abies Dataset
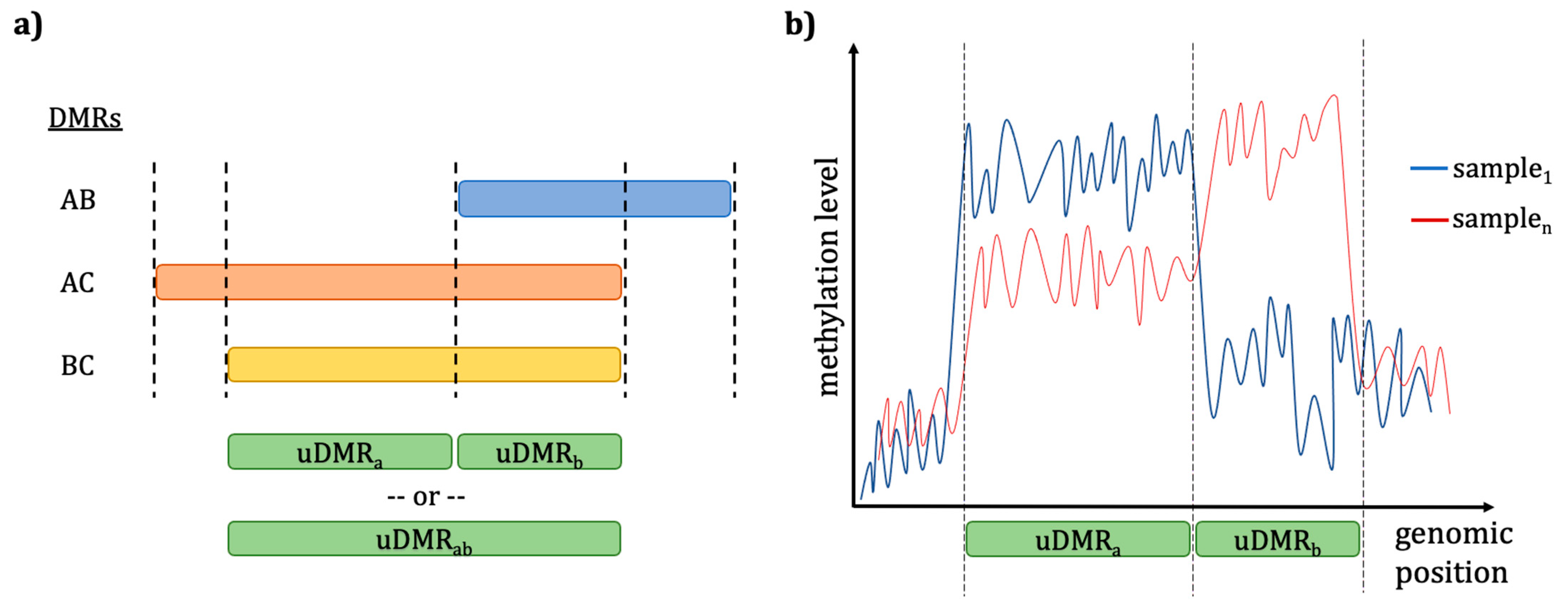
DMP/DMR Analysis Considerations Using Different Callers
Filtering Missing Data after Uniting Individual Methylomes
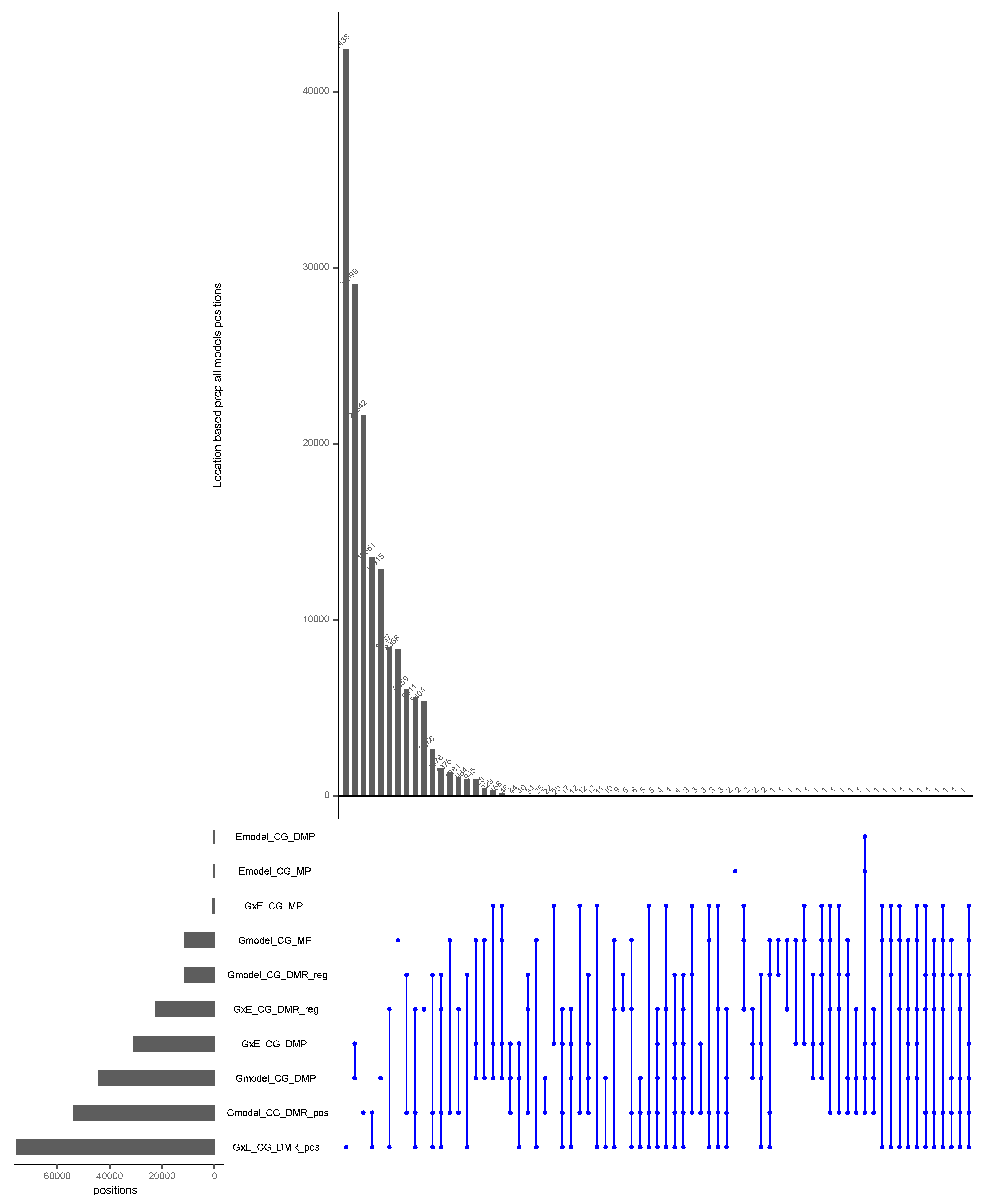
The Intersection of Positions with All Inputs and Models for the CG Context
Removal of Genetic Variants That Might Be Interpreted as Significant Epigenetic Marks
Emodel Output Gene Ontology (GO) Analysis
CG Context G and GxE GO Analysis
2.3. Conclusions
3. Materials and Methods
3.1. The EpiDiverse EWAS Pipeline
3.2. Analysis of Q. lobata Data
3.3. Analysis of P. abies Data
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fuchs, J.; Demidov, D.; Houben, A.; Schubert, I. Chromosomal histone modification patterns—From conservation to diversity. Trends Plant Sci. 2006, 11, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Weinhold, B. Epigenetics: The Science of Change. Environ. Heal Perspect. 2006, 114, A160–A167. [Google Scholar] [CrossRef]
- Sudan, J.; Raina, M.; Singh, R. Plant epigenetic mechanisms: Role in abiotic stress and their generational heritability. Biotech 2018, 8, 172. [Google Scholar] [CrossRef] [PubMed]
- Quadrana, L.; Colot, V. Plant Transgenerational Epigenetics. Annu. Rev. Genet. 2016, 50, 467–491. [Google Scholar] [CrossRef]
- Weigel, D.; Colot, V. Epialleles in plant evolution. Genome Biol. 2012, 13, 249. [Google Scholar] [CrossRef]
- Cubas, P.; Vincent, C.; Coen, E. An epigenetic mutation responsible for natural variation in floral symmetry. Nature 1999, 401, 157–161. [Google Scholar] [CrossRef]
- McClintock, B. Genetic Control of Differentiation, Brookhaven Symposia in Biology. In The Control of Gene Action in Maize; Royal Society: London, UK, 1965; Volume 18, pp. 162–184. [Google Scholar]
- Manning, K.; Tör, M.; Poole, M.; Hong, Y.; Thompson, A.J.; King, G.J.; Giovannoni, J.J.; Seymour, G.B. A naturally occurring epigenetic mutation in a gene encoding an SBP-box transcription factor inhibits tomato fruit ripening. Nat. Genet. 2006, 38, 948–952. [Google Scholar] [CrossRef]
- Mirouze, M.; Paszkowski, J. Epigenetic contribution to stress adaptation in plants. Curr. Opin. Plant Biol. 2011, 14, 267–274. [Google Scholar] [CrossRef]
- McCue, A.D.; Nuthikattu, S.; Reeder, S.H.; Slotkin, R.K. Gene Expression and Stress Response Mediated by the Epigenetic Regulation of a Transposable Element Small RNA. PLoS Genet. 2012, 8, e1002474. [Google Scholar] [CrossRef] [PubMed]
