Targeting EZH2 in Multiple Myeloma—Multifaceted Anti-Tumor Activity

Abstract
1. Introduction
2. Deregulation of Chromatin Regulators in Multiple Myeloma
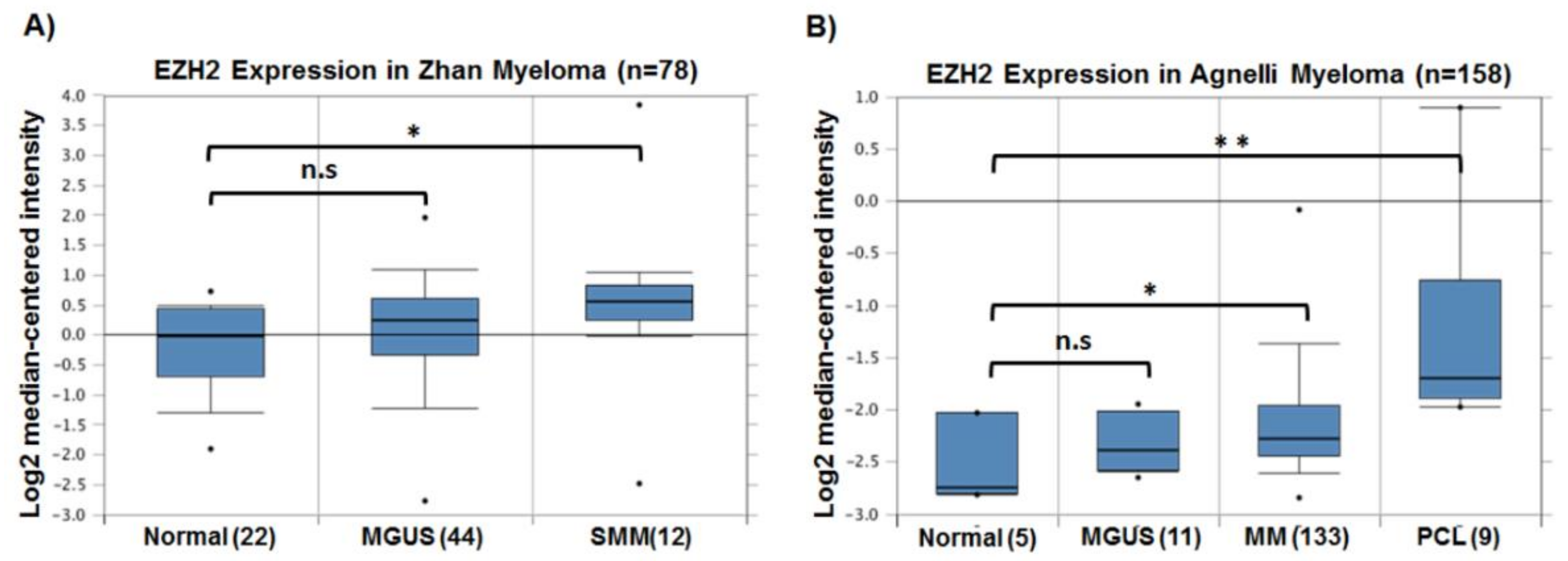
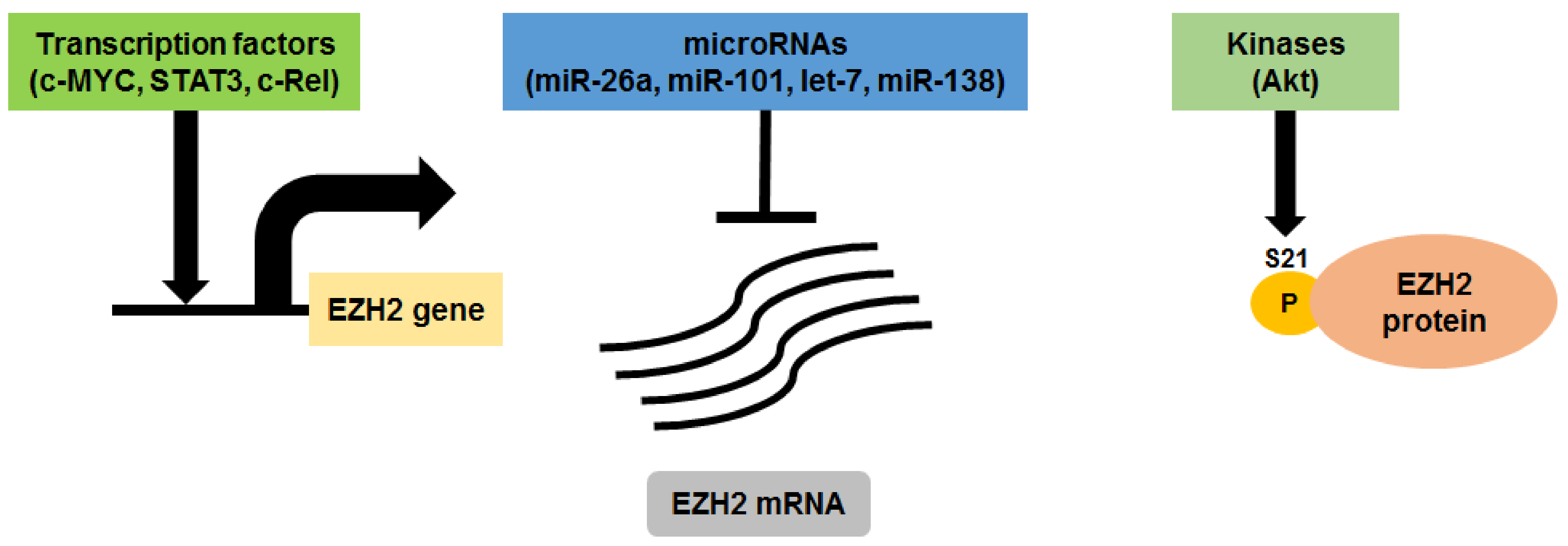
3. EZH2 in Multiple Myeloma
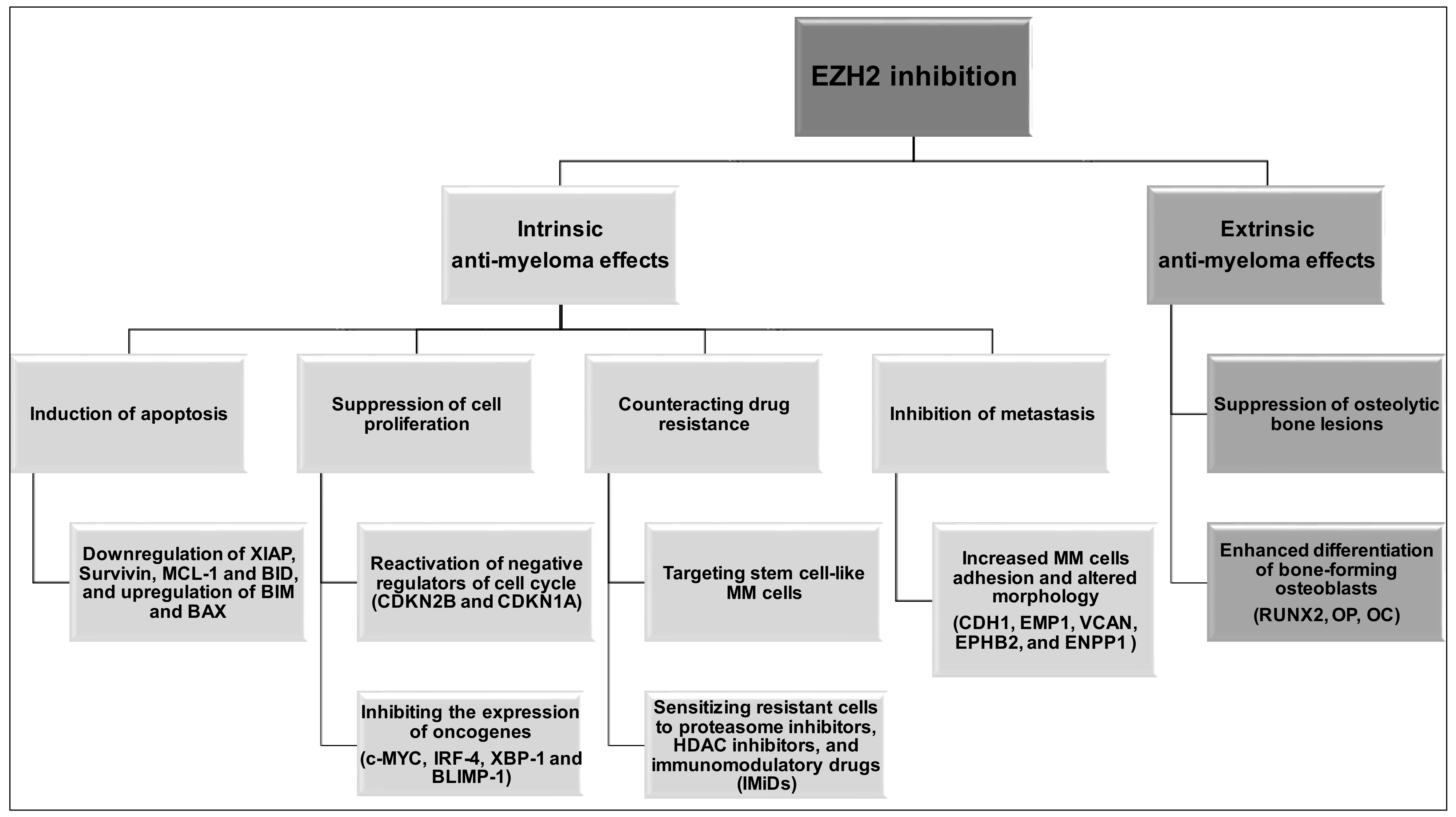
4. EZH2 Inhibition in Multiple Myeloma
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Waddington, C.H. The epigenotype. 1942. Int. J. Epidemiol. 2012, 41, 10–13. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.E.; Meissner, A.; Lander, E.S. The mammalian epigenome. Cell 2007, 128, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.D.; Allis, C.D.; Bernstein, E. Epigenetics: A landscape takes shape. Cell 2007, 128, 635–638. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, A.; Yamada, Y.; Yamanaka, S. Epigenetic regulation in pluripotent stem cells: A key to breaking the epigenetic barrier. Philos. Trans. R. Soc. B. 2013, 368, 20120292. [Google Scholar] [CrossRef] [PubMed]
- Thiagarajan, R.D.; Morey, R.; Laurent, L.C. The epigenome in pluripotency and differentiation. Epigenomics 2014, 6, 121–137. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Jones, P.A. Epigenetic determinants of cancer. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: From mechanism to therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.; Luo, H.; Lee, S.; Jin, F.; Yang, J.S.; Montellier, E.; Buchou, T.; Cheng, Z.; Rousseaux, S.; Rajagopal, N.; et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 2011, 146, 1016–1028. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Sabari, B.R.; Garcia, B.A.; Allis, C.D.; Zhao, Y. Snapshot: Histone modifications. Cell 2014, 159, 458–458. [Google Scholar] [CrossRef] [PubMed]
- Verdone, L.; Caserta, M.; di Mauro, E. Role of histone acetylation in the control of gene expression. Biochem. Cell Biol. 2005, 83, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Rossetto, D.; Avvakumov, N.; Cote, J. Histone phosphorylation: A chromatin modification involved in diverse nuclear events. Epigenetics 2012, 7, 1098–1108. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.; Zhang, Y. The diverse functions of histone lysine methylation. Nat. Rev. Mol. Cell Biol. 2005, 6, 838–849. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Carey, M.; Workman, J.L. The role of chromatin during transcription. Cell 2007, 128, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Litt, M.; Qiu, Y.; Huang, S. Histone arginine methylations: Their roles in chromatin dynamics and transcriptional regulation. Biosci. Rep. 2009, 29, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Di Lorenzo, A.; Bedford, M.T. Histone arginine methylation. FEBS Lett. 2011, 585, 2024–2031. [Google Scholar] [CrossRef] [PubMed]
- Latham, J.A.; Dent, S.Y. Cross-regulation of histone modifications. Nat. Struct. Mol. Biol. 2007, 14, 1017–1024. [Google Scholar] [CrossRef] [PubMed]
- Lennartsson, A.; Ekwall, K. Histone modification patterns and epigenetic codes. Biochim. Biophys. Acta 2009, 1790, 863–868. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, T.B.; Veland, N.; Chen, T. Writers, Readers, and Erasers of Epigenetic Marks. In Epigenetic Cancer Therapy; Gray, S.G., Ed.; Academic Press: Boston, MA, USA, 2015; pp. 31–66. [Google Scholar]
- Hyun, K.; Jeon, J.; Park, K.; Kim, J. Writing, erasing and reading histone lysine methylations. Exp. Mol. Med. 2017, 49, e324. [Google Scholar] [CrossRef] [PubMed]
- Kuehl, W.M.; Bergsagel, P.L. Multiple myeloma: Evolving genetic events and host interactions. Nat. Rev. Cancer 2002, 2, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Kyle, R.A.; Therneau, T.M.; Rajkumar, S.V.; Offord, J.R.; Larson, D.R.; Plevak, M.F.; Melton, L.J., 3rd. A long-term study of prognosis in monoclonal gammopathy of undetermined significance. N. Engl. J. Med. 2002, 346, 564–569. [Google Scholar] [CrossRef] [PubMed]
