Divergent DNA Methylation Signatures of Juvenile Seedlings, Grafts and Adult Apple Trees

 ,
, 
Abstract
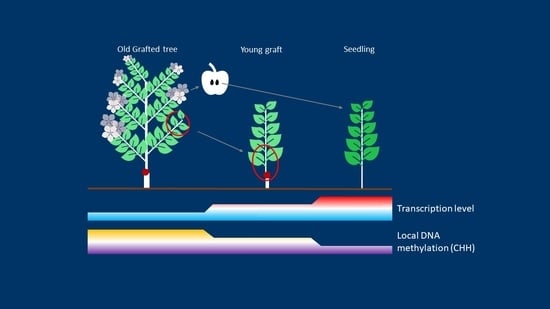
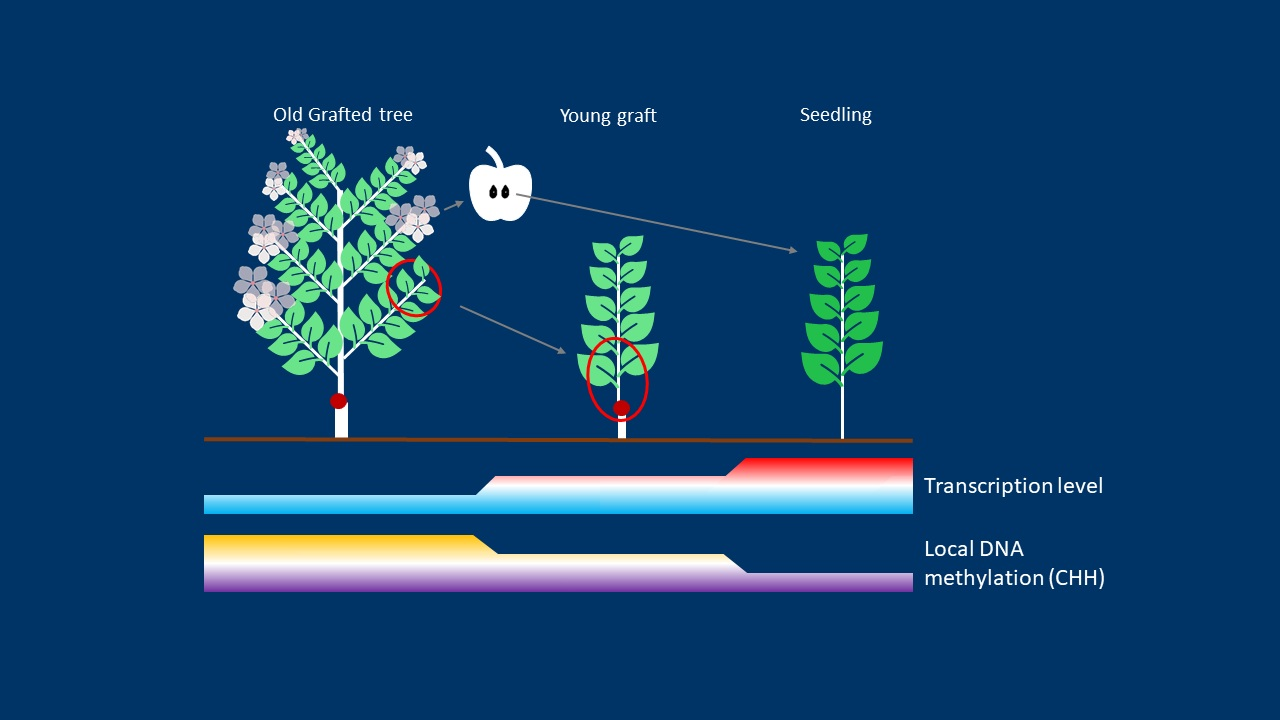
1. Introduction
2. Results
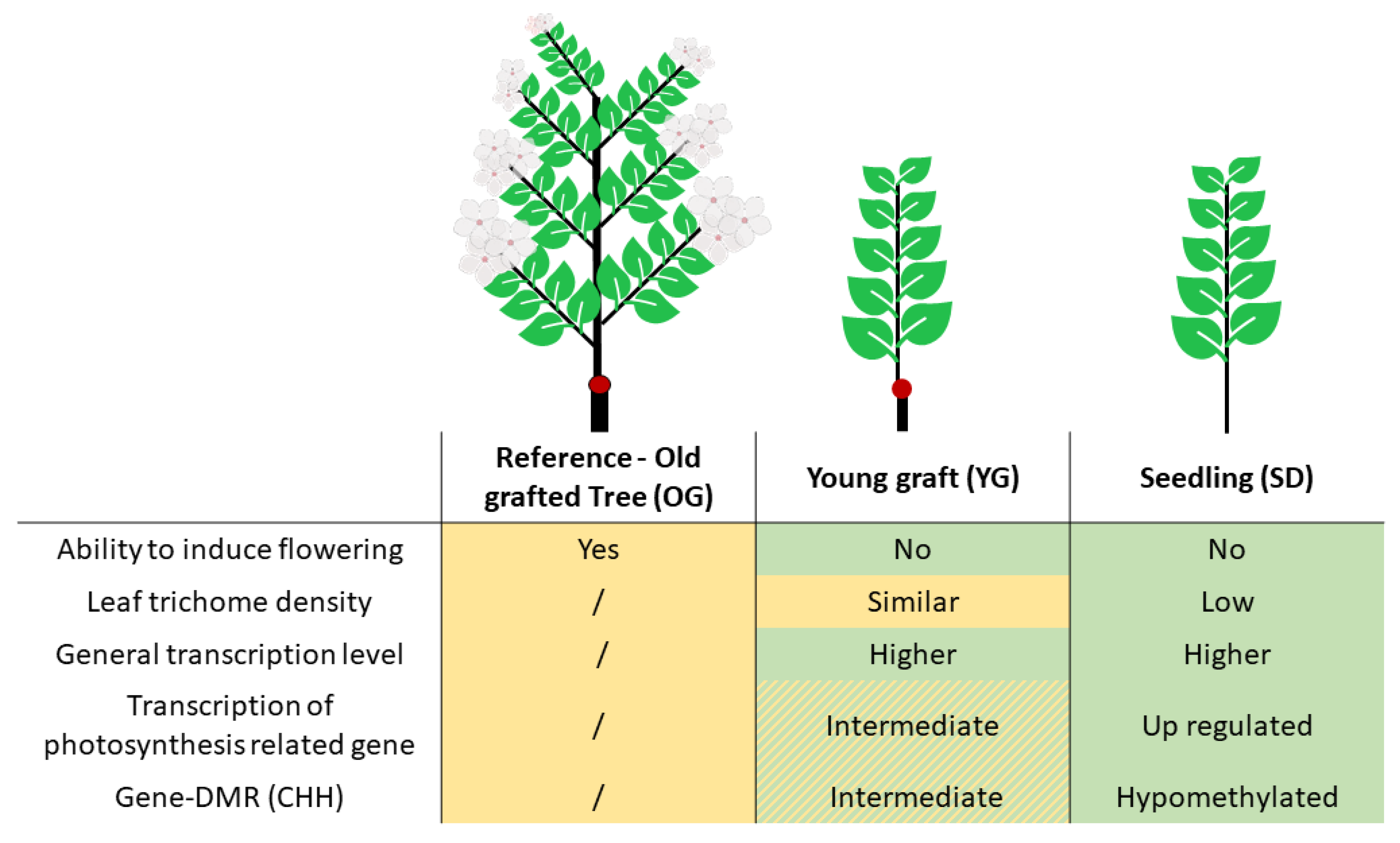
2.1. Phenotypic Comparison of Seedlings, Young Grafts and Adult Trees
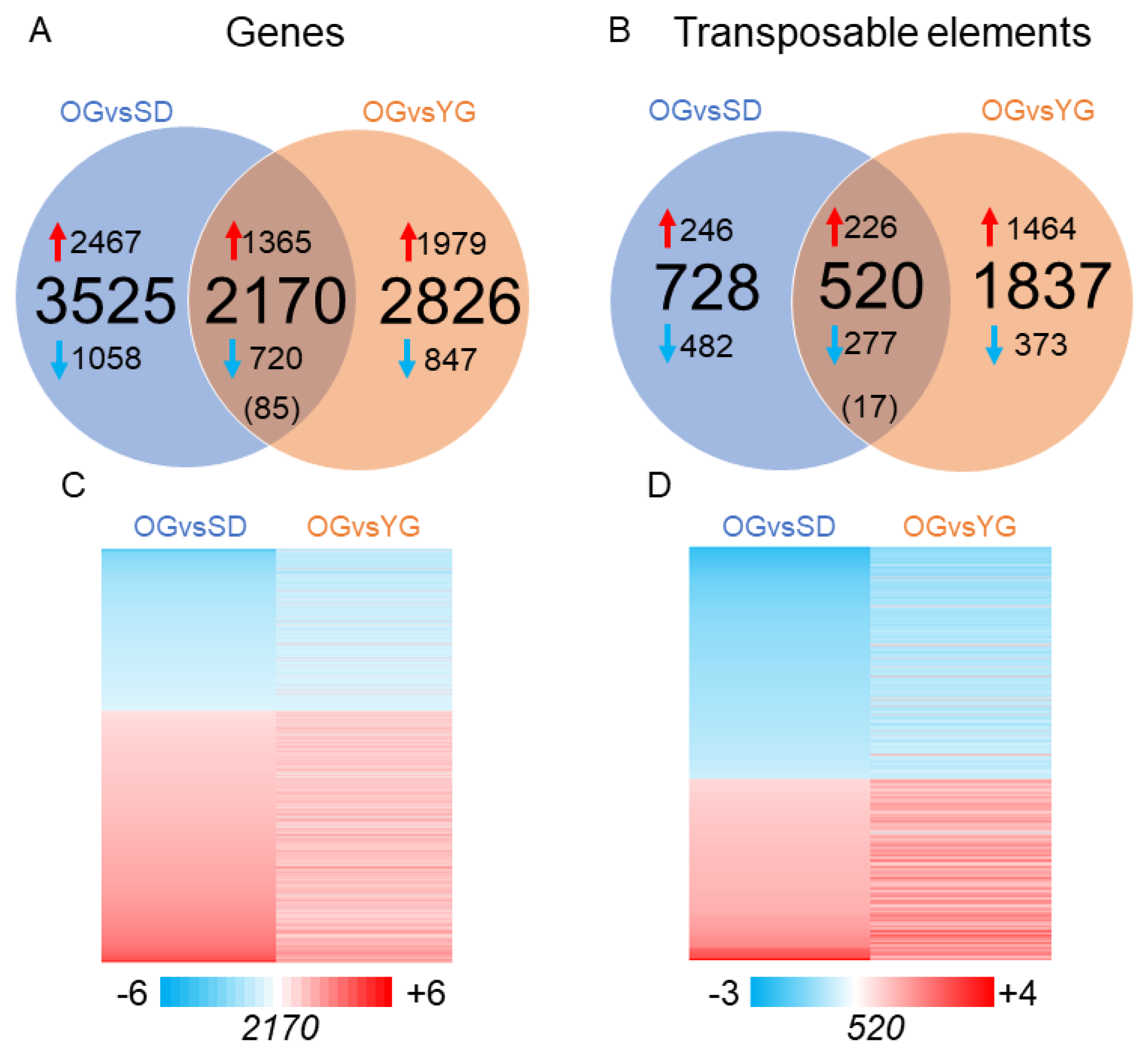
2.2. Transcriptional Profiles of Seedlings, Young Grafts and Adult Trees
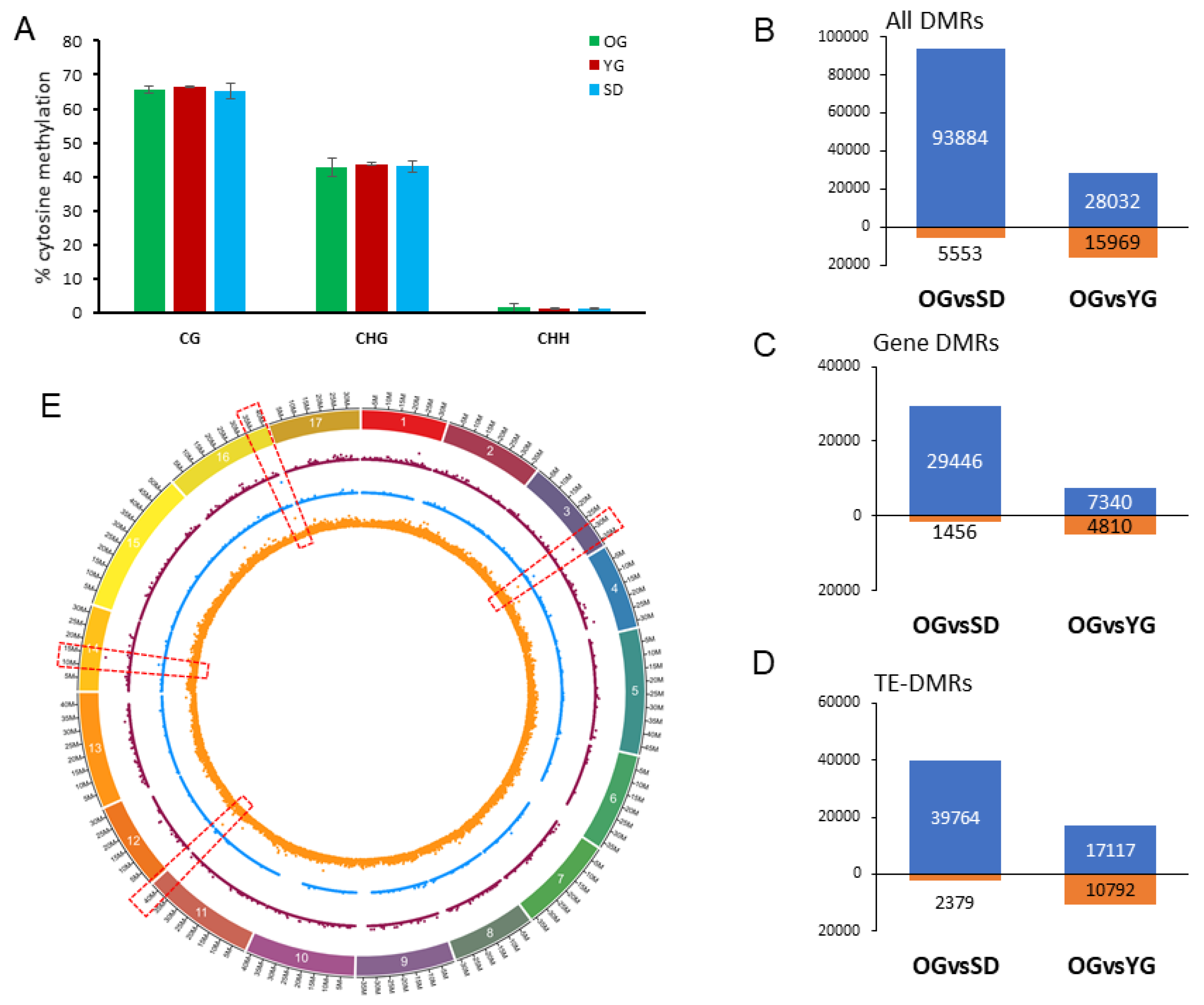
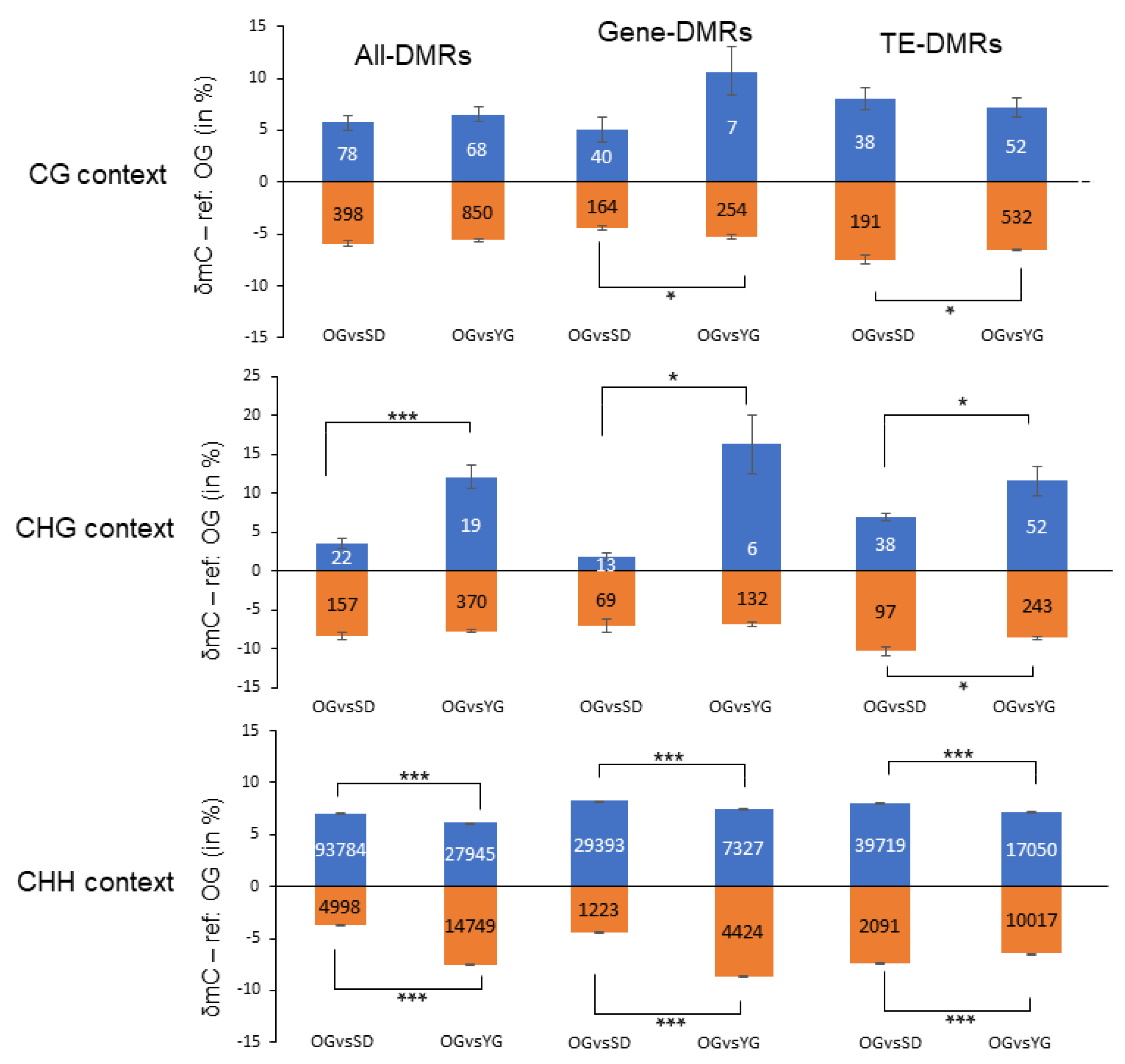
2.3. Global DNA Methylation Analysis of Seedlings, Young Grafts and Adult Trees
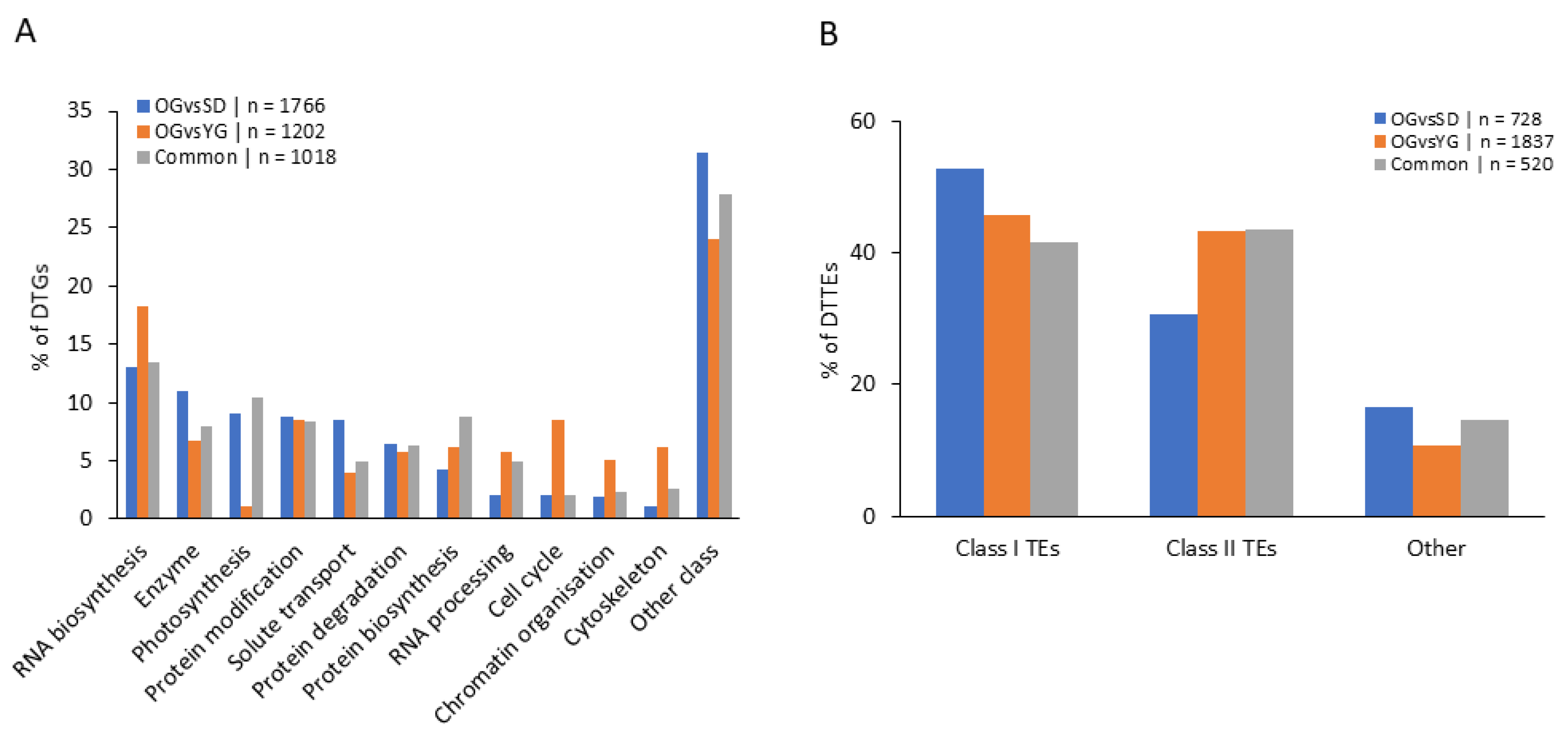
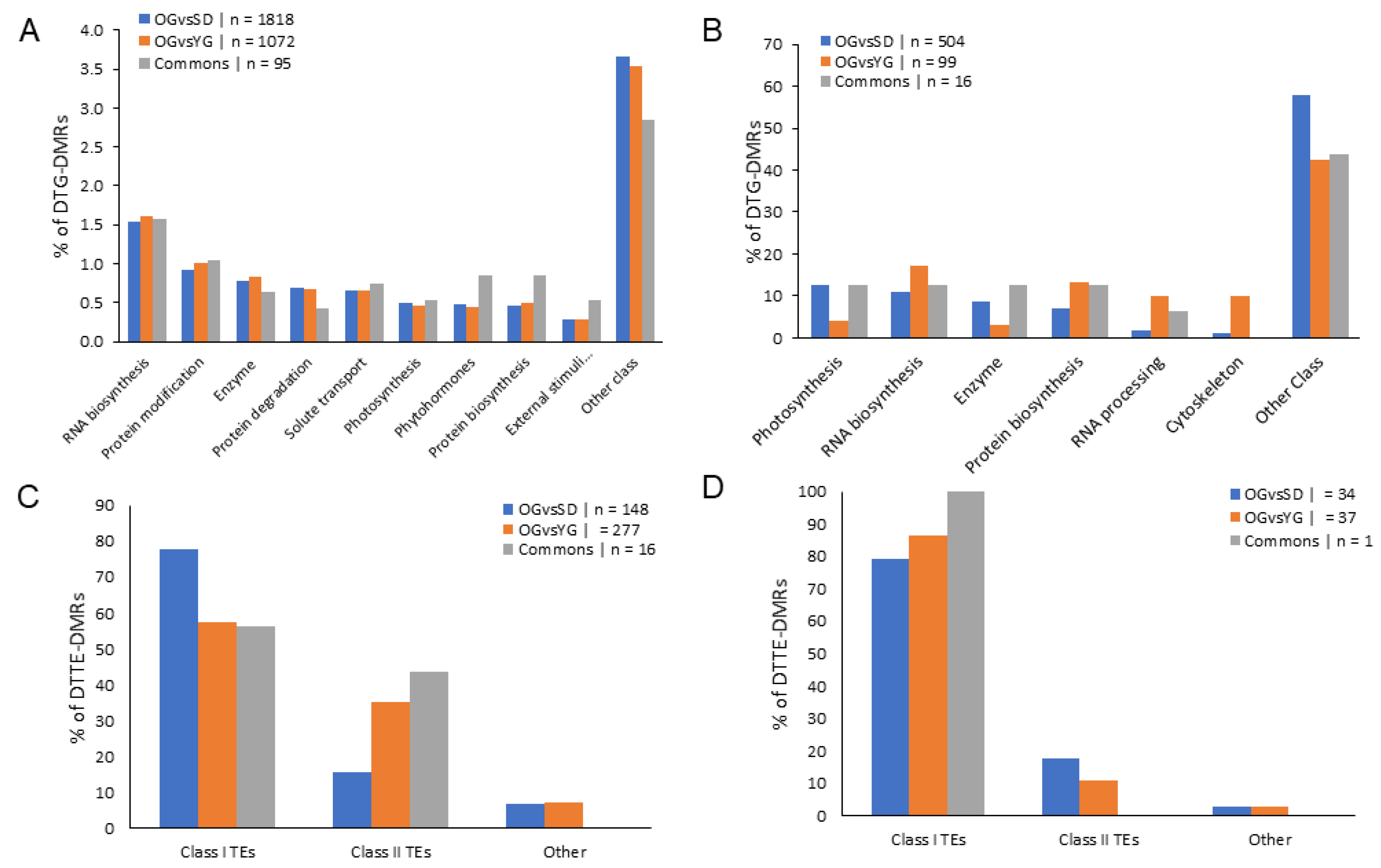
2.4. Classes of Genes Enriched with Differentially Methylated Regions (DMRs)
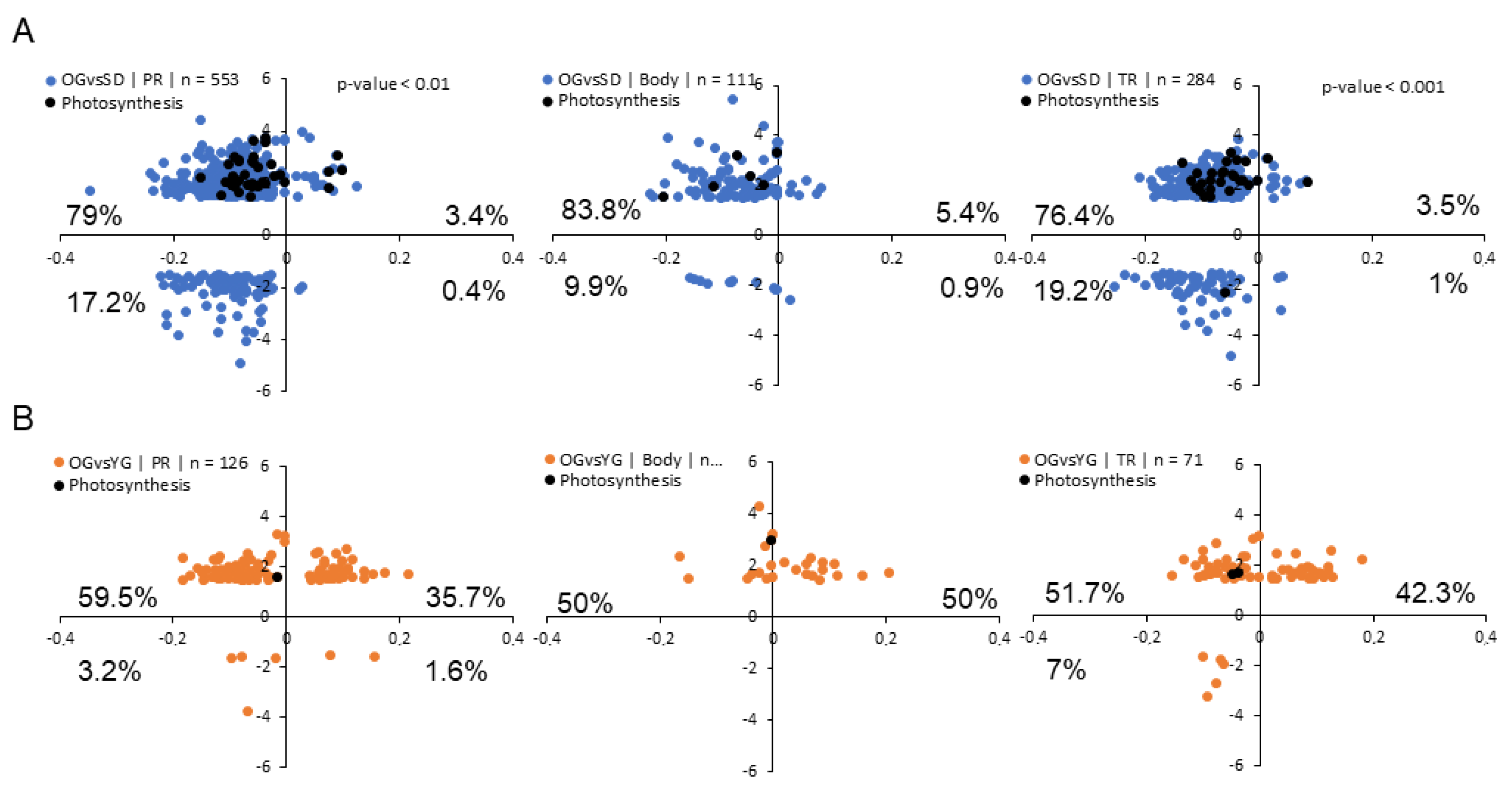
2.5. Relationship between DNA Methylation and Transcription
3. Discussion
3.1. Newly Grafted Plants Are at an Intermediate State between Adult Tree and Juvenile Seedling
3.2. DMRs Correlate with Neighboring Gene Transcription
4. Materials and Methods
4.1. Plant Material
4.2. Phenotyping
4.3. DNA and RNA Extraction
4.4. Bisulfite Sequencing and DMRs Calling
4.5. Microarray
4.6. Reverse Transcriptase Quantitative Polymerase Chain Reaction (RT-qPCR) Microarray Validation
4.7. Differentially Expressed Transcript (DET) Analysis
4.8. Association between DMR and Differentially Transcribed Gene (DTG) or Differentially Transcribed Transposable Element (DTTE)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Holliday, R.; Pugh, J.E. DNA Modification Mechanisms and Gene Activity during Development. Source Sci. New Ser. 1975, 187, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Hauser, M.T.; Aufsatz, W.; Jonak, C.; Luschnig, C. Transgenerational epigenetic inheritance in plants. Biochim. Biophys. Acta Gene Regul. Mech. 2011, 1809, 459–468. [Google Scholar] [CrossRef]
- Gutierrez-Marcos, J.F.; Dickinson, H.G. Epigenetic Reprogramming in Plant Reproductive Lineages. Plant Cell Physiol. 2012, 53, 817–823. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kawashima, T.; Berger, F. Epigenetic reprogramming in plant sexual reproduction. Nat. Rev. Genet. 2014, 15, 613–624. [Google Scholar] [CrossRef]
- Quadrana, L.; Colot, V. Plant Transgenerational Epigenetics. Annu. Rev. Genet. 2016, 50, 467–491. [Google Scholar] [CrossRef]
- Madlung, A.; Comai, L. The effect of stress on genome regulation and structure. Ann. Bot. 2004, 94, 481–495. [Google Scholar] [CrossRef]
- Mirouze, M.; Paszkowski, J. Epigenetic contribution to stress adaptation in plants. Curr. Opin. Plant Biol. 2011, 14, 267–274. [Google Scholar] [CrossRef]
