Abstract
In the pathophysiologic setting of acute and chronic kidney injury, the excessive activation and recruitment of blood-borne monocytes prompts their differentiation into inflammatory macrophages, a process that leads to progressive glomerulosclerosis and interstitial fibrosis. Importantly, this differentiation of monocytes into macrophages requires the meticulous coordination of gene expression at both the transcriptional and post-transcriptional level. The transcriptomes of these cells are ultimately determined by RNA-binding proteins such as QUAKING (QKI), that define their pre-mRNA splicing and mRNA transcript patterns. Using two mouse models, namely (1) quaking viable mice (qkv) and (2) the conditional deletion in the myeloid cell lineage using the lysozyme 2-Cre (QKIFL/FL;LysM-Cre mice), we demonstrate that the abrogation of QKI expression in the myeloid cell lineage reduces macrophage infiltration following kidney injury induced by unilateral urethral obstruction (UUO). The qkv and QKIFL/FL;LysM-Cre mice both showed significant diminished interstitial collagen deposition and fibrosis in the UUO-damaged kidney, as compared to wild-type littermates. We show that macrophages isolated from QKIFL/FL;LysM-Cre mice are associated with defects in pre-mRNA splicing. Our findings demonstrate that reduced expression of the alternative splice regulator QKI in the cells of myeloid lineage attenuates renal interstitial fibrosis, suggesting that inhibition of this splice regulator may be of therapeutic value for certain kidney diseases.
1. Introduction
The QUAKING RNA-binding proteins belong to the family of KH-type RNA-binding proteins [1,2]. The qkI gene expresses three major alternatively spliced mRNAs (five, six and seven kb) encoding QKI-5, QKI-6 and QKI-7 that differ in their C-terminal 30 amino acids [3]. The QKI-5 isoform contains a unique stretch of basic amino acids at its C-terminus that functions as a nuclear localization signal [4]. QKI belongs to the heteronuclear ribonucleoprotein particle K (hnRNP K) homology (KH) domain family of RNA-binding proteins. QKI binds specific RNA sequences with high affinity termed a QKI response element (QRE) with sequence an ACUAAY (1–20) UAAY (Y; C/U) [5]. The QKI isoforms dynamically alter the post-transcriptional landscape in many cell types including oligodendrocytes, endothelial cells, (smooth) muscle cells, monocytes and macrophages by regulating RNA processing including pre-mRNA splicing [3,6,7,8].
Whole body knockout of qkI is embryonic lethal [9]. However, a mouse model termed the quaking viable mouse (qkv) contains a 1 Mb deletion in the qkI promoter region [10]. When recessive it leads to ubiquitous reduced levels of qkI mRNAs and proteins, subtly reducing QKI-5, and almost completely ablating QKI-6 and -7 expression [3,11]. Bone marrow transplanted from qkv mice into LDLR-/- mice fed a high-fat diet had reduced infiltrating macrophages in their atherosclerotic lesions [12]. Our previous study reported QKI proteins as key players in the post-transcriptional process defining the identity of monocyte and macrophages and their proper function including cell adhesion, migration and phagocytosis [12]. Interestingly, a patient was identified that harbors a deletion within qkI causing QKI haploinsufficiency [13]. Monocytes from this patient had reduced adhesion and migration potential, demonstrating a role for QKI in monocyte/macrophage function [12]. The altered expression of the QKI RNA-binding protein has also been shown to be associated with cancer including angiocentric gliomas, as well as schizophrenia, atherosclerosis and vascular stenosis [11,14,15,16]. Whether QKI plays a role in kidney disease has hitherto not been investigated.
In the pathophysiologic setting of acute and chronic kidney injury, the recruitment of circulating monocytes has been extensively described to play a central role in driving glomerulosclerosis and interstitial fibrosis [17,18,19]. The chronic and progressive sclerosis of the kidney, commonly known as chronic kidney disease (CKD) can lead to end-stage renal disease caused by e.g., diabetes, hypertension, ischemia reperfusion injury following kidney transplantation and acute or chronic allograft rejection [20,21]. A fibrotic process is directly associated with decreased renal function, necessitating renal-replacement therapies such as dialysis or organ transplantation [22,23]. Therefore, gaining a better understanding of macrophage-induced kidney injury, and their role in the initiation, maintenance and progression of CKD to end-stage renal disease could provide new insights for generating therapeutic strategies to prevent loss of renal function.
Circulating monocytes serve as potent mediators of inflammation and organ damage by their homing, extravasation and differentiation into tissue macrophages at sites of injury [24,25]. Depending on the local milieu, monocytes differentiate into macrophages that are characterized by distinct capacities to generate cytokines, phagocytose dead cells and debris, and instruct adaptive immune responses by presenting antigens. A subset of these circulating monocytes, namely non-classical monocytes, has been implicated in directing processes that are geared toward resolving detrimental organ dysfunction by promoting angiogenesis, tissue remodeling and scar-tissue formation [26]. This well-orchestrated reparative response ideally restores tissue architecture and organ function.
Previously, we demonstrated that the RNA-binding protein QKI serves an essential role in orchestrating the transcriptome of monocytes and macrophages by governing pre-mRNA splicing and gene expression [12]. Given that monocyte recruitment and infiltration into the damaged kidney leading to local inflammation and phagocytosis by macrophages is associated with kidney injury and fibrotic responses, we sought to determine if the specific abrogation of QKI in monocytes could attenuate tissue injury and interstitial fibrosis. Herein, we report that QKI-deficiency in monocytes in vivo, using both quaking viable mice and a conditional allele of qkI in mice, led to a reduction in injury, inflammation and renal interstitial fibrosis upon unilateral urethral obstruction. We postulate that the modulation of the cellular transcriptome by interfering with QKI RNA-binding proteins represents a new and potent means of altering monocyte/macrophage responses and could aid in the prevention of kidney diseases.
2. Results
2.1. Abundant Expression of the QKI RNA-Binding Protein in the Kidney
To determine the expression of the QKI isoforms within the kidney, we performed immunohistochemistry on cryosections of healthy mouse kidneys using antibodies that specifically detect the individual QKI-5, QKI-6 and QKI-7 isoforms, together with the endothelial cell marker MECA-32. As illustrated in Figure 1A, all three QKI isoforms were expressed in tubular epithelial cells, although some tubuli appeared void of QKI-7. Endothelial cells (arrowheads) and a few striking intraglomerular cells (arrows) that were MECA-32 negative, did express QKI. The latter are too few in number to represent either mesangial cells or podocytes. QKI-5 and QKI-6 display nuclear enrichment within renal cells, but were also abundantly detected in the cytoplasm of the tubular epithelium, consistent with their known cellular localizations [15]. In contrast, QKI-7 was primarily localized in the peri-nuclear compartment and the cytoplasm (Figure 1A; arrows lower panels). Given that QKI is regarded as a global regulator of pre-mRNA splicing [11] and thus affecting the total cellular transcriptome, the expression of QKI in individual cell types within the kidney could impact their transcriptome and thereby cellular function in a disease setting.