- Chinnusamy, V.; Zhu, J.K. Epigenetic regulation of stress responses in plants. Curr. Opin. Plant Biol. 2009, 12, 133–139. [Google Scholar] [CrossRef]
- Kim, D.H.; Sung, S. Accelerated vernalization response by an altered PHD-finger protein in Arabidopsis. Plant Signal. Behav. 2017, 12, e1308619. [Google Scholar] [CrossRef]
- Yi, S.V. Insights into Epigenome Evolution from Animal and Plant Methylomes. Genome Biol. Evol. 2017, 9, 3189–3201. [Google Scholar] [CrossRef]
- Colot, V.; Rossignol, J.L. Eukaryotic DNA methylation as an evolutionary device. Bioessays 1999, 21, 402–411. [Google Scholar] [CrossRef]
- Feng, S.; Jacobsen, S.E.; Reik, W. Epigenetic Reprogramming in Plant and Animal Development. Science 2010, 330, 622–627. [Google Scholar] [CrossRef] [PubMed]
- Gent, J.I.; Dong, Y.; Jiang, J.; Dawe, R.K. Strong epigenetic similarity between maize centromeric and pericentromeric regions at the level of small RNAs, DNA methylation and H3 chromatin modifications. Nucleic Acids Res. 2011, 40, 1550–1560. [Google Scholar] [CrossRef]
- Lister, R.; O’Malley, R.C.; Tonti-Filippini, J.; Gregory, B.D.; Berry, C.C.; Millar, A.H.; Ecker, J.R. Highly Integrated Single-Base Resolution Maps of the Epigenome in Arabidopsis. Cell 2008, 133, 523–536. [Google Scholar] [CrossRef] [PubMed]
- Law, J.A.; Jacobsen, S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010, 11, 204–220. [Google Scholar] [CrossRef]
- Gu, H.; Smith, Z.D.; Bock, C.; Boyle, P.; Gnirke, A.; Meissner, A. Preparation of reduced representation bisulfite sequencing libraries for genome-scale DNA methylation profiling. Nat. Protoc. 2011, 6, 468–481. [Google Scholar] [CrossRef]
- Kishore, K.; Pelizzola, M. Identification of Differentially Methylated Regions in the Genome of Arabidopsis thaliana. Methods Mol. Biol. 2018, 1675, 61–69. [Google Scholar]
- Tsai, P.C.; Bell, J.T. Power and sample size estimation for epigenome-wide association scans to detect differential DNA methylation. Int. J. Epidemiol. 2015, 44, 1429–1441. [Google Scholar]
- Rakyan, V.K.; Down, T.A.; Balding, D.J.; Beck, S. Epigenome-wide association studies for common human diseases. Nat. Rev. Genet. 2011, 12, 529–541. [Google Scholar] [CrossRef]
- Tam, V.; Patel, N.; Turcotte, M.; Bossé, Y.; Paré, G.; Meyre, D. Benefits and limitations of genome-wide association studies. Nat. Rev. Genet. 2019, 20, 467–484. [Google Scholar] [CrossRef] [PubMed]
- Bush, W.S.; Moore, J.H. Chapter 11: Genome-wide association studies. PLoS Comput. Biol. 2012, 8, e1002822. [Google Scholar]
