Epigenetic Modulation of Self-Renewal Capacity of Leukemic Stem Cells and Implications for Chemotherapy


Abstract
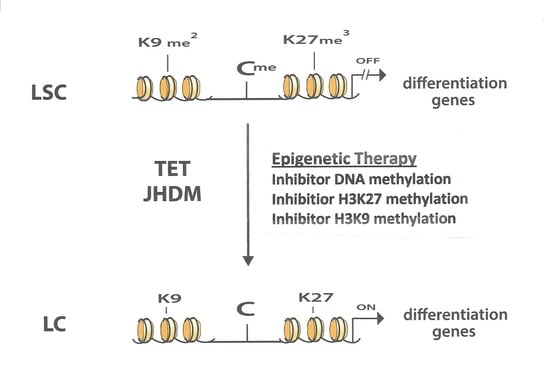
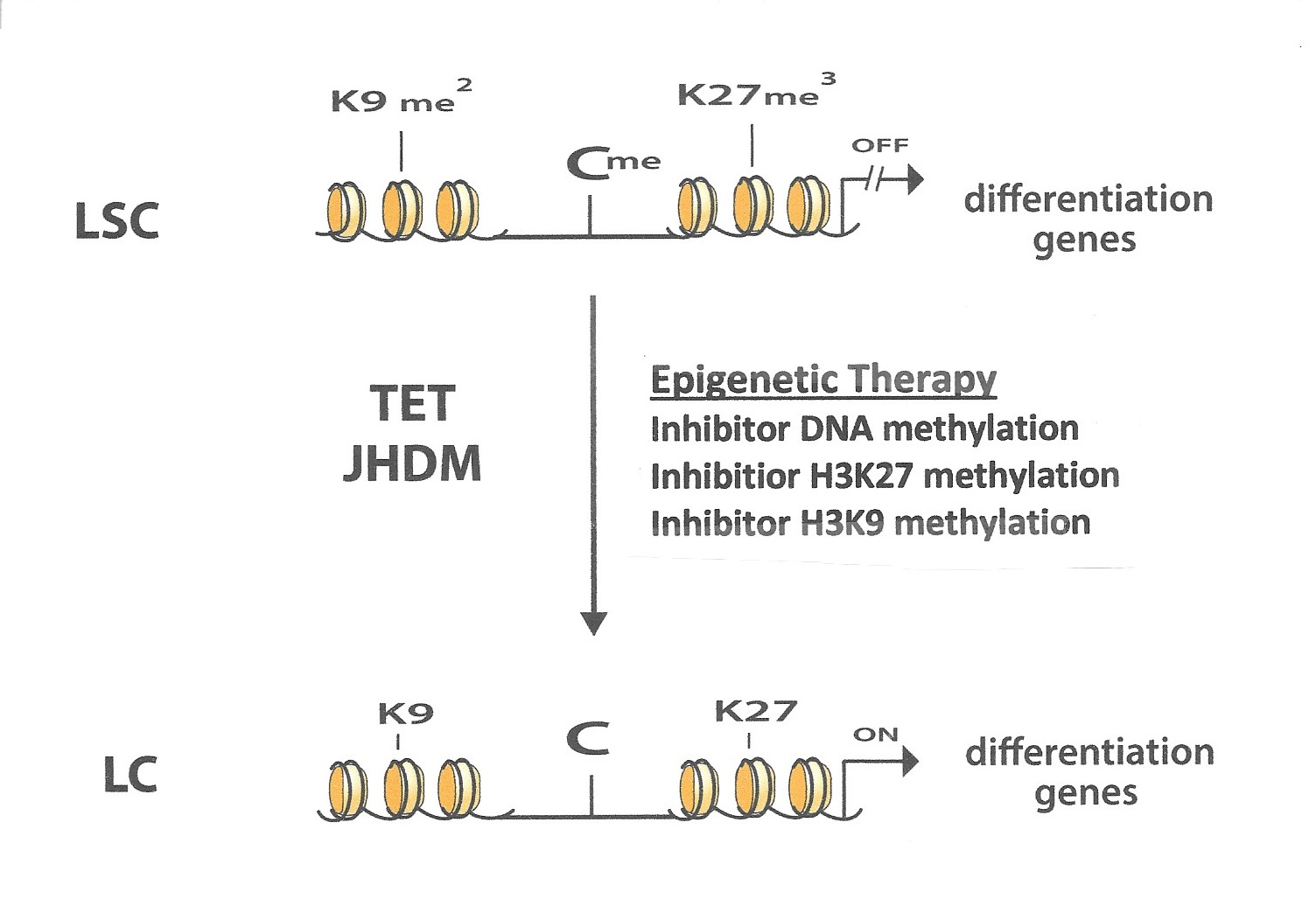{kind=link}
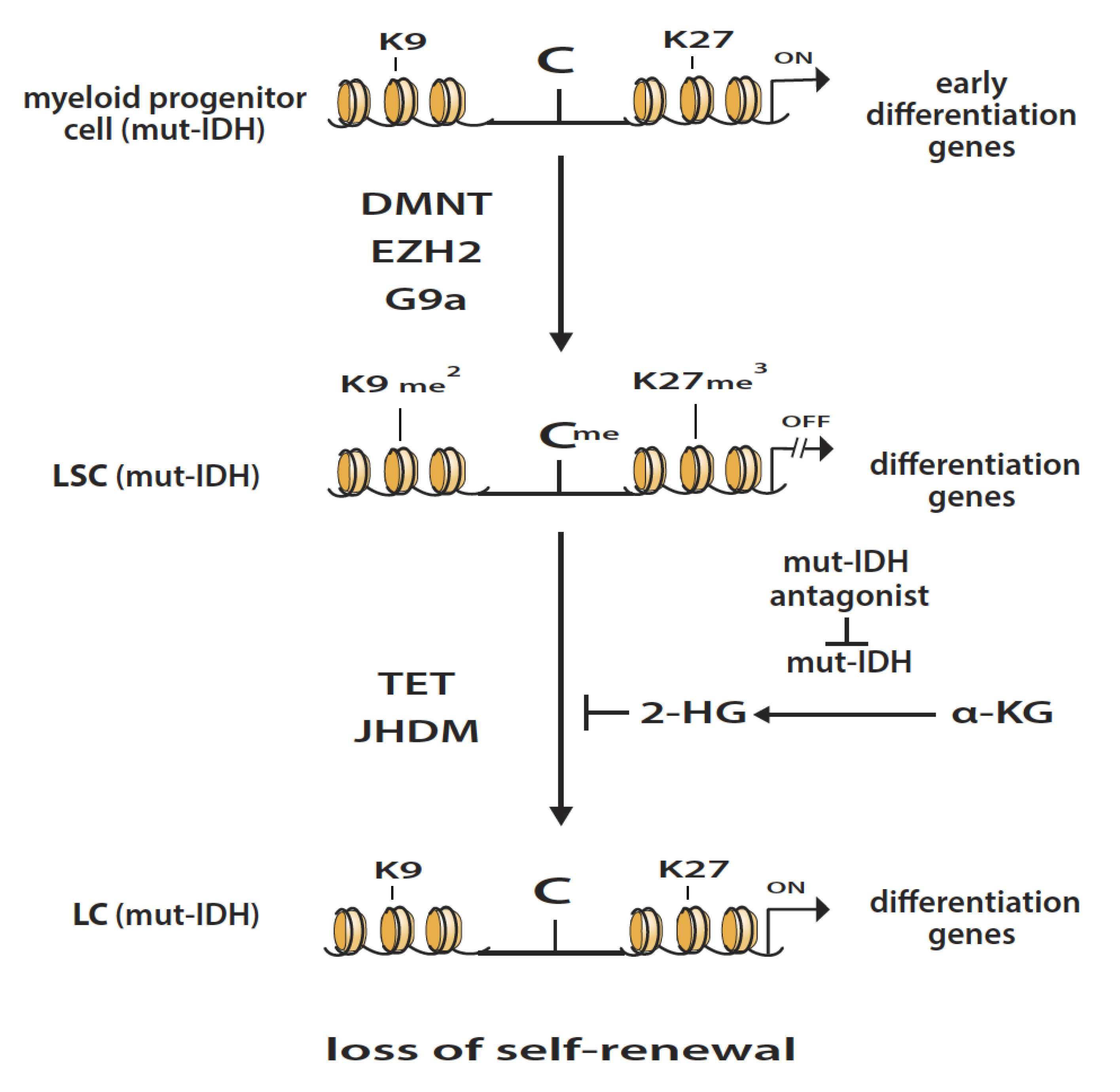{kind=link}
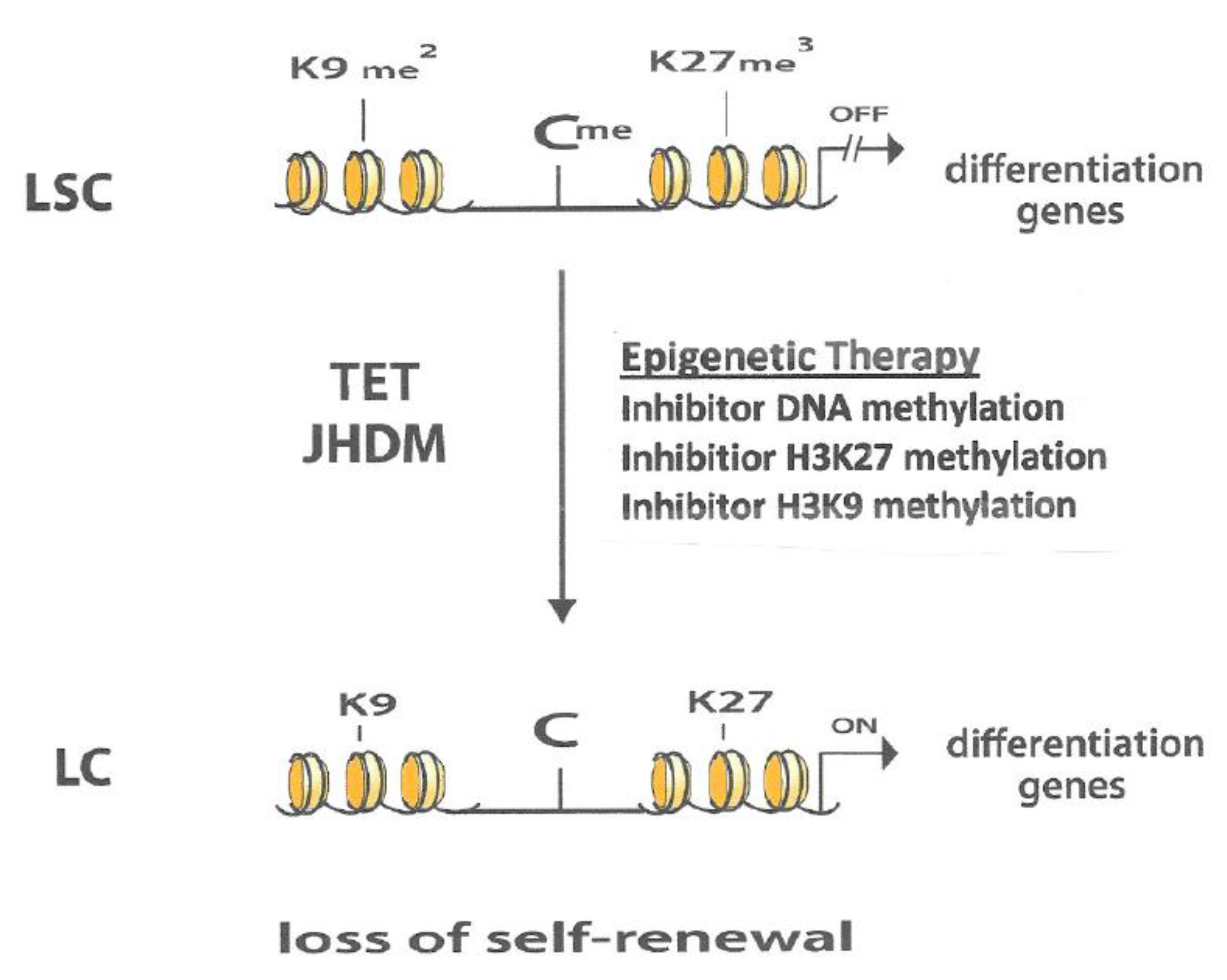{kind=link}
1. Introduction
2. Proliferative Potential of Leukemic Stem Cells
3. Isocitrate Dehydrogenase Mutations in AML
4. Epigenetic Enzymatic Functions That Favor the Development of Leukemogenesis
5. Epigenetic Enzymatic Functions That Can Suppress Leukemogenesis
6. Interaction between Different Epigenetic Gene-Silencing Mechanisms
7. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| AML | Acute myeloid leukemia |
| LSC | Leukemic stem cell |
| IDH | Isocitrate dehydrogenase |
| α-KG | Alpha-keto-glutarate |
| 2-HG | 2-Hydroxyglutarate |
| JHDM | Jumonji-C domain histone demethylase |
| mut-IDH | Mutated Isocitrate dehydrogenase |
| TET | Ten-Eleven-Translocation |
| PRC2 | Polycomb repressive complex 2 |
| EZH2 | Enhancer of zeste 2 polycomb repressive complex 2 subunit |
| DNMT | DNA methyltransferase |
| 5AZA-CdR | 5-Aza-2′-deoxycytidine |
| DZNep | 3-Deazaneplanocin-A |
| H3K27me3 | Histone 3-lysine 27-trimethylated |
| H3K9me2 | Histone 3-lysine 9-diimethylated |
References
- Dohner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute myeloid leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef] [PubMed]