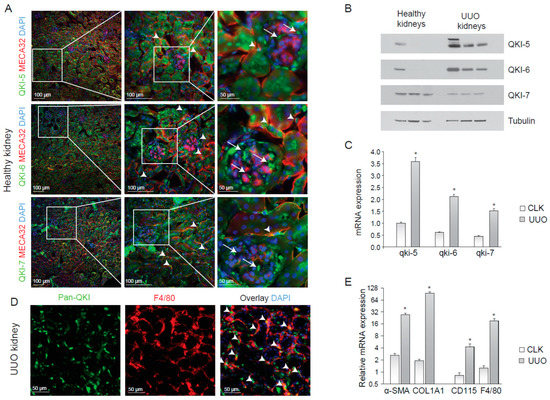
Figure 1.
QUAKING (QKI) is expressed in infiltrating macrophages upon unilateral urethral obstruction. (A) Immunostaining of mouse kidney cryosections for QKI-5 (green, upper panels), QKI-6 (green, middle panels) and QKI-7 (green, lower panels). Endothelial cells are stained using the MECA-32 antibody in red. Nuclei are stained blue using DAPI. (B) Western blot analysis using whole kidney lysates prepared from (n = 3) healthy contralateral kidneys (healthy kidneys) and unilateral urethral obstruction (UUO) kidneys were performed using anti-QKI-5, -QKI-6, -QKI-7 and -tubulin antibodies. (C) Whole kidney lysates of either contralateral kidneys (CLK) or UUO kidneys were assessed for mRNA levels of fibrosis markers (α-SMA, COL1A1) and macrophage markers (CD115, F4/80). (D) Whole kidney lysates were assessed for QKI mRNA levels using qRT-PCR in healthy contralateral kidneys (CLK) as compared to fibrotic kidneys 10 days after UUO. (E) Immunostaining for Pan-QKI (green) and F4/80 (macrophage marker in red) on cryosections of fibrotic kidneys 10 days after UUO. * p ≤ 0.05 by Students’ t-test, error bars represent the standard error of the mean (SEM).
2.2. Kidney Injury Induces an Influx of QKI-Expressing Macrophages
Macrophage infiltration during kidney disease is known to contribute to inflammation and fibrosis [24,25]. To examine whether QKI-expressing macrophages were involved in the inflammatory response during renal injury, we performed unilateral urethral obstruction (UUO) in wild-type C57BL6 mice, a kidney injury model that is associated with tubular cell injury, inflammation and elaborate interstitial fibrosis [27]. After 10 days, we harvested protein and RNA and tissue from the damaged (UUO) kidney and contralateral kidney (CLK). The antibodies were specific for each QKI isoform, as determined by immunoblotting (Figure 1B). Interestingly, the expression of both isoform, QKI-5 and QKI-6, which have access to the nucleus, were increased during UUO (Figure 1B). Furthermore, the mRNA expression levels of all three qkI isoforms significantly increased in fibrotic kidneys, as compared to healthy contralateral kidneys (Figure 1C, gray bars). This increase is likely the result of an influx of inflammatory cells such as bone marrow-derived monocytes and macrophages in the UUO-damaged kidneys. Indeed, immunohistochemical analysis revealed expression of the distinct QKI isoforms in infiltrating macrophages, as evidenced by pan anti-QKI antibody staining (detects all three QKI isoforms) and F4/80 co-localization in UUO-damaged kidneys (Figure 1E, arrowheads). As expected, we detected increased production of the extracellular matrix protein collagen 1A1 (COL1A1), as well as enhanced intracellular expression of the fibroblast-marker smooth-muscle alpha actin (α-SMA) in UUO kidneys (Figure 1D), confirming the induction of a vast fibrotic response. An increase in mRNA expression of F4/80 and CD115, markers of bone marrow-derived monocytes and macrophages, confirmed an influx of inflammatory cells during injury (Figure 1E, gray bars).
2.3. QKI Viable Mice (qkv) Show Decreased Interstitial Fibrosis upon UUO
We next investigated whether reduced QKI expression in monocytes/macrophages offered protection against kidney injury induced by UUO. In vitro M-CSF stimulated bone marrow-derived mouse macrophages isolated from wild-type mice expressed abundant levels of all three of the qkI mRNAs with the nuclear isoform qkI-5 being the most elevated (Figure 2A, open bars). This expression pattern was reduced in qkv mice (Figure 2A, closed bars), a mouse model of reduced QKI expression [10]. This partial maintenance of QKI-5 expression is expected for qkv mice, as the qkI promoter/enhancer deletion has been shown to affect all isoforms within a particular prominent effect on the expression of the cytoplasmic QKI-6 and QKI-7 isoforms [15]. We next induced kidney damage using UUO and compared the damaged kidney with its healthy counterpart (CLK). Importantly, fibrotic kidneys harvested from wild-type littermate controls displayed abundant expression of the macrophage-specific markers F4/80 and CD115 at day 5 and 10 following UUO, consistent with macrophage infiltration (Figure 2B). In contrast, the qkv mouse fibrotic kidneys were characterized by significantly lower mRNA levels of macrophage-specific markers F4/80 and CD115 (Figure 2B). These results suggested that reduced QKI expression in monocytes diminished their kidney influx and subsequent differentiation into macrophages. The reduction in macrophage content was validated by Western blot analysis of whole kidney lysates for the macrophage marker CD206, in which decreased expression was observed in qkv UUO-damaged kidneys (Figure 2C, quantitation of n = 7 mice is presented below). Given the attributing role of infiltrating macrophages in inducing renal fibrosis, we assessed whether mRNA levels of the fibrosis markers α-SMA and COL1A1 were similarly reduced in qkv UUO kidneys. Indeed, these markers were substantially less abundant in qkv UUO kidneys on the mRNA (Figure 2D) and protein level, as assessed by Western blot (Figure 2C, upper blot, quantitation of n = 7 mice is presented below). To further substantiate these anti-fibrotic effects observed in qkv UUO kidneys, we performed Sirius red staining of paraffin-embedded kidney sections to visualize and quantitate all interstitially deposited collagens. These studies clearly showed decreased interstitial fibrosis, as evidenced by less collagen staining in qkv UUO kidneys as compared to wild-type littermate control UUO kidneys (representative photomicrographs are presented in Figure 2E and colorimetric quantitation in Figure 2F). Moreover, a strong correlation between F4/80 and COL1A1 mRNA expression existed (Pearson R = 0.68, p < 0.0001, Figure 2G). Taken together, these data suggest that reduction of QKI expression leads to reduced macrophage influx and subsequently reduced interstitial fibrosis of the kidney.