- Visscher, P.M.; Brown, M.A.; McCarthy, M.I.; Yang, J. Five Years of GWAS Discovery. Am. J. Hum. Genet. 2012, 90, 7–24. [Google Scholar] [CrossRef] [PubMed]
- Visscher, P.M.; Wray, N.R.; Zhang, Q.; Sklar, P.; McCarthy, M.I.; Brown, M.A.; Yang, J. 10 Years of GWAS Discovery: Biology, Function, and Translation. Am. J. Hum. Genet. 2017, 101, 5–22. [Google Scholar] [CrossRef]
- Cortes, L.T.; Zhang, Z.; Yu, J. Status and prospects of genome-wide association studies in plants. Plant Genome 2021, 14, e20077. [Google Scholar] [CrossRef]
- Ersoz, E.S.; Yu, J.; Buckler, E. Applications of Linkage Disequilibrium and Association Mapping in Crop Plants. In Genomics-Assisted Crop Improvement; Varshney, R.K., Tuberosa, R., Eds.; Springer: Dordrecht, The Netherlands, 2007; pp. 97–119. [Google Scholar]
- Liu, H.-J.; Yan, J. Crop genome-wide association study: A harvest of biological relevance. Plant J. 2019, 97, 8–18. [Google Scholar] [CrossRef]
- Sukumaran, S.; Yu, J. Association Mapping of Genetic Resources: Achievements and Future Perspectives. Genom. Plant Genet. Resour. 2013, 207–235. [Google Scholar] [CrossRef]
- Varshney, R.K.; Ribaut, J.-M.; Buckler, E.S.; Tuberosa, R.; Rafalski, J.A.; Langridge, P. Can genomics boost productivity of orphan crops? Nat. Biotechnol. 2012, 30, 1172–1176. [Google Scholar] [CrossRef]
- Gupta, P.K.; Kulwal, P.L.; Jaiswal, V. Association mapping in plants in the post-GWAS genomics era. Adv. Genet. 2019, 104, 75–154. [Google Scholar] [CrossRef]
- Chen, E.; Huang, X.; Tian, Z.; Wing, R.A.; Han, B. The genomics of Oryza species provides insights into rice domestication and heterosis. Annu. Rev. Plant Biol. 2019, 70, 639–665. [Google Scholar] [CrossRef]
- Wang, S.-B.; Feng, J.-Y.; Ren, W.-L.; Huang, B.; Zhou, L.; Wen, Y.-J.; Zhang, J.; Dunwell, J.M.; Xu, S.; Zhang, Y.-M. Improving power and accuracy of genome-wide association studies via a multi-locus mixed linear model methodology. Sci. Rep. 2016, 6, 19444. [Google Scholar] [CrossRef]
- Zhang, Y.; Massel, K.; Godwin, I.D.; Gao, C. Applications and potential of genome editing in crop improvement. Genome Biol. 2018, 19, 210. [Google Scholar] [CrossRef]
- Hindorff, L.A.; Sethupathy, P.; Junkins, H.A.; Ramos, E.M.; Mehta, J.P.; Collins, F.S.; Manolio, T.A. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl. Acad. Sci. USA 2009, 106, 9362–9367. [Google Scholar] [CrossRef] [PubMed]
- Lappalainen, T.; Greally, J.M. Associating cellular epigenetic models with human phenotypes. Nat. Rev. Genet. 2017, 18, 441–451. [Google Scholar] [CrossRef]