- Köhler, C.; Wolff, P.; Spillane, C. Epigenetic Mechanisms Underlying Genomic Imprinting in Plants. Annu. Rev. Plant Biol. 2012, 63, 331–352. [Google Scholar] [CrossRef]
- Song, J.; Irwin, J.; Dean, C. Minireview Remembering the Prolonged Cold of Winter. Curr. Biol. 2013, 23, 807–811. [Google Scholar] [CrossRef]
- Manning, K.; Tör, M.; Poole, M.; Hong, Y.; Thompson, A.J.; King, G.J.; Giovannoni, J.J.; Seymour, G.B. A naturally occurring epigenetic mutation in a gene encoding an SBP-box transcription factor inhibits tomato fruit ripening. Nat. Genet. 2006, 38, 948–952. [Google Scholar] [CrossRef]
- Schmitz, R.J.; Schultz, M.D.; Urich, M.A.; Nery, J.R.; Pelizzola, M.; Libiger, O.; Alix, A.; McCosh, R.B.; Chen, H.; Schork, N.J.; et al. Patterns of population epigenomic diversity. Nature 2013, 495, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.Y.; Zilberman, D. DNA methylation as a system of plant genomic immunity. Trends Plant Sci. 2014, 19, 320–326. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.M.; Bird, A. DNA methylation landscapes: Provocative insights from epigenomics. Nat. Rev. Genet. 2008, 9, 465–476. [Google Scholar] [CrossRef]
- Miura, A.; Yonebayashi, S.; Watanabe, K.; Toyama, T.; Shimada, H.; Kakutani, T. Mobilization of transposons by a mutation abolishing full DNA methylation in Arabidopsis. Nature 2001, 411, 212–214. [Google Scholar] [CrossRef]
- Mirouze, M.; Reinders, J.; Bucher, E.; Nishimura, T.; Schneeberger, K.; Ossowski, S.; Cao, J.; Weigel, D.; Paszkowski, J.; Mathieu, O. Selective epigenetic control of retrotransposition in Arabidopsis. Nature 2009, 461, 427–430. [Google Scholar] [CrossRef]
- Ito, H.; Gaubert, H.; Bucher, E.; Mirouze, M.; Vaillant, I.; Paszkowski, J. An siRNA pathway prevents transgenerational retrotransposition in plants subjected to stress. Nature 2011, 472, 115–119. [Google Scholar] [CrossRef]
- Gruenbaum, Y.; Naveh-Many, T.; Cedar, H.; Razin, A. Sequence specificity of methylation in higher plant DNA. Nature 1981, 292, 860–862. [Google Scholar] [CrossRef]
- Meyer, P.; Niedenhof, I.; Ten Lohuis, M. Evidence for Cytosine Methylation of Non-Symmetrical Sequences in Transgenic Petunia Hybrida. EMBO J. 1994, 13, 2084–2088. [Google Scholar] [CrossRef]
- Finnegan, E.J.; Genger, R.K.; Peacock, W.J.; Dennis, E.S. DNA Methylation in plants. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1998, 49, 223–247. [Google Scholar] [CrossRef]
- Chan, S.W.-L.; Henderson, I.R.; Jacobsen, S.E. Gardening The Genome: DNA Methylation in Arabidopsis thaliana. Nat. Rev. Genet. 2005, 6, 351. [Google Scholar] [CrossRef]