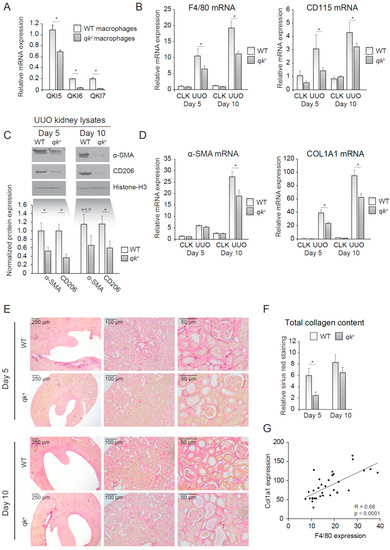
Figure 2.
Quaking viable mice (qkv) display decreased macrophage influx and decreased interstitial fibrosis upon unilateral urethral obstruction (UUO). (A) mRNA expression of QKI mRNA levels in cultured macrophages of either qkv or wild-type littermate controls. (B) mRNA levels of macrophage markers (F4/80, CD115) in whole kidney lysates derived from either qkv mice (gray bars) or wild-type littermate controls on day 5 and 10 after UUO. (C) Western blot analysis of whole UUO kidney lysates for α-SMA, CD206 and histone H3. Quantitation using ImageJ software is provided below. (D) mRNA levels of fibrosis markers (α-SMA and COL1A1) in whole kidney lysates derived from either qkv mice (gray bars) or wild-type littermate controls (open bars) on day 5 and 10 after UUO. (E) Representative photomicrographs of Sirius Red staining for collagen on 4 μm sections of 5- or 10-day UUO kidneys from either qkv or wild-type littermate controls. (F) Quantitation of Sirius red staining is provided. (G) Pearson R correlation is plotted for COL1A1 and F4/80 expression. * p ≤ 0.05 by Students’ t-test, error bars represent the SEM.
2.4. QKIFL/FL;LysM-Cre Mice Show Decreased Interstitial Fibrosis upon UUO Kidney Damage
To test whether the observed effects were cell autonomous to monocytes and macrophages and not the result of a reduction of QKI in e.g., interstitial fibroblasts or renal epithelial cells, we generated QKIFL/FL;LysM-Cre mice, yielding a monocyte/macrophage-specific reduction of QKI (Figure 3A). These genetically modified QKIFL/FL;LysM-Cre mice constitutively express a Cre-recombinase driven by the LysM promoter (also described as Lyz2), yielding monocyte/macrophage-specific Cre-recombinase expression [28]. The presence of loxP sites flanking the genomic DNA sequence encoding exon 2 of the qkI gene induces an exonic frame-shift that generates a premature stop codon in exon 3 [3], leading to the monocyte-specific abrogation of all QKI protein isoforms that are generated by extensive alternative splicing of their 3′ end (as illustrated in Figure 3A). Although the LysM-driven LoxP-recombination event does not terminate transcription of the qkI locus in the non-myeloid cells of the kidney, mRNA levels of the floxed exon 2 were significantly reduced in whole kidney lysates of QKIFL/FL;LysM-Cre UUO kidneys (Figure 3B), suggesting that the residual exon 2 expression is likely contributed by non-macrophage infiltrating cells. Moreover, this demonstrates that the recombination event is specifically induced in infiltrating cells in UUO kidneys and not in contralateral kidneys that are void of infiltrating cells. We subsequently assessed whether these mice are protected from UUO-induced kidney fibrosis, similar to the qkv mice. Indeed, QKIFL/FL;LysM-Cre mice also showed reduced interstitial fibrosis as evidenced by decreased Sirius red staining (representative photomicrographs are shown in Figure 3C and quantitation is provided in Figure 3D). Furthermore, mRNA levels of the macrophage markers F4/80 and CD115 were significantly reduced in QKIFL/FL;LysM-Cre fibrotic kidneys (Figure 3E), whereas the fibrosis genes α-SMA and COL1A1 only showed a trend toward reduced expression levels in QKIFL/FL;LysM-Cre UUO kidneys (Figure 3F). These data further support our hypothesis that QKI expression levels in monocytes and macrophages contribute to the formation of interstitial fibrosis upon UUO kidney damage.
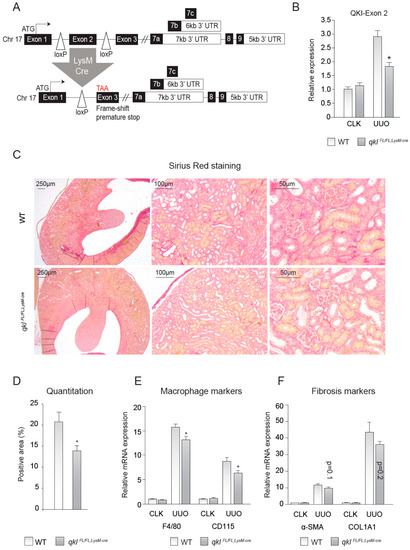
Figure 3.
Monocyte-specific knockout of QKI ameliorates interstitial fibrosis upon unilateral urethral obstruction (UUO). (A) Schematic representing the genomic architecture of the conditional QKI knockout mouse (not to scale). (B) Relative expression level of the mRNA levels of the floxed exon 2 of the QKI mRNA in 10-day CLK and UUO kidneys normalized to the mRNA of β-actin is shown. (C) Sirius red staining for collagen in 4 μm sections of 10-day UUO kidneys derived from qkIFL/FL;LysM-cre or wild-type littermate controls. (D) Colorimetric quantitation of Sirius Red staining using ImageJ software. (E) mRNA levels of macrophage markers (CD115, F4/80) in whole kidney lysates derived from either qkIFL/FL;LysM-cre mice (gray bars) or wild-type littermate controls (open bars) on day 10 after UUO. (F) mRNA levels of fibrosis markers (α-SMA, COL1A1) in whole kidney lysates derived from either qkIFL/FL;LysM-cre mice (gray bars) or wild-type littermate controls (open bars) on day 10 after UUO. * p ≤ 0.05 by Students’ t-test, error bars represent SEM.