- Heard, E.; Martienssen, R.A. Transgenerational epigenetic inheritance: Myths and mechanisms. Cell 2014, 157, 95–109. [Google Scholar] [CrossRef] [PubMed]
- Kalisz, S.; Purugganan, M.D. Epialleles via DNA methylation: Consequences for plant evolution. Trends Ecol. Evol. 2004, 19, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Boyko, A.; Blevins, T.; Yao, Y.; Golubov, A.; Bilichak, A.; Ilnytskyy, Y.; Hollander, J.; Meins, F., Jr.; Kovalchuk, I. Transgenerational adaptation of Arabidopsis to stress requires DNA methylation and the function of Dicer-like proteins. PLoS ONE 2010, 5, e9514. [Google Scholar] [CrossRef]
- Lang-Mladek, C.; Popova, O.; Kiok, K.; Berlinger, M.; Rakic, B.; Aufsatz, W.; Jonak, C.; Hauser, M.-T.; Luschnig, C. Transgenerational Inheritance and Resetting of Stress-Induced Loss of Epigenetic Gene Silencing in Arabidopsis. Mol. Plant 2010, 3, 594–602. [Google Scholar] [CrossRef] [PubMed]
- Latzel, V.; Gonzalez, A.P.R.; Rosenthal, J. Epigenetic Memory as a Basis for Intelligent Behavior in Clonal Plants. Front. Plant Sci. 2016, 7, 1354. [Google Scholar] [CrossRef]
- Paul, D.S.; Beck, S. Advances in epigenome-wide association studies for common diseases. Trends Mol. Med. 2014, 20, 541–543. [Google Scholar] [CrossRef] [PubMed]
- Verma, M. Genome-wide association studies and epigenome-wide association studies go together in cancer control. Future Oncol. 2016, 12, 1645–1664. [Google Scholar] [CrossRef] [PubMed]
- Gugger, P.F.; Fitz-Gibbon, S.; PellEgrini, M.; Sork, V.L. Species-wide patterns of DNA methylation variation in Quercus lobata and their association with climate gradients. Mol. Ecol. 2016, 25, 1665–1680. [Google Scholar] [CrossRef]
- Ong-Abdullah, M.; Ordway, J.M.; Jiang, N.; Ooi, S.-E.; Kok, S.-Y.; Sarpan, N.; Azimi, N.; Hashim, A.T.; Ishak, Z.; Rosli, S.K.; et al. Loss of Karma transposon methylation underlies the mantled somaclonal variant of oil palm. Nature 2015, 525, 533–537. [Google Scholar] [CrossRef]
- Sáez-Laguna, E.; Guevara, M.-Á.; Díaz, L.-M.; Sánchez-Gómez, D.; Collada, C.; Aranda, I.; Cervera, M.-T. Epigenetic Variability in the Genetically Uniform Forest Tree Species Pinus pinea L. PLoS ONE 2014, 9, e103145. [Google Scholar] [CrossRef]