- Hanekamp, D.; Cloos, J.; Schuurhuis, G. Leukemic stem cells: Identification and clinical application. Int. J. Hematol. 2017, 105, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Wouters, B.J.; Delwel, R. Epigenetics and approaches to targeted epigenetic therapy in acute myeloid leukemia. Blood 2016, 127, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Schlesinger, Y.; Straussman, R.; Keshet, I.; Farkash, S.; Hecht, M.; Zimmerman, J.; Eden, E.; Yakhini, Z.; Ben-Shushan, E.; Reubinoff, B.E.; et al. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat. Genet 2007, 39, 232–236. [Google Scholar] [CrossRef]
- Widschwendter, M.; Fiegl, H.; Egle, D.; Mueller-Holzner, E.; Spizzo, G.; Marth, C.; Weisenberger, D.J.; Campan, M.; Young, J.; Jacobs, I.; et al. Epigenetic stem cell signature in cancer. Nat. Genet 2007, 39, 157–158. [Google Scholar] [CrossRef]
- Kim, K.H.; Roberts, C.W.M. Targeting EZH2 in cancer. Nat. Med. 2016, 22, 128–134. [Google Scholar] [CrossRef]
- Easwaran, H.; Johnstone, S.E.; Van Neste, L.; Ohm, J.; Mosbruger, T.; Wang, Q.; Aryee, M.J.; Joyce, P.; Ahuja, N.; Weisenberger, D.; et al. A DNA hypermethylation module for the stem/progenitor cell signature of cancer. Genome Res. 2012, 5, 837–849. [Google Scholar] [CrossRef]
- Ho, T.C.; LaMere, M.; Stevens, B.M.; Ashton, J.M.; Myers, J.R.; O’Dwyer, K.M.; Liesveld, J.L.; Mendler, J.H.; Guzman, M.; Morrissette, J.D.; et al. Evolution of acute myelogenous leukemia stem cell properties after treatment and progression. Blood 2016, 128, 1671–1678. [Google Scholar] [CrossRef]