2.5. QKI Mediates Alternative Splicing in Mouse Macrophages
Previously, we provided evidence that QKI governs alternative splicing of pre-mRNA transcripts in human macrophages that affect their adhesion and ability to migrate [12]. To confirm whether QKI regulates alternative splicing in mouse macrophages, we isolated total RNA from wild-type and QKIFL/FL;LysM-Cre cultured bone marrow-derived macrophages (Figure 4A) and performed RT-PCR. We initially examined the ability of the QKIFL/FL;LysM-Cre to fully remove the QKI-5, QKI-6 and QKI-7 isoforms. Indeed deletion of qkI exon 2 deletes all three isoforms [3,29], as detected using a pan-QKI antibody. This antibody was generated against the common region of the QKI isoforms and also recognizes, at a low level, some other unknown RNA-binding proteins [30] remaining in the QKIFL/FL;LysM-Cre macrophages. We chose genes for the cytoskeletal protein γ-adducin (ADD3), protein tyrosine phosphatase receptor type O (PTPRO), fibroblast growth factor receptor 1 oncogene partner 2 (FGFR1OP2) and RalBP1-associated Eps domain-containing protein 1 (REPS1), as each gene is known to harbor a QRE neighboring the splicing event [12]. Indeed QKI-deficient macrophages displayed exon inclusion for ADD3, PTPRO and REPS1, and exon exclusion for FGFR1OP2 (Figure 4C,D, quantitation in Figure 4E). These findings show that QKI-deficiency in mouse macrophages influences the transcriptome by regulating alternative splicing.
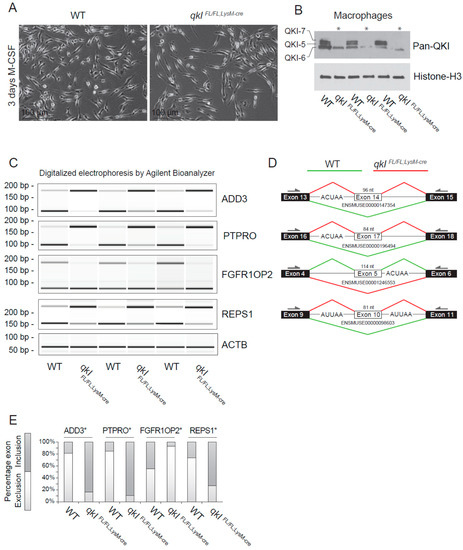
Figure 4.
QKI mediates alternative splicing in macrophages. (A) Photomicrographs of cultured bone marrow-derived macrophages 3 days after M-CSF stimulation. (B) Western blot analysis of cultured mouse macrophages from qkIFL/FL;LysM-cre or wild-type littermate controls using a pan-QKI (N-terminal) antibody and histone H3 loading control. (C) PCR analysis of cassette exon alternative splicing in 5 days M-CSF stimulated cultured macrophages derived from either wild-type or qkIFL/FL;LysM-cre mouse bone marrow. Gel electrophoresis of PCR products generated using primers in flanking exons are shown as digitalized electrophoresis by Agilent Bioanalyzer, illustrating the abundance of alternative mRNA isoforms expressed. (D) In silico analysis of the particular exon assessed illustrates the predicted alternative splicing event based on QRE position (defined as an ACUAA/AUUAA motif) and denotes the predominant splicing event in wild-type (green line) or qkIFL/FL;LysM-cre (red line) macrophages. * p ≤ 0.05 by Students’ t-test. (E) Densitometric quantification of bands in panel (C) using ImageJ software and statistical testing is provided for the assessed splicing events.
3. Discussion
In the present manuscript, we show that the abrogation of QKI expression in the myeloid cell lineage using the quaking viable mice (qkv) and lysozyme 2-Cre (QKIFL/FL;LysM-Cre mice) diminished macrophage infiltration into the UUO-damaged kidney. The qkv and QKIFL/FL;LysM-Cre mice showed a significant diminished interstitial collagen deposition and fibrosis, as compared to wild-type littermates. Furthermore, we show that macrophages isolated from QKIFL/FL;LysM-Cre mice are associated with defects in pre-mRNA splicing that affect the macrophage transcriptome. Previously, we have shown that perturbation of splicing in monocytes impacts their capacity to become bona fide macrophages [12]. Here, we show that reduced expression of the alternative splice regulator QKI in cells of myeloid lineage limits renal interstitial fibrosis. Thus, QKI RNA-binding proteins may be of therapeutic value for certain kidney diseases.
At present, abrogating the expression or activity of RNA-binding proteins does not yet belong to the list of therapeutic modalities utilized for limiting disease progression in general. This could potentially be due to their upstream hierarchical position in guiding cellular transcriptomes in health and disease. Nonetheless, our data suggest that targeting QKI in the monocyte could be an effective means of limiting their adhesion, extravasation and differentiation at sites of kidney injury. To achieve this goal, several technologies could be implemented, namely: (1) naked or lipid-encapsulated siRNAs to target individual RNA-binding proteins (reviewed in [31]); (2) pRNAs and RNA aptamer-based approaches [32,33,34]; and (3) RNA-based antisense oligonucleotides. Importantly, QKI does not represent the sole RNA-binding protein that is active in monocytes and macrophages, as a diverse repertoire of such proteins are active and meticulously determining the transcriptome, including AU-rich element-binding proteins (ARE-BPs) that in particular interact with cytokine mRNAs [35,36] and Roquins, which also drive mRNA decay processes [37]. These RNA-binding proteins, in concert with RNA-binding proteins such as QKI [11], CELF1 [38] and MBNL1 [39], are responsible for triggering both changes in monocyte/macrophage function and their mediation of pathophysiological processes.
ADD3, a structural cytoskeletal protein that influences the cortical actin layer [40], could very well influence the homing of monocytes to the injured kidney. Moreover, PTPRO, a tyrosine phosphatase, has been shown to localize to the cellular “integrin adhesome” [41], which is essential in monocyte adhesion and migration [42]. PTPRO has furthermore been implicated in ischemia reperfusion injury of the liver by activating Nuclear factor-κB (NF-κB) [43] and thereby the classical or non-classical activation of monocytes/macrophages [44,45]. Interestingly, FGFR1OP2 has been extensively investigated in the context of hematopoietic malignancies including acute myeloid leukemia and myelomonocytic leukemia where it is involved in monocyte precursor differentiation in the bone marrow [46,47,48]. The conclusion that the described alternative splicing events and their resulting protein isoforms contribute to proper physiological monocyte/macrophage function is likely, but remains to be fully elucidated.