- Rahmani, E.; Yedidim, R.; Shenhav, L.; Schweiger, R.; Weissbrod, O.; Zaitlen, N.; Halperin, E. GLINT: A user-friendly toolset for the analysis of high-throughput DNA-methylation array data. Bioinformatics 2017, 33, 1870–1872. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhao, L.; Liu, D.; Hu, S.; Song, X.; Li, J.; Lv, H.; Duan, L.; Zhang, M.; Jiang, Q.; et al. EWAS: Epigenome-wide association study software 2.0. Bioinformatics 2018, 34, 2657–2658. [Google Scholar] [CrossRef]
- Pan, H.; Holbrook, J.D.; Karnani, N.; Kwoh, C.K. Gene, Environment and Methylation (GEM): A tool suite to efficiently navigate large scale epigenome wide association studies and integrate genotype and interaction between genotype and environment. BMC Bioinform. 2016, 17, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Richards, C.L.; Alonso, C.; Becker, C.; Bossdorf, O.; Bucher, E.; Colomé-Tatché, M.; Durka, W.; Engelhardt, J.; Gaspar, B.; Gogol-Döring, A.; et al. Ecological plant epigenetics: Evidence from model and non-model species, and the way forward. Ecol. Lett. 2017, 20, 1576–1590. [Google Scholar] [CrossRef]
- Nunn, A.; Otto, C.; Stadler, P.F.; Langenberger, D. Comprehensive benchmarking of software for mapping whole genome bisulfite data: From read alignment to DNA methylation analysis. Briefings Bioinform. 2021, 10. [Google Scholar] [CrossRef]
- Kreutz, C.; Can, N.S.; Bruening, R.S.; Meyberg, R.; Mérai, Z.; Fernandez-Pozo, N.; Rensing, S.A. A blind and independent benchmark study for detecting differentially methylated regions in plants. Bioinformatics 2020, 36, 3314–3321. [Google Scholar] [CrossRef]
- Di Tommaso, P.; Chatzou, M.; Floden, E.W.; Barja, P.P.; Palumbo, E.; Notredame, C. Nextflow enables reproducible computational workflows. Nat. Biotechnol. 2017, 35, 316–319. [Google Scholar] [CrossRef] [PubMed]
- Kurtzer, G.M.; Sochat, V.; Bauer, M.W. Singularity: Scientific containers for mobility of compute. PLoS ONE 2017, 12, e0177459. [Google Scholar] [CrossRef]
- Merkel, D. Docker: Lightweight Linux containers for consistent development and deployment. Linux. J. 2014, 2014, 1075–3583. [Google Scholar]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Raineri, E.; Dabad, M.; Heath, S. A Note on Exact Differences between Beta Distributions in Genomic (Methylation) Studies. PLoS ONE 2014, 9, e97349. [Google Scholar] [CrossRef]
- Heer, K.; Ullrich, K.K.; Hiss, M.; Liepelt, S.; Brüning, R.S.; Zhou, J.; Opgenoorth, L.; Rensing, S.A. Detection of somatic epigenetic variation in Norway spruce via targeted bisulfite sequencing. Ecol. Evol. 2018, 8, 9672–9682. [Google Scholar] [CrossRef] [PubMed]
