Abstract
Cytoplasmic components and their interactions with the nuclear genome may mediate patterns of phenotypic expression to form a joint inheritance system. However, proximate mechanisms underpinning these interactions remain elusive. To independently assess nuclear genetic and epigenetic cytoplasmic effects, we created a full-factorial design in which representative cytoplasms and nuclear backgrounds from each of two geographically disjunct populations of Drosophila melanogaster were matched together in all four possible combinations. To capture slowly-accumulating epimutations in addition to immediately occurring ones, these constructed populations were examined one year later. We found the K4 methylation of histone H3, H3K4me3, an epigenetic marker associated with transcription start-sites had diverged across different cytoplasms. The loci concerned mainly related to metabolism, mitochondrial function, and reproduction. We found little overlap (<8%) in sites that varied genetically and epigenetically, suggesting that epigenetic changes have diverged independently from any cis-regulatory sequence changes. These results are the first to show cytoplasm-specific effects on patterns of nuclear histone methylation. Our results highlight that experimental nuclear-cytoplasm mismatch may be used to provide a platform to identify epigenetic candidate loci to study the molecular mechanisms of cyto-nuclear interactions.
1. Introduction
The idea that gene–environment interactions (GxE), not the genotype (G) alone, underpin the expression of the phenotype is now well established [1,2,3]. Based on this GxE→P idea, we and others developed a more general concept based on systems biology stating that G is an inheritance system that includes other interacting and heritable components including the epigenome and cytoplasmic components such as mitochondria, Wolbachia, and viruses [4,5,6,7]. It is the entirety of this system, and not its individual components, which produces phenotypic variation. This systems approach has the advantage of being able to incorporate the possibility that variation in one part of the inheritance system can have consequences on other parts. For example, sequence differences in the genomes of cytoplasmic elements such as mitochondria, which interact with the nucleus, may drive variation in the nuclear genome and the epigenome. Considering these heritable interaction effects on the phenotype is important because they can be as strong as the effects by the individual cytoplasmic components [8].
Mitochondrial genomes have been widely reported to interact with nuclear genomes to affect phenotypic expression [6,7,8,9,10,11,12,13,14,15,16,17]. Nuclear and mitochondrial genomes also routinely coevolve within populations as suggested from studies reporting hybrid disadvantages observed when coadapted combinations of mito-nuclear genotypes have been experimentally disrupted by placing the mt genotype of one population alongside the nuclear genome of another population [13,18,19,20]. Mito-nuclear interactions, therefore, are part of the same inheritance system. These heritable mito-nuclear interactions can ultimately underpin the emergence of new species [18,21,22,23] and, at a less severe level, have been implicated in shaping health outcomes and the penetrance of mitochondrial and other common diseases in humans [10,11,15,24,25].
The proximate and epigenetic mechanisms of these mito-nuclear interactions are currently only superficially understood. Tight coordination and bidirectional crosstalk between mitochondrial and nuclear genomes are necessary for mitochondrial function, and overall cellular homeostasis [18,22,26,27]. Mitochondrial function is largely dependent on the nuclear genome, first and foremost due to the fact that the vast majority of proteins present in mitochondria are nuclear-encoded. However, studies have also shown or suggested that mitochondria can influence nuclear gene expression and might be involved in the epigenetic regulation of the nuclear genome [9,12,17,28,29,30,31,32]. For instance, depleting mitochondria in cultured human kidney cells caused changes in nuclear DNA methylation, an important bearer of epigenetic information [33]. It is therefore conceivable that different mito-nuclear combinations might be under different epigenetic regulations and might entail the reported changes in life-history traits or disease [10,16,17,22,34]. Negative effects could arise from mito-nuclear mismatch (i.e., if a mitochondrial genotype is placed alongside a nuclear genomic background to which it has no close coevolutionary history and hence lacks the evolutionary ‘optimization’). These negative effects, in turn, may cause intrinsic selection and favor the evolution of compensatory nuclear alleles to restore optimal mito-nuclear function [18,22,35]. To date, it is not clear whether mitochondrial variation can cause evolutionary changes in the nuclear genome and epigenome and if so, whether they are independent from one another.
A second important cytoplasmic component that may shape patterns of inheritance are heritable bacteria, such as endosymbionts. Like mitochondria, some of them are vertically transmitted through the female germline, and they also have profound and heritable effects on host physiology and life history, such as changes in reproductive incompatibility between individuals that differ in cytoplasm [36,37,38,39]. Perhaps the most well-known representative is Wolbachia pipientis, an intracellular α-Proteobacterium commonly found to form an association with a wide range of insects and other invertebrates, and which is the most widespread and main endosymbiotic bacteria species in Drosophila melanogaster [38]. Just as for the mitochondria, the proximate mechanisms underpinning interactions between endosymbiont genomes, such as Wolbachia, with the nuclear genome of their host, are not well understood, including any epigenetic alterations.