It is tempting to speculate that a reduction of QKI expression levels not only affects the phenotype of monocytes and infiltrated macrophages (for review see [11]), but could also skew these cells toward a tissue-regenerative and fibrosis-ameliorating function. Although some controversy still exists about the exact role of monocytes and macrophages in organ fibrosis, it has been suggested that macrophages can also contribute to the resolution of fibrosis by affecting myofibroblast function and enhancing matrix metalloproteinase (MMP) and collagenase expression, while concomitantly decreasing the expression of tissue inhibitors of MMPs (TIMPs) [49]. While numerous studies have provided evidence that organ fibrosis can be reversed [50,51,52,53,54,55,56], a limited number of studies have actually shown regression or resolution of fibrotic lesions in the renal compartment [57,58,59]. Recently, Sugimoto and colleagues provided evidence of effective renal fibrosis resolution following the administration of an activator of Activin-like kinase 3 (ALK3/BMPR1A), namely THR-123 [60]. Similar to our observation that a reduction of QKI in a monocyte-specific fashion reduces monocyte infiltration, inflammation and kidney fibrosis, with the THR-123 treatment regimen employed, lead to a decrease in monocyte influx and a reversal of kidney fibrosis in vivo. Similarly, we showed previously that modifying the transcriptome of bone marrow cells by hematopoietic overexpression of microRNA-126, exerts anti-inflammatory and anti-fibrotic effects in vivo [61]. We, therefore, propose that monocytes and macrophages are the ideal cell type of which the cellular phenotype can be tailored to ameliorate or even direct reversal of kidney fibrosis.
In conclusion, we have identified that the RNA-binding protein QKI influences the phenotype of infiltrating macrophages and their capacity to drive kidney injury. Thus, diminishing expression or reducing QKI activity in monocytes and macrophages could serve as an effective means of limiting renal fibrosis in the damaged kidney.
4. Material and Methods
4.1. Cell Culture
Mouse macrophages were derived from whole bone marrow samples of mouse femurs. Cells were plated in 6-well plates at a density of 2 × 106 cells per well in RPMI medium containing 100 U/mL penicillin, 100 U/mL streptomycin, 300 μg/mL glutamine (Thermo Fisher Scientific, Waltham, MA, USA) and 10% fetal calf serum (Cambrex, East Rutherford, NJ, USA). Additionally, 10 ng/mL recombinant mouse M-CSF (Peprotech, Rocky Hill, CT, USA) was added to differentiate the cells into macrophages in 7 days. Medium was changed once after 3 days. Monocytes were isolated using MACS-technology using the CD115 positive selection kit for mouse monocytes (Miltenyi, Bergisch Gladbach, Germany).
4.2. Antibodies
The commercial antibodies purchased in this study are depicted in Table 1.

Table 1.
Antibodies.
4.3. Western Blot Analysis
A quarter of a kidney was homogenized using a IKA® tissue homogenizer in 500 μL RIPA buffer (Sigma Aldrich, St. Louis, MO, USA) containing Complete® protease inhibitors (Roche, Basel, Switerland). Subsequently, a Pierce BCA protein assay (Thermo Fisher Scientific, Waltham, MA, USA) was performed to assess the absolute quantity of protein in each sample. Next 10–40 μg of total protein lysate was loaded per lane on precast Any-kD acrylamide gels (Bio-Rad, Hercules, CA, USA) for gel electrophoresis. Using the Trans-Blot® Turbo™ Transfer system (Bio-Rad), the protein was transferred to Nitrocellulose (Bio-Rad) membranes for further analysis. Membranes were blocked and antibodies incubated o/n at 4 °C in 5% skim milk powder (Nutricia, Zoetermeer, Netherlands) in PBS. The appropriate HRP-labeled secondary antibodies (Dako, Glostrup, Denmark) were incubated for 1 h at room temperature, followed by extensive washing with PBS. Band-visualization was achieved using SuperSignal West-Dura® Extended Duration Substrate (Thermo Fisher Scientific, Waltham, MA, USA) for HRP. Next, exposure on UltraCruz® Autoradiography Film or KODAK-XAR film and development by a Konica developer followed. Finally, band intensities were quantified using ImageJ 1.x [62].
4.4. RNA Isolation, cDNA Synthesis and qRT-PCR Analysis
A quarter of a kidney was lysed in Trizol reagent (Invitrogen, Carlsbad, CA, USA) using an electrical tissue homogenizer (IKA®). Subsequently, chloroform and ethanol were used to phase-separate the RNA and subsequently RNeasy mini-columns (Qiagen, Hilden, Germany) were used to further purify the RNA according to manufacturers’ guidelines. The on-column DNase step was also performed to eliminate any contaminating genomic DNA. cDNA synthesis was performed using 1 μg RNA, random primers, FS-buffer, DNTPs, M-MLV-RT in the presence of RNAsin according to the manufacturers’ guidelines (all Promega products, Madison, WI, USA). RT-qPCR was performed using SYBR-select (Invitrogen) and PCR was run on a Bio-Rad CFX-384. Relative mRNA expression was calculated using the ΔΔCT method, and was normalized to GAPDH mRNA levels. PCR products generated by the splicing primers were run on an Agilent Bioanalyzer for quantitative analysis. Primer pairs used are listed below in Table 2.
Table 2.
Primers for reverse transcription PCR.
4.5. Conditional Mouse Design and Genotyping
The mouse qkI conditional null allele was reported previously [3]. The final genomic organization is depicted in Figure 3A with two loxP sequences flanking exon 2 of the qkI gene. Ear biopsies were used to isolate gDNA using the AccuStart II PCR Genotyping Kit (Quanta-Bio, Beverly, MA, USA) for genotyping using primers listed in Table 3.
Table 3.
QKI genotyping primers.
4.6. Immunohistochemistry
Fluorescence immunostainings were performed on mouse kidney cryosections that were obtained by freezing tissue samples embedded in O.C.T. (TissueTek®, Torrance, CA, USA) and placing the mold in an isopentane solution cooled with liquid nitrogen. Next, 4 μm sections were cut using a cryostat (Leica, Wetzler, Germany), slides were air-dried for 30 min, then fixed in acetone for 10 min at room temperature, air-dried again for 30 min and stored at −20 °C. Before staining, slides were washed three times in PBS at room temperature and incubated in blocking buffer (PBS + 1%BSA + 1% FCS) for at least 1 h at room temperature. Slides were incubated with primary antibodies in blocking buffer for at least 3 h at room temperature or at 4 °C overnight, where after extensive washing in PBS followed and subsequently incubated with the appropriate Alexa® (Invitrogen) labeled secondary antibodies in blocking buffer. Embedding the slides was performed in Prolong gold (Invitrogen) containing DAPI. Imaging was performed on a Leica DM5500 or a Leica SP5 confocal imaging system.