- Sork, V.L.; Fitz-Gibbon, S.T.; Puiu, D.; Crepeau, M.; Gugger, P.F.; Sherman, R.; Stevens, K.; Langley, C.H.; Pellegrini, M.; Salzberg, S.L. First Draft Assembly and Annotation of the Genome of a California Endemic OakQuercus lobataNée (Fagaceae). G3 Genes Genomes Genet. 2016, 6, 3485–3495. [Google Scholar] [CrossRef]
- Nystedt, B.; Street, N.R.; Wetterbom, A.; Zuccolo, A.; Lin, Y.-C.; Scofield, D.G.; Vezzi, F.; Delhomme, N.; Giacomello, S.; Alexeyenko, A.; et al. The Norway spruce genome sequence and conifer genome evolution. Nature 2013, 497, 579–584. [Google Scholar] [CrossRef]
- Murray, K.D.; Webers, C.; Ong, C.S.; Borevitz, J.; Warthmann, N. kWIP: The k-mer weighted inner product, a de novo estimator of genetic similarity. PLoS Comput. Biol. 2017, 13, e1005727. [Google Scholar] [CrossRef]
- Jühling, F.; Kretzmer, H.; Bernhart, S.H.; Otto, C.; Stadler, P.F.; Hoffmann, S.D. Metilene: Fast and sensitive calling of differentially methylated regions from bisulfite sequencing data. Genome Res. 2016, 26, 256–262. [Google Scholar] [CrossRef]
- Akalin, A.; Kormaksson, M.; Li, S.; E Garrett-Bakelman, F.; E Figueroa, M.; Melnick, A.; E Mason, C. methylKit: A comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012, 13, R87. [Google Scholar] [CrossRef] [PubMed]
- Condon, D.E.; Tran, P.V.; Lien, Y.-C.; Schug, J.; Georgieff, M.K.; Simmons, R.A.; Won, K.-J. Defiant: (DMRs: Easy, fast, identification and ANnoTation) identifies differentially Methylated regions from iron-deficient rat hippocampus. BMC Bioinform. 2018, 19, 1–12. [Google Scholar] [CrossRef]
- Ito, T.; Nishio, H.; Tarutani, Y.; Emura, N.; Honjo, M.N.; Toyoda, A.; Fujiyama, A.; Kakutani, T.; Kudoh, H. Seasonal Stability and Dynamics of DNA Methylation in Plants in a Natural Environment. Genes 2019, 10, 544. [Google Scholar] [CrossRef]
- Conway, J.R.; Lex, A.; Gehlenborg, N. UpSetR: An R package for the visualization of intersecting sets and their properties. Bioinformatics 2017, 33, 2938–2940. [Google Scholar] [CrossRef] [PubMed]
- Karger, D.N.; Conrad, O.; Böhner, J.; Kawohl, T.; Kreft, H.; Soria-Auza, R.W.; Zimmermann, N.E.; Linder, H.P.; Kessler, M. Climatologies at high resolution for the earth’s land surface areas. Sci. Data 2017, 4, 170122. [Google Scholar] [CrossRef] [PubMed]
- Wilhelmsson, P.K.I.; Chandler, J.O.; Fernandez-Pozo, N.; Graeber, K.; Ullrich, K.K.; Arshad, W.; Khan, S.; Hofberger, J.A.; Buchta, K.; Edger, P.P.; et al. Usability of reference-free transcriptome assemblies for detection of differential expression: A case study on Aethionema arabicum dimorphic seeds. BMC Genom. 2019, 20, 1–19. [Google Scholar] [CrossRef]