If indeed cytoplasmic heritable elements, the nuclear genome, and the nuclear epigenome are part of the inheritance system, then any change in the cytoplasmic elements is expected to exert a forward action on the nuclear elements and entail modifications in the genome and/or epigenome. Here we tested this hypothesis and asked whether replacement of cytoplasmic components cause heritable nuclear changes and whether or not epigenetic changes are independent from sequence changes. We used a full-factorial design of nuclear genomes and cytoplasmic components from two geographically distant populations of D. melanogaster (Australia and Canada, A and C, respectively). We generated experimental populations that consisted of nuclear genomes with either originally coadapted, or experimentally mismatched cytoplasms, so called cyto-nuclear genotypes [22] or, in our approach, cyto-nuclear inheritance systems. Population A carried Wolbachia, which was, therefore, inherited alongside the mitochondrial genome. Population C was clear of Wolbachia. Following their creation, all cyto-nuclear inheritance systems were left free to adapt for one year; i.e., 27 generations (Figure 1) in order to capture both immediately and slowly-accumulating epimutations, if any. ChIP-seq assays of a histone mark associated with transcriptional activation (H3K4me3) were performed to investigate potential differences in the nuclear epigenome after experimental mismatching of the cytoplasm. We choose this mark since it was shown before that the mitochondrial content can influence H3K4 methylation in HeLa cells [40]. Below we detail our results, using the notation of ‘cyto-nuclear’ interaction to mark the identity of the cytoplasm first and the nuclear background second for each of the experimental populations; i.e., AC denotes Australian cytoplasm and Canadian nuclear (epi)genomes.
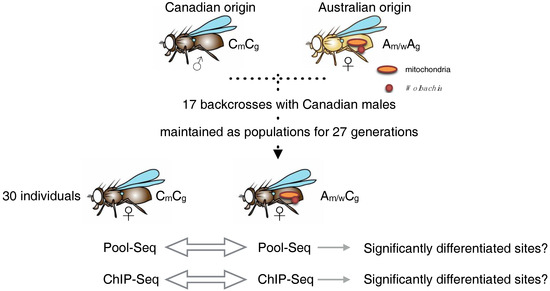
Figure 1.
Schematic representation of the design to create Drosophila lines that differ in either cytoplasm or nuclear background. Males carrying one background (Canadian) were repeatedly crossed with females from the same, or the other (Australian) population. After 17 generations of crossing, this resulted in cyto-nuclear ‘hybrids’ carrying either the original coevolved or the geographically separate cytoplasm. The Australian cytoplasmic component included mitochondria and Wolbachia, the Canadian only mitochondria. Then, 30 female individuals each were pooled and subjected to genomic or epigenomic analysis.
2. Results
2.1. Cytoplasms Differ in Mitochondrial Genomes and Wolbachia Presence
Differentiation between Australian and Canadian mitochondrial (mt) genomes were assessed using two measures of genetic differentiation, the fixation index (FST) and Fisher’s exact test for allele frequency differences (as implemented in Popoolation2 [41]). We combined mitochondrial sequence data from the two lines containing Australian mt genomes (AA and AC) and from the two containing Canadian mt genomes (CC and CA) into single Australian and Canadian pools, respectively. FST and allele frequency differences were calculated per SNP. Sites considered significantly differentiated between Australian and Canadian mt genomes had to differ significantly in Fisher’s exact test (p < 0.05; FDR of 5%) and fall under the TOP 1% of FST values. We found seven such sites distributed across five protein-coding genes, of which six were synonymous and one nonsynonymous (Table 1). The nonsynonymous change concerned a change from isoleucine (Ile) in the Canadian ATPase6 to methionine (Met) in the Australian ATPase6 at position 185.

Table 1.
Significantly differentiated SNPs between the Australian and Canadian mitochondrial genomes of Drosophila melanogaster populations used in this study.
Diagnostic PCR confirmed that both lines with Australian cytoplasms (AA and AC) were infected with Wolbachia, and both lines with Canadian cytoplasms were uninfected (CC and CA). In the following we set out to find potential epigenetic nuclear differences between CC and AC. Diagnostic SNPs in the mtDNA COI show strong non-random association with different Wolbachia genotypes [42,43,44], suggesting the Wolbachia found in our population were likely of the wMel genotype of the Wolbachia.
2.2. Nuclear CC and AC Epigenomes Show Epiallele Frequency Distortion in Loci Associated with Mitochondrial Inner Membrane Presequence Translocase Complex
C nuclear genomes did not reveal global changes in H3K4me3 profiles between CC and AC (coadapted C compared to mismatched A cytoplasms), or between technical replicates (Figure 2A). However, specific loci with differential trimethylation of H3K4 between AC and CC were detected in 441 regions by ChromstaR [45] (Table S1), 424 of which were along the major chromosomes. Examples of differentially enriched regions are shown in Figure 2B.
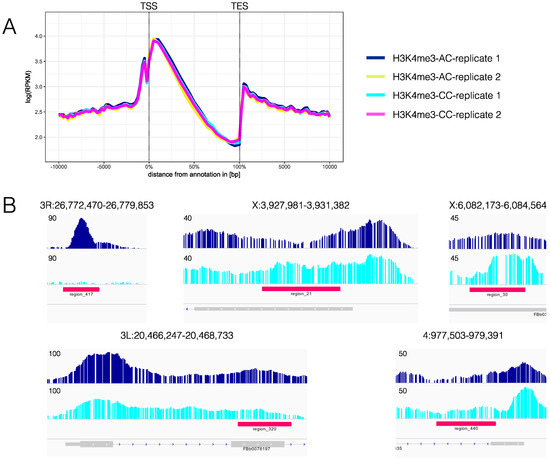
Figure 2.