- Morgan, G.J.; Walker, B.A.; Davies, F.E. The genetic architecture of multiple myeloma. Nat. Rev. Cancer 2012, 12, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.A.; Wardell, C.P.; Chiecchio, L.; Smith, E.M.; Boyd, K.D.; Neri, A.; Davies, F.E.; Ross, F.M.; Morgan, G.J. Aberrant global methylation patterns affect the molecular pathogenesis and prognosis of multiple myeloma. Blood 2011, 117, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Keats, J.J.; Chesi, M.; Egan, J.B.; Garbitt, V.M.; Palmer, S.E.; Braggio, E.; van Wier, S.; Blackburn, P.R.; Baker, A.S.; Dispenzieri, A.; et al. Clonal competition with alternating dominance in multiple myeloma. Blood 2012, 120, 1067–1076. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.A.; Wardell, C.P.; Melchor, L.; Brioli, A.; Johnson, D.C.; Kaiser, M.F.; Mirabella, F.; Lopez-Corral, L.; Humphray, S.; Murray, L.; et al. Intraclonal heterogeneity is a critical early event in the development of myeloma and precedes the development of clinical symptoms. Leukemia 2014, 28, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Bolli, N.; Avet-Loiseau, H.; Wedge, D.C.; van Loo, P.; Alexandrov, L.B.; Martincorena, I.; Dawson, K.J.; Iorio, F.; Nik-Zainal, S.; Bignell, G.R.; et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat. Commun. 2014, 5, 2997. [Google Scholar] [CrossRef] [PubMed]
- Rasche, L.; Chavan, S.S.; Stephens, O.W.; Patel, P.H.; Tytarenko, R.; Ashby, C.; Bauer, M.; Stein, C.; Deshpande, S.; Wardell, C.; et al. Spatial genomic heterogeneity in multiple myeloma revealed by multi-region sequencing. Nat. Commun. 2017, 8, 268. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, A.; Anderson, K. Multiple myeloma. N. Engl. J. Med. 2011, 364, 1046–1060. [Google Scholar] [CrossRef] [PubMed]
- Raab, M.S.; Podar, K.; Breitkreutz, I.; Richardson, P.G.; Anderson, K.C. Multiple myeloma. Lancet 2009, 374, 324–339. [Google Scholar] [CrossRef]
- Kumar, S. Multiple myeloma—Current issues and controversies. Cancer Treat. Rev. 2010, 36, S3–S11. [Google Scholar] [CrossRef]
- Bergsagel, P.L.; Kuehl, W.M. Chromosome translocations in multiple myeloma. Oncogene 2001, 20, 5611–5622. [Google Scholar] [CrossRef] [PubMed]
- Bergsagel, P.L.; Kuehl, W.M. Molecular pathogenesis and a consequent classification of multiple myeloma. J. Clin. Oncol. 2005, 23, 6333–6338. [Google Scholar] [CrossRef] [PubMed]
- Zhan, F.; Huang, Y.; Colla, S.; Stewart, J.P.; Hanamura, I.; Gupta, S.; Epstein, J.; Yaccoby, S.; Sawyer, J.; Burington, B.; et al. The molecular classification of multiple myeloma. Blood 2006, 108, 2020–2028. [Google Scholar] [CrossRef] [PubMed]
- Broyl, A.; Hose, D.; Lokhorst, H.; de Knegt, Y.; Peeters, J.; Jauch, A.; Bertsch, U.; Buijs, A.; Stevens-Kroef, M.; Beverloo, H.B.; et al. Gene expression profiling for molecular classification of multiple myeloma in newly diagnosed patients. Blood 2010, 116, 2543–2553. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, R.; Bergsagel, P.L.; Drach, J.; Shaughnessy, J.; Gutierrez, N.; Stewart, A.K.; Morgan, G.; van Ness, B.; Chesi, M.; Minvielle, S.; et al. International myeloma working group molecular classification of multiple myeloma: Spotlight review. Leukemia 2009, 23, 2210–2221. [Google Scholar] [CrossRef] [PubMed]
- Ross, F.M.; Chiecchio, L.; Dagrada, G.; Protheroe, R.K.; Stockley, D.M.; Harrison, C.J.; Cross, N.C.; Szubert, A.J.; Drayson, M.T.; Morgan, G.J. The t(14;20) is a poor prognostic factor in myeloma but is associated with long–term stable disease in monoclonal gammopathies of undetermined significance. Haematologica 2010, 95, 1221–1225. [Google Scholar] [CrossRef] [PubMed]
- Joao, C.; Costa, C.; Coelho, I.; Vergueiro, M.J.; Ferreira, M.; da Silva, M.G. Long-term survival in multiple myeloma. Clin. Case Rep. 2014, 2, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.K.; Dispenzieri, A.; Lacy, M.Q.; Gertz, M.A.; Buadi, F.K.; Pandey, S.; Kapoor, P.; Dingli, D.; Hayman, S.R.; Leung, N.; et al. Continued improvement in survival in multiple myeloma: Changes in early mortality and outcomes in older patients. Leukemia 2014, 28, 1122–1128. [Google Scholar] [CrossRef] [PubMed]
- Kourelis, T.V.; Kumar, S.K.; Srivastava, G.; Gertz, M.A.; Lacy, M.Q.; Buadi, F.K.; Kyle, R.A.; Dispenzieri, A. Long–term response to lenalidomide in patients with newly diagnosed multiple myeloma. Leukemia 2014, 28, 455–457. [Google Scholar] [CrossRef] [PubMed]
