
Large-Scale Integrative Analysis of Epigenetic Modifications Induced by Isotretinoin, Doxycycline and Metronidazole in Murine Colonic Intestinal Epithelial Cells

 , , ,
, , ,
Abstract
1. Introduction
2. Results
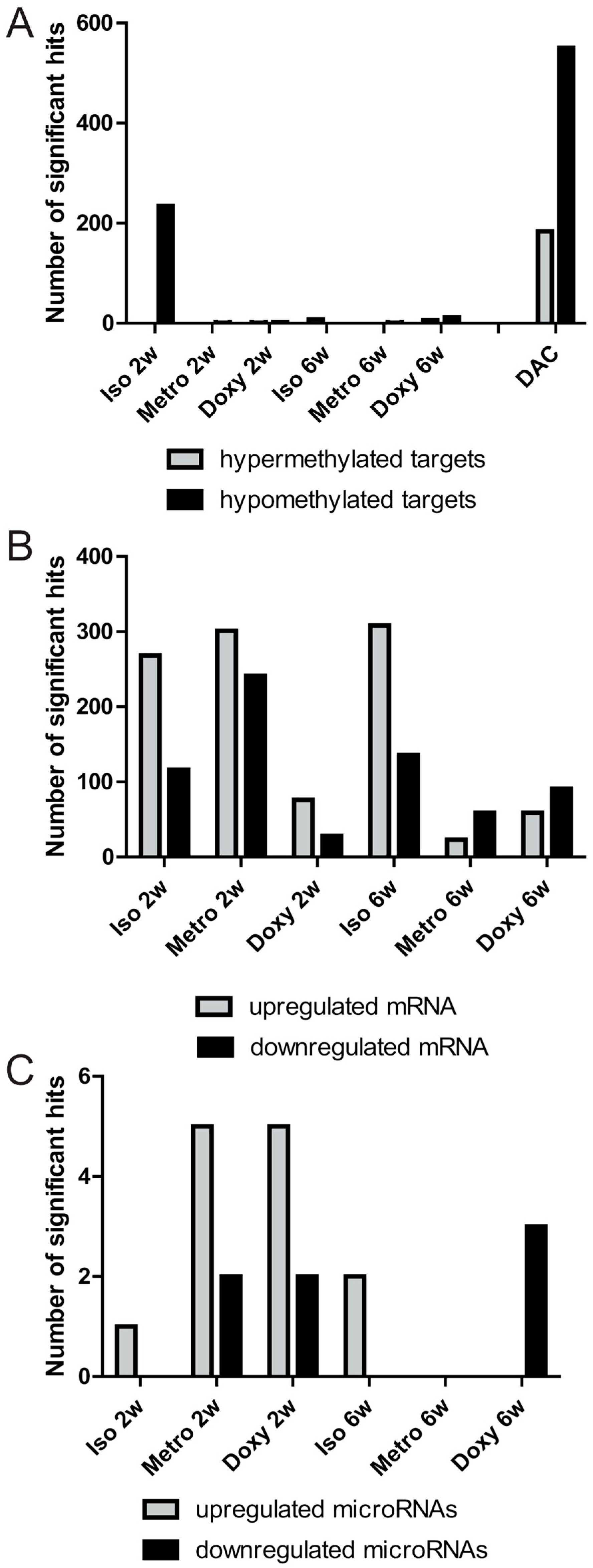
2.1. Quantitative Overview of Epigenetic Modifications after Treatment Courses per Time Point
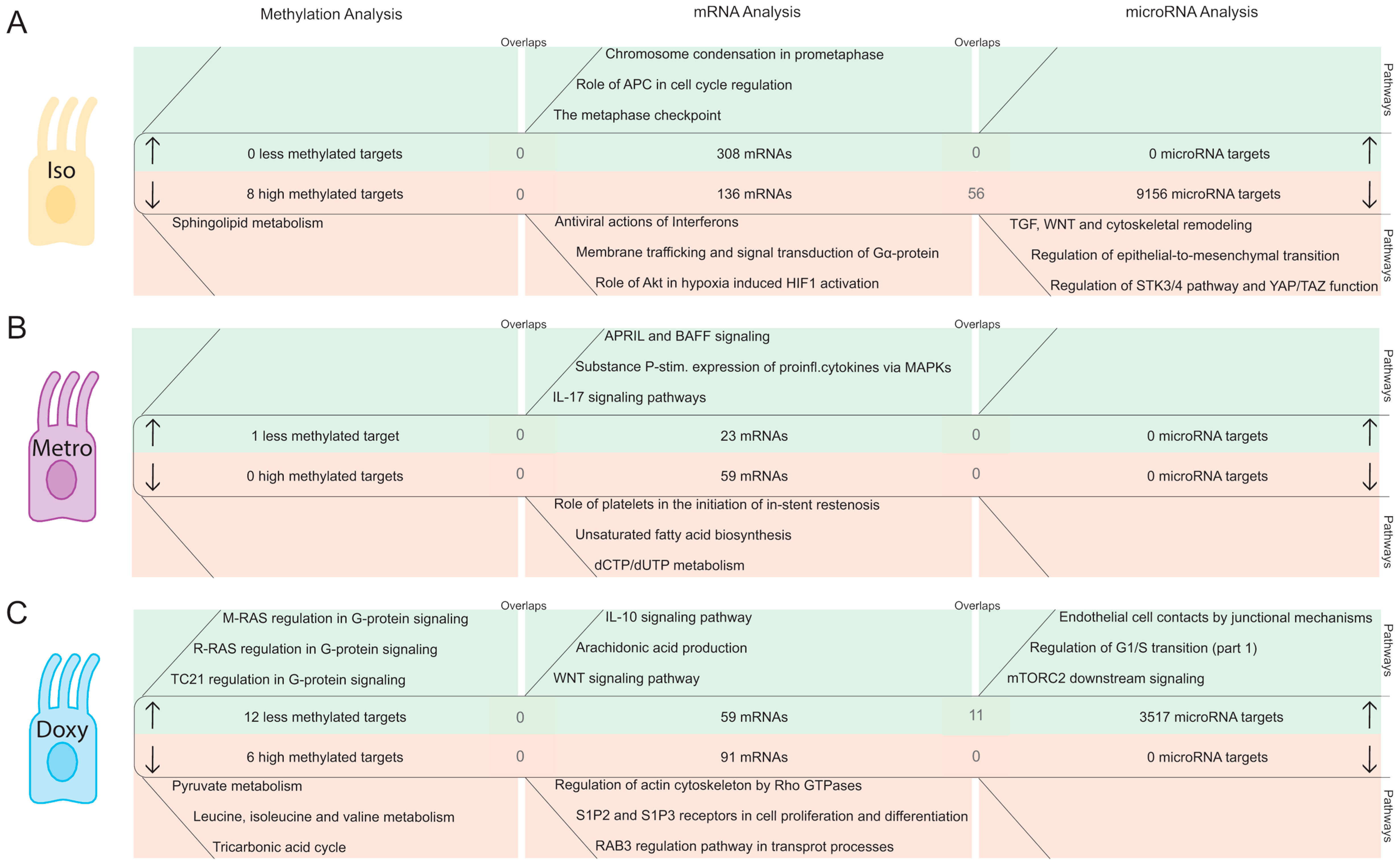
2.2. Integrative Pathway Analysis of Epigenetic Modifications in Iintestinal Epithelial Cells after Isotretinoin or Antibiotic Treatment
2.2.1. DNA Methylation Pattern in Intestinal Epithelial Cells Directly after Isotretinoin or Antibiotic Treatment (Direct Effect)
2.2.2. MicroRNA Expression Pattern in Intestinal Epithelial Cells Directly after Isotretinoin or Antibiotic Treatment (Direct Effect)
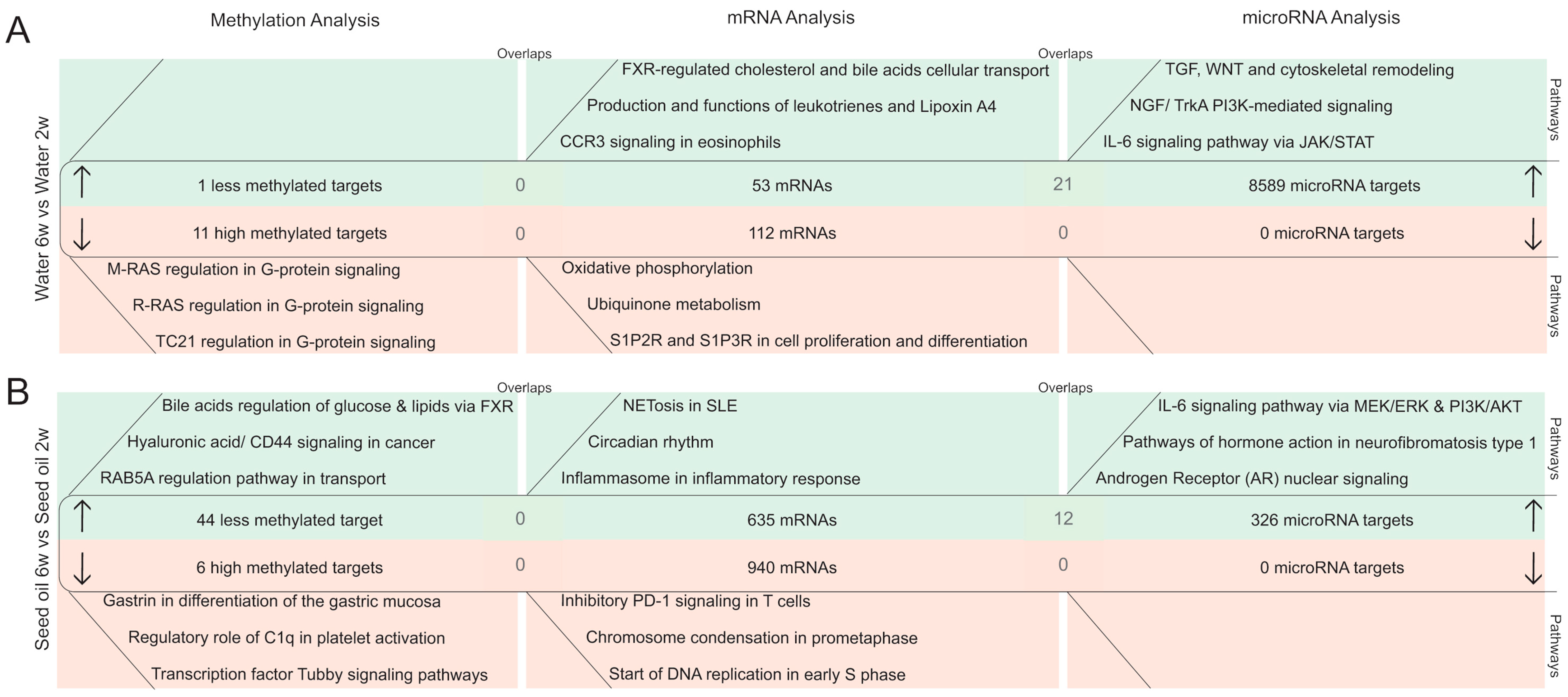
2.2.3. DNA Methylation Pattern in Intestinal Epithelial Cells after the Washout Phase of Isotretinoin or Antibiotic Treated Groups (Long-Term Effects)
2.2.4. MicroRNA Expression Pattern in Intestinal Epithelial Cells after the Washout Phase of Isotretinoin or Antibiotic Treated Groups (Long-Term Effect)
2.3. Time-Dependent Changes in Epigenetic Mechanisms in Murine Colonic Intestinal Epithelial Cells
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Ethics Approval
