Salt Stress Induces Non-CG Methylation in Coding Regions of Barley Seedlings (Hordeum vulgare)

 , , ,
, , , 
Abstract
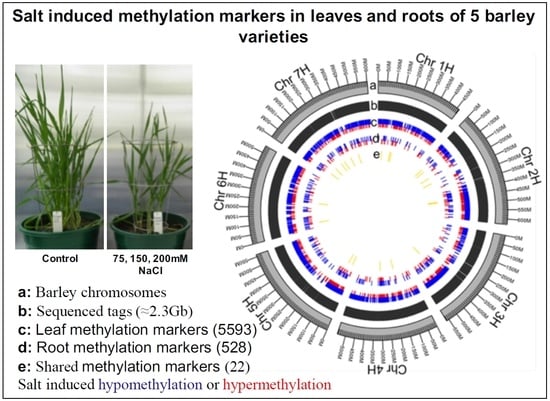
:
1. Introduction
2. Results
2.1. Methylation-Sensitive Genotyping-By-Sequencing (ms-GBS)
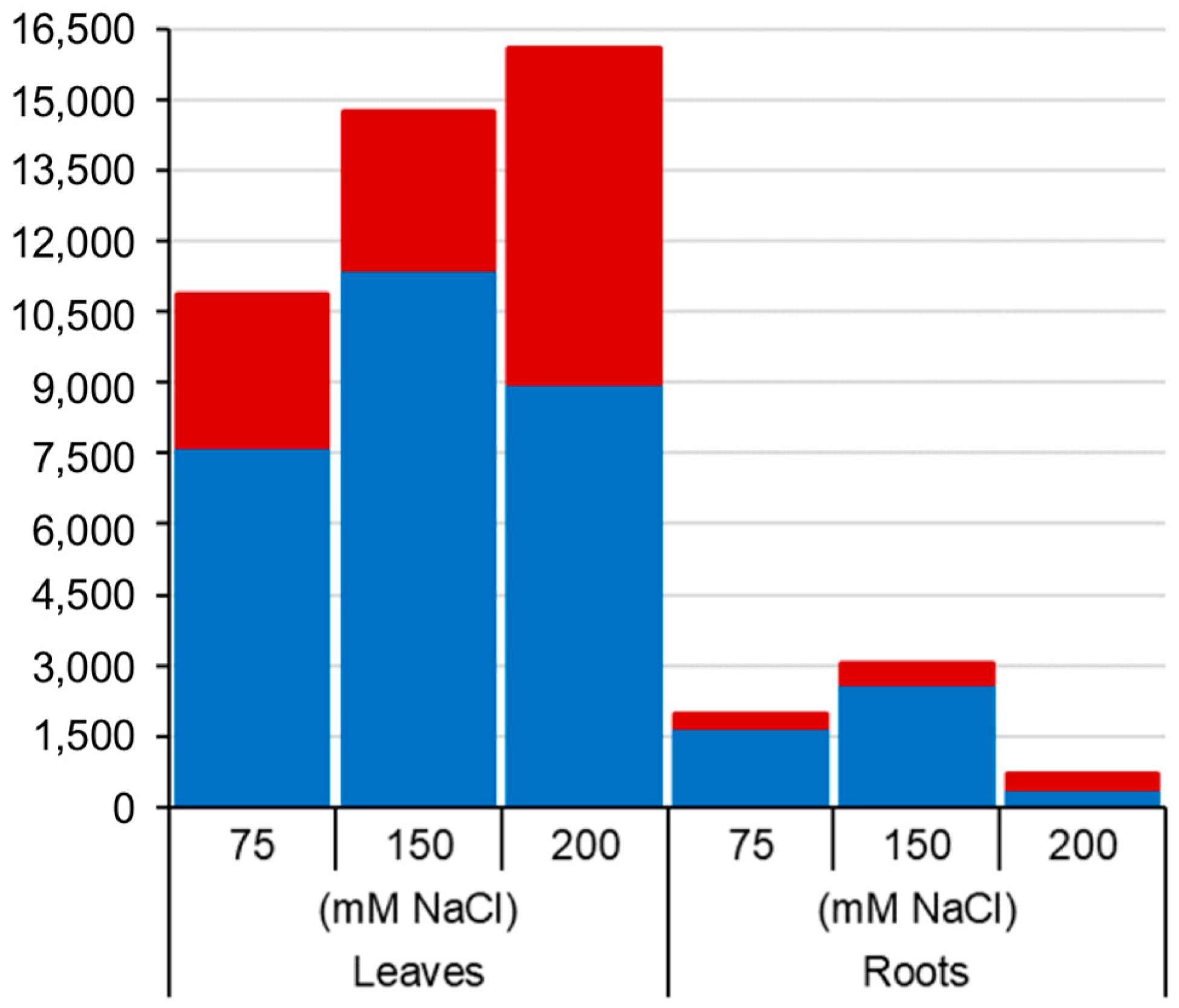
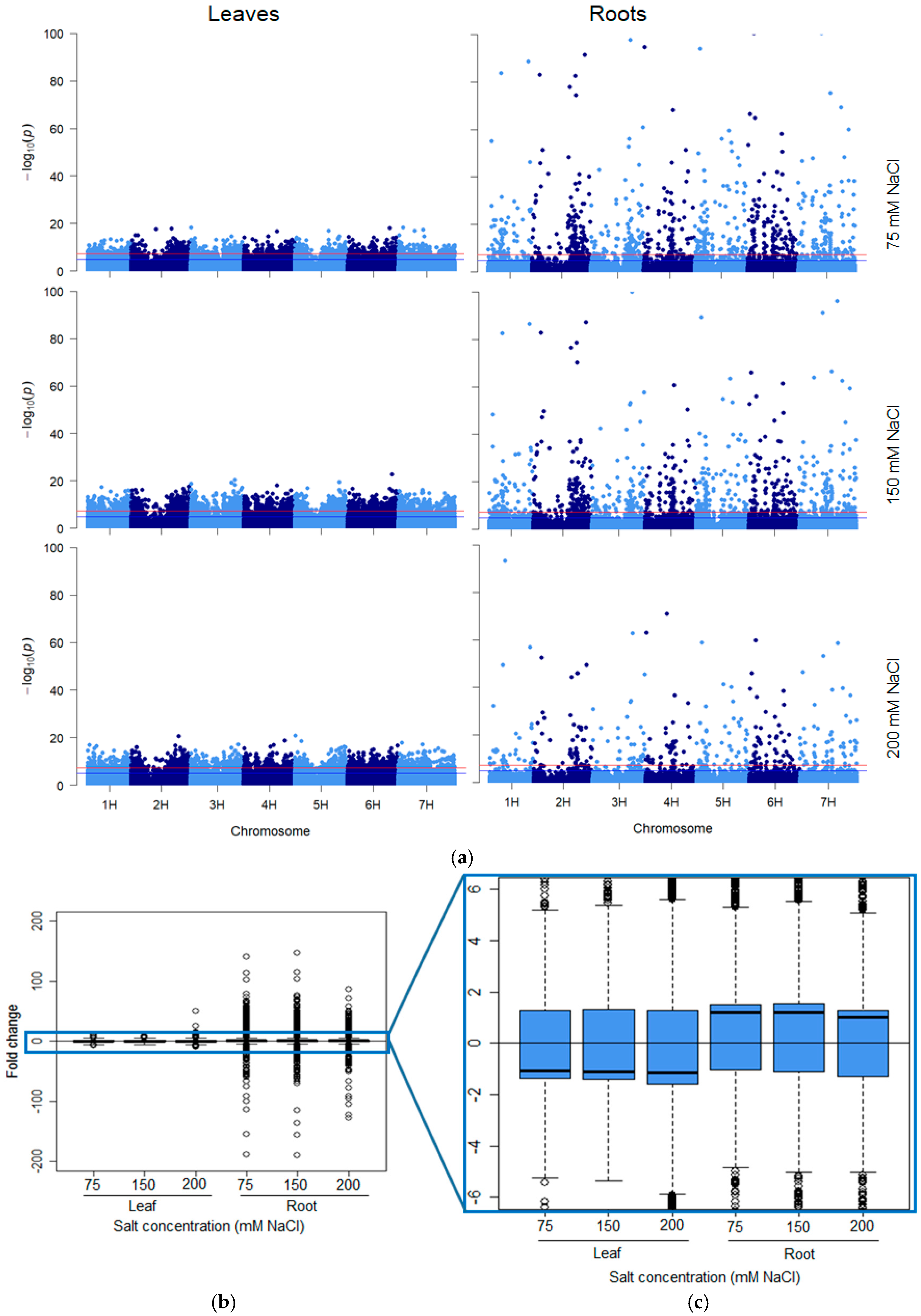
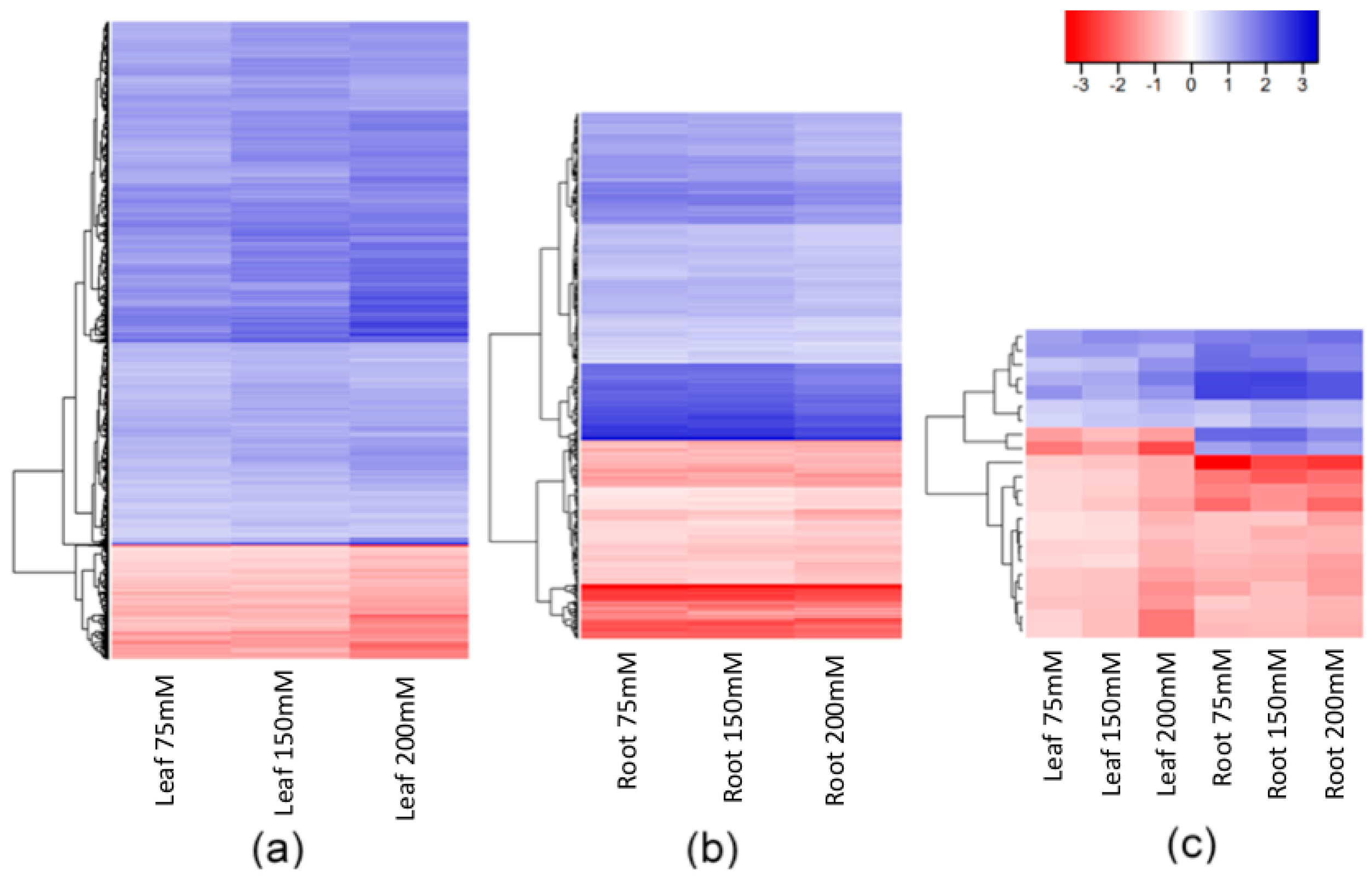
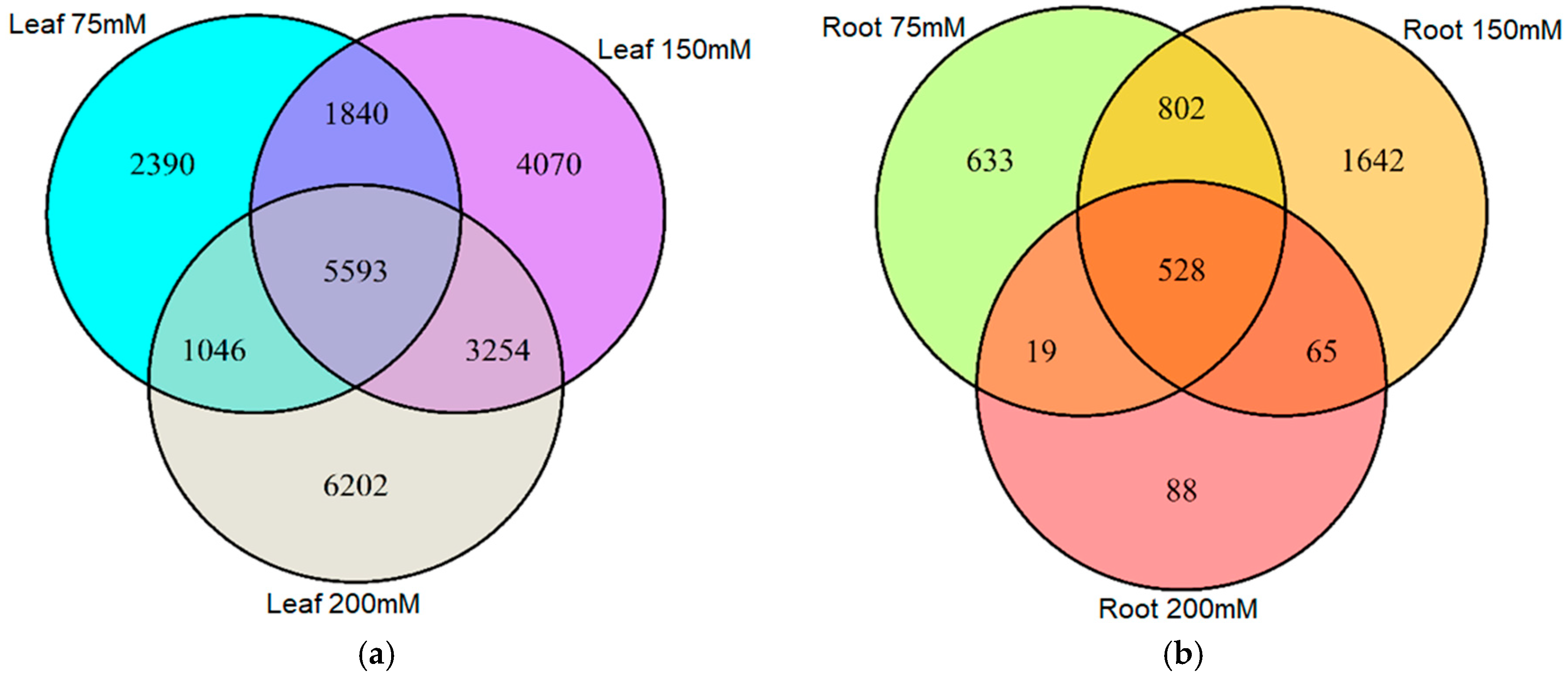
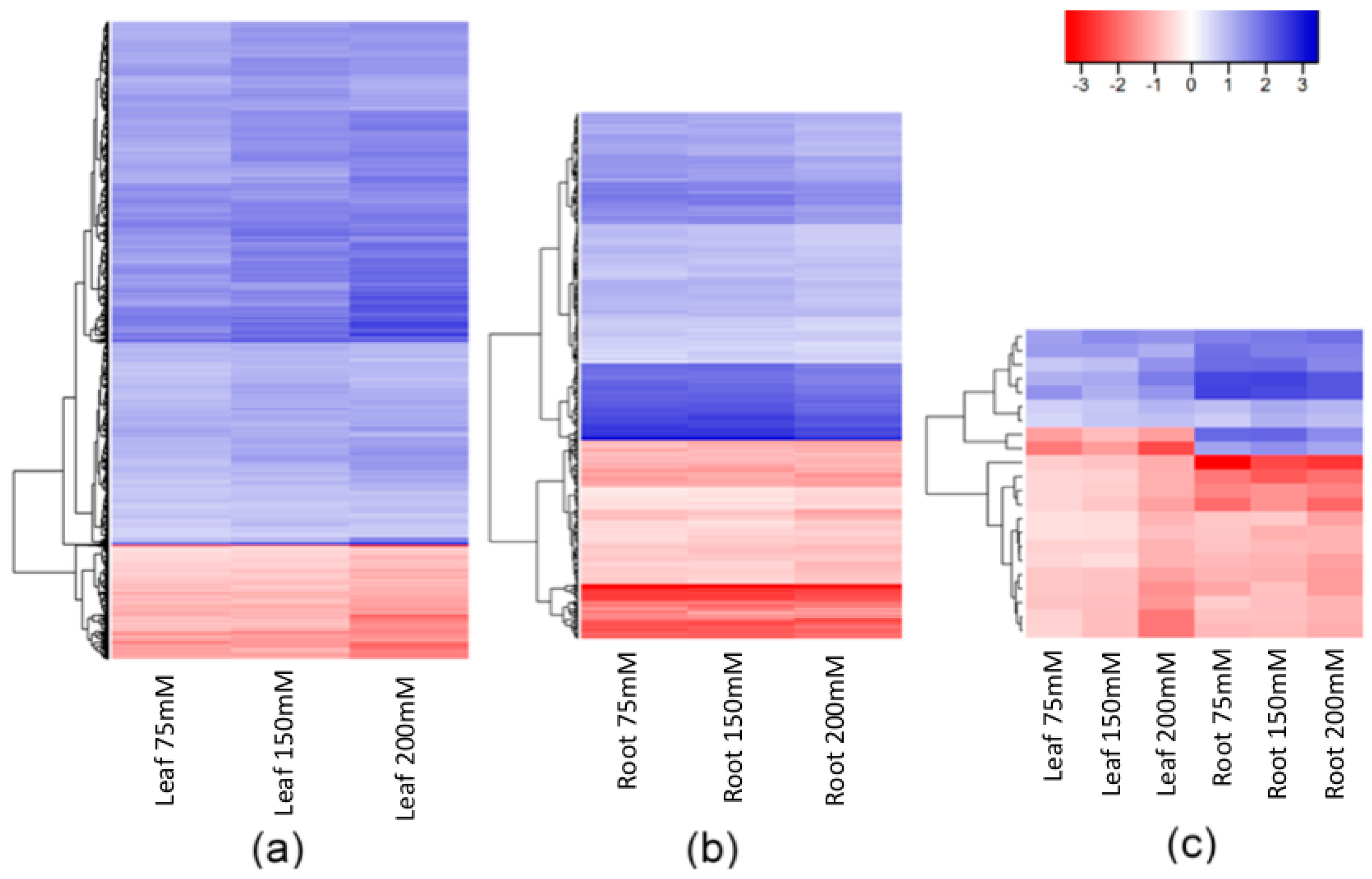
2.2. Salt-Induced DNA Methylation Changes Are Organ and Concentration Specific
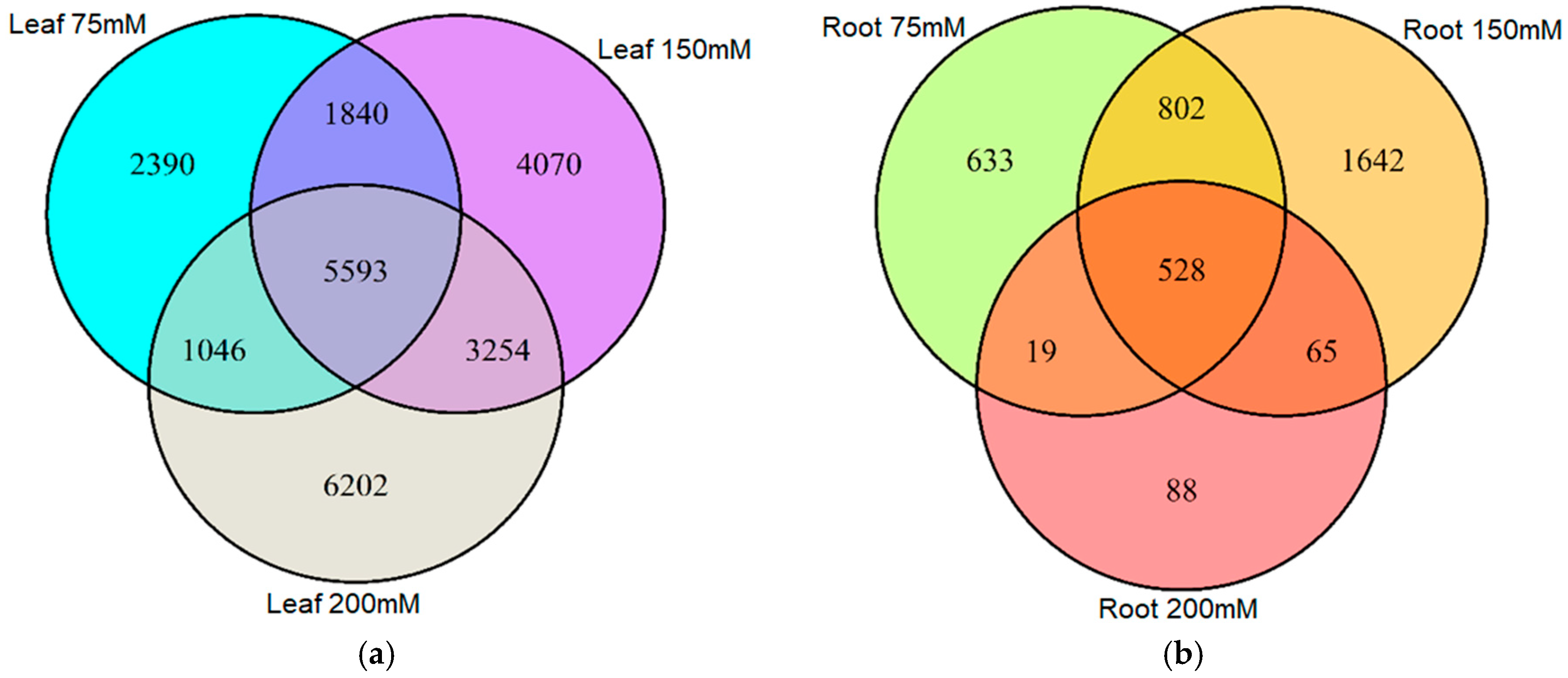
2.3. Stability of Salt-Induced DMMs across Treatments
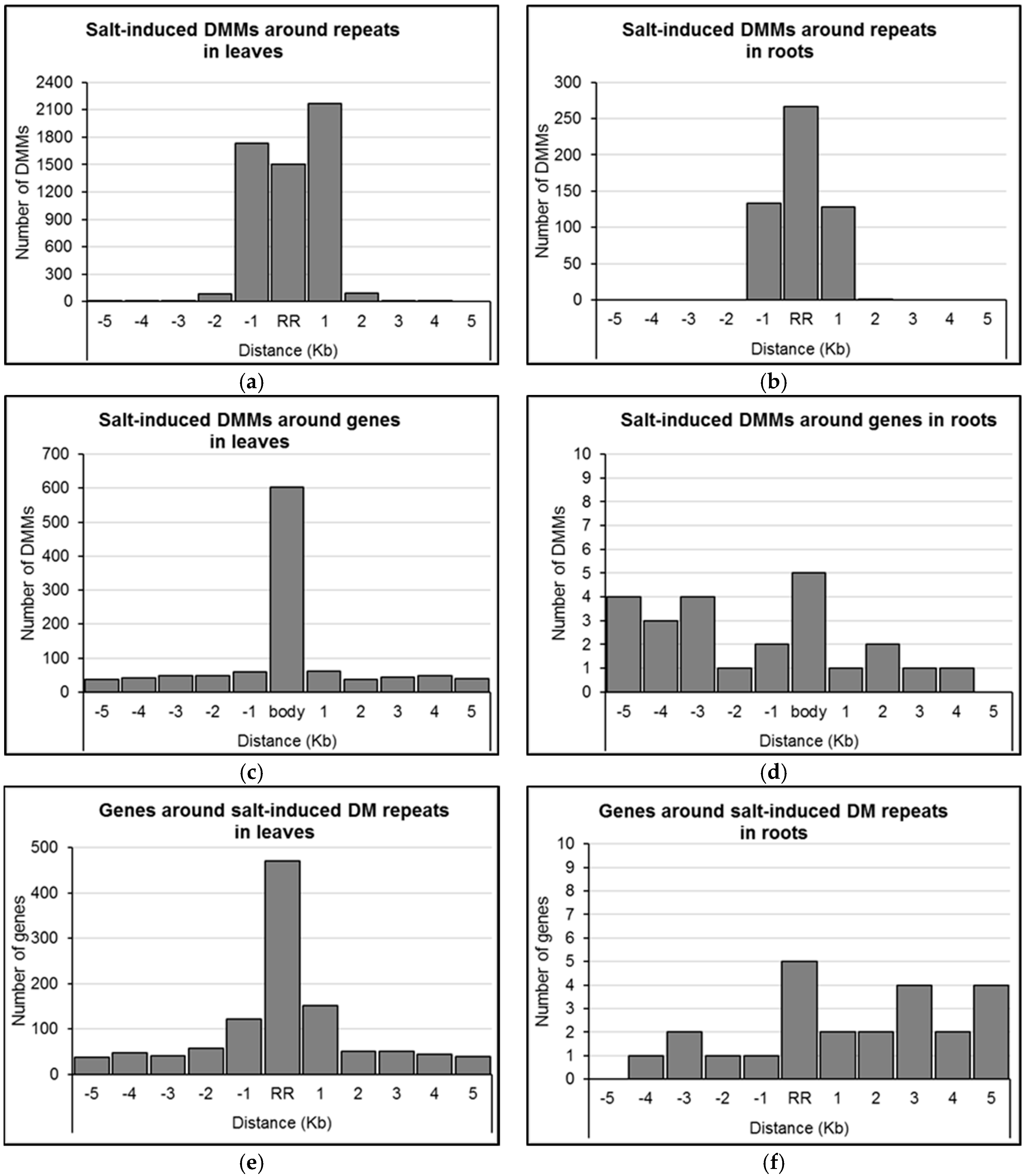
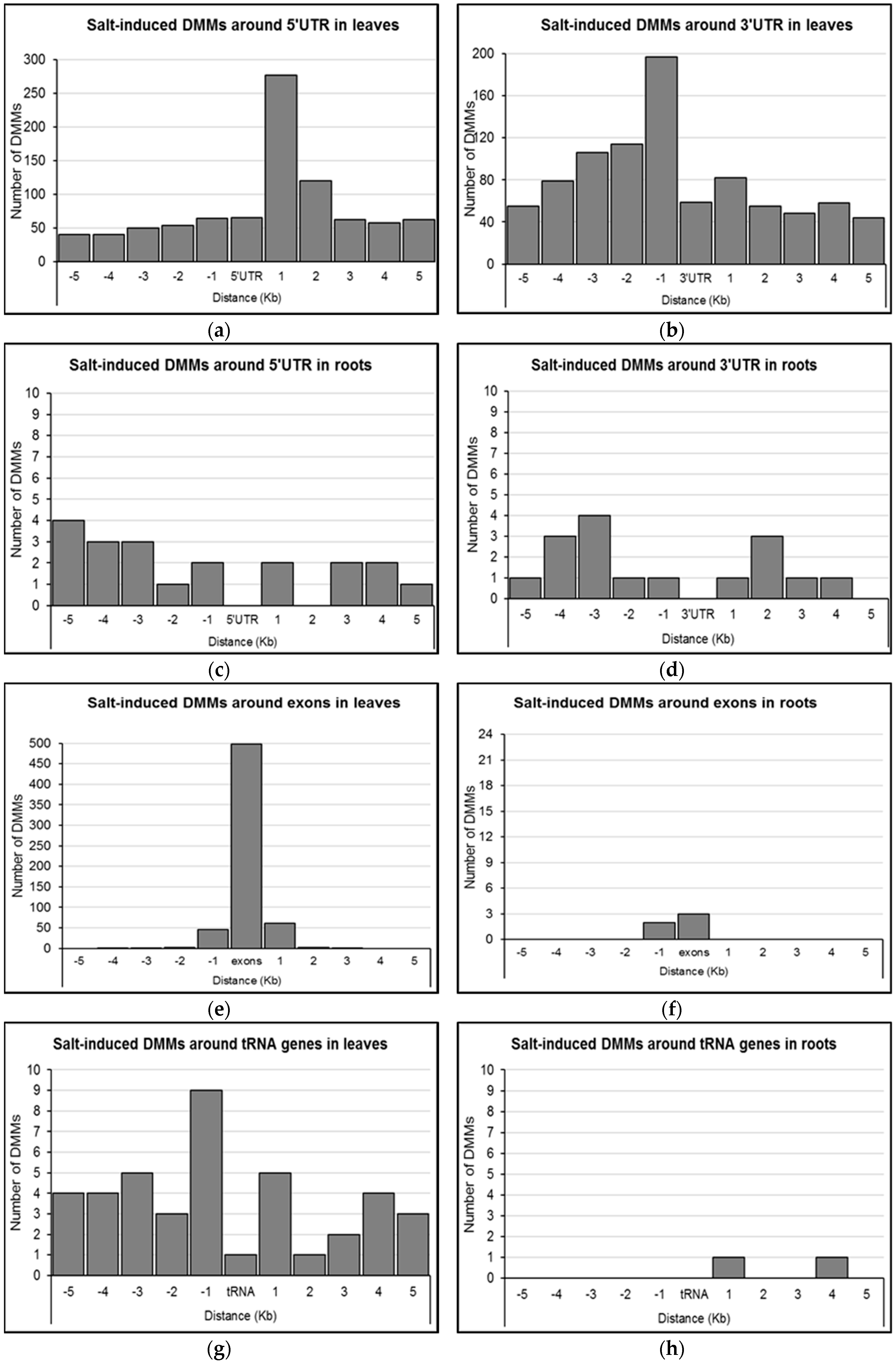
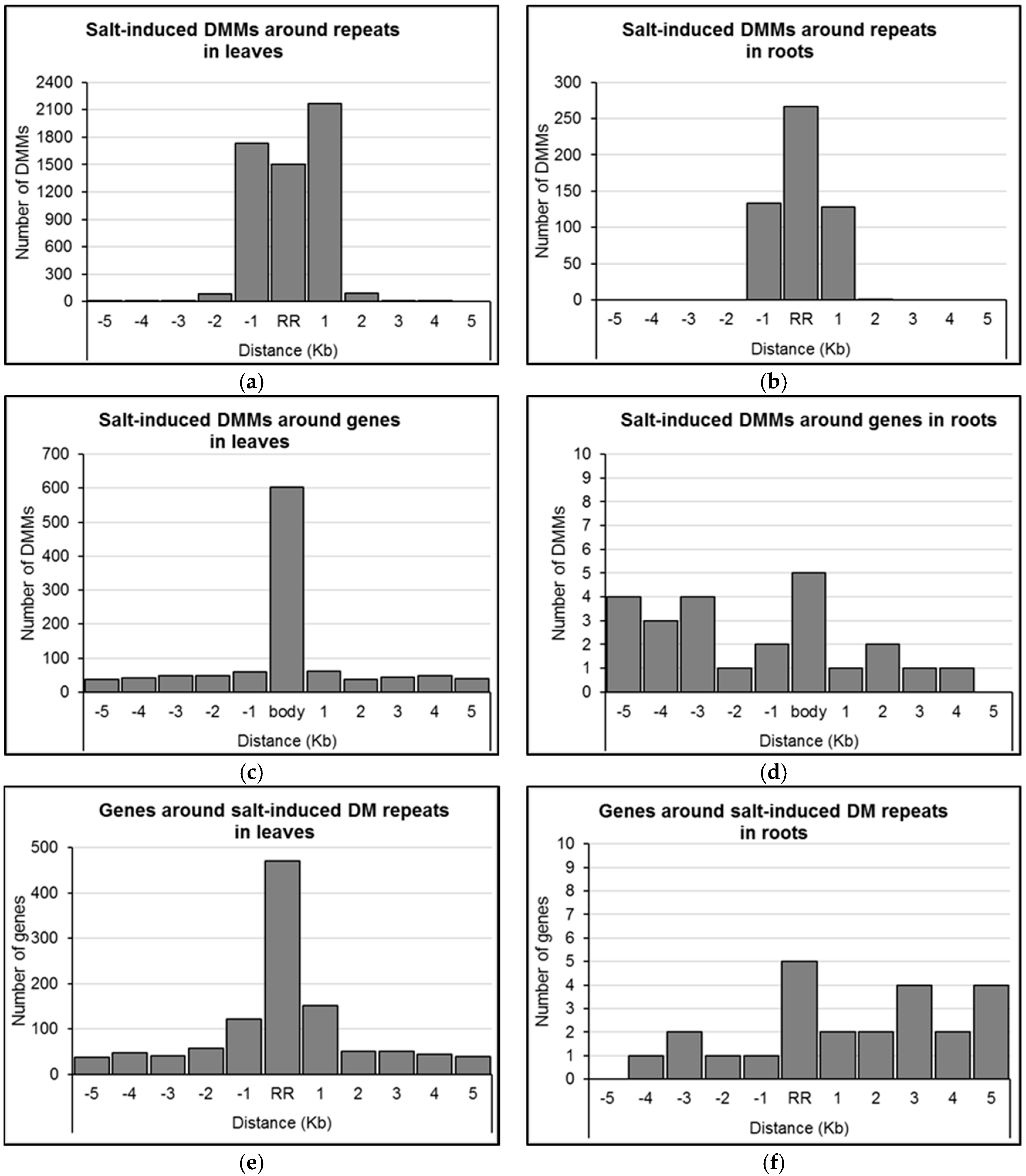
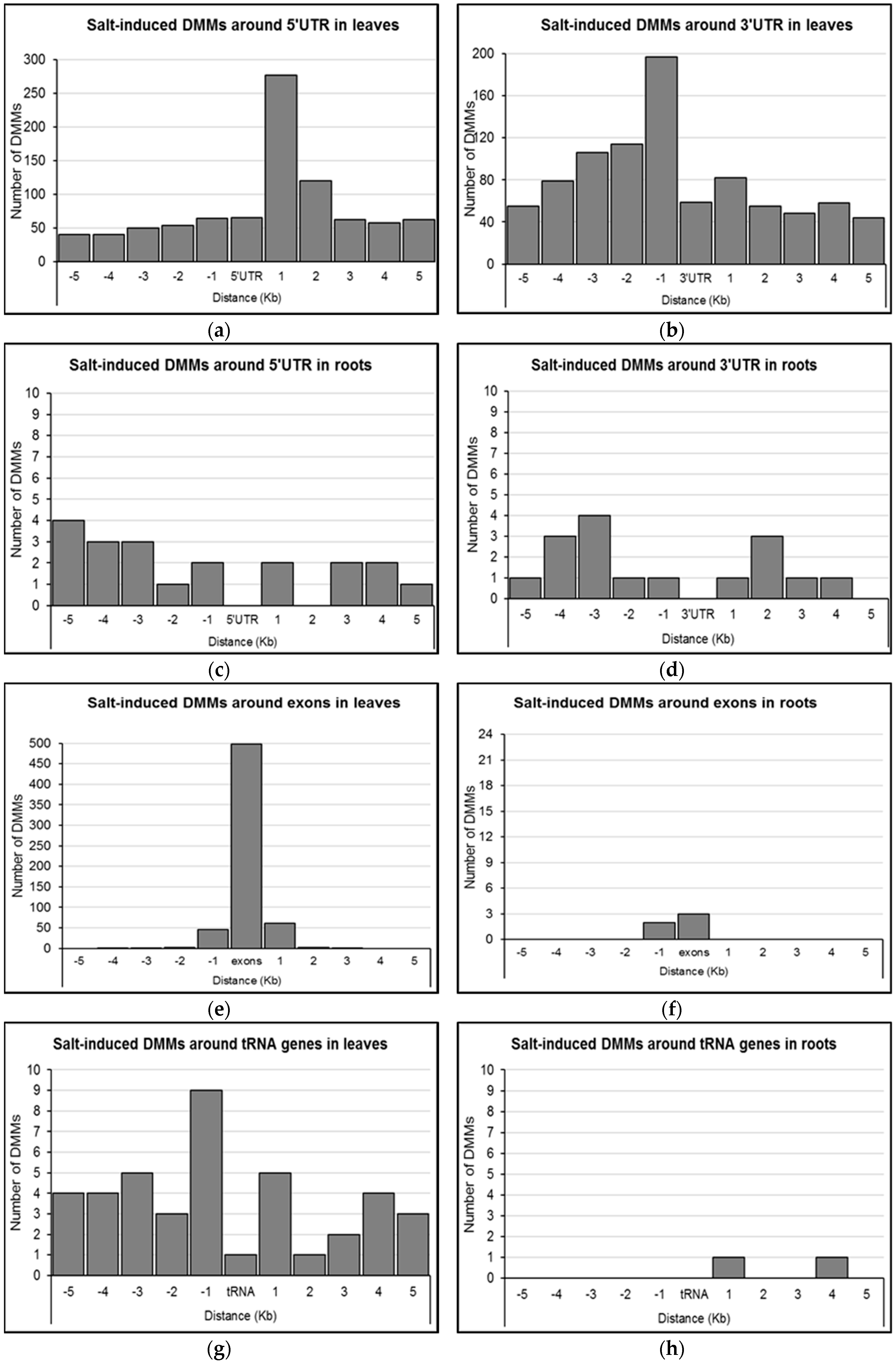
2.4. Distribution of Salt-Induced DMMs around Annotated Genomic Features
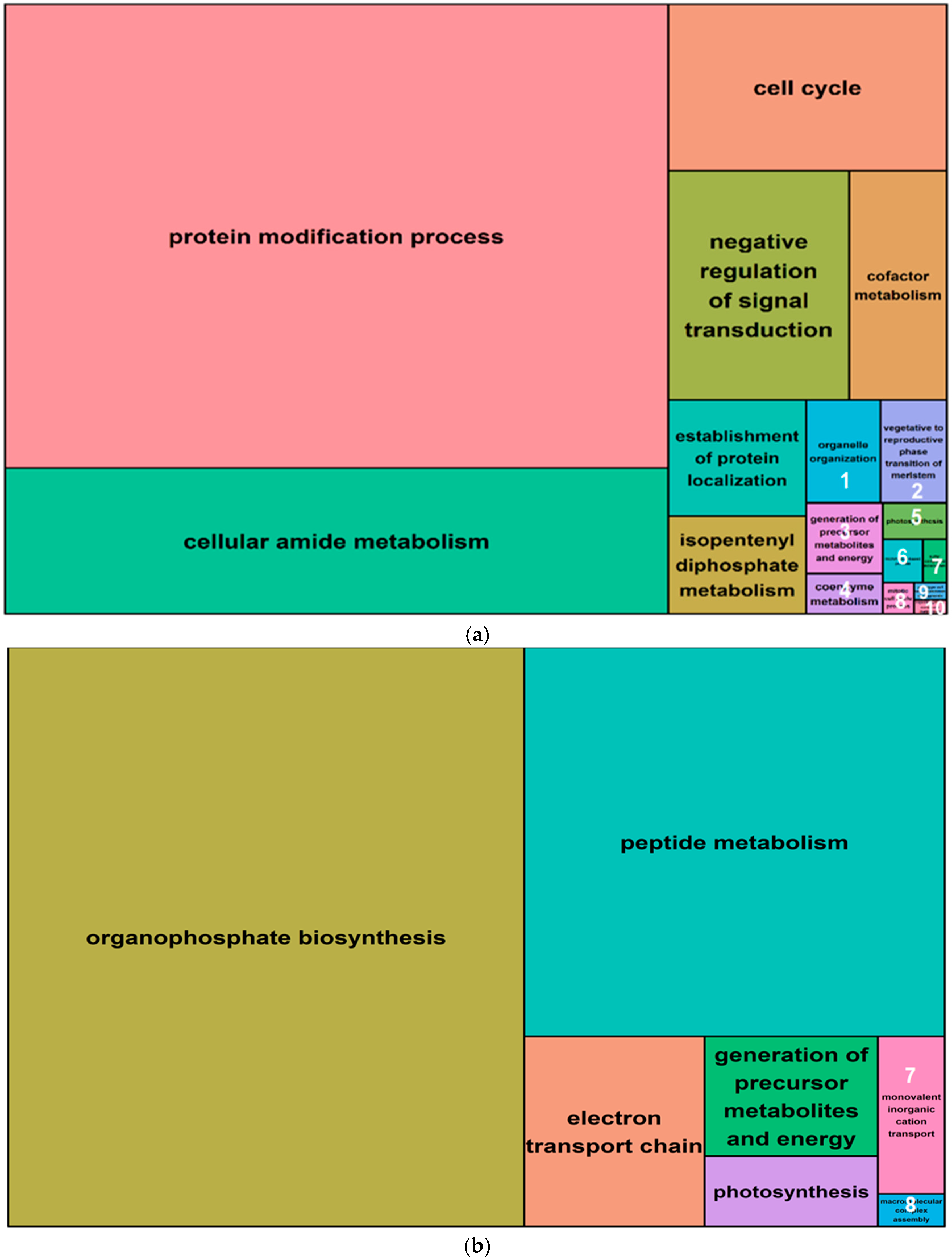
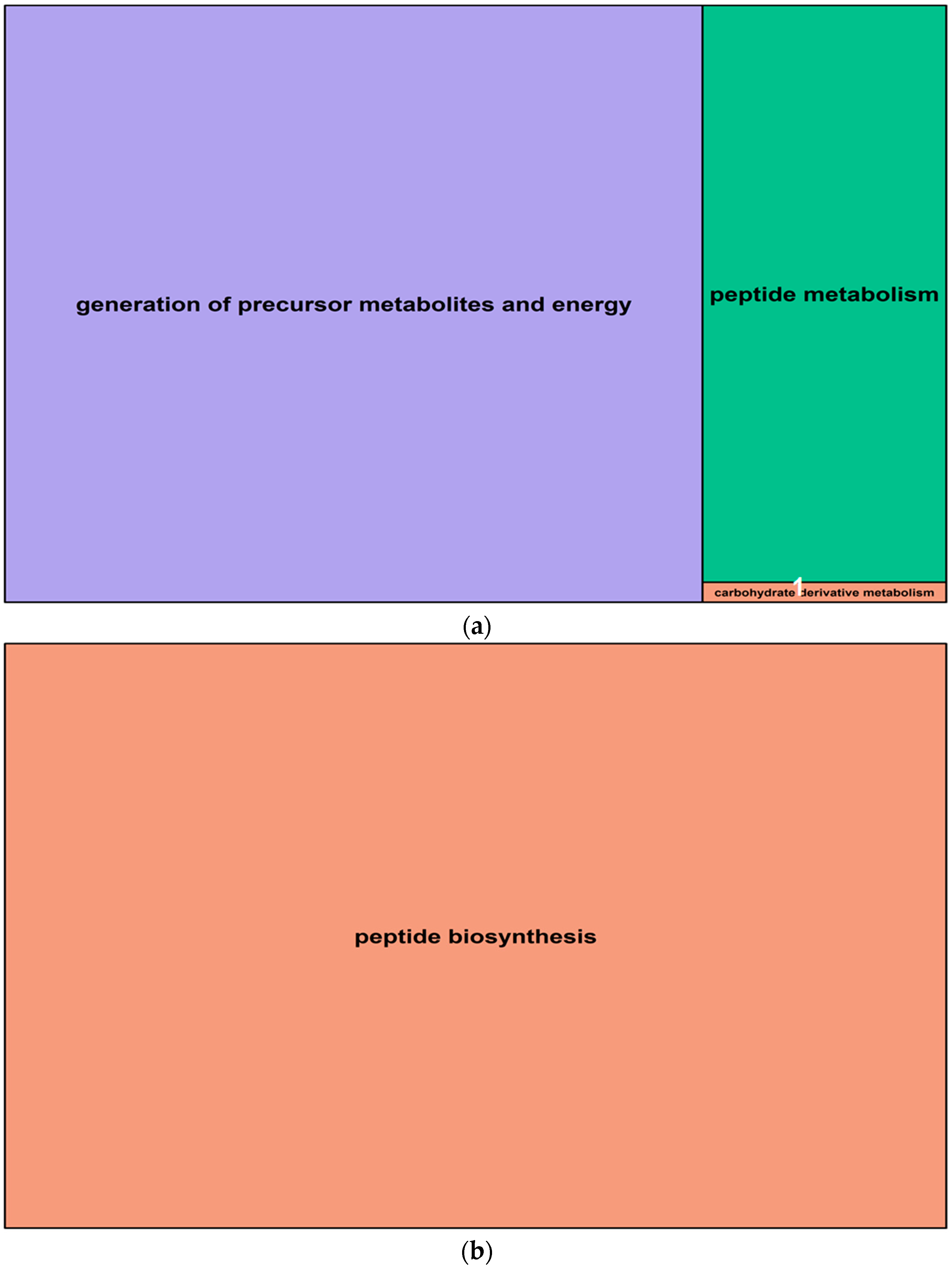
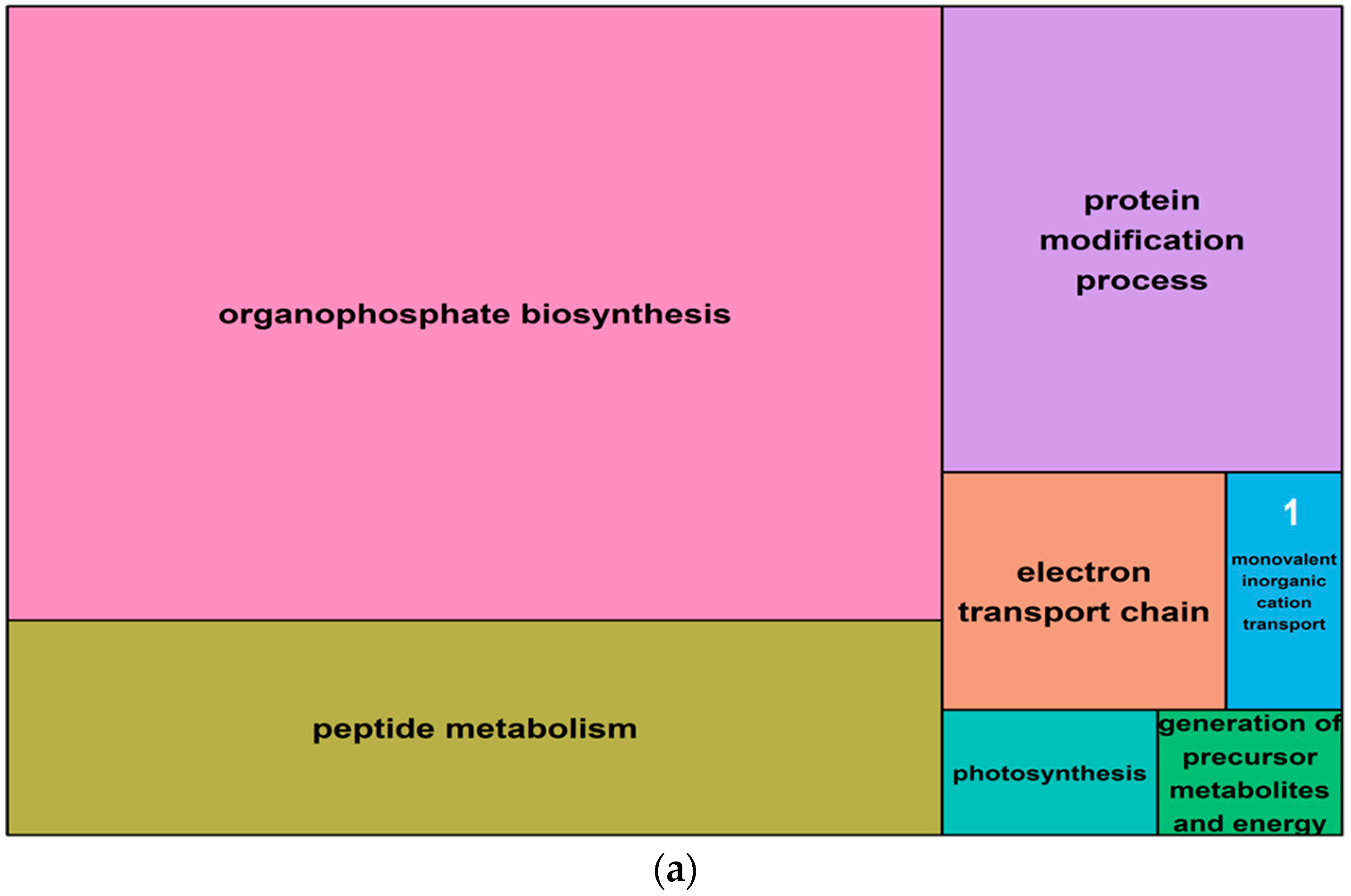
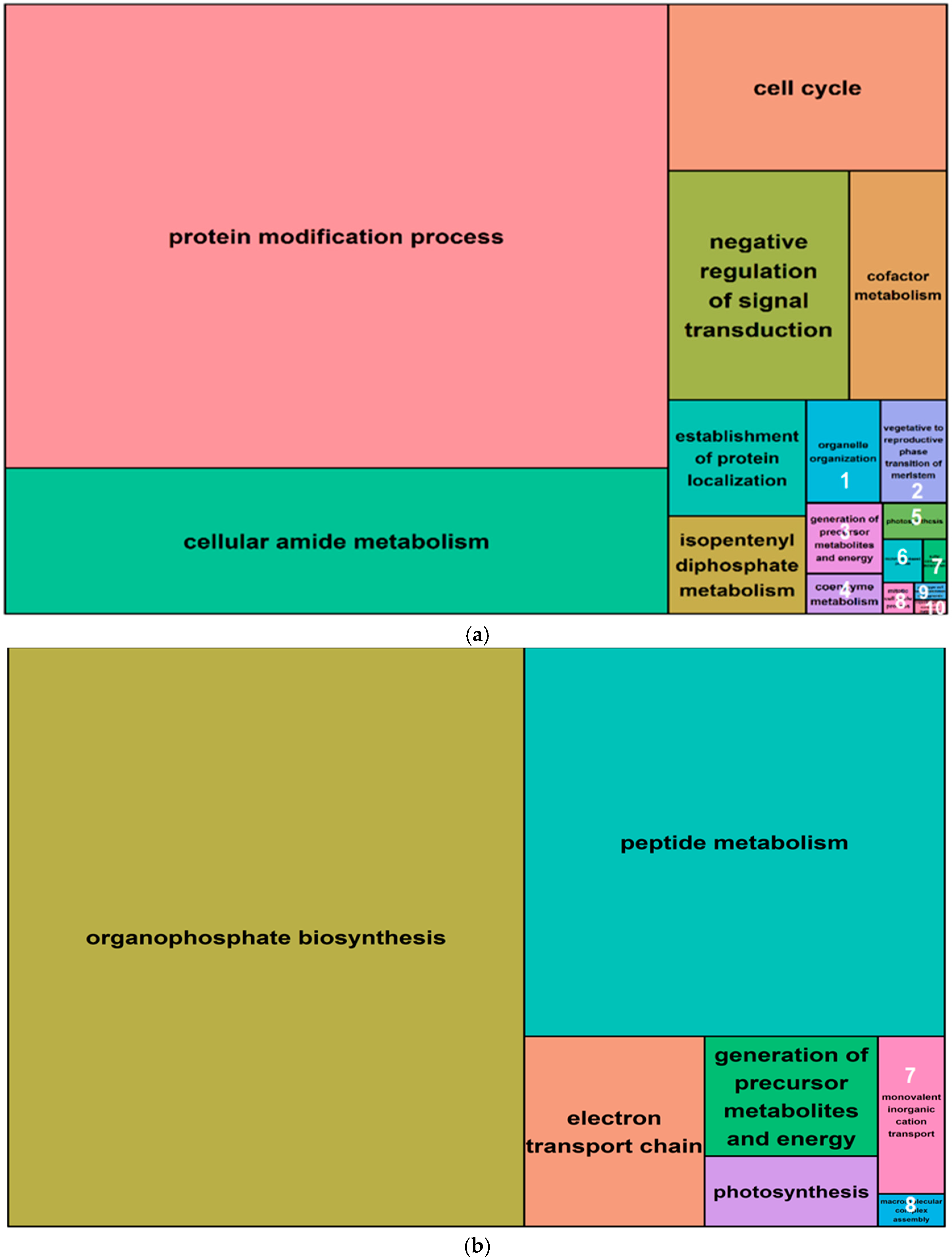
2.5. Gene Ontology Analysis of Salt-Induced DMMs
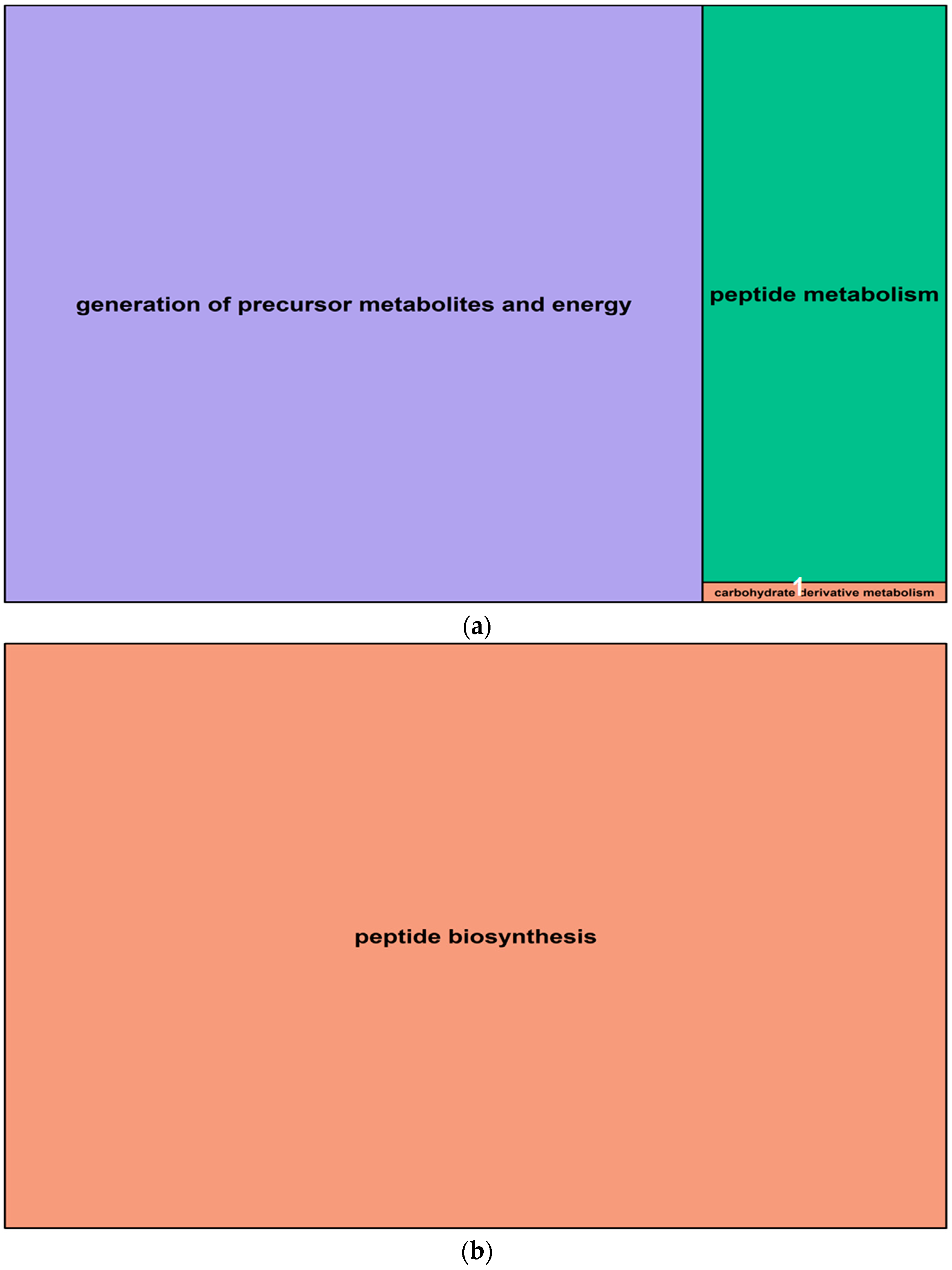
2.6. Differentially Expressed Genes in Barley Roots
3. Discussion
3.1. Consistency of Salt-Induced DMMs
3.2. Salt Induces Different Changes to the DNA Methylation of Leaves and Roots
3.3. Salt-Induced Changes in DNA Methylation May Influence Gene Regulation
3.4. Salt-Induced DMMs Correlate with Stress Related Genes
4. Materials and Methods
4.1. Plant Material and Stress Treatment
4.2. DNA Extraction
4.3. Methylation Sensitive Genotyping by Sequencing (ms-GBS)
4.4. Data Analysis
4.5. Salinity Induced Differentially Methylated Markers in Barley
4.6. Distribution of Salt-Induced DMMs around Genomic Features
4.7. Gene Ontology of Differentially Methylated Genes
4.8. Gene Expression and Ontology Analysis of Root Transcriptome
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- FAO. Barley, Malt, Beer. In Agribusiness; FAO: Rome, Italy, 2009. [Google Scholar]
- Zhou, M.X. Barley production and consumption. In Genetics and Improvement of Barley Malt Quality; Zhang, G., Li, C., Eds.; Springer: Berlin/Heidelberg, Germany, 2010; pp. 1–17. [Google Scholar]
- Hill, C.B.; Cassin, A.; Keeble-Gagnère, G.; Doblin, M.S.; Bacic, A.; Roessner, U. De novo transcriptome assembly and analysis of differentially expressed genes of two barley genotypes reveal root-zone-specific responses to salt exposure. Sci. Rep. 2016, 6, 31558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayer, K.F.X.; Nussbaumer, T.; Gundlach, H.; Martis, M.; Spannagl, M.; Pfeifer, M. A physical, genetic and functional sequence assembly of the barley genome. Nature 2012, 491, 711–716. [Google Scholar] [PubMed] [Green Version]
- Munns, R.; Tester, M. Mechanisms of salinity tolerance. Annu. Rev. Plant Biol. 2008, 59, 651–681. [Google Scholar] [CrossRef] [PubMed]
- Glenn, E.P.; Brown, J.J.; Blumwald, E. Salt tolerance and crop potential of halophytes. Crit. Rev. Plant Sci. 1999, 18, 227–255. [Google Scholar] [CrossRef]
- Wang, B.; Fu, R.; Zhang, M.; Ding, Z.; Chang, L.; Zhu, X.; Wang, Y.; Fan, B.; Ye, W.; Yuan, Y. Analysis of methylation-sensitive amplified polymorphism in different cotton accessions under salt stress based on capillary electrophoresis. Genes Genom. 2015, 37, 713–724. [Google Scholar] [CrossRef]