- Medeiros, B.C.; Fathi, A.T.; DiNardo, C.D.; Pollyea, D.A.; Chan, S.M.; Swords, R. Isocitrate dehydrogenase mutations in myeloid malignancies. Leukemia 2017, 31, 272–281. [Google Scholar] [CrossRef]
- Golub, D.; Iyengar, N.; Dogra, S.; Wong, T.; Bready, D.; Tang, K.; Modrek, A.S.; Placantonakis, D.G. Mutant Isocitrate Dehydrogenase Inhibitors as Targeted Cancer Therapeutics. Front. Oncol. 2019, 9, 417. [Google Scholar] [CrossRef]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Jones, P.A. Epigenetic Determinants of Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019505. [Google Scholar] [CrossRef] [PubMed]
- Morera, L.; Lubbert, M.; Jung, M. Targeting histone methyltransferases and demethylases in clinical trials for cancer therapy. Clin. Epigenetics 2016, 8, 57. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Eng. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef] [PubMed]
- Ushijima, T.; Watanabe, N.; Okochi, E.; Kaneda, A.; Sugimura, T.; Miyamoto, K. Fidelity of the methylation pattern and its variation in the genome. Genome Res. 2003, 13, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, J.; Huang, Y.H.; Muise, A.; Goodell, M.A.; Reich, N.O. Mutations in the DNMT3A DNA methyltransferase in acute myeloid leukemia patients cause both loss and gain of function and differential regulation by protein partners. J. Biol. Chem. 2019, 294, 4898–4910. [Google Scholar] [CrossRef]
- Lübbert, M.; Rüter, B.H.; Claus, R.; Schmoor, C.; Schmid, M.; Germing, U.; Kuendgen, A.; Rethwisch, V.; Ganser, A.; Platzbecker, U.; et al. A multicenter phase II trial of decitabine as first-line treatment for older patients with acute myeloid leukemia judged unfit for induction chemotherapy. Haematologica 2012, 97, 393–401. [Google Scholar] [CrossRef]
- Wilson, V.L.; Jones, P.A.; Momparler, R.L. Inhibition of DNA methylation in L1210 leukemic cells by 5-aza-2′-deoxycytidine as a possible mechanism for chemotherapeutic action. Cancer Res. 1983, 43, 3493–3497. [Google Scholar]
- Fiskus, W.; Wang, Y.; Sreekumar, A.; Buckley, K.M.; Shi, H.; Jillella, A.; Ustun, C.; Rao, R.; Fernandez, P.; Chen, J.; et al. Combined epigenetic therapy with the histone methyltransferase EZH2 inhibitor 3-deazaneplanocin A and the histone deacetylase inhibitor panobinostat against human AML cells. Blood 2009, 114, 2732–4743. [Google Scholar] [CrossRef]
- Xu, F.; Li, X.; Wu, L.; Zhang, Q.; Yang, R.; Yang, Y.; Zhang, Z.; He, Q.; Chang, C. Overexpression of the EZH2, RING1 and BMI1 genes is common in myelodysplastic syndromes: Relation to adverse epigenetic alteration and poor prognostic scoring. Ann. Hematol. 2011, 90, 643–653. [Google Scholar] [CrossRef]
- Ntziachristos, P. Genetic inactivation of the polycomb repressive complex 2 in T cell acute lymphoblastic leukemia. Nat. Med. 2012, 18, 298–301. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.S.; Kelly, J.D.; Hayami, S.; Toyokawa, G.; Takawa, M.; Yoshimatsu, M.; Tsunoda, T.; Field, H.I.; Neal, D.E.; Ponder, B.A.J.; et al. Enhanced expression of EHMT2 is involved in the proliferation of cancer cells through negative regulation of SIAH1. Neoplasia 2011, 13, 676–684. [Google Scholar] [CrossRef] [PubMed]
- Vitkeviciene, A.; Baksiene, S.; Borutinskaite, V.; Navakauskiene, R. Epigallocatechin-3-gallate and BIX-01294 have different impact on epigenetics and senescence modulation in acute and chronic myeloid leukemia cells. Eur. J. Pharmacol. 2018, 838, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, K.D.; Jia, G.; Johansen, J.V.; Pedersen, M.T.; Rapin, N.; Bagger, F.O.; Porse, B.T.; Bernard, O.A.; Christensen, J.; Helin, K. Loss of TET2 in hematopoietic cells leads to DNA hypermethylation of active enhancers and induction of leukemogenesis. Genes Dev. 2015, 29, 910–922. [Google Scholar] [CrossRef]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef]
- Stief, S.M.; Hanneforth, A.; Weser, S.; Mattes, R.; Carlet, M.; Liu, W.-H.; Domínguez Moreno, H.; Oettle, M.; Kempf, J. Loss of KDM6A confers drug resistance in acute myeloid leukemia. Leukemia 2020, 34, 50–62. [Google Scholar] [CrossRef]
- Wang, X.; Fan, H.; Xu, C.; Jiang, G.; Wang, H.; Zhang, J. KDM3B suppresses APL progression by restricting chromatin accessibility and facilitating the ATRA-mediated degradation of PML/RARα. Cancer Cell Int. 2019, 19, 256. [Google Scholar] [CrossRef]
- Xu, X.; Nagel, S.; Quentmeier, H.; Wang, Z.; Pommerenke, C.; Dirks, W.G.; Dirks, W.G.; Macleod, R.A.F.; Drexler, H.G.; Hu, Z. KDM3B shows tumor-suppressive activity and transcriptionally regulates HOXA1 through retinoic acid response elements in acute myeloid leukemia. Leuk. Lymphoma. 2018, 59, 204–213. [Google Scholar] [CrossRef]
- Si, J.; Boumber, Y.A.; Shu, J.; Qin, T.; AhMed, S.; He, R.; Jelinek, J.; Issa, J.P. Chromatin remodeling is required for gene reactivation after decitabine-mediated DNA hypomethylation. Cancer Res. 2010, 70, 6968–6977. [Google Scholar] [CrossRef]
- Momparler, R.L.; Idaghdour, Y.; Marquez, V.E.; Momparler, L.F. Synergistic antileukemic action of inhibitors of DNA methylation and histone methylation. Leuk. Res. 2012, 36, 1049–1054. [Google Scholar] [CrossRef]
- Momparler, R.L.; Côté, S.; Momparler, L.F.; Idaghdour, Y. Inhibition of DNA and histone methylation by 5-aza-2′-deoxycytidine (Decitabine) and 3-deazaneplanocin-A on antineoplastic action and gene expression in myeloid leukemic cells. Front. Oncol. 2017, 7, 19. [Google Scholar] [CrossRef] [PubMed]
- Takeshima, H.; Wakabayashi, M.; Hattori, N.; Yamashita, S.; Ushijima, T. Identification of coexistence of DNA methylation and H3K27me3 specifically in cancer cells as a promising target for epigenetic therapy. Carcinogenesis 2015, 36, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Tian, Y.; Salwen, H.R.; Chlenski, A.; Godley, L.A.; Yang, Q. Histone-lysine methyltransferase EHMT2 is involved in proliferation, apoptosis, cell invasion, and DNA methylation of human neuroblastoma cells. Anticancer Drugs 2013, 24, 484–493. [Google Scholar] [CrossRef]
- Momparler, R.L.; Côté, S.; Momparler, L.F. Enhancement of the Antileukemic Action of 5-aza-2′-Deoxycytidine and 3-Deazaneplanocin-A by Inhibition of Histone Methyltransferase G9a (EHMT2) and by Vitamin C. (Unpublished; Manuscript in Preparation).
- Sato, T.; Cesaroni, M.; Chung, W.; Panjarian, S.; Tran, A.; Madzom, J.; Okamoto, Y.; Zhang, H.; Chen, X.; Jelinek, J.; et al. Transcriptional Selectivity of Epigenetic Therapy in Cancer. Cancer Res. 2017, 77, 470–481. [Google Scholar] [CrossRef] [PubMed]
- Curry, E.; Green, I.; Chapman-Rothe, N.; Shamsaei, E.; Kandil, S.; Cherblanc, F.L.; Payne, L.; Bell, E.; Ganesh, T.; Srimongkolpithak, N.; et al. Dual EZH2 and EHMT2 histone methyltransferase inhibition increases biological efficacy in breast cancer cells. Clin. Epigenetics 2015, 7, 84. [Google Scholar] [CrossRef]
- Momparler, R.L.; Momparler, L.F.; Samson, J. Comparison of the antileukemic activity of 5-aza-2′-deoxycytidine, 1-ß-D-arabinofuranosyl-cytosine and 5-azacytidine against L1210 leukemia. Leuk. Res. 1984, 8, 1043–1049. [Google Scholar] [CrossRef]
- Lemaire, M.; Chabot, G.G.; Raynal, N.J.; Momparler, L.F.; Hurtubise, A.; Bernstein, M.L.; Momparler, R.L. Importance of dose-schedule of 5-aza-2′-deoxycytidine for epigenetic therapy of cancer. BMC Cancer. 2008, 8, 128. [Google Scholar] [CrossRef]
- Momparler, R.L.; Samson, J.; Momparler, L.F.; Rivard, G.E. Cell cycle effects and cellular pharmacology of 5-aza-2′-deoxycytidine. Cancer Chemother. Pharm. 1984, 13, 191–194. [Google Scholar] [CrossRef]
- Momparler, R.L.; Côté, S.; Marquez, V.E.; Momparler, L.F. Comparison of the antineoplastic action of 3-deazaneplanocin-A and inhibitors that target the catalytic site of EZH2 histone methyltransferase. Cancer Rep. Rev. 2020, 3, 1–4. [Google Scholar]
- Fernandes do Nascimento, A.S.; Côté, S.; Jeong, L.S.; Yu, J.; Momparler, R.L. Synergistic antineoplastic action of 5-aza-2′deoxycytidine (decitabine) in combination with different inhibitors of enhancer of zeste homolog 2 (EZH2) on human lung carcinoma cells. J. Cancer Res. Ther. 2016, 4, 42–49. [Google Scholar]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Momparler, R.L.; Côté, S.; Momparler, L.F. Epigenetic Modulation of Self-Renewal Capacity of Leukemic Stem Cells and Implications for Chemotherapy. Epigenomes 2020, 4, 3. https://doi.org/10.3390/epigenomes4010003
Momparler RL, Côté S, Momparler LF. Epigenetic Modulation of Self-Renewal Capacity of Leukemic Stem Cells and Implications for Chemotherapy. Epigenomes. 2020; 4(1):3. https://doi.org/10.3390/epigenomes4010003
Chicago/Turabian StyleMomparler, Richard L., Sylvie Côté, and Louise F. Momparler. 2020. "Epigenetic Modulation of Self-Renewal Capacity of Leukemic Stem Cells and Implications for Chemotherapy" Epigenomes 4, no. 1: 3. https://doi.org/10.3390/epigenomes4010003
APA StyleMomparler, R. L., Côté, S., & Momparler, L. F. (2020). Epigenetic Modulation of Self-Renewal Capacity of Leukemic Stem Cells and Implications for Chemotherapy. Epigenomes, 4(1), 3. https://doi.org/10.3390/epigenomes4010003