- Law, J.A.; Jacobsen, S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010, 11, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Lindroth, A.M. Requirement of CHROMOMETHYLASE3 for Maintenance of CpXpG Methylation. Science (80-) 2001, 292, 2077–2080. [Google Scholar] [CrossRef] [PubMed]
- Schermelleh, L.; Haemmer, A.; Spada, F.; Rö Sing, N.; Meilinger, D.; Rothbauer, U.; Cardoso, M.C.; Leonhardt, H. Dynamics of Dnmt1 interaction with the replication machinery and its role in postreplicative maintenance of DNA methylation. Nucleic Acids Res. 2007, 35, 4301–4312. [Google Scholar] [CrossRef] [PubMed]
- Wassenegger, M.; Heimes, S.; Riedel, L.; Sänger, H.L. RNA-directed de novo methylation of genomic sequences in plants. Cell 1994, 76, 567–576. [Google Scholar] [CrossRef]
- Chedin, F.; Lieber, M.R.; Hsieh, C.-L. The DNA methyltransferase-like protein DNMT3L stimulates de novo methylation by Dnmt3a. Proc. Natl. Acad. Sci. USA 2002, 99, 16916–16921. [Google Scholar] [CrossRef]
- Herr, A.J.; Jensen, M.B.; Dalmay, T.; Baulcombe, D.C. RNA polymerase IV directs silencing of endogenous DNA. Science (80) 2005, 308, 118–120. [Google Scholar] [CrossRef]
- Kanno, T.; Huettel, B.; Mette, F.; Aufsatz, W.; Jaligot, E.; Daxinger, L.; Kreil, D.P.; Matzke, M.; Matzke, A.J.M. Atypical RNA polymerase subunits required for RNA-directed DNA methylation. Nat. Genet. 2005, 37, 761–765. [Google Scholar] [CrossRef]
- Hummer, K.E.; Janick, J. Rosaceae: Taxonomy, Economic Importance, Genomics. In Genetics and Genomics of Rosaceae; Springer: New York, NY, USA, 2009; pp. 1–17. [Google Scholar]
- Verhoeven, K.J.F.; Preite, V. Epigenetic variation in asexually reproducing organisms. Evolution (N. Y.) 2014, 68, 644–655. [Google Scholar] [CrossRef]
- Choi, Y.; Gehring, M.; Johnson, L.; Hannon, M.; Harada, J.J.; Goldberg, R.B.; Jacobsen, S.E.; Fischer, R.L.; California, B. DEMETER, a DNA Glycosylase Domain Protein, Is Required for Endosperm Gene Imprinting and Seed Viability in Arabidopsis Replicates to form a Syncytium, and Following Cellulariza-tion, Produces storage Proteins, Lipids, and Starch. Cell 2002, 110, 33–42. [Google Scholar] [CrossRef]
- Ibarra, C.A.; Feng, X.; Schoft, V.K.; Hsieh, T.F.; Uzawa, R.; Rodrigues, J.A.; Zemach, A.; Chumak, N.; Machlicova, A.; Nishimura, T.; et al. Active DNA demethylation in plant companion cells reinforces transposon methylation in gametes. Science (80) 2012, 337, 1360–1364. [Google Scholar] [CrossRef]