Histochemistry analysis for collagen content was done using Picosirius red staining on formalin-fixed, paraffin-embedded tissues. CLK and UUO kidneys were fixed in 3.7% formalin in PBS for 2 h then put on 70% ethanol and subsequently dehydrated overnight followed by paraffin embedding according to standard protocols. For analyses, 4 μm sections were prepared. Prior to staining, slides were brought to water by first “de-paraffinizing” in Xylene, then taken through a series of solutions decreasing in ethanol percentage (100% to 50%) in 10% steps taking 5 min at a time, finally incubation in H2O for 30 min. Slides were submerged in Sirius red F3B solution (0.1% Direct Red 80, Sigma Aldrich) in a saturated aqueous solution of picric acid for 1 h at room temperature. Slides were washed in 3 stages of acidified water consisting of 5 mL glacial acetic acid (Millipore-Sigma, Carlsbad, CA, USA) in 1 L of water. After this, slides were dehydrated to 100% ethanol and thereafter Xylene, then embedded in Entellan. Images were taken using a Leica DMI4400B microscope and collagen deposition was quantified using ImageJ [62]. Outliers were removed using a statistical outlier test.
4.7. Statistical Analysis
All data were statistically tested using a two-tailed Students’ t-test. Error bars in all figures represent SEM.
4.8. Mouse Studies
All mouse studies were approved and performed according to the guidelines of the relevant authorities of the Leiden University Medical Center, Leiden University in The Netherlands or the Lady Davis Institute, McGill University, Montreal, Canada. Unilateral Urethral Obstruction was performed as described earlier [27].
Author Contributions
R.G.d.B., G.V., J.M.J.G.D., J.P., R.B., E.P.v.d.V., J.M.v.G. and H.J.P.v.d.Z. performed experiments included in the manuscript. R.G.d.B, G.V., A.J.v.Z., S.R., E.P.v.d.V. designed the experiments and edited the manuscript. R.G.d.B. composed all the figures. J.M.v.G., H.D.d.B. and T.J.R. contributed through in depth discussions, experimental advice and revision of the manuscript. A.J.v.Z., S.R. and E.P.v.d.V. provided funding. All authors have read and agreed to the published version of the manuscript.
Funding
Funding was provided by the Netherlands Institute for Regenerative Medicine grant no: FES0908 to E.P.v.d.V. and A.J.v.Z. and from the Canadian Institute of Health Research FDN-154303 to S.R.
Acknowledgments
We thank Koren Mann and Lama Darbelli for in depth discussions, materials and technical advice. We also acknowledge the staff of the small animal research facilities of both the Lady Davis Institute and the Leiden University Medical Center.
Conflicts of Interest
All authors have no competing financial interests related to this research
References
- Bielli, P.; Busà, R.; Paronetto, M.P.; Sette, C. The RNA-binding protein sam68 is a multifunctional player in human cancer. Endocr. Relat Cancer 2011, 18, R91–R102. [Google Scholar] [CrossRef]
- Lukong, K.E.; Chang, K.W.; Khandjian, E.W.; Richard, S. Rna-binding proteins in human genetic disease. Trends Genet. 2008, 24, 416–425. [Google Scholar] [CrossRef]
- Darbelli, L.; Vogel, G.; Almazan, G.; Richard, S. Quaking regulates neurofascin 155 expression for myelin and axoglial junction maintenance. J. Neurosci. 2016, 36, 4106–4120. [Google Scholar] [CrossRef]
- Wu, J.; Zhou, L.; Tonissen, K.; Tee, R.; Artzt, K. The quaking i-5 (qki-5) has a novel nuclear localization signal and shuttles between the nucleus and the cytoplasm. J. Biol. Chem. 1999, 274, 29202–29210. [Google Scholar] [CrossRef]
- Galarneau, A.; Richard, S. Target RNA motif and target mRNAs of the quaking star protein. Nat. Struct. Mol. Biol. 2005, 12, 691–698. [Google Scholar] [CrossRef]
- van der Veer, E.P.; de Bruin, R.G.; Kraaijeveld, A.O.; de Vries, M.R.; Bot, I.; Pera, T.; Segers, F.M.; Trompet, S.; van Gils, J.M.; Roeten, M.K.; et al. Quaking, an RNA-binding protein, is a critical regulator of vascular smooth muscle cell phenotype. Circ. Res. 2013, 113, 1065–1075. [Google Scholar] [CrossRef]
- Hall, M.P.; Nagel, R.J.; Fagg, W.S.; Shiue, L.; Cline, M.S.; Perriman, R.J.; Donohue, J.P.; Ares, M. Quaking and ptb control overlapping splicing regulatory networks during muscle cell differentiation. RNA 2013, 19, 627–638. [Google Scholar] [CrossRef]
- de Bruin, R.G.; van der Veer, E.P.; Prins, J.; Lee, D.H.; Dane, M.J.; Zhang, H.; Roeten, M.K.; Bijkerk, R.; de Boer, H.C.; Rabelink, T.J. The RNA-binding protein quaking maintains endothelial barrier function and affects ve-cadherin and β-catenin protein expression. Sci. Rep. 2016, 6, 21643. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Takakura, N.; Oike, Y.; Imanaka, T.; Araki, K.; Suda, T.; Kaname, T.; Kondo, T.; Abe, K.; Yamamura, K. Defective smooth muscle development in qki-deficient mice. Dev. Growth Differ. 2003, 45, 449–462. [Google Scholar] [CrossRef] [PubMed]
- Ebersole, T.A.; Chen, Q.; Justice, M.J.; Artzt, K. The quaking gene unites signal transduction and RNA-binding in the developing nervous system. Nat. Genet. 1996, 12, 260–265. [Google Scholar] [CrossRef] [PubMed]
- de Bruin, R.G.; Rabelink, T.J.; van Zonneveld, A.J.; van der Veer, E.P. Emerging roles for RNA-binding proteins as effectors and regulators of cardiovascular disease. Eur. Heart J. 2017, 38, 1380–1388. [Google Scholar] [CrossRef] [PubMed]
- de Bruin, R.G.; Shiue, L.; Prins, J.; de Boer, H.C.; Singh, A.; Fagg, W.S.; van Gils, J.M.; Duijs, J.M.; Katzman, S.; Kraaijeveld, A.O.; et al. Quaking promotes monocyte differentiation into pro-atherogenic macrophages by controlling pre-mRNA splicing and gene expression. Nat. Commun. 2016, 7, 10846. [Google Scholar] [CrossRef]