- Mathieu, O.; Reinders, J.; Čaikovski, M.; Smathajitt, C.; Paszkowski, J. Transgenerational Stability of the Arabidopsis Epigenome Is Coordinated by CG Methylation. Cell 2007, 130, 851–862. [Google Scholar] [CrossRef]
- Belt, T.; Altgen, M.; Mäkelä, M.; Hänninen, T.; Rautkari, L. Cellular level chemical changes in Scots pine heartwood during incipient brown rot decay. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Välimaa, A.-L.; Honkalampi-Hämäläinen, U.; Pietarinen, S.; Willför, S.; Holmbom, B.; Von Wright, A. Antimicrobial and cytotoxic knotwood extracts and related pure compounds and their effects on food-associated microorganisms. Int. J. Food Microbiol. 2007, 115, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Kähkönen, M.P.; Hopia, A.I.; Vuorela, H.J.; Rauha, J.P.; Pihlaja, K.; Kujala, T.S.; Heinonen, M. Antioxidant activity of plant extracts containing phenolic compounds. J. Agric. Food Chem. 1999, 47, 3954–3962. [Google Scholar] [CrossRef] [PubMed]
- Ganthaler, A.; Stöggl, W.; Mayr, S.; Kranner, I.; Schüler, S.; Wischnitzki, E.; Sehr, E.M.; Fluch, S.; Trujillo-Moya, C. Association genetics of phenolic needle compounds in Norway spruce with variable susceptibility to needle bladder rust. Plant Mol. Biol. 2017, 94, 229–251. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Krokene, P.; Björklund, N.; Långström, B.; Solheim, H.; Christiansen, E.; Borg-Karlson, A.-K. The influence of Ceratocystis polonica inoculation and methyl jasmonate application on terpene chemistry of Norway spruce, Picea abies. Phytochemistry 2010, 71, 1332–1341. [Google Scholar] [CrossRef] [PubMed]
- Kohler, M.; Kunz1, J.; Herrmann, J.; Hartmann, P.; Jansone, L.; Puhlmann, H.; Wilpert, K.V.; Bauhus, J. The Potential of Liming to Improve Drought Tolerance of Norway Spruce [Picea abies (L.) Karst.]. Front. Plant Sci. 2019, 10, 382. [Google Scholar] [CrossRef]
- Kivimäenpää, M.; Sutinen, S.; Karlsson, P.E.; Selldén, G. Cell Structural Changes in the Needles of Norway Spruce Exposed to Long-term Ozone and Drought. Ann. Bot. 2003, 92, 779–793. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Input | Description | File(s) Formats | Required for Which Runs? | Required for Which Model? |
|---|---|---|---|---|
| sample sheet | Sample list, which includes sample names as key variables, single environment/phenotype data, and covariate(s). | txt | Required for all runs | Required for all models |