- Roy, S.J.; Negrão, S.; Tester, M. Salt resistant crop plants. Curr. Opin. Biotechnol. 2014, 26, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Ziemann, M.; Kamboj, A.; Hove, R.M.; Loveridge, S.; El-Osta, A.; Bhave, M. Analysis of the barley leaf transcriptome under salinity stress using mRNA-Seq. Acta Physiol. Plant. 2013, 35, 1915–1924. [Google Scholar] [CrossRef]
- Moose, S.P.; Mumm, R.H. Molecular plant breeding as the foundation for 21st century crop improvement. Plant Physiol. 2008, 147, 969–977. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez Lopez, C.M.; Wilkinson, M.J. Epi-fingerprinting and epi-interventions for improved crop production and food quality. Front. Plant Sci. 2015, 6, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Bender, J. DNA methylation and epigenetics. Annu. Rev. Plant Biol. 2004, 55, 41–68. [Google Scholar] [CrossRef] [PubMed]
- Bossdorf, O.; Arcuri, D.; Richards, C.; Pigliucci, M. Experimental alteration of DNA methylation affects the phenotypic plasticity of ecologically relevant traits in Arabidopsis thaliana. Evol. Ecol. 2010, 24, 541–553. [Google Scholar] [CrossRef]
- Boyko, A.; Kovalchuk, I. Epigenetic control of plant stress response. Environ. Mol. Mutagen. 2008, 49, 61–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Fu, X.W.; Peng, X.; Xiao, Z.; Li, Z.G.; Chen, G.J.; Wang, X.F. DNA methylation profiling reveals correlation of differential methylation patterns with gene expression in human epilepsy. J. Mol. Neurosci. 2016, 59, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Zilberman, D.; Gehring, M.; Tran, R.K.; Ballinger, T.; Henikoff, S. Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nat. Genet. 2007, 39, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Causevic, A.; Delaunay, A.; Ounnar, S.; Righezza, M.; Delmotte, F.; Brignolas, F.; Hagège, D.; Maury, S. DNA methylating and demethylating treatments modify phenotype and cell wall differentiation state in sugarbeet cell lines. Plant Physiol. Biochem. 2005, 43, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Wada, Y.; Miyamoto, K.; Kusano, T.; Sano, H. Association between up-regulation of stress-responsive genes and hypomethylation of genomic DNA in tobacco plants. Mol. Genet. Genom. 2004, 271, 658–666. [Google Scholar] [CrossRef] [PubMed]
- Aceituno, F.; Moseyko, N.; Rhee, S.; Gutiérrez, R. The rules of gene expression in plants: Organ identity and gene body methylation are key factors for regulation of gene expression in Arabidopsis thaliana. BMC Genom. 2008, 9, 438. [Google Scholar] [CrossRef] [PubMed]
- Bird, A.; Jaenisch, R. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat. Genet. 2003, 33, 245–254. [Google Scholar]
- Li, Q.; Song, J.; West, P.T.; Zynda, G.; Eichten, S.R.; Vaughn, M.W.; Springer, N.M. Examining the causes and consequences of context-specific differential DNA methylation in maize. Plant Physiol. 2015, 168, 1262–1274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.T.; Hong-Qi, W.U.; Hai-Bin, G.U.; Guang-Long, F.E.N.G.; Ze, W.A.N.G.; Sheng, J.D. Variability of soil salinity at multiple spatio-temporal scales and the related driving factors in the oasis areas of Xinjiang, China. Pedosphere 2014, 24, 753–762. [Google Scholar] [CrossRef]
- Guo, Y.; Huang, J.; Shi, Z.; Li, H. Mapping spatial variability of soil salinity in a coastal paddy field based on electromagnetic sensors. PLoS ONE 2015, 10, e0127996. [Google Scholar] [CrossRef] [PubMed]