- Li, Y.; Kumar, S.; Qian, W. Active DNA demethylation: Mechanism and role in plant development. Plant Cell Rep. 2018, 37, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Slotkin, R.K.; Vaughn, M.; Borges, F.; Tanurdzić, M.; Becker, J.D.; Feijó, J.A.; Martienssen, R.A. Epigenetic reprogramming and small RNA silencing of transposable elements in pollen. Cell 2009, 136, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.; Gao, H.; Zhang, J.; Aldridge, B.; Vickers, M.; Higgins, J.D.; Feng, X. Sexual-lineage-specific DNA methylation regulates meiosis in Arabidopsis. Nat. Genet. 2018, 50, 130–137. [Google Scholar] [CrossRef]
- Olmedo-Monfil, V.; Durán-Figueroa, N.; Arteaga-Vandázquez, M.; Demesa-Arévalo, E.; Autran, D.; Grimanelli, D.; Slotkin, K.; Martienssen, R.A.; Vielle-Calzada, J.-P. Control of female gamete formation by a small RNA pathway in Arabidopsis HHS Public Access. Nature 2010, 464, 628–632. [Google Scholar] [CrossRef]
- Han, Y.Z.; Huang, B.Q.; Zee, S.Y.; Yuan, M. Symplastic communication between the central cell and the egg apparatus cells in the embryo sac of Torenia fournieri Lind. before and during fertilization. Planta 2000, 211, 158–162. [Google Scholar] [CrossRef]
- Saze, H.; Scheid, O.M.; Paszkowski, J. Maintenance of CpG methylation is essential for epigenetic inheritance during plant gametogenesis. Nat. Genet. 2003, 34, 65–69. [Google Scholar] [CrossRef]
- Jablonka, E.; Raz, G. Transgenerational Epigenetic Inheritance: Prevalence, Mechanisms, and Implications for the Study of Heredity and Evolution. Q. Rev. Biol. 2009, 84, 131–176. [Google Scholar] [CrossRef]
- Feng, S.; Jacobsen, S.E.; Reik, W. Epigenetic reprogramming in plant and animal development. Science (80-) 2010, 330, 622–627. [Google Scholar] [CrossRef]
- Herrera, C.M.; Medrano, M.; Bazaga, P. Comparative spatial genetics and epigenetics of plant populations: Heuristic value and a proof of concept. Mol. Ecol. 2016, 25, 1653–1664. [Google Scholar] [CrossRef]
- Niederhuth, C.E.; Bewick, A.J.; Ji, L.; Alabady, M.; Kim, K.D.; Page, J.T.; Li, Q.; Rohr, N.A.; Rambani, A.; Burke, J.M.; et al. Widespread natural variation of DNA methylation within angiosperms. Genome Biol. 2016, 17, 194. [Google Scholar] [CrossRef]
- Wilschut, R.A.; Oplaat, C.; Snoek, L.B.; Kirschner, J.; Verhoeven, K.J.F. Natural epigenetic variation contributes to heritable flowering divergence in a widespread asexual dandelion lineage. Mol. Ecol. 2016, 25, 1759–1768. [Google Scholar] [CrossRef] [PubMed]
- Herman, J.J.; Sultan, S.E. Adaptive transgenerational plasticity in plants: Case studies, mechanisms, and implications for natural populations. Front. Plant Sci. 2011, 2, 102. [Google Scholar] [CrossRef] [PubMed]