- Mimura, N.; Hideshima, T.; Anderson, K.C. Novel therapeutic strategies for multiple myeloma. Exp. Hematol. 2015, 43, 732–741. [Google Scholar] [CrossRef] [PubMed]
- Pawlyn, C.; Kaiser, M.; Heuck, C.; Melchor, L.; Wardell, C.; Murison, A.; Chavan, S.; Johnson, D.C.; Begum, D.B.; Dahir, N.; et al. The spectrum and clinical impact of epigenetic modifier mutations in myeloma. Clin. Cancer Res. 2016, 22, 5783–5794. [Google Scholar] [CrossRef] [PubMed]
- Stewart, A.K.; Fonseca, R. Prognostic and therapeutic significance of myeloma genetics and gene expression profiling. J. Clin. Oncol. 2005, 23, 6339–6344. [Google Scholar] [CrossRef] [PubMed]
- Abdi, J.; Chen, G.; Chang, H. Drug resistance in multiple myeloma: Latest findings and new concepts on molecular mechanisms. Oncotarget 2013, 4, 2186–2207. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, K.; Gimsing, P.; Gronbaek, K. The role of epigenetics in the biology of multiple myeloma. Blood Cancer J. 2014, 4, e207. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, Y.; Kikuchi, J. Epigenetic mechanisms of cell adhesion-mediated drug resistance in multiple myeloma. Int. J. Hematol 2016, 104, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Dupere-Richer, D.; Licht, J.D. Epigenetic regulatory mutations and epigenetic therapy for multiple myeloma. Curr. Opin. Hematol. 2017, 24, 336–344. [Google Scholar] [CrossRef] [PubMed]
- Alzrigat, M.; Párraga, A.A.; Jernberg-Wiklund, H. Epigenetics in multiple myeloma: From mechanisms to therapy. Semin. Cancer Biol. 2018, 51, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Chapman, M.A.; Lawrence, M.S.; Keats, J.J.; Cibulskis, K.; Sougnez, C.; Schinzel, A.C.; Harview, C.L.; Brunet, J.P.; Ahmann, G.J.; Adli, M.; et al. Initial genome sequencing and analysis of multiple myeloma. Nature 2011, 471, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Lohr, J.G.; Stojanov, P.; Carter, S.L.; Cruz-Gordillo, P.; Lawrence, M.S.; Auclair, D.; Sougnez, C.; Knoechel, B.; Gould, J.; Saksena, G.; et al. Widespread genetic heterogeneity in multiple myeloma: Implications for targeted therapy. Cancer Cell 2014, 25, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.A.; Boyle, E.M.; Wardell, C.P.; Murison, A.; Begum, D.B.; Dahir, N.M.; Proszek, P.Z.; Johnson, D.C.; Kaiser, M.F.; Melchor, L.; et al. Mutational spectrum, copy number changes, and outcome: Results of a sequencing study of patients with newly diagnosed myeloma. J. Clin. Oncol. 2015, 33, 3911–3920. [Google Scholar] [CrossRef] [PubMed]
- Lagana, A.; Perumal, D.; Melnekoff, D.; Readhead, B.; Kidd, B.A.; Leshchenko, V.; Kuo, P.Y.; Keats, J.; DeRome, M.; Yesil, J.; et al. Integrative network analysis identifies novel drivers of pathogenesis and progression in newly diagnosed multiple myeloma. Leukemia 2018, 32, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Jagani, Z.; Wiederschain, D.; Loo, A.; He, D.; Mosher, R.; Fordjour, P.; Monahan, J.; Morrissey, M.; Yao, Y.M.; Lengauer, C.; et al. The polycomb group protein bmi–1 is essential for the growth of multiple myeloma cells. Cancer Res. 2010, 70, 5528–5538. [Google Scholar] [CrossRef] [PubMed]
- Bolomsky, A.; Schlangen, K.; Schreiner, W.; Zojer, N.; Ludwig, H. Targeting of BMI-1 with PTC-209 shows potent anti-myeloma activity and impairs the tumor microenvironment. J. Hematol. Oncol. 2016, 9, 17. [Google Scholar] [CrossRef] [PubMed]
- Keats, J.J.; Reiman, T.; Maxwell, C.A.; Taylor, B.J.; Larratt, L.M.; Mant, M.J.; Belch, A.R.; Pilarski, L.M. In multiple myeloma, t(4;14)(p16;q32) is an adverse prognostic factor irrespective of FGFR3 expression. Blood 2003, 101, 1520–1529. [Google Scholar] [CrossRef] [PubMed]