| MPs | Context-specific methylation calls per sample. | bedGraph | Required for all runs | Required for all models |
| DMPs | Context-specific differentially methylated positions. | bed | Required to run the pipeline with DMPs | Allowed for all models |
| DMRs | Context-specific differentially methylated regions. | bed | Required to run the pipeline with DMRs | Allowed for all models |
| Genetic variants | Genetic markers either in single or multisample formats. | vcf or vcf.gz | Required to run the G and GxE models | Required for G and GxE models |
| CG | tmax 1 | tmin 2 | GSDD5 3 | CWD 4 |
|---|---|---|---|---|
| Gugger et al., 2016 | 38 | 1 | 0 | 0 |
| EpiDiverse EWAS pipeline | 47 | 2 | 0 | 0 |
| shared amount | 33 | 1 | not applicable | not applicable |
| Shared % based on Gugger et al., 2016 | 86.42% | 100% | 100% | 100% |
| CHG | ||||
| Gugger et al., 2016 | 1 | 0 | 1 | 0 |
| EpiDiverse EWAS pipeline | 1 | 0 | 0 | 1 |
| shared amount | 1 | Not applicable | 0 | 0 |
| Shared % based on Gugger et al., 2016 | 100% | 100% | 0% | 0% |
| CHH | ||||
| Gugger et al., 2016 | 1 | 0 | 1 | 0 |
| EpiDiverse EWAS pipeline | 3 | 16 | 0 | 0 |
| shared amount | 1 | not applicable | 0 | not applicable |
| Shared % based on Gugger et al., 2016 | 100% | 0% | 0% | 100% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Can, S.N.; Nunn, A.; Galanti, D.; Langenberger, D.; Becker, C.; Volmer, K.; Heer, K.; Opgenoorth, L.; Fernandez-Pozo, N.; Rensing, S.A. The EpiDiverse Plant Epigenome-Wide Association Studies (EWAS) Pipeline. Epigenomes 2021, 5, 12. https://doi.org/10.3390/epigenomes5020012
Can SN, Nunn A, Galanti D, Langenberger D, Becker C, Volmer K, Heer K, Opgenoorth L, Fernandez-Pozo N, Rensing SA. The EpiDiverse Plant Epigenome-Wide Association Studies (EWAS) Pipeline. Epigenomes. 2021; 5(2):12. https://doi.org/10.3390/epigenomes5020012
Chicago/Turabian StyleCan, Sultan Nilay, Adam Nunn, Dario Galanti, David Langenberger, Claude Becker, Katharina Volmer, Katrin Heer, Lars Opgenoorth, Noe Fernandez-Pozo, and Stefan A. Rensing. 2021. "The EpiDiverse Plant Epigenome-Wide Association Studies (EWAS) Pipeline" Epigenomes 5, no. 2: 12. https://doi.org/10.3390/epigenomes5020012
APA StyleCan, S. N., Nunn, A., Galanti, D., Langenberger, D., Becker, C., Volmer, K., Heer, K., Opgenoorth, L., Fernandez-Pozo, N., & Rensing, S. A. (2021). The EpiDiverse Plant Epigenome-Wide Association Studies (EWAS) Pipeline. Epigenomes, 5(2), 12. https://doi.org/10.3390/epigenomes5020012