- Herrera, C.M.; Bazaga, P. Epigenetic correlates of plant phenotypic plasticity: DNA methylation differs between prickly and nonprickly leaves in heterophyllous Ilex aquifolium (Aquifoliaceae) trees. Bot. J. Linn. Soc. 2013, 171, 441–452. [Google Scholar] [CrossRef]
- Medrano, M.; Herrera, C.M.; Bazaga, P. Epigenetic variation predicts regional and local intraspecific functional diversity in a perennial herb. Mol. Ecol. 2014, 23, 4926–4938. [Google Scholar] [CrossRef]
- Colicchio, J.M.; Miura, F.; Kelly, J.K.; Ito, T.; Hileman, L.C. DNA methylation and gene expression in Mimulus guttatus. BMC Genom. 2015, 16, 507. [Google Scholar] [CrossRef][Green Version]
- Agrawal, A.A.; Strauss, S.Y.; Stout, M.J. Costs of induced responses and tolerance to herbivore in male and female fitness components of wild radish. Evolution (N. Y.) 1999, 53, 1093–1104. [Google Scholar]
- Bilichak, A.; Kovalchuk, I. Transgenerational response to stress in plants and its application for breeding. J. Exp. Bot. 2016, 67, 2081–2092. [Google Scholar] [CrossRef]
- Ramírez-Carrasco, G.; Martínez-Aguilar, K.; Alvarez-Venegas, R. Transgenerational defense priming for crop protection against plant pathogens: A hypothesis. Front. Plant Sci. 2017, 8, 696. [Google Scholar] [CrossRef]
- Latzel, V.; Rendina González, A.P.; Rosenthal, J. Epigenetic memory as a basis for intelligent behavior in clonal plants. Front. Plant Sci. 2016, 7, 1–7. [Google Scholar] [CrossRef]
- Dewan, S.; De Frenne, P.; Broeck, A.V.; Steenackers, M.; Mijnsbrugge, K.V.; Verheyen, K. Transgenerational effects in asexually reproduced offspring of Populus. PLoS ONE 2018, 13, 1–16. [Google Scholar] [CrossRef]
- FAOSTAT. 2017. FAOSTAT. In FOASTAT. Available online: http://www.fao.org/faostat/fr/#data/QC (accessed on 4 May 2019).
- Lane, W.D. Micropropagation of Apple (Malus Domestica Barkh.); Springer: Berlin/Heidelberg, Germany, 1992; pp. 229–243. [Google Scholar]
- Visser, T. Juvenile Phase and Growth of Apple and Pear Seedlings; Springer: Berlin, Germany, 1964; Volume 13. [Google Scholar]
- Lavee, S.; Avidan, N.; Haskal, A.; Ogrodovich, A. Juvenility Period Reduction in Olive Seedlings—A Tool for Enhancement of Breeding. Olivae 1996, 60, 33–41. [Google Scholar]
- Hackett, W.P.; Murray, J.R. Maturation and Rejuvenation in Woody Plants; Springer: Berlin, Germany, 2015; ISBN 0870554921. [Google Scholar]
- Rugini, E. Olive (Olea eu- ropaea L.). In Biotechnology in Agri-Culture and Forestry; Springer: Berlin, Germany, 1986; pp. 253–267. [Google Scholar]
- Bellini, E. Variabilidad genetica heredabilidad de algunos caracteres en plantas de semillas de olive obtenidas por cruzamiento. Olivae Rev. Of. del Cons. Oleícola Int. 1993, 49, 21–34. [Google Scholar]