Epigenetic analysis of cyto-nuclear mismatch. (A) Average profiles for 9076 genes for the four replicates AC 1 and 2, and CC 1 and 2. On the X-axis, there is the position on the genome with transcription start site (TSS) at 0% and transcription end site (TES) at 100%. Upstream and downstream positions are in bp. On the Y-axis, RPKM is shown, which is ‘reads per kilobase and million’. As expected, H3K4me3 is enriched around the TSS. No strong differences were found between the biological and technical replicates; and (B) Examples for regions in which ChromstaR [45] detects differences in H3K4me3 profiles (red bars). Base pair number is on the X-axis, Y-axis shows the coverage. Profiles obtained using PeakRanger [47] are depicted above the profiles. ChromstaR identifies only differences as significant that are supported by both replicates. For the sake of clarity, however, only H3K4me3 profiles for AC (replicate 1) (dark blue) and for CC (replicate 1) (light blue) are shown. Maximum y values are shown on the left. The panel below the profiles, in grey, shows gene annotations.
Of the 424 significantly differentiated sites along the major chromosomes, 392 (92.5%) did not overlap with significantly differentiated nucleotide sites between AC and CC. Differentially enriched regions contained 155 genes (Table S2), which were subjected to GO enrichment analysis using ChIP-Enrich [46]. A single significantly enriched GO category (FDR 20%) was identified: “mitochondrial inner membrane presequence translocase complex”.
3. Discussion
Our results provide evidence that cytoplasmic components affected the nuclear epigenome, and hence form part of the functional inheritance system in D. melanogaster. We are not aware of a previous demonstration of this kind. We found the overlap between sites of significant genetic and epigenetic differentiation to be small, suggesting cis regulatory DNA sequence changes are likely to play a minor role in the differential H3K4me3 enrichments observed here. While the result is replicated across two replicate lines and so is relatively robust against individual variation, we note that our data do not allow any conclusions as to which tissues are responsible of the observed overall epigenetic changes. If mitochondrial variation in energy metabolism were responsible for the observed epigenetic changes, as suggested e.g., by [48], the differences in energy metabolism of different tissues would actually predict tissue-specific epigenetic profiles.
The cytoplasms we used in our study and that caused the effects differed starkly—in Wolbachia infection status as well as in synonymous and nonsynonymous base pair changes in the mt genome. Specifically, CA differed from AA in both the mt DNA haplotype and the absence of Wolbachia, whereas AC differed from CC in mt DNA haplotype and the presence of Wolbachia. Our lines showed one nonsynonymous change in the ATPase6 gene resulting in a substitution from methionine in A to isoleucine in C, at position 185 of the ATPase6. Amino acid position 185 is located between the two final transmembrane helices of the ATPase6 in the proximity of the conserved arginine residue that is essential for ATPase6 function [49,50]. Given the central position of ATPase6 in shaping mitochondrial membrane morphology [51], organismal ageing [52] as well as energy production [53], the nonsynonymous change in ATPase6 may lead to slight changes in energy coupling and, therefore, affect the initiation of the Krebs cycle and glycolysis. Future studies should test the causal nature of this potential link of ATPase6 variation and chromatin structure that we identified here. As glycolysis is not as efficient as oxidative phosphorylation, less energy may be available but stored as fat. In addition, methionine, in contrast to isoleucine, is a sulfur-containing amino acid. Like the other sulfur-containing amino acid, cysteine, methionine residues can serve as cellular antioxidants, stabilize protein structure and act as regulatory switches through reversible oxidation and reduction [54]. It would, therefore, be very interesting to score the long-term survival phenotypes of our populations differing in ATPase6 in the presence and absence of an oxidative challenge.
Both mitochondria and Wolbachia have been implicated to affect the nuclear epigenome [12,32,55]. While both could, therefore, be causing the differential H3K4me3 enrichment we observed, it is interesting to note that the only significantly enriched GO category we found was related to mitochondrial function. The mitochondrial inner membrane presequence translocase complex is essential in importing nuclear-encoded proteins across the inner mitochondrial membrane into mitochondria [56]. This is an important process for the biogenesis and maintenance of healthy mitochondria since the vast majority of mitochondrial proteins are encoded by nuclear genes. Previous research has also found that variation in the mt genotype can be associated with variation in mtDNA copy number [57,58]. Suggestions that epigenetic nuclear variation may be related to mtDNA copy number per cell [28,29,59] may therefore be an attractive hypothesis to be tested in future research.
We know of no other study addressing the impact of cytoplasmic variation on patterns of histone methylation. One study that is, however, broadly comparable to ours is a recent study in mice [32]. These authors examined differences in DNA methylation in individuals of strains with identical nuclear DNA but of different cytoplasms. After 8 generations of coevolution, these authors found differences in in hypo- or hypermethylation at several hundred loci. However, these authors [32] did not control for epigenetic changes caused by the nuclear transfer procedure itself.
4. Materials and Methods
4.1. Fly Lines
Cyto-nuclear hybrid populations of D. melanogaster were generated from two large mass-bred population samples originating from two natural populations: Coffs Harbour, Australia (A) [60], and Dundas near Hamilton, Canada (C) [61]. By repeated crossing (introgression) of 45 virgin females of the cyto-nuclear hybrid population to be created with 45 males of the population whose nuclear DNA was required, cytoplasmic components, and mitochondrial (mt) genomes of each population were placed alongside the nuclear genomes of the other population. Introgression took place over 17 generations, after which theoretically more than 99.999% of the original nuclear genome has been replaced by the introgressed one. As a control, mitochondrial genomes were introgressed in the same way into their native nuclear background (Figure 1). Thus, in total four different cyto-nuclear combinations, each in two replicates, were generated: Australian cytoplasm–Canadian nuclear genome (AC), Canadian cytoplasm–Australian nuclear genome (CA), Australian cytoplasm–Australian nuclear genome (AA), and Canadian cytoplasm–Canadian nuclear genome (CC). After 17 generations of introgression, the different mito-nuclear combinations were allowed to coevolve over 27 generations. Each generation, 45 females, aged 4 to 6 days and hence, multiply mated, were randomly chosen from each line and allowed to oviposit overnight. Flies were kept on a standard cornmeal-sucrose-yeast medium at 25 °C under a 12 h/12 h light/dark cycle with non-overlapping 14-day generations.
4.2. Wolbachia Diagnostic PCR
For each line, DNA was extracted from pools of approximately 15 adult flies per replicate population using the innuPREP DNA Mini Kit (Analytik Jena, Jena, Germany). The success of each DNA extraction was confirmed with a PCR [44]. Wolbachia infection status for each line was determined using primers for the Wolbachia wsp gene [44]. Fly lines from the Drosophila Population Genomics Project (DPGP; [62] with known infection status [44] served as positive and negative controls (FR14 and FR59, respectively). PCR conditions were as described in [44] and each PCR was repeated twice to confirm that the results were consistent. PCRs were performed using the DreamTaq Green DNA Polymerase (Thermo Fisher Scientific, Waltham, MA, USA).
4.3. Sequence Analysis
Input DNA controls from the ChIP-Seq experiment (see ‘Epigenetic analysis’) were used for whole genome sequence analysis. One pool of 30 female flies for each cyto-nuclear combination (AA, AC, CA, CC) was sequenced as single-end libraries of 75 bp length on an Illumina HiSeq 2500 to obtain a mean sequencing depth of 20×. Our yields of precipitated DNA were too low to enable estimations in 3 of the 8 line replicates (both Australian background-Australian haplotype lines, and one Canadian background-Australian haplotype line). Prior to mapping, reads were trimmed from the 3′ end to remove low-quality bases using Popoolation1 v1.2.2 [63] using default settings and a quality threshold of 20. Then reads were mapped to the D. melanogaster reference genome (Release 6; http://flybase.org) using BWA v.7.12-5 [64] using default settings followed by mapping of all unmapped reads using Stampy v1.0.28 [65]. Reads with mapping quality scores below 20 were excluded and optical duplicates were removed using Picard v2.1.0 (http://picard.sourceforge.net/). Repeat sequences, identified by RepeatMasker v4.0.6 (http://www.repeatmasker.org), together with 5-bp windows around indels were masked in the subsequent analysis. Differences in allele frequencies per SNP were assessed by FST and Fisher’s exact test as implemented in Popoolation2 v1.201 [41], controlled for false discovery rate (FDR) [66].
4.4. Epigenetic Analysis
A standard native ChIP protocol [67] was adapted to D. melanogaster adults. We used an anti-H3K4me3 monoclonal antibody (Millipore cat. # 04-745, Lot # 2392291, Temecula, USA). Chromatin for each sample was obtained from thirty female adult flies aged 4–6 days. One input control for each cyto-nuclear combination was derived from the unbound chromatin fraction of the “no antibody” control experiments. Immunoprecipitated DNA and input controls were sequenced (as described in ‘Sequence analysis’), quality checked and aligned to the D. melanogaster reference genome (Release 6; http://flybase.org) with Bowtie2.1 using parameters ‘end-to-end’, ‘sensitive’, ‘gbar 4’ and filtered for unique match with samtools [68] view -Sh -q 40 -F 0x0004 - | grep -v XS:I. Contigs ≤5 kb were removed to reduce complexity. BAM files were converted into BED and 17 million aligned reads were subsampled for each replicate. In our hands, this non-parametric bootstrapping approach provides superior normalization compared to “build-in” normalization tools implemented in downstream analyses. The “Ranger” method for narrow peaks of PeakRanger v1.16 [47] with p-value cut off 0.0001, FDR cut off 0.01, Read extension length 200, Smoothing bandwidth 99 and Delta 1 was used for peak identification. Input samples were used as negative control (−c). Wiggle files were generated for visualization under Integrated Genome Viewer (IGV) [69]. Peak region positions were saved as BED files (Ranger-BED). For comparison of chromatin profiles of each mar, the HMM-based software ChromstaR v1.3.1 [45] was used. Its parameters were optimized using the PeakRanger peak call as reference. The reason for this was that PeakRanger is very good in peak detection with default parameters while ChromstaR needs exhaustive testing of its parameter space. Best matches between peak calls of PeakRanger and ChromstaR were obtained with downsampling to 0.5 Mio aligned reads, read.cutoff.absolute = 2000, and binsize = 500 at default mapq > 10 filter. While PeakRanger output served then only for visual inspection, ChromstaR was then used to identify differentially enriched H3K4me3 regions between AC and CC using differential.score > 0.99 and a minimum length of differential regions of 249 bp. The rationale behind this was that this DNA length corresponds roughly to what is wound around a nucleosome and that smaller regions have no biological significance. ChromstaR only considers differences significant that are supported by both replicates. Average H3K4 trimethylation over genes were calculated using the plotEnrichment function for ‘counts’ over 9076 genes of the FlyBase annotation Release 6 (http://flybase.org). The identified differentially enriched regions were analyzed for GO term enrichment using ChIP-Enrich [46] an application for gene set enrichment testing for ChIP-seq data. ChIP-Enrich was run on the D. melanogaster reference genome Release 6 (http://flybase.org), and genes with differentially enriched regions within ±1 kb of transcription start sites were considered. All sequence data were submitted to the public repository NCBI Sequence Read Archive (SRA) and are available through the BioProject ID: PRJNA312490.
5. Conclusions
We show here that cyto-nuclear mismatch was sufficient in D. melanogaster to cause alterations in the nuclear epigenome after replacement of the cytoplasmic component. Cyto-nuclear interactions are, therefore, part of the inheritance system in D. melanogaster. A haplotype-specific influence of mitochondrial metabolism on nuclear gene expression has been outlined for ageing, cancer, and other diseases [9] as well as for evolutionary processes [18,20,22,34]. Our study now indicates that cytoplasmatically mediated modifications of the nuclear epigenome should also be considered as possible regulators of these effects on downstream components of health [12,29,30,31]. The previously elusive molecular mechanisms uncovered here are likely candidates to mediate the documented mito-nuclear effects on the expression of health and life history traits such as longevity and reproductive success. Our findings support previous suggestions [9,12,29,30,31,32,48] that epigenetic changes may be involved in the phenotypic effects of mito-nuclear interactions and provide a hypothesis as to why histone methylation was correlated to altered development in cattle embryos after mitochondrial replacement [29].
In addition to isolating mitochondrial from other cytoplasmic influences on the phenotype, we see three future challenges: (i) to reveal whether the observed changes represent nuclear modifier (epi) alleles that compensate for costs arising from mitochondria defects or Wolbachia infection or arise from other sources, (ii) to examine whether the epimutations identified here are environment-dependent, genotype-dependent or random, and (iii) to examine whether the observed epimutations are immediate environmental effects, or arise after some evolutionary time. Our current work is a first step and further work will aim to establish the chromatin structure of individual flies and, in the longer term, tissue specificity in individual flies. This will ultimately enable researchers to dissect within-individual, between-individual, and within-population variation and to quantify the relative contribution of genetic, epigenetic, cytoplasmic and environmental factors to phenotypic variance.
Supplementary Materials
The following are available online at http://www.mdpi.com/2075-4655/2/3/17/s1, Table S1. Differentially enriched H3K4me3 regions between the AC and CC cyto-nuclear Drosophila melanogaster lines; Table S2. List of genes that show differential H3K4me3 enrichment between AC and CC cyto-nuclear lines.
Author Contributions
Conceptualization, C.G., R.D. and K.R.; Methodology, C.G., S.V., R.D.; Formal Analysis, C.G., S.V.; Data Curation, C.G., S.V.; Writing-Original Draft Preparation, C.G., S.V., K.R.; Writing-Review & Editing, C.G., S.V., R.D., D.K.D., K.R.; Visualization, C.G.; Project Administration, C.G. and K.R.
Funding
This research was funded by The Deutsche Forschungsgemeinschaft (Excellence Initiative to TU Dresden) (S.V., R.D., K.R.) and grant number Do1776/1-1 (R.D.), the Australian Research Council (grant numbers FT160100022 and DP170100165) (D.K.D.), and JSPS (grant number S17087) (C.G.).
Acknowledgments
We thank Andreas Dahl and the Deep Sequencing Group SFB 655 at the BIOTEChnology Center TU Dresden, Grit Mehnert for support in performing the ChIP-Seq, Ünal Coskun from the Paul-Langerhans Institute Dresden, for discussion regarding ATPase6.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, and in the decision to publish the results.
References
- Bowman, J.C. Genotype × environment interactions. Ann. Genet. Sel. Anim. 1972, 4, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Ingleby, F.C.; Junt, J.; Hosken, D.J. The role of genotype-by-environment interactions in sexual selection. J. Evol. Biol. 2010, 23, 2031–2045. [Google Scholar] [CrossRef] [PubMed]
- Favé, M.-J.; Lamaze, F.C.; Soave, D.; Hodgkinson, A.; Gauvin, H.; Bruat, V.; Grenier, J.-C.; Gbeha, E.; Skead, K.; Smargiassi, A.; et al. Gene-by-environment interactions in urban populations modulate risk phenotypes. Nat. Commun. 2018, 9, 827. [Google Scholar] [CrossRef] [PubMed]
- Bonduriansky, R.; Day, T. Nongenetic inheritance and its evolutionary implications. Rev. Ecol. Evol. Syst. 2009, 40, 103–125. [Google Scholar] [CrossRef]
- Danchin, É.; Charmantier, A.; Champagne, F.A.; Mesoudi, A.; Pujol, B.; Blanchet, S. Beyond DNA: Integrating inclusive inheritance into an extended theory of evolution. Nat. Rev. Gen. 2011, 12, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Bonduriansky, R. Rethinking heredity, again. Trends Ecol. Evol. 2012, 27, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Cosseau, C.; Wolkenhauer, O.; Padalino, G.; Geyer, K.K.; Hoffmann, K.F.; Grunau, C. (Epi)genetic Inheritance in Schistosoma mansoni: A Systems Approach. Trends Parasitol. 2016, 33, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Dobler, R.; Rogell, B.; Budar, F.; Dowling, D.K. A meta-analysis of the strength and nature of cytoplasmic genetic effects. J. Evol. Biol. 2014, 27, 2021–2034. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C.; Fan, W. Energetics, epigenetics, mitochondrial genetics. Mitochondrion 2010, 10, 12–31. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, K.; Dowling, D.K.; Morrow, E.H. Mitochondrial Replacement, Evolution, and the Clinic. Science 2013, 341, 1345–1346. [Google Scholar] [CrossRef] [PubMed]
- Dowling, D.K. Evolutionary perspectives on the links between mitochondrial genotype and disease phenotype. Biochim. Biophys. Acta 2014, 1840, 1393–1403. [Google Scholar] [CrossRef] [PubMed]
- Castegna, A.; Iacobazzi, V.; Infantino, V. The mitochondrial side of epigenetics. Physiol. Genom. 2015, 47, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Morrow, E.H.; Reinhardt, K.; Wolff, J.N.; Dowling, D.K. Risks inherent to mitochondrial replacement. EMBO Rep. 2015, 16, 541–544. [Google Scholar] [CrossRef] [PubMed]
- Jhuang, H.-Y.; Lee, H.-Y.; Leu, J.-Y. Mitochondrial-nuclear co-evolution leads to hybrid incompatibility through pentatricopeptide repeat proteins. EMBO Rep. 2016, 18, 87–101. [Google Scholar] [CrossRef] [PubMed]
- Baris, T.Z.; Wagner, D.N.; Dayan, D.I.; Du, X.; Blier, P.U.; Pichaud, N.; Oleksiak, M.F.; Crawford, D.L. Evolved genetic and phenotypic differences due to mitochondrial-nuclear interactions. PLoS Genet. 2017, 13, E1006517. [Google Scholar] [CrossRef] [PubMed]
- Marom, S.; Friger, M.; Mishmar, D. MtDNA meta-analysis reveals both phenotype specificity and allele heterogeneity: A model for differential association. Sci. Rep. 2017, 7, 43449. [Google Scholar] [CrossRef] [PubMed]
- Parikh, V.S.; Conrad-Webb, H.; Docherty, R.; Butow, R.A. Interaction between the yeast mitochondrial and nuclear genomes influences the abundance of novel transcripts derived from the spacer region of the nuclear ribosomal DNA repeat. Mol. Cell. Biol. 1989, 9, 1897–1907. [Google Scholar] [CrossRef] [PubMed]
- Burton, R.S.; Barreto, F.S. A disproportionate role for mtDNA in Dobzhansky–Muller incompatibilities? Mol. Ecol. 2012, 21, 4942–4957. [Google Scholar] [CrossRef] [PubMed]
- Yee, W.K.W.; Sutton, K.L.; Dowling, D.K. In vivo male fertility is affected by naturally occurring mitochondrial haplotypes. Curr. Biol. 2013, 23, R55–R56. [Google Scholar] [CrossRef] [PubMed]
- Wolff, J.N.; Pichaud, N.; Camus, M.F.; Côté, G.; Blier, P.U.; Dowling, D.K. Evolutionary implications of mitochondrial genetic variation: Mitochondrial genetic effects on OXPHOS respiration and mitochondrial quantity change with age and sex in fruit flies. J. Evol. Biol. 2016, 29, 736–747. [Google Scholar] [CrossRef] [PubMed]
- Gershoni, M.; Templeton, A.R.; Mishmar, D. Mitochondrial bioenergetics as a major motive force of speciation. Bioessays 2009, 31, 642–650. [Google Scholar] [CrossRef] [PubMed]
- Burton, R.S.; Pereira, R.J.; Barreto, F.S. Cytonuclear Genomic Interactions and Hybrid Breakdown. Rev. Ecol. Evol. Syst. 2013, 44, 281–302. [Google Scholar] [CrossRef]
- Hill, G.E. Mitonuclear Ecology. Mol. Biol. Evol. 2015, 32, 1917–1927. [Google Scholar] [CrossRef] [PubMed]
- Hudson, G.; Gomez-Duran, A.; Wilson, I.J.; Chinnery, P.F. Recent Mitochondrial DNA Mutations Increase the Risk of Developing Common Late-Onset Human Diseases. PLoS Genet. 2014, 10, E1004369. [Google Scholar] [CrossRef] [PubMed]
- Dobler, R.; Dowling, D.K.; Morrow, E.H.; Reinhardt, K. A systematic review and meta-analysis reveals pervasive effects of germline mitochondrial replacement on components of health. Hum. Repr. Update 2018. [Google Scholar] [CrossRef] [PubMed]
- Ballard, J.W.; Melvin, R.G. Linking the mitochondrial genotype to the organismal phenotype. Mol. Ecol. 2010, 19, 1523–1539. [Google Scholar] [CrossRef] [PubMed]
- Quirós, P.M.; Mottis, A.; Auwerx, J. Mitonuclear communication in homeostasis and stress. Nat. Rev. Mol. Cell Biol. 2016, 17, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Innocenti, P.; Morrow, E.H.; Dowling, D.K. Experimental Evidence Supports a Sex-Specific Selective Sieve in Mitochondrial Genome Evolution. Science 2011, 332, 845–848. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Yan, Z.; Ma, Q.; Jiao, F.; Huang, S.; Zeng, F.; Zeng, Y. Association between mitochondrial DNA haplotype compatibility and increased efficiency of bovine intersubspecies cloning. J. Genet. Genom. 2011, 38, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Bellizzi, D.; D’Aquila, P.; Giordano, M.; Montesanto, A.; Passarino, G. Global DNA methylation levels are modulated by mitochondrial DNA variants. Epigenomics 2012, 4, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.D.W.; Rodda, A.E.; Dickinson, A.; Mahmud, A.; Nefzger, C.M.; Lee, W.; Forsythe, J.S.; Polo, J.M.; Trounce, I.A.; McKenzie, M. Mitochondrial DNA Haplotypes Define Gene Expression Patterns in Pluripotent and Differentiating Embryonic Stem Cells. Stem Cells 2013, 31, 703–716. [Google Scholar] [CrossRef] [PubMed]
- Vivian, C.J.; Brinker, A.E.; Graw, S.; Koestler, D.C.; Legendre, C.; Gooden, G.C.; Salhia, B.; Welch, D.R. Mitochondrial genomic backgrounds affect nuclear DNA methylation and gene expression. Cancer Res. 2017, 77, 6202–6214. [Google Scholar] [CrossRef] [PubMed]
- Lozoya, O.A.; Martinez-Reyes, I.; Wang, T.; Grenet, D.; Bushel, P.; Li, J.; Chandel, N.; Woychik, R.P.; Santos, J.H. Mitochondrial nicotinamide adenine dinucleotide reduced (NADH) oxidation links the tricarboxylic acid (TCA) cycle with methionine metabolism and nuclear DNA methylation. PLoS Biol. 2018, 16, e2005707. [Google Scholar] [CrossRef] [PubMed]
- Morrow, E.H.; Camus, F. Mitonuclear epistasis and mitochondrial disease. Mitochondrion 2017, 35, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Havird, J.C.; Hall, M.D.; Dowling, D.K. The evolution of sex: A new hypothesis based on mitochondrial mutational erosion. BioEssays 2015, 37, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Mateos, M.; Castrezana, S.J.; Nankivell, B.J.; Estes, A.M.; Markow, T.A.; Moran, N.A. Heritable endosymbionts of Drosophila. Genetics 2006, 174, 363–376. [Google Scholar] [CrossRef] [PubMed]
- Niehuis, O.; Judson, A.K.; Gadau, J. Cytonuclear genic incompatibilities cause increased mortality in male F2 hybrids of Nasonia giraulti and N. vitripennis. Genetics 2008, 178, 413–426. [Google Scholar] [CrossRef] [PubMed]
- Werren, J.H.; Baldo, L.; Clark, M.E. Wolbachia: Master manipulators of invertebrate biology. Nat. Rev. Microbiol. 2008, 6, 741–751. [Google Scholar] [CrossRef] [PubMed]
- Zug, R.; Hammerstein, P. Bad guys turned nice? A critical assessment of Wolbachia mutualisms in arthropod hosts: Wolbachia mutualisms in arthropods. Biol. Rev. 2015, 90, 89–111. [Google Scholar] [CrossRef] [PubMed]
- Guantes, R.; Rastrojo, A.; Neves, R.; Lima, A.; Aguado, B.; Iborra, F.J.K. Global variability in gene expression and alternative splicing is modulated by mitochondrial content. Genome Res. 2015, 25, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Kofler, R.; Pandey, R.V.; Schlötterer, C. PoPoolation2: Identifying differentiation between populations using sequencing of pooled DNA samples (Pool-Seq). Bioinformatics 2011, 27, 3435–3436. [Google Scholar] [CrossRef] [PubMed]
- Ilinsky, Y.; Zakharov, I.K. Genetic correlation between types of mtDNA of Drosophila melanogaster and genotypes of its primary endosymbiont, Wolbachia. Dros. Inf. Serv. 2006, 89, 89–90. [Google Scholar]
- Nunes, M.D.; Nolte, V.; Schlötterer, C. Nonrandom Wolbachia infection status of Drosophila melanogaster strains with different mtDNA haplotypes. Mol. Biol. Evol. 2006, 25, 2493–2498. [Google Scholar] [CrossRef] [PubMed]
- Richardson, M.F.; Weinert, L.A.; Welch, J.J.; Linheiro, R.S.; Magwire, M.M.; Jiggins, F.M.; Bergman, C.M. Population genomics of the Wolbachia endosymbiont in Drosophila melanogaster. PLoS Genet. 2012, 8, E1003129. [Google Scholar] [CrossRef] [PubMed]
- Taudt, A.; Nguyen, M.A.; Heinig, M.; Johannes, F.; Colome-Tatche, M. chromstaR: Tracking combinatorial chromatin state dynamics in space and time. bioRxiv 2016. [Google Scholar] [CrossRef]
- Welch, R.P.; Lee, C.; Imbriano, P.M.; Patil, S.; Weymouth, T.E.; Smith, R.A.; Scott, L.J.; Sartor, M.A. ChIP-Enrich: Gene set enrichment testing for ChIP-seq data. Nucl. Acid Res. 2014, 42, e105. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Grossman, R.; Stein, L. PeakRanger: A cloud-enabled peak caller for ChIP-seq data. BMC Bioinform. 2011, 12, 139. [Google Scholar] [CrossRef] [PubMed]
- Chinnery, P.F.; Elliott, H.R.; Hudson, G.; Samuels, D.C.; Relton, C.L. Epigenetics, epidemiology and mitochondrial DNA diseases. Int. J. Epidemiol. 2012, 41, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Angevine, C.M.; Herold, K.A.G.; Vincent, O.D.; Fillingame, R.H. Aqueous access pathways in ATP Synthase subunit a. J. Biol. Chem. 2007, 282, 9001–9007. [Google Scholar] [CrossRef] [PubMed]
- Niedzwiecka, K.; Kabala, A.M.; Lasserre, J.-P.; Tribouillard-Tanvier, D.; Golik, P.; Dautant, A.; di Rago, J.-P.; Kucharczyk, R. Yeast models of mutations in the mitochondrial ATP6 gene found in human cancer cells. Mitochondrion 2016, 29, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Hahn, A.; Parey, K.; Bublitz, M.; Mills, D.J.; Zickermann, V.; Vonck, J.; Kühlbrandt, W.; Meier, T. Structure of a complete ATP Synthase dimer reveals the molecular basis of inner mitochondrial membrane morphology. Mol. Cell 2016, 63, 445–456. [Google Scholar] [CrossRef] [PubMed]
- Daum, B.; Walter, A.; Horst, A.; Osiewacz, H.D.; Kühlbrandt, W. Age-dependent dissociation of ATP synthase dimers and loss of inner-membrane cristae in mitochondria. PNAS 2013, 110, 15301–15306. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Pagadala, V.; Mueller, D.M. Understanding structure, function, and mutations in the mitochondrial ATP synthase. Microb. Cell 2015, 2, 105–125. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.; Weiss, S.J.; Levine, R.L. Methionine oxidation and reduction in proteins. Biochim. Biophys. Acta 2014, 1840, 901–905. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Díaz, E.; Jordà, M.; Peinado, M.A.; Rivero, A. Epigenetics of host–pathogen interactions: The road ahead and the road behind. PLoS Pathog. 2012, 8, E1003007. [Google Scholar] [CrossRef] [PubMed]
- Dudek, J.; Rehling, P.; van der Laan, M. Mitochondrial protein import: Common principles and physiological networks. Biochim. Biophys. Acta 2013, 1833, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Camus, M.F.; Wolf, J.B.W.; Morrow, E.H.; Dowling, D.K. Single nucleotides in the mtDNA sequence modify mitochondrial molecular function and are associated with sex-specific effects on fertility and aging. Curr. Biol. 2015, 25, 2717–2722. [Google Scholar] [CrossRef] [PubMed]
- Wolff, J.N.; Ladoukakis, E.D.; Enríquez, J.A.; Dowling, D.K. Mitonuclear interactions: Evolutionary consequences over multiple biological scales. Phil. Trans. R. Soc. B 2014, 369, 20130443. [Google Scholar] [CrossRef] [PubMed]
- Guha, M.; Srinivasan, S.; Guja, K.; Mejia, E.; Garcia-Diaz, M.; Johnson, F.B.; Ruthel, G.; Kaufman, B.A.; Rappaport, E.F.; Glineburg, M.R.; et al. HnRNPA2 is a novel histone acetyltransferase that mediates mitochondrial stress-induced nuclear gene expression. Cell Discov. 2016, 2, 16045. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.R.; Van Heerwaarden, B.; Dowling, D.K.; Sgrò, C.M. A multivariate test of evolutionary constraints for thermal tolerance in Drosophila melanogaster. J. Evol. Biol. 2012, 25, 1415–1426. [Google Scholar] [CrossRef] [PubMed]
- MacLellan, K.; Whitlock, M.C.; Rundle, H.D. Sexual selection against deleterious mutations via variable male search success. Biol. Lett. 2009, 5, 795–797. [Google Scholar] [CrossRef] [PubMed]
- Pool, J.E.; Corbett-Detig, R.B.; Sugino, R.P.; Stevens, K.A.; Cardeno, C.M.; Crepeau, M.W.; Duchen, P.; Emerson, J.J.; Saelao, P.; Begun, D.J.; et al. Population Genomics of Sub-Saharan Drosophila melanogaster: African Diversity and Non-African Admixture. PLoS Genet. 2012, 8, E1003080. [Google Scholar] [CrossRef] [PubMed]
- Kofler, R.; Orozco-terWengel, P.; De Maio, N.; Pandey, R.V.; Nolte, V.; Futschik, A.; Kosiol, C.; Schlötterer, C. PoPoolation: A Toolbox for Population Genetic Analysis of Next Generation Sequencing Data from Pooled Individuals. PLoS ONE 2011, 6, E15925. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Lunter, G.; Goodson, M. Stampy: A statistical algorithm for sensitive and fast mapping of Illumina sequence reads. Genome Res. 2011, 21, 936–939. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, M. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. B 1995, 57, 289–300. [Google Scholar]
- Cosseau, C.; Azzi, A.; Smith, K.; Freitag, M.; Mitta, G.; Grunau, C. Native chromatin immunoprecipitation (N-ChIP) and ChIP-Seq of Schistosoma mansoni: Critical experimental parameters. Mol. Biochem. Parasitol. 2009, 166, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative Genomics Viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).