- Backx, L.; Fryns, J.P.; Marcelis, C.; Devriendt, K.; Vermeesch, J.; Van Esch, H. Haploinsufficiency of the gene quaking (qki) is associated with the 6q terminal deletion syndrome. Am. J. Hum. Genet. 2010, 152A, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Zong, F.-Y.; Fu, X.; Wei, W.-J.; Luo, Y.-G.; Heiner, M.; Cao, L.-J.; Fang, Z.; Fang, R.; Lu, D.; Ji, H. The RNA-binding protein qki suppresses cancer-associated aberrant splicing. PLoS Genet. 2014, 10, e1004289. [Google Scholar] [CrossRef]
- Darbelli, L.; Richard, S. Emerging functions of the quaking RNA-binding proteins and link to human diseases. Wiley Interdiscip. Rev. RNA 2016, 7, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Bandopadhayay, P.; Ramkissoon, L.A.; Jain, P.; Bergthold, G.; Wala, J.; Zeid, R.; Schumacher, S.E.; Urbanski, L.; O’Rourke, R.; Gibson, W.J. Myb-qki rearrangements in angiocentric glioma drive tumorigenicity through a tripartite mechanism. Nat. Genet. 2016, 48, 273–282. [Google Scholar] [CrossRef]
- Lin, S.L.; Castaño, A.P.; Nowlin, B.T.; Lupher, M.L.; Duffield, J.S. Bone marrow ly6chigh monocytes are selectively recruited to injured kidney and differentiate into functionally distinct populations. J. Immunol. 2009, 183, 6733–6743. [Google Scholar] [CrossRef]
- Anders, H.-J.; Ryu, M. Renal microenvironments and macrophage phenotypes determine progression or resolution of renal inflammation and fibrosis. Kidney Int. 2011, 80, 915–925. [Google Scholar] [CrossRef]
- Wada, T.; Sakai, N.; Sakai, Y.; Matsushima, K.; Kaneko, S.; Furuichi, K. Involvement of bone-marrow-derived cells in kidney fibrosis. Clin. Exp. Nephrol. 2011, 15, 8–13. [Google Scholar] [CrossRef][Green Version]
- Rogers, N.M.; Ferenbach, D.A.; Isenberg, J.S.; Thomson, A.W.; Hughes, J. Dendritic cells and macrophages in the kidney: A spectrum of good and evil. Nat. Rev. Nephrol. 2014, 10, 625–643. [Google Scholar] [CrossRef]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. Inflammatory processes in renal fibrosis. Nat. Rev. Nephrol. 2014, 10, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, M.; Neilson, E.G. Mechanisms of tubulointerstitial fibrosis. J. Am. Soc. Nephrol. 2010, 21, 1819–1834. [Google Scholar] [CrossRef] [PubMed]
- Levey, A.S.; Coresh, J. Chronic kidney disease. Lancet 2012, 379, 165–180. [Google Scholar] [CrossRef]
- Geissmann, F.; Manz, M.G.; Jung, S.; Sieweke, M.H.; Merad, M.; Ley, K. Development of monocytes, macrophages, and dendritic cells. Science 2010, 327, 656–661. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Pamer, E.G. Monocyte recruitment during infection and inflammation. Nat. Rev. Immunol. 2011, 11, 762–774. [Google Scholar] [CrossRef]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef]
- Bijkerk, R.; de Bruin, R.G.; van Solingen, C.; van Gils, J.M.; Duijs, J.M.; van der Veer, E.P.; Rabelink, T.J.; Humphreys, B.D.; van Zonneveld, A.J. Silencing of microRNA-132 reduces renal fibrosis by selectively inhibiting myofibroblast proliferation. Kidney Int. 2016, 89, 1268–1280. [Google Scholar] [CrossRef]
- Odegaard, J.I.; Ricardo-Gonzalez, R.R.; Goforth, M.H.; Morel, C.R.; Subramanian, V.; Mukundan, L.; Eagle, A.R.; Vats, D.; Brombacher, F.; Ferrante, A.W. Macrophage-specific ppar&ggr; controls alternative activation and improves insulin resistance. Nature 2007, 447, 1116–1120. [Google Scholar]
- Darbelli, L.; Choquet, K.; Richard, S.; Kleinman, C.L. Transcriptome profiling of mouse brains with qki-deficient oligodendrocytes reveals major alternative splicing defects including self-splicing. Sci. Rep. 2017, 7, 7554. [Google Scholar] [CrossRef]
- Chen, T.; Richard, S. Structure-function analysis of qk1: A lethal point mutation in mouse quaking prevents homodimerization. Mol. Cell Biol. 1998, 18, 4863–4871. [Google Scholar] [CrossRef]
- Kanasty, R.; Dorkin, J.R.; Vegas, A.; Anderson, D. Delivery materials for siRNA therapeutics. Nat. Mater. 2013, 12, 967–977. [Google Scholar] [CrossRef] [PubMed]
- Shu, D.; Shu, Y.; Haque, F.; Abdelmawla, S.; Guo, P. Thermodynamically stable RNA three-way junction for constructing multifunctional nanoparticles for delivery of therapeutics. Nat. Nanotechnol. 2011, 6, 658–667. [Google Scholar] [CrossRef] [PubMed]
- Shu, Y.; Cinier, M.; Shu, D.; Guo, P. Assembly of multifunctional phi29 pRNA nanoparticles for specific delivery of siRNA and other therapeutics to targeted cells. Methods 2011, 54, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, P.; Kurniawan, H.; Byrne, M.E.; Wower, J. Therapeutic RNA aptamers in clinical trials. European J. Pharm. Sci. 2013, 48, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Anderson, P. Post-transcriptional control of cytokine production. Nat. Immunol. 2008, 9, 353–359. [Google Scholar] [CrossRef]
- Chen, C.-Y.; Gherzi, R.; Ong, S.-E.; Chan, E.L.; Raijmakers, R.; Pruijn, G.J.; Stoecklin, G.; Moroni, C.; Mann, M.; Karin, M. Au binding proteins recruit the exosome to degrade are-containing mRNAs. Cell 2001, 107, 451–464. [Google Scholar] [CrossRef]
- Leppek, K.; Schott, J.; Reitter, S.; Poetz, F.; Hammond, M.C.; Stoecklin, G. Roquin promotes constitutive mRNA decay via a conserved class of stem-loop recognition motifs. Cell 2013, 153, 869–881. [Google Scholar] [CrossRef]
- Lin, J.; Hu, Y.; Nunez, S.; Foulkes, A.S.; Cieply, B.; Xue, C.; Gerelus, M.; Li, W.; Zhang, H.; Rader, D.J. Transcriptome-wide analysis reveals modulation of human macrophage inflammatory phenotype through alternative splicing. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1434–1447. [Google Scholar] [CrossRef]