- Meilan, R. Floral Induction in Woody Angiosperms; Kluwer Academic Publishers: Berlin, Germany, 1997; Volume 14. [Google Scholar]
- Telfer, A.; Bollman, K.M.; Poethig, R.S. Phase change and the regulation of trichome distribution in Arabidopsis thaliana. Development 1997, 124, 645–654. [Google Scholar] [PubMed]
- Poethig, R.S. Phase Change and the Regulation of Developmental Timing in Plants. Science (80) 2003, 301, 334–336. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-W.; Park, M.Y.; Wang, L.-J.; Koo, Y.; Chen, X.-Y.; Weigel, D.; Poethig, R.S. MiRNA Control of Vegetative Phase Change in Trees. PLoS Genet. 2011, 7, e1002012. [Google Scholar] [CrossRef]
- Critchfield, W.B. Leaf dimorphism in Populus trichocarpa. Am. J. Bot. 1960, 47, 699–711. [Google Scholar] [CrossRef]
- Lohse, M.; Nagel, A.; Herter, T.; May, P.; Schroda, M.; Zrenner, R.; Tohge, T.; Fernie, A.R.; Stitt, M.; Usadel, B. Mercator: A fast and simple web server for genome scale functional annotation of plant sequence data. Plant Cell Environ. 2014, 37, 1250–1258. [Google Scholar] [CrossRef]
- Daccord, N.; Celton, J.-M.; Linsmith, G.; Becker, C.; Choisne, N.; Schijlen, E.; van de Geest, H.; Bianco, L.; Micheletti, D.; Velasco, R.; et al. High-quality de novo assembly of the apple genome and methylome dynamics of early fruit development. Nat. Genet. 2017, 49, 1099–1106. [Google Scholar] [CrossRef]
- Basheer-Salimia, R. Juvenility, Maturation, and Rejuvenation in Woody Plants. Hortic. Rev. (Am. Soc. Hortic. Sci.) 2007, 109–155. [Google Scholar]
- Zimmerman, R.H. Juvenility and flowering of fruit trees. In Proceedings of the Acta Horticulturae 34: Symposium on Growth Regulators in Fruit Production, Beltsville, MD, USA, 1 December 1973; pp. 139–142. [Google Scholar]
- Hanke, M.-V.; Flachowsky, H.; Peil, A.; Hättasch, C. No flower no fruit—Genetic potentials to trigger flowering in fruit trees. Genes Genomes Genom. 2007, 1, 1–20. [Google Scholar]
- Zhang, X.Z.; Zhao, Y.B.; Li, C.M.; Chen, D.M.; Wang, G.P.; Chang, R.F.; Shu, H.R. Potential polyphenol markers of phase change in apple (Malus domestica). J. Plant Physiol. 2007, 164, 574–580. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.R.; Smith, A.G.; Hackett, W.P. Differential dihydroflavonol reductase transcription and anthocyanin pigmentation in the juvenile and mature phases of ivy (Hedera helix L.). Planta 1994, 194, 102–109. [Google Scholar] [CrossRef]
- Hand, P.; Besford, R.T.; Richardson, C.M.; Peppitt, S.D. Antibodies to phase related proteins in juvenile and mature Prunus avium. Plant Growth Regul. 1996, 20, 25–29. [Google Scholar] [CrossRef]