- Keats, J.J.; Maxwell, C.A.; Taylor, B.J.; Hendzel, M.J.; Chesi, M.; Bergsagel, P.L.; Larratt, L.M.; Mant, M.J.; Reiman, T.; Belch, A.R.; et al. Overexpression of transcripts originating from the MMSET locus characterizes all t(4;14)(p16;q32)-positive multiple myeloma patients. Blood 2005, 105, 4060–4069. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Garcia, E.; Popovic, R.; Min, D.J.; Sweet, S.M.; Thomas, P.M.; Zamdborg, L.; Heffner, A.; Will, C.; Lamy, L.; Staudt, L.M.; et al. The MMSET histone methyl transferase switches global histone methylation and alters gene expression in t(4;14) multiple myeloma cells. Blood 2011, 117, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Popovic, R.; Martinez-Garcia, E.; Giannopoulou, E.G.; Zhang, Q.; Ezponda, T.; Shah, M.Y.; Zheng, Y.; Will, C.M.; Small, E.C.; Hua, Y.; et al. Histone methyltransferase MMSET/NSD2 alters EZH2 binding and reprograms the myeloma epigenome through global and focal changes in H3K36 and H3K27 methylation. PLoS Genet. 2014, 10, e1004566. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.Y.; Martinez-Garcia, E.; Phillip, J.M.; Chambliss, A.B.; Popovic, R.; Ezponda, T.; Small, E.C.; Will, C.; Phillip, M.P.; Neri, P.; et al. MMSET/WHSC1 enhances DNA damage repair leading to an increase in resistance to chemotherapeutic agents. Oncogene 2016, 35, 5905–5915. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, J.R.; Tian, E.; Heuck, C.J.; Johann, D.J.; Epstein, J.; Swanson, C.M.; Lukacs, J.L.; Binz, R.L.; Johnson, M.; Sammartino, G.; et al. Evidence of an epigenetic origin for high-risk 1q21 copy number aberrations in multiple myeloma. Blood 2015, 125, 3756–3759. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Chen, L.; Barlogie, B.; Stephens, O.; Wu, X.; Williams, D.R.; Cartron, M.A.; van Rhee, F.; Nair, B.; Waheed, S.; et al. High-risk myeloma is associated with global elevation of miRNAs and overexpression of EIF2C2/AGO2. Proc. Natl. Acad. Sci. USA 2010, 107, 7904–7909. [Google Scholar] [CrossRef] [PubMed]
- Lionetti, M.; Biasiolo, M.; Agnelli, L.; Todoerti, K.; Mosca, L.; Fabris, S.; Sales, G.; Deliliers, G.L.; Bicciato, S.; Lombardi, L.; et al. Identification of microRNA expression patterns and definition of a microRNA/mRNA regulatory network in distinct molecular groups of multiple myeloma. Blood 2009, 114, e20–e26. [Google Scholar] [CrossRef] [PubMed]
- Chi, J.; Ballabio, E.; Chen, X.H.; Kusec, R.; Taylor, S.; Hay, D.; Tramonti, D.; Saunders, N.J.; Littlewood, T.; Pezzella, F.; et al. MicroRNA expression in multiple myeloma is associated with genetic subtype, isotype and survival. Biol. Direct 2011, 6, 23. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.M.; Boyd, K.; Davies, F.E. The potential role of epigenetic therapy in multiple myeloma. Br. J. Haematol. 2010, 148, 702–713. [Google Scholar] [CrossRef] [PubMed]
- Maes, K.; Menu, E.; van Valckenborgh, E.; van Riet, I.; Vanderkerken, K.; de Bruyne, E. Epigenetic modulating agents as a new therapeutic approach in multiple myeloma. Cancers 2013, 5, 430–461. [Google Scholar] [CrossRef] [PubMed]
- Issa, M.E.; Takhsha, F.S.; Chirumamilla, C.S.; Perez-Novo, C.; Vanden Berghe, W.; Cuendet, M. Epigenetic strategies to reverse drug resistance in heterogeneous multiple myeloma. Clin. Epigenetics 2017, 9, 17. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, G.; Richardson, P.G.; Anderson, K.C. Promising therapies in multiple myeloma. Blood 2015, 126, 300–310. [Google Scholar] [CrossRef] [PubMed]
- San-Miguel, J.F.; Hungria, V.T.; Yoon, S.S.; Beksac, M.; Dimopoulos, M.A.; Elghandour, A.; Jedrzejczak, W.W.; Gunther, A.; Nakorn, T.N.; Siritanaratkul, N.; et al. Overall survival of patients with relapsed multiple myeloma treated with panobinostat or placebo plus bortezomib and dexamethasone (the panorama 1 trial): A randomised, placebo-controlled, phase 3 trial. Lancet Haematol. 2016, 3, e506–e515. [Google Scholar] [CrossRef]
- Richardson, P.G.; Harvey, R.D.; Laubach, J.P.; Moreau, P.; Lonial, S.; San-Miguel, J.F. Panobinostat for the treatment of relapsed or relapsed/refractory multiple myeloma: Pharmacology and clinical outcomes. Expert Rev. Clin. Pharmacol. 2016, 9, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Wahaib, K.; Beggs, A.E.; Campbell, H.; Kodali, L.; Ford, P.D. Panobinostat: A histone deacetylase inhibitor for the treatment of relapsed or refractory multiple myeloma. Am. J. Health Syst. Pharm. 2016, 73, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Margueron, R.; Reinberg, D. The polycomb complex PRC2 and its mark in life. Nature 2011, 469, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Wang, L.; Wang, H.; Xia, L.; Erdjument–Bromage, H.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone H3 lysine 27 methylation in polycomb–group silencing. Science 2002, 298, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Di Croce, L.; Helin, K. Transcriptional regulation by polycomb group proteins. Nat. Struct. Mol. Biol. 2013, 20, 1147–1155. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Roberts, C.W. Targeting EZH2 in cancer. Nat. Med. 2016, 22, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Konze, K.D.; Jin, J.; Wang, G.G. Targeting EZH2 and PRC2 dependence as novel anticancer therapy. Exp. Hematol. 2015, 43, 698–712. [Google Scholar] [CrossRef] [PubMed]
- Melnick, A. Epigenetic therapy leaps ahead with specific targeting of EZH2. Cancer Cell 2012, 22, 569–570. [Google Scholar] [CrossRef] [PubMed]
- Chase, A.; Cross, N.C. Aberrations of EZH2 in cancer. Clin. Cancer Res. 2011, 17, 2613–2618. [Google Scholar] [CrossRef] [PubMed]
- Simon, J.A.; Lange, C.A. Roles of the EZH2 histone methyltransferase in cancer epigenetics. Mutat. Res. 2008, 647, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Italiano, A.; Soria, J.C.; Toulmonde, M.; Michot, J.M.; Lucchesi, C.; Varga, A.; Coindre, J.M.; Blakemore, S.J.; Clawson, A.; Suttle, B.; et al. Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: A first-in-human, open-label, phase 1 study. Lancet. Oncol. 2018, 19, 649–659. [Google Scholar] [CrossRef]
- Chang, C.J.; Hung, M.C. The role of EZH2 in tumour progression. Br. J. Cancer 2012, 106, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Ezponda, T.; Licht, J.D. Molecular pathways: Deregulation of histone H3 lysine 27 methylation in cancer-different paths, same destination. Clin. Cancer Res. 2014, 20, 5001–5008. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.G.; Konze, K.D.; Tao, J. Polycomb genes, miRNA, and their deregulation in B-cell malignancies. Blood 2015, 125, 1217–1225. [Google Scholar] [CrossRef] [PubMed]
- Herviou, L.; Cavalli, G.; Cartron, G.; Klein, B.; Moreaux, J. EZH2 in normal hematopoiesis and hematological malignancies. Oncotarget 2016, 7, 2284–2296. [Google Scholar] [CrossRef] [PubMed]
- Zhan, F.; Hardin, J.; Kordsmeier, B.; Bumm, K.; Zheng, M.; Tian, E.; Sanderson, R.; Yang, Y.; Wilson, C.; Zangari, M.; et al. Global gene expression profiling of multiple myeloma, monoclonal gammopathy of undetermined significance, and normal bone marrow plasma cells. Blood 2002, 99, 1745–1757. [Google Scholar] [CrossRef] [PubMed]
- Zhan, F.; Tian, E.; Bumm, K.; Smith, R.; Barlogie, B.; Shaughnessy, J., Jr. Gene expression profiling of human plasma cell differentiation and classification of multiple myeloma based on similarities to distinct stages of late-stage B-cell development. Blood 2003, 101, 1128–1140. [Google Scholar] [CrossRef] [PubMed]
- Croonquist, P.A.; van Ness, B. The polycomb group protein enhancer of zeste homolog 2 (EZH2) is an oncogene that influences myeloma cell growth and the mutant RAS phenotype. Oncogene 2005, 24, 6269–6280. [Google Scholar] [CrossRef] [PubMed]
- Kalushkova, A.; Fryknas, M.; Lemaire, M.; Fristedt, C.; Agarwal, P.; Eriksson, M.; Deleu, S.; Atadja, P.; Osterborg, A.; Nilsson, K.; et al. Polycomb target genes are silenced in multiple myeloma. PLoS ONE 2010, 5, e11483. [Google Scholar] [CrossRef] [PubMed]
- Pawlyn, C.; Bright, M.D.; Buros, A.F.; Stein, C.K.; Walters, Z.; Aronson, L.I.; Mirabella, F.; Jones, J.R.; Kaiser, M.F.; Walker, B.A.; et al. Overexpression of EZH2 in multiple myeloma is associated with poor prognosis and dysregulation of cell cycle control. Blood Cancer J. 2017, 7, e549. [Google Scholar] [CrossRef] [PubMed]
- Neo, W.H.; Lim, J.F.; Grumont, R.; Gerondakis, S.; Su, I.H. c-Rel regulates EZH2 expression in activated lymphocytes and malignant lymphoid cells. J. Biol. Chem. 2014, 289, 31693–31707. [Google Scholar] [CrossRef] [PubMed]
- Pichiorri, F.; Suh, S.S.; Ladetto, M.; Kuehl, M.; Palumbo, T.; Drandi, D.; Taccioli, C.; Zanesi, N.; Alder, H.; Hagan, J.P.; et al. MicroRNAs regulate critical genes associated with multiple myeloma pathogenesis. Proc. Natl. Acad. Sci. USA 2008, 105, 12885–12890. [Google Scholar] [CrossRef] [PubMed]
- Seckinger, A.; Meissner, T.; Moreaux, J.; Benes, V.; Hillengass, J.; Castoldi, M.; Zimmermann, J.; Ho, A.D.; Jauch, A.; Goldschmidt, H.; et al. miRNAs in multiple myeloma—A survival relevant complex regulator of gene expression. Oncotarget 2015, 6, 39165–39183. [Google Scholar] [CrossRef] [PubMed]
- Rastgoo, N.; Pourabdollah, M.; Abdi, J.; Reece, D.; Chang, H. Dysregulation of EZH2/miR-138 axis contributes to drug resistance in multiple myeloma by downregulating RBPMS. Leukemia 2018. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, J.; Koyama, D.; Wada, T.; Izumi, T.; Hofgaard, P.O.; Bogen, B.; Furukawa, Y. Phosphorylation-mediated EZH2 inactivation promotes drug resistance in multiple myeloma. J. Clin. Invest. 2015, 125, 4375–4390. [Google Scholar] [CrossRef] [PubMed]
- Morin, R.D.; Johnson, N.A.; Severson, T.M.; Mungall, A.J.; An, J.; Goya, R.; Paul, J.E.; Boyle, M.; Woolcock, B.W.; Kuchenbauer, F.; et al. Somatic mutations altering EZH2 (tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat. Genet. 2010, 42, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Sneeringer, C.J.; Scott, M.P.; Kuntz, K.W.; Knutson, S.K.; Pollock, R.M.; Richon, V.M.; Copeland, R.A. Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone h3 (H3K27) in human B-cell lymphomas. Proc. Natl. Acad. Sci. USA 2010, 107, 20980–20985. [Google Scholar] [CrossRef] [PubMed]
- Majer, C.R.; Jin, L.; Scott, M.P.; Knutson, S.K.; Kuntz, K.W.; Keilhack, H.; Smith, J.J.; Moyer, M.P.; Richon, V.M.; Copeland, R.A.; et al. A687V EZH2 is a gain-of-function mutation found in lymphoma patients. FEBS Lett. 2012, 586, 3448–3451. [Google Scholar] [CrossRef] [PubMed]
- McCabe, M.T.; Ott, H.M.; Ganji, G.; Korenchuk, S.; Thompson, C.; van Aller, G.S.; Liu, Y.; Graves, A.P.; Della Pietra, A., 3rd.; Diaz, E.; et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 2012, 492, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Ntziachristos, P.; Tsirigos, A.; van Vlierberghe, P.; Nedjic, J.; Trimarchi, T.; Flaherty, M.S.; Ferres–Marco, D.; da Ros, V.; Tang, Z.; Siegle, J.; et al. Genetic inactivation of the polycomb repressive complex 2 in t cell acute lymphoblastic leukemia. Nat. Med. 2012, 18, 298–301. [Google Scholar] [CrossRef] [PubMed]
- Nikoloski, G.; Langemeijer, S.M.; Kuiper, R.P.; Knops, R.; Massop, M.; Tonnissen, E.R.; van der Heijden, A.; Scheele, T.N.; Vandenberghe, P.; de Witte, T.; et al. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat. Genet. 2010, 42, 665–667. [Google Scholar] [CrossRef] [PubMed]
- Sashida, G.; Harada, H.; Matsui, H.; Oshima, M.; Yui, M.; Harada, Y.; Tanaka, S.; Mochizuki-Kashio, M.; Wang, C.; Saraya, A.; et al. EZH2 loss promotes development of myelodysplastic syndrome but attenuates its predisposition to leukaemic transformation. Nat. Commun. 2014, 5, 4177. [Google Scholar] [CrossRef] [PubMed]
- Ernst, T.; Chase, A.J.; Score, J.; Hidalgo-Curtis, C.E.; Bryant, C.; Jones, A.V.; Waghorn, K.; Zoi, K.; Ross, F.M.; Reiter, A.; et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat. Genet. 2010, 42, 722–726. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.; Alzrigat, M.; Parraga, A.A.; Enroth, S.; Singh, U.; Ungerstedt, J.; Osterborg, A.; Brown, P.J.; Ma, A.; Jin, J.; et al. Genome-wide profiling of histone H3 lysine 27 and lysine 4 trimethylation in multiple myeloma reveals the importance of polycomb gene targeting and highlights EZH2 as a potential therapeutic target. Oncotarget 2016, 7, 6809–6823. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, D.R.; Yu, J.; Shanker, K.; Deshpande, N.; Varambally, R.; Ghosh, D.; Barrette, T.; Pandey, A.; Chinnaiyan, A.M. Oncomine: A cancer microarray database and integrated data-mining platform. Neoplasia 2004, 6, 1–6. [Google Scholar] [CrossRef]
- Zhan, F.; Barlogie, B.; Arzoumanian, V.; Huang, Y.; Williams, D.R.; Hollmig, K.; Pineda-Roman, M.; Tricot, G.; van Rhee, F.; Zangari, M.; et al. Gene-expression signature of benign monoclonal gammopathy evident in multiple myeloma is linked to good prognosis. Blood 2007, 109, 1692–1700. [Google Scholar] [CrossRef] [PubMed]
- Agnelli, L.; Mosca, L.; Fabris, S.; Lionetti, M.; Andronache, A.; Kwee, I.; Todoerti, K.; Verdelli, D.; Battaglia, C.; Bertoni, F.; et al. A SNP microarray and fish-based procedure to detect allelic imbalances in multiple myeloma: An integrated genomics approach reveals a wide gene dosage effect. Genes Chromosomes Cancer 2009, 48, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Kuo, A.J.; Cheung, P.; Chen, K.; Zee, B.M.; Kioi, M.; Lauring, J.; Xi, Y.; Park, B.H.; Shi, X.; Garcia, B.A.; et al. NSD2 links dimethylation of histone H3 at lysine 36 to oncogenic programming. Mol. Cell 2011, 44, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Ezponda, T.; Dupere-Richer, D.; Will, C.M.; Small, E.C.; Varghese, N.; Patel, T.; Nabet, B.; Popovic, R.; Oyer, J.; Bulic, M.; et al. UTX/KDM6A loss enhances the malignant phenotype of multiple myeloma and sensitizes cells to EZH2 inhibition. Cell Rep. 2017, 21, 628–640. [Google Scholar] [CrossRef] [PubMed]
- Rius, M.; Lyko, F. Epigenetic cancer therapy: Rationales, targets and drugs. Oncogene 2012, 31, 4257–4265. [Google Scholar] [CrossRef] [PubMed]
- Gaudichon, J.; Milano, F.; Cahu, J.; DaCosta, L.; Martens, A.C.; Renoir, J.M.; Sola, B. Deazaneplanocin a is a promising drug to kill multiple myeloma cells in their niche. PLoS ONE 2014, 9, e107009. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Bi, C.; Cheong, L.L.; Liu, S.C.; Huang, G.; Zhou, J.; Yu, Q.; Chen, C.S.; Chng, W.J. Determinants of sensitivity to DZNep induced apoptosis in multiple myeloma cells. PLoS ONE 2011, 6, e21583. [Google Scholar] [CrossRef] [PubMed]
- Neri, P.; Bahlis, N.J.; Lonial, S. Panobinostat for the treatment of multiple myeloma. Expert Opin. Investig. Drugs 2012, 21, 733–747. [Google Scholar] [CrossRef] [PubMed]
- Alzrigat, M.; Parraga, A.A.; Agarwal, P.; Zureigat, H.; Osterborg, A.; Nahi, H.; Ma, A.; Jin, J.; Nilsson, K.; Oberg, F.; et al. EZH2 inhibition in multiple myeloma downregulates myeloma associated oncogenes and upregulates microRNAs with potential tumor suppressor functions. Oncotarget 2017, 8, 10213–10224. [Google Scholar] [CrossRef] [PubMed]
- Rizq, O.; Mimura, N.; Oshima, M.; Saraya, A.; Koide, S.; Kato, Y.; Aoyama, K.; Nakajima-Takagi, Y.; Wang, C.; Chiba, T.; et al. Dual inhibition of EZH2 and EZH1 sensitizes PRC2-dependent tumors to proteasome inhibition. Clin. Cancer Res. 2017, 23, 4817–4830. [Google Scholar] [CrossRef] [PubMed]
- Zeng, D.; Liu, M.; Pan, J. Blocking EZH2 methylation transferase activity by GSK126 decreases stem cell-like myeloma cells. Oncotarget 2017, 8, 3396–3411. [Google Scholar] [CrossRef] [PubMed]
- Harding, T.; Swanson, J.; van Ness, B. EZH2 inhibitors sensitize myeloma cell lines to panobinostat resulting in unique combinatorial transcriptomic changes. Oncotarget 2018, 9, 21930–21942. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, K.; Sogaard Helbo, A.; Fibiger Munch-Petersen, H.; Sjo, L.; Christensen, J.; Sommer Kristensen, L.; Asmar, F.; Hermansen, N.E.U.; O’Connel, C.; Gimsing, P.; et al. Dual inhibition of DNMTs and EZH2 can overcome both intrinsic and acquired resistance of myeloma cells to IMiDs in a cereblon-independent manner. Mol. Oncol. 2018, 12, 180–195. [Google Scholar] [CrossRef] [PubMed]
- Hernando, H.; Gelato, K.A.; Lesche, R.; Beckmann, G.; Koehr, S.; Otto, S.; Steigemann, P.; Stresemann, C. EZH2 inhibition blocks multiple myeloma cell growth through upregulation of epithelial tumor suppressor genes. Mol. Cancer Ther. 2016, 15, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Honma, D.; Kanno, O.; Watanabe, J.; Kinoshita, J.; Hirasawa, M.; Nosaka, E.; Shiroishi, M.; Takizawa, T.; Yasumatsu, I.; Horiuchi, T.; et al. Novel orally bioavailable EZH1/2 dual inhibitors with greater antitumor efficacy than an EZH2 selective inhibitor. Cancer Sci. 2017, 108, 2069–2078. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Johnson, P.W.M.; Winter, J.; Leonard, J.; Giulino-Roth, L.; Horner, T.; Radswillas, K.; Carver, J.; Dhar, A. A phase I, open-label study of GSK2816126, an enhancer of zeste homolog 2 (EZH2) inhibitor, in patients with relapsed/refractory diffuse large B-cell lymphoma (DLBCL), transformed follicular lymphoma (tFL), other non-Hodgkin’s lymphomas (NHL), multiple myeloma (MM) and solid tumor. J. Clin. Oncol. 2016, 34. [Google Scholar] [CrossRef]
- Alzrigat, M.; Jernberg-Wiklund, H. The miR–125a and miR-320c are potential tumor suppressor microRNAs epigenetically silenced by the polycomb repressive complex 2 in multiple myeloma. RNA Dis. 2017, 4, e1529. [Google Scholar] [PubMed]
- Baughn, L.B.; di Liberto, M.; Niesvizky, R.; Cho, H.J.; Jayabalan, D.; Lane, J.; Liu, F.; Chen-Kiang, S. CDK2 phosphorylation of Smad2 disrupts TGF-beta transcriptional regulation in resistant primary bone marrow myeloma cells. J. Immunol. 2009, 182, 1810–1817. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Ding, L.; Zhang, H.; Han, J.; Yang, X.; Yan, J.; Zhu, Y.; Li, J.; Song, H.; Ye, Q. Potentiation of Smad-mediated transcriptional activation by the RNA-binding protein RBPMS. Nucleic Acids Res. 2006, 34, 6314–6326. [Google Scholar] [CrossRef] [PubMed]
- Daly, A.C.; Vizán, P.; Hill, C.S. Smad3 protein levels are modulated by RAS activity and during the cell cycle to dictate transforming growth factor-beta responses. J. Biol. Chem. 2010, 285, 6489–6497. [Google Scholar] [CrossRef] [PubMed]
- Alzrigat, M.; Parraga, A.A.; Majumder, M.M.; Ma, A.; Jin, J.; Osterborg, A.; Nahi, H.; Nilsson, K.; Heckman, C.A.; Oberg, F.; et al. The polycomb group protein BMI-1 inhibitor PTC-209 is a potent anti–myeloma agent alone or in combination with epigenetic inhibitors targeting EZH2 and the BET bromodomains. Oncotarget 2017, 8, 103731–103743. [Google Scholar] [CrossRef] [PubMed]
- Hemming, S.; Cakouros, D.; Vandyke, K.; Davis, M.J.; Zannettino, A.C.; Gronthos, S. Identification of novel EZH2 targets regulating osteogenic differentiation in mesenchymal stem cells. Stem Cells Dev. 2016, 25, 909–921. [Google Scholar] [CrossRef] [PubMed]
- Hemming, S.; Cakouros, D.; Isenmann, S.; Cooper, L.; Menicanin, D.; Zannettino, A.; Gronthos, S. EZH2 and KDM6A act as an epigenetic switch to regulate mesenchymal stem cell lineage specification. Stem Cells 2014, 32, 802–815. [Google Scholar] [CrossRef] [PubMed]
- D'Souza, S.; del Prete, D.; Jin, S.; Sun, Q.; Huston, A.J.; Kostov, F.E.; Sammut, B.; Hong, C.S.; Anderson, J.L.; Patrene, K.D.; et al. Gfi1 expressed in bone marrow stromal cells is a novel osteoblast suppressor in patients with multiple myeloma bone disease. Blood 2011, 118, 6871–6880. [Google Scholar] [CrossRef] [PubMed]
- Adamik, J.; Jin, S.; Sun, Q.; Zhang, P.; Weiss, K.R.; Anderson, J.L.; Silbermann, R.; Roodman, G.D.; Galson, D.L. EZH2 or HDAC1 inhibition reverses multiple myeloma–induced epigenetic suppression of osteoblast differentiation. Mol. Cancer Res. 2017, 15, 405–417. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| EZH2 Inhibitor | Anti-MM Activity | Treatment Type | Reference |
|---|---|---|---|
| UNC1999 | In vitro and in vivo | Single agent treatment or in combination with Bortezomib | [102,112,113] |
| GSK343 | In vitro | Single agent treatment | [58,102,107] |
| GSK126 | In vitro and in vivo | Single agent treatment or in combination with Bortezomib and Panobinostat | [107,114,115] |
| EPZ-7438 | In vitro and in vivo | Single agent treatment or in combination with Lenalidomide, Pomalidomide, Bortezomib and Panobinostat | [115,116,117] |
| EPZ005687 | In vitro | Single agent treatment | [88] |
| OR-S1 and OR-S2 | In vitro | Single agent treatment | [118] |
| GSK2816126 | Phase I clinical trial (NCT02082977)–Terminated | Single agent treatment | [119] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alzrigat, M.; Jernberg-Wiklund, H.; Licht, J.D. Targeting EZH2 in Multiple Myeloma—Multifaceted Anti-Tumor Activity. Epigenomes 2018, 2, 16. https://doi.org/10.3390/epigenomes2030016
Alzrigat M, Jernberg-Wiklund H, Licht JD. Targeting EZH2 in Multiple Myeloma—Multifaceted Anti-Tumor Activity. Epigenomes. 2018; 2(3):16. https://doi.org/10.3390/epigenomes2030016
Chicago/Turabian StyleAlzrigat, Mohammad, Helena Jernberg-Wiklund, and Jonathan D. Licht. 2018. "Targeting EZH2 in Multiple Myeloma—Multifaceted Anti-Tumor Activity" Epigenomes 2, no. 3: 16. https://doi.org/10.3390/epigenomes2030016
APA StyleAlzrigat, M., Jernberg-Wiklund, H., & Licht, J. D. (2018). Targeting EZH2 in Multiple Myeloma—Multifaceted Anti-Tumor Activity. Epigenomes, 2(3), 16. https://doi.org/10.3390/epigenomes2030016