- Karan, R.; DeLeon, T.; Biradar, H.; Subudhi, P.K. Salt stress induced variation in DNA methylation pattern and its influence on gene expression in contrasting rice genotypes. PLoS ONE 2012, 7, e40203. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhao, X.; Pan, Y.; Zhu, L.; Fu, B.; Li, Z. DNA methylation changes detected by methylation-sensitive amplified polymorphism in two contrasting rice genotypes under salt stress. J. Genet. Genom. 2011, 38, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Xu, Y.H.; Wang, J.B. DNA-methylation changes induced by salt stress in wheat Triticum aestivum. Afr. J. Biotechnol. 2009, 8, 6201–6207. [Google Scholar]
- Vogt, G. Stochastic developmental variation, an epigenetic source of phenotypic diversity with far-reaching biological consequences. J. Biosci. 2015, 40, 159–204. [Google Scholar] [CrossRef] [PubMed]
- Baek, D.; Jiang, J.; Chung, J.-S.; Wang, B.; Chen, J.; Xin, Z.; Shi, H. Regulated AtHKT1 gene expression by a distal enhancer element and DNA methylation in the promoter plays an important role in salt tolerance. Plant Cell Physiol. 2011, 52, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, T.; Seki, M. Epigenetic memory for stress response and adaptation in plants. Plant Cell Physiol. 2014, 55, 1859–1863. [Google Scholar] [CrossRef] [PubMed]
- Chinnusamy, V.; Zhu, J.-K. Epigenetic regulation of stress responses in plants. Curr. Opin. Plant Biol. 2009, 12, 133–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Qin, L.; Xie, C.; Li, W.; Yuan, J.; Kong, L.; Yu, W.; Xia, G.; Liu, S. Induced and constitutive DNA methylation in a salinity-tolerant wheat introgression line. Plant Cell Physiol. 2014, 55, 1354–1365. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Wu, X.; Chen, B.; Gao, G.; Xu, K. Evaluation of genetic and epigenetic modification in rapeseed (Brassica napus) induced by salt stress. J. Integr. Plant Biol. 2007, 49, 1599–1607. [Google Scholar]
- Tan, M.-P. Analysis of DNA methylation of maize in response to osmotic and salt stress based on methylation-sensitive amplified polymorphism. Plant Physiol. Biochem. 2010, 48, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Karim, K.; Amani Ben, N.; M’Barek Ben, N. Transcriptional changes in salt-responsive genes of barley subjected to salt stress. Int. J. Innov. Appl. Stud. 2014, 7, 85–94. [Google Scholar]
- Kumar, S. Epigenomics of Plant Responses to Environmental Stress. Epigenomes 2018, 2, 6. [Google Scholar] [CrossRef]
- Laird, P.W. Principles and challenges of genome-wide DNA methylation analysis. Nat. Rev. Genet. 2010, 11, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Tricker, P.J.; Gibbings, J.G.; Rodríguez López, C.M.; Hadley, P.; Wilkinson, M.J. Low relative humidity triggers RNA-directed de novo DNA methylation and suppression of genes controlling stomatal development. J. Exp. Bot. 2012, 63, 3799–3813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez López, C.M.; Morán, P.; Lago, F.; Espiñeira, M.; Beckmann, M.; Consuegra, S. Detection and quantification of tissue of origin in salmon and veal products using methylation sensitive AFLPs. Food Chem. 2012, 131, 1493–1498. [Google Scholar] [CrossRef]
- Rois, A.; Rodriguez Lopez, C.; Cortinhas, A.; Erben, M.; Espirito-Santo, D.; Wilkinson, M.; Caperta, A. Epigenetic rather than genetic factors may explain phenotypic divergence between coastal populations of diploid and tetraploid Limonium spp. (Plumbaginaceae) in Portugal. BMC Plant Biol. 2013, 13, 205. [Google Scholar] [CrossRef] [PubMed]
- Paun, O.; Schönswetter, P. Amplified fragment length polymorphism: An invaluable fingerprinting technique for genomic, transcriptomic, and epigenetic studies. In Plant DNA Fingerprinting and Barcoding; Sucher, N.J., Hennell, J.R., Carles, M.C., Eds.; Humana Press: New York, NY, USA, 2012; Volume 862, pp. 75–87. [Google Scholar]
- Fulnecek, J.; Kovarik, A. How to interpret methylation sensitive amplified polymorphism (MSAP) profiles? BMC Genet. 2014, 15, 2. [Google Scholar] [CrossRef] [PubMed]
- Walder, R.Y.; Langtimm, C.J.; Chatterjee, R.; Walder, J.A. Cloning of the MspI modification enzyme. The site of modification and its effects on cleavage by MspI and HpaII. J. Biol. Chem. 1983, 258, 1235–1241. [Google Scholar] [PubMed]
- Xia, Z.; Zou, M.; Zhang, S.; Feng, B.; Wang, W. AFSM sequencing approach: A simple and rapid method for genome-wide SNP and methylation site discovery and genetic mapping. Sci. Rep. 2014, 4, 7300. [Google Scholar] [CrossRef] [PubMed]
- Kitimu, S.R.; Taylor, J.; March, T.J.; Tairo, F.; Wilkinson, M.J.; Rodriguez Lopez, C.M. Meristem micropropagation of cassava (Manihot esculenta) evokes genome-wide changes in DNA methylation. Front. Plant Sci. 2015, 6, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.-S.; Sano, H. Abiotic-stress induces demethylation and transcriptional activation of a gene encoding a glycerophosphodiesterase-like protein in tobacco plants. Mol. Genet. Genom. 2007, 277, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Walia, H.; Wilson, C.; Zeng, L.; Ismail, A.M.; Condamine, P.; Close, T.J. Genome-wide transcriptional analysis of salinity stressed japonica and indica rice genotypes during panicle initiation stage. Plant Mol. Biol. 2007, 63, 609–623. [Google Scholar] [CrossRef] [PubMed]
- Tilbrook, J.; Schilling, R.K.; Berger, B.; Garcia, A.F.; Trittermann, C.; Coventry, S.; Rabie, H.; Brien, C.; Nguyen, M.; Tester, M.; Roy, S.J. Variation in shoot tolerance mechanisms not related to ion toxicity in barley. Funct. Plant Biol. 2017, 44, 1194–1206. [Google Scholar] [CrossRef] [Green Version]
- Soen, Y.; Knafo, M.; Elgart, M. A principle of organization which facilitates broad Lamarckian-like adaptations by improvisation. Biol. Direct 2015, 10, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Shelden, M.C.; Roessner, U.; Sharp, R.E.; Tester, M.; Bacic, A. Genetic variation in the root growth response of barley genotypes to salinity stress. Funct. Plant Biol. 2013, 40, 516–530. [Google Scholar] [CrossRef]
- Genc, Y.; McDonald, G.K.; Tester, M. Reassessment of tissue Na+ concentration as a criterion for salinity tolerance in bread wheat. Plant Cell Environ. 2007, 30, 1486–1498. [Google Scholar] [CrossRef] [PubMed]
- Baraket, M.; Sahli, A.; Amara, H.; Nasr, H.; Selmi, H.; Trifa, Y. Introduction and evaluation of salt-tolerant barley genotypes. Ann. Biol. Res. 2014, 5, 64–69. [Google Scholar]
- Zhu, M.; Zhou, M.; Shabala, L.; Shabala, S. Linking osmotic adjustment and stomatal characteristics with salinity stress tolerance in contrasting barley accessions. Funct. Plant Biol. 2015, 42, 252–263. [Google Scholar] [CrossRef]
- Boyko, A.; Golubov, A.; Bilichak, A.; Kovalchuk, I. Chlorine ions but not sodium ions alter genome stability of Arabidopsis thaliana. Plant Cell Physiol. 2010, 51, 1066–1078. [Google Scholar] [CrossRef] [PubMed]
- Genc, Y.; Oldach, K.; Taylor, J.; Lyons, G.H. Uncoupling of sodium and chloride to assist breeding for salinity tolerance in crops. New Phytol. 2016, 210, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Ding, G.H.; Fang, K.; Zhao, F.G.; Qin, P. New perspectives on the mechanism of alleviating salt stress by spermidine in barley seedlings. Plant Growth Regul. 2006, 49, 147–156. [Google Scholar] [CrossRef]
- Gao, X.; Cao, D.; Liu, J.; Wang, X.; Geng, S.; Liu, B.; Shi, D. Tissue-specific and cation/anion-specific DNA methylation variations occurred in C. virgata in response to salinity stress. PLoS ONE 2013, 8, e78426. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.J.; Azevedo, V.; Maroco, J.; Margarida Oliveira, M.; Santos, A.P. Salt tolerant and sensitive rice varieties display differential methylome flexibility under salt stress. PLoS ONE 2015, 10, e0124060. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Huang, F.; Qin, Q.; Zhao, X.; Li, Z.; Fu, B. Comparative analysis of DNA methylation changes in two rice genotypes under salt stress and subsequent recovery. Biochem. Biophys. Res. Commun. 2015, 465, 790–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demirkiran, A.; Marakli, S.; Temel, A.; Gozukirmizi, N. Genetic and epigenetic effects of salinity on in vitro growth of barley. Genet. Mol. Biol. 2013, 36, 566–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munns, R.; Gilliham, M. Salinity tolerance of crops—What is the cost? New Phytol. 2015, 208, 668–673. [Google Scholar] [CrossRef] [PubMed]
- Tavakkoli, E.; Fatehi, F.; Coventry, S.; Rengasamy, P.; McDonald, G.K. Additive effects of Na(+) and Cl(−) ions on barley growth under salinity stress. J. Exp. Bot. 2011, 62, 2189–2203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Feng, L.; Li, J.; He, Z. Genetic and epigenetic control of plant heat responses. Front. Plant Sci. 2015, 6, 267. [Google Scholar] [CrossRef] [PubMed]
- Amoah, S.; Kurup, S.; Rodriguez Lopez, C.; Welham, S.; Powers, S.; Hopkins, C.; Wilkinson, M.; King, G. A hypomethylated population of Brassica rapa for forward and reverse Epi-genetics. BMC Plant Biol. 2012, 12, 193. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Maury, L.; Marguerat, S.; Bahler, J. Tuning gene expression to changing environments: From rapid responses to evolutionary adaptation. Nat. Rev. Genet. 2008, 9, 583–593. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Huang, W.; Xiong, J.; Yan, S.; Tao, T.; Li, J.; Wu, J.; Luo, L. Differentially methylated epiloci generated from numerous genotypes of contrasting tolerances are associated with osmotic-tolerance in rice seedlings. Front. Plant Sci. 2017, 8, 11. [Google Scholar] [CrossRef] [PubMed]
- Greaves, I.K.; Groszmann, M.; Ying, H.; Taylor, J.M.; Peacock, W.J.; Dennis, E.S. Trans chromosomal methylation in Arabidopsis hybrids. Proc. Natl. Acad. Sci. USA 2012, 109, 3570–3575. [Google Scholar] [CrossRef] [PubMed]
- Bewick, A.J.; Ji, L.; Niederhuth, C.E.; Willing, E.-M.; Hofmeister, B.T.; Shi, X.; Wang, L.; Lu, Z.; Rohr, N.A.; Hartwig, B.; et al. On the origin and evolutionary consequences of gene body DNA methylation. Proc. Natl. Acad. Sci. USA 2016, 113, 9111–9116. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yazaki, J.; Sundaresan, A.; Cokus, S.; Chan, S.W.L.; Chen, H.; Henderson, I.R.; Shinn, P.; Pellegrini, M.; Jacobsen, S.E.; et al. Genome-wide high-resolution mapping and functional analysis of DNA methylation in Arabidopsis. Cell 2006, 126, 1189–1201. [Google Scholar] [CrossRef] [PubMed]
- Tomek, W.; Wollenhaupt, K. The “closed loop model” in controlling mRNA translation during development. Anim. Reprod. Sci. 2012, 134, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Archer, S.K.; Shirokikh, N.E.; Hallwirth, C.V.; Beilharz, T.H.; Preiss, T. Probing the closed-loop model of mRNA translation in living cells. RNA Biol. 2015, 12, 248–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bicknell, A.A.; Cenik, C.; Chua, H.N.; Roth, F.P.; Moore, M.J. Introns in UTRs: Why we should stop ignoring them. Bioessays 2012, 34, 1025–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Illingworth, R.; Kerr, A.; DeSousa, D.; Jørgensen, H.; Ellis, P.; Stalker, J.; Jackson, D.; Clee, C.; Plumb, R.; Rogers, J.; et al. A novel CpG island set identifies tissue-specific methylation at developmental gene loci. PLoS Biol. 2008, 6, e22. [Google Scholar] [CrossRef] [PubMed]
- Deaton, A.M.; Bird, A. CpG islands and the regulation of transcription. Genes Dev. 2011, 25, 1010–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cokus, S.J.; Feng, S.; Zhang, X.; Chen, Z.; Merriman, B.; Haudenschild, C.D.; Pradhan, S.; Nelson, S.F.; Pellegrini, M.; Jacobsen, S.E. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 2008, 452, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Konate, M.; Sai, N.; Tesfamicael, K.G.; Cavagnaro, T.; Gilliham, M.; Breen, J.; Metcalfe, A.; Stephen, J.; DeBei, R.; et al. Global DNA Methylation Patterns Can Play a Role in Defining Terroir in Grapevine (Vitis vinifera cv. Shiraz). Front. Plant Sci. 2017, 8, 1860. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Xu, C.; von Wettstein, D.; Liu, B. Tissue-specific differences in cytosine methylation and their association with differential gene expression in sorghum. Plant Physiol. 2011, 156, 1955–1966. [Google Scholar] [CrossRef] [PubMed]
- Tuorto, F.; Liebers, R.; Musch, T.; Schaefer, M.; Hofmann, S.; Kellner, S.; Frye, M.; Helm, M.; Stoecklin, G.; Lyko, F. RNA cytosine methylation by Dnmt2 and NSun2 promotes tRNA stability and protein synthesis. Nat. Struct. Mol. Biol. 2012, 19, 900–905. [Google Scholar] [CrossRef] [PubMed]
- Besser, D.; Götz, F.; Schulze-Forster, K.; Wagner, H.; Kröger, H.; Simon, D. DNA methylation inhibits transcription by RNA polymerase III of a tRNA gene, but not of a 5S rRNA gene. FEBS Lett. 1990, 269, 358–362. [Google Scholar] [CrossRef] [Green Version]
- Hori, H. Methylated nucleosides in tRNA and tRNA methyltransferases. Front. Genet. 2014, 5, 144. [Google Scholar] [CrossRef] [PubMed]
- Munns, R.; James, R.A.; Xu, B.; Athman, A.; Conn, S.J.; Jordans, C.; Byrt, C.S.; Hare, R.A.; Tyerman, S.D.; Tester, M.; et al. Wheat grain yield on saline soils is improved by an ancestral Na+ transporter gene. Nat. Biotechnol. 2012, 30, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Mian, A.; Oomen, R.J.F.J.; Isayenkov, S.; Sentenac, H.; Maathuis, F.J.M.; Véry, A.-A. Over-expression of an Na+- and K+-permeable HKT transporter in barley improves salt tolerance. Plant J. 2011, 68, 468–479. [Google Scholar] [CrossRef] [PubMed]
- Byrt, C.S.; Xu, B.; Krishnan, M.; Lightfoot, D.J.; Athman, A.; Jacobs, A.K.; Watson-Haigh, N.S.; Plett, D.; Munns, R.; Tester, M.; et al. The Na+ transporter, TaHKT1; 5-D, limits shoot Na+ accumulation in bread wheat. Plant J. 2014, 80, 516–526. [Google Scholar] [CrossRef] [PubMed]
- Drøbak, B.K.; Watkins, P.A.C. Inositol(1,4,5)trisphosphate production in plant cells: An early response to salinity and hyperosmotic stress. Fed. Eur. Biochem. Soc. Lett. 2000, 481, 240–244. [Google Scholar] [CrossRef]
- Raychaudhuri, A.; Majumder, A.L. Salinity-induced enhancement of L-myo-inositol 1-phosphate synthase in rice (Oryza sativa L.). Plant Cell Environ. 1996, 19, 1437–1442. [Google Scholar] [CrossRef]
- Grattan, S.R.; Maas, E.V. Effect of salinity on phosphate accumulation and injury in soybean. Plant Soil 1988, 109, 65–71. [Google Scholar] [CrossRef]
- Aslam, M.; Flowers, T.J.; Qureshi, R.H.; Yeo, A.R. Interaction of Phosphate and Salinity on the Growth and Yield of Rice (Oryza sativa L.). J. Agron. Crop Sci. 1996, 176, 249–258. [Google Scholar] [CrossRef]
- Kawakatsu, T.; Stuart, T.; Valdes, M.; Breakfield, N.; Schmitz, R.J.; Nery, J.R.; Urich, M.A.; Han, X.; Lister, R.; Benfey, P.N.; et al. Unique cell-type specific patterns of DNA methylation in the root meristem. Nat. Plants 2016, 2, 16058. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, J.; Majumder, A.L. Salt-induced abnormalities on root tip mitotic cells of Allium cepa: Prevention by inositol pretreatment. Protoplasma 2010, 245, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Katsuhara, M.; Kawasaki, T. Salt stress induced nuclear and DNA degradation in meristematic cells of barley roots. Plant Cell Physiol. 1996, 37, 169–173. [Google Scholar] [CrossRef]
- Berger, B.; Regt, B.; Tester, M. Trait dissection of salinity tolerance with plant phenomics. In Plant Salt Tolerance; Shabala, S., Cuin, T.A., Eds.; Humana Press: New York, NY, USA, 2012; Volume 913, pp. 399–413. [Google Scholar]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple Genotyping-by-Sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [PubMed]
- Poland, J.; Endelman, J.; Dawson, J.; Rutkoski, J.; Wu, S.; Manes, Y.; Dreisigacker, S.; Crossa, J.; Sánchez-Villeda, H.; Sorrells, M.; et al. Genomic selection in wheat breeding using Genotyping-by-Sequencing. Plant Genome 2012, 5, 103–113. [Google Scholar] [CrossRef]
- Mayne, B.T.; Leemaqz, S.Y.; Buckberry, S.; Rodriguez Lopez, C.M.; Roberts, C.T.; Bianco-Miotto, T.; Breen, J. msgbsR: An R package for analysing methylation-sensitive restriction enzyme sequencing data. Sci. Rep. 2018, 8, 2190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eichten, S.R.; Vaughn, M.W.; Hermanson, P.J.; Springer, N.M. Variation in DNA methylation patterns is more common among maize inbreds than among tissues. Plant Genome 2013, 6, 1–10. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Carlson, M. GO.db: A Set of Annotation Maps Describing the Entire Gene Ontology. R Package Version 3.4.0. Available online: http://bioconductor.org/packages/release/data/annotation/html/GO.db.html (accessed on 4 March 2017).
- Gentleman, R. Annotate: Annotation for Microarrays. R Package Version 1.52.0. Available online: https://www.bioconductor.org/packages/release/bioc/html/annotate.html (accessed on 4 March 2017).
- Dunn, O.J. Multiple comparisons among means. J. Am. Stat. Assoc. 1961, 56, 52–64. [Google Scholar] [CrossRef]
- Supek, F.; Bošnjak, M.; Škunca, N.; Šmuc, T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef] [PubMed]
- Schubert, M.; Lindgreen, S.; Orlando, L. AdapterRemoval v2: Rapid adapter trimming, identification, and read merging. BMC Res. Notes 2016, 9, 88. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Meth. 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. FeatureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Law, C.W.; Alhamdoosh, M.; Su, S.; Smyth, G.K.; Ritchie, M.E. RNA-seq analysis is easy as 1-2-3 with limma, Glimma and edgeR. F1000Research 2016, 5, 1408. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sequencing Results | Yield |
|---|---|
| Raw reads | 1,015,703,602 |
| Reads that matched barcodes | 1,004,318,258 |
| Reads aligned to barley reference genome | 496,960,365 |
| Samples | 200 |
| Average reads per sample | 2,484,801 |
| Total unique tags | 892,859 |
| Polymorphic tags | 645,297 |
| Methylation Change | DMGs | GO Terms per GO Group | Total GO Terms | ||
|---|---|---|---|---|---|
| Biological Process | Cellular Component | Molecular Function | |||
| Leaf hypo | 1017 | 315 | 40 | 73 | 433 |
| Leaf hyper | 53 | 64 | 21 | 14 | 99 |
| Root hypo | 15 | 19 | 10 | 0 | 29 |
| Root hyper | 9 | 13 | 11 | 0 | 24 |
| Expression Change | DE Genes | GO Terms per GO Group | Total GO Terms | |||
|---|---|---|---|---|---|---|
| Total Transcripts | Annotated | Biological Process | Cellular Component | Molecular Function | ||
| Upregulated | 124 | 76 | 94 | 22 | 29 | 145 |
| Downregulated | 34 | 18 | 23 | 12 | 0 | 53 |
| DE Genes | DMMs | Statistics | Annotation | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Gene ID | Range | Chrom | Position | Methylation | logFC | p-Value | FDR | dist2Gene | |
| MSTRG.4246 | 1:435681474–435731845 | 1H | 435,689,351 | hyper | −1.76 | 0.000 | 0.053 | 0 | - |
| MSTRG.31525 | 5:507135444–507397451 | 5H | 507,332,872 | hypo | −1.47 | 0.002 | 0.083 | 0 | MLOC_2917 |
| MSTRG.43260 | 7:427906474–427974581 | 7H | 427,925,930 | hyper | −1.05 | 0.006 | 0.093 | 0 | MLOC_73155 |
| MSTRG.43261 | 7:427906474–427974581 | 7H | 427,948,871 | hypo | −1.05 | 0.006 | 0.093 | 0 | MLOC_73155 |
| MSTRG.10572 | 2:543673444–543674117 | 2H | 543,678,039 | hypo | −1.05 | 0.006 | 0.095 | 3922 | - |
| MSTRG.6485 | 2:17425326–17624569 | 2H | 17,517,122 | hypo | 1.39 | 0.007 | 0.095 | 0 | - |
| MSTRG.6418 | 2:15418194–15419914 | 2H | 15,414,469 | hypo | 1.53 | 0.004 | 0.089 | 3725 | MLOC_24124 |
| MSTRG.10644 | 2:545135370–545135958 | 2H | 545,131,372 | hyper | 3.43 | 0.003 | 0.086 | 3998 | MLOC_48766 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Konate, M.; Wilkinson, M.J.; Mayne, B.T.; Pederson, S.M.; Scott, E.S.; Berger, B.; Rodriguez Lopez, C.M. Salt Stress Induces Non-CG Methylation in Coding Regions of Barley Seedlings (Hordeum vulgare). Epigenomes 2018, 2, 12. https://doi.org/10.3390/epigenomes2020012
Konate M, Wilkinson MJ, Mayne BT, Pederson SM, Scott ES, Berger B, Rodriguez Lopez CM. Salt Stress Induces Non-CG Methylation in Coding Regions of Barley Seedlings (Hordeum vulgare). Epigenomes. 2018; 2(2):12. https://doi.org/10.3390/epigenomes2020012
Chicago/Turabian StyleKonate, Moumouni, Michael J. Wilkinson, Benjamin T. Mayne, Stephen M. Pederson, Eileen S. Scott, Bettina Berger, and Carlos M. Rodriguez Lopez. 2018. "Salt Stress Induces Non-CG Methylation in Coding Regions of Barley Seedlings (Hordeum vulgare)" Epigenomes 2, no. 2: 12. https://doi.org/10.3390/epigenomes2020012
APA StyleKonate, M., Wilkinson, M. J., Mayne, B. T., Pederson, S. M., Scott, E. S., Berger, B., & Rodriguez Lopez, C. M. (2018). Salt Stress Induces Non-CG Methylation in Coding Regions of Barley Seedlings (Hordeum vulgare). Epigenomes, 2(2), 12. https://doi.org/10.3390/epigenomes2020012