- Ryan, M.G.; Binkley, D.; Fownes, J.H. Age related decline in forest productivity: Pattern and process. Adv. Ecol. Res. 1997, 27, 213–262. [Google Scholar]
- Day, M.E.; Greenwood, M.S.; Diaz-Sala, C. Age- and size-related trends in woody plant shoot development: Regulatory pathways and evidence for genetic control. Tree Physiol. 2002, 22, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Bond, B.J. Age-related changes in photosynthesis of woody plants. Trends Plant Sci. 2000, 5, 349–353. [Google Scholar] [CrossRef]
- Greenwood, M.S. Juvenility and maturation in conifers. Curr. Concepts 1995, 15, 433–438. [Google Scholar]
- Greenwood, M.S. Phase change in loblolly pine:Shoot development as a function of age. Physiol. Plant. 1984, 61, 518–522. [Google Scholar] [CrossRef]
- Hutchison, K.W.; Sherman, C.D.; Weber, J.; Smith, S.S.; Singer, P.B.; Greenwood, M.S. Maturation in Larch: II. Effects of Age on Photosynthesis and Gene Expression in Developing Foliage. Plant Physiol. 1990, 94, 1308–1315. [Google Scholar] [CrossRef][Green Version]
- Rebbeck, J.; Jensen, K.F.; Greenwood, M.S. Ozone effects on grafted mature and juvenile red spruce: Photosynthesis, stomatal conductance, and chlorophyll concentration. Can. J. For. Res. 1993, 23, 450–456. [Google Scholar] [CrossRef]
- McGowran, E.; Douglas, G.C.; Parkinson, M. Morphological and physiological markers of juvenility and maturity in shoot cultures of oak (Quercus robur and Q. petraea). Tree Physiol. 1998, 18, 251–257. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, X.; Yazaki, J.; Sundaresan, A.; Cokus, S.; Chan, S.W.L.; Chen, H.; Henderson, I.R.; Shinn, P.; Pellegrini, M.; Jacobsen, S.E.; et al. Genome-wide High-Resolution Mapping and Functional Analysis of DNA Methylation in Arabidopsis. Cell 2006, 126, 1189–1201. [Google Scholar] [CrossRef] [PubMed]
- Zilberman, D.; Gehring, M.; Tran, R.K.; Ballinger, T.; Henikoff, S. Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nat. Genet. 2007, 39, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Lespinasse, Y.; Bouvier, L.; Djulbic, M.; Chevreau, E. Haploidy in apple and pear. Acta Hortic. 1998, 484, 49–54. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Xi, Y.; Li, W. BSMAP: Whole genome bisulfite sequence MAPping program. BMC Bioinformatics 2009, 10, 232. [Google Scholar] [CrossRef]
- R Core Team R. A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2016. [Google Scholar]
- Buels, R.; Yao, E.; Diesh, C.M.; Hayes, R.D.; Munoz-Torres, M.; Helt, G.; Goodstein, D.M.; Elsik, C.G.; Lewis, S.E.; Stein, L.; et al. JBrowse: A dynamic web platform for genome visualization and analysis. Genome Biol. 2016, 17, 66. [Google Scholar] [CrossRef]
- Depuydt, S.; Trenkamp, S.; Fernie, A.R.; Elftieh, S.; Renou, J.-P.; Vuylsteke, M.; Holsters, M.; Vereecke, D. An Integrated Genomics Approach to Define Niche Establishment by Rhodococcus fascians. Plant Physiol. 2009, 149, 1366–1386. [Google Scholar] [CrossRef]
- Smyth, G.K.; Michaud, J.; Scott, H.S. Use of within-array replicate spots for assessing differential expression in microarray experiments. Bioinformatics 2005, 21, 2067–2075. [Google Scholar] [CrossRef]
- Untergasser, A.; Nijveen, H.; Rao, X.; Bisseling, T.; Geurts, R.; Leunissen, J.A.M. Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids 2007, 35, W71–W74. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef] [PubMed]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3, RESEARCH0034. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A pratical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. 1995, 57, 289–300. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Old Graft vs. Seedling | Old Graft vs. Young Graft | |||||
|---|---|---|---|---|---|---|
| Context | Hypomethylated | Hypermethylated | ∑ | Hypomethylated | Hypermethylated | ∑ |
| Number (%) | Number (%) | Number (%) | Number (%) | Number (%) | Number (%) | |
| CHH | 93.784 (94,9) | 4998 (5,1) | 98.782 (99,3) | 27.945 (65,5) | 14.749 (34,5) | 42.694 (97,0) |
| CHG | 22 (12,3) | 157 (87,7) | 179 (0,2) | 19 (4,9) | 370 (95,1) | 389 (0,9) |
| CG | 78 (16,4) | 398 (83,6) | 476 (0,5) | 68 (7,4) | 850 (92,6) | 918 (2,1) |
| ∑ | 93.884 (94,4) | 5553 (5,6) | 99.437 | 28.032 (63,7) | 15.969 (36,3) | 44.001 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perrin, A.; Daccord, N.; Roquis, D.; Celton, J.-M.; Vergne, E.; Bucher, E. Divergent DNA Methylation Signatures of Juvenile Seedlings, Grafts and Adult Apple Trees. Epigenomes 2020, 4, 4. https://doi.org/10.3390/epigenomes4010004
Perrin A, Daccord N, Roquis D, Celton J-M, Vergne E, Bucher E. Divergent DNA Methylation Signatures of Juvenile Seedlings, Grafts and Adult Apple Trees. Epigenomes. 2020; 4(1):4. https://doi.org/10.3390/epigenomes4010004
Chicago/Turabian StylePerrin, Adrien, Nicolas Daccord, David Roquis, Jean-Marc Celton, Emilie Vergne, and Etienne Bucher. 2020. "Divergent DNA Methylation Signatures of Juvenile Seedlings, Grafts and Adult Apple Trees" Epigenomes 4, no. 1: 4. https://doi.org/10.3390/epigenomes4010004
APA StylePerrin, A., Daccord, N., Roquis, D., Celton, J.-M., Vergne, E., & Bucher, E. (2020). Divergent DNA Methylation Signatures of Juvenile Seedlings, Grafts and Adult Apple Trees. Epigenomes, 4(1), 4. https://doi.org/10.3390/epigenomes4010004