- Liu, H.; Lorenzini, P.A.; Zhang, F.; Xu, S.; Wong, M.S.M.; Zheng, J.; Roca, X. Alternative splicing analysis in human monocytes and macrophages reveals mbnl1 as major regulator. Nucleic Acids Res. 2018, 46, 6069–6086. [Google Scholar] [CrossRef]
- Matsuoka, Y.; Li, X.; Bennett, V. Adducin: Structure, function and regulation. Cell Mol. Life Sci. 2000, 57, 884–895. [Google Scholar] [CrossRef]
- Winograd-Katz, S.E.; Fassler, R.; Geiger, B.; Legate, K.R. The integrin adhesome: From genes and proteins to human disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 273–288. [Google Scholar] [CrossRef]
- Imhof, B.A.; Aurrand-Lions, M. Adhesion mechanisms regulating the migration of monocytes. Nat. Rev. Immunol. 2004, 4, 432–444. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Xia, Y.; Jiang, R.; Chen, D.; Xu, J.; Deng, L.; Huang, X.; Wang, X.; Sun, B. Ptpro plays a dual role in hepatic ischemia reperfusion injury through feedback activation of nf-kappab. J. Hepatol. 2014, 60, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Gordon, S. The m1 and m2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [PubMed]
- Waddell, S.J.; Popper, S.J.; Rubins, K.H.; Griffiths, M.J.; Brown, P.O.; Levin, M.; Relman, D.A. Dissecting interferon-induced transcriptional programs in human peripheral blood cells. PLoS ONE 2010, 5, e9753. [Google Scholar] [CrossRef] [PubMed]
- Bossi, D.; Carlomagno, F.; Pallavicini, I.; Pruneri, G.; Trubia, M.; Raviele, P.R.; Marinelli, A.; Anaganti, S.; Cox, M.C.; Viale, G.; et al. Functional characterization of a novel fgfr1op-ret rearrangement in hematopoietic malignancies. Mol. Oncol. 2014, 8, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Wu, Q.; Cowell, J.K.; Ren, M. Fgfr1op2-fgfr1 induced myeloid leukemia and t-cell lymphoma in a mouse model. Haematologica 2016, 101, e91–e94. [Google Scholar] [CrossRef] [PubMed]
- Ballerini, P.; Struski, S.; Cresson, C.; Prade, N.; Toujani, S.; Deswarte, C.; Dobbelstein, S.; Petit, A.; Lapillonne, H.; Gautier, E.F.; et al. Ret fusion genes are associated with chronic myelomonocytic leukemia and enhance monocytic differentiation. Leukemia 2012, 26, 2384–2389. [Google Scholar] [CrossRef]
- Giannandrea, M.; Parks, W.C. Diverse functions of matrix metalloproteinases during fibrosis. Dis. Model Mech. 2014, 7, 193–203. [Google Scholar] [CrossRef]
- Duffield, J.S.; Forbes, S.J.; Constandinou, C.M.; Clay, S.; Partolina, M.; Vuthoori, S.; Wu, S.; Lang, R.; Iredale, J.P. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J. Clin. Investig. 2005, 115, 56–65. [Google Scholar] [CrossRef]
- Atabai, K.; Jame, S.; Azhar, N.; Kuo, A.; Lam, M.; McKleroy, W.; DeHart, G.; Rahman, S.; Xia, D.D.; Melton, A.C. Mfge8 diminishes the severity of tissue fibrosis in mice by binding and targeting collagen for uptake by macrophages. J. Clin. Investig. 2009, 119, 3713–3722. [Google Scholar] [CrossRef] [PubMed]
- Lucattelli, M.; Cavarra, E.; De Santi, M.; Tetley, T.; Martorana, P.; Lungarella, G. Collagen phagocytosis by lung alveolar macrophages in animal models of emphysema. Eur. Respir. J. 2003, 22, 728–734. [Google Scholar] [CrossRef] [PubMed]
- Madsen, D.H.; Leonard, D.; Masedunskas, A.; Moyer, A.; Jürgensen, H.J.; Peters, D.E.; Amornphimoltham, P.; Selvaraj, A.; Yamada, S.S.; Brenner, D.A. M2-like macrophages are responsible for collagen degradation through a mannose receptor–mediated pathway. J. Cell Biol. 2013, 202, 951–966. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Kwon, J.; Popov, Y.; Gajdos, G.B.; Ordog, T.; Brekken, R.A.; Mukhopadhyay, D.; Schuppan, D.; Bi, Y.; Simonetto, D.; et al. Vascular endothelial growth factor promotes fibrosis resolution and repair in mice. Gastroenterology 2014, 146, 1339–1350 e1331. [Google Scholar] [CrossRef]
- Fallowfield, J.A.; Mizuno, M.; Kendall, T.J.; Constandinou, C.M.; Benyon, R.C.; Duffield, J.S.; Iredale, J.P. Scar-associated macrophages are a major source of hepatic matrix metalloproteinase-13 and facilitate the resolution of murine hepatic fibrosis. J. Immunol. 2007, 178, 5288–5295. [Google Scholar] [CrossRef]
- Ramachandran, P.; Pellicoro, A.; Vernon, M.A.; Boulter, L.; Aucott, R.L.; Ali, A.; Hartland, S.N.; Snowdon, V.K.; Cappon, A.; Gordon-Walker, T.T. Differential ly-6c expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc. Natl. Acad. Sci. USA 2012, 109, E3186–E3195. [Google Scholar] [CrossRef]
- Aldigier, J.C.; Kanjanbuch, T.; Ma, L.-J.; Brown, N.J.; Fogo, A.B. Regression of existing glomerulosclerosis by inhibition of aldosterone. J. Am. Soc. Nephrol. 2005, 16, 3306–3314. [Google Scholar] [CrossRef]
- Fioretto, P.; Steffes, M.W.; Sutherland, D.E.; Goetz, F.C.; Mauer, M. Reversal of lesions of diabetic nephropathy after pancreas transplantation. N. Engl. J. Med. 1998, 339, 69–75. [Google Scholar] [CrossRef]
- Pichaiwong, W.; Hudkins, K.L.; Wietecha, T.; Nguyen, T.Q.; Tachaudomdach, C.; Li, W.; Askari, B.; Kobayashi, T.; O’Brien, K.D.; Pippin, J.W.; et al. Reversibility of structural and functional damage in a model of advanced diabetic nephropathy. J. Am. Soc. Nephrol. 2013, 24, 1088–1102. [Google Scholar] [CrossRef]
- Sugimoto, H.; LeBleu, V.S.; Bosukonda, D.; Keck, P.; Taduri, G.; Bechtel, W.; Okada, H.; Carlson, W.; Bey, P.; Rusckowski, M. Activin-like kinase 3 is important for kidney regeneration and reversal of fibrosis. Nat. Med. 2012, 18, 396–404. [Google Scholar] [CrossRef]
- Bijkerk, R.; van Solingen, C.; de Boer, H.C.; van der Pol, P.; Khairoun, M.; de Bruin, R.G.; van Oeveren-Rietdijk, A.M.; Lievers, E.; Schlagwein, N.; van Gijlswijk, D.J. Hematopoietic microRNA-126 protects against renal ischemia/reperfusion injury by promoting vascular integrity. J. Am. Soc. Nephrol. 2014, 25, 1710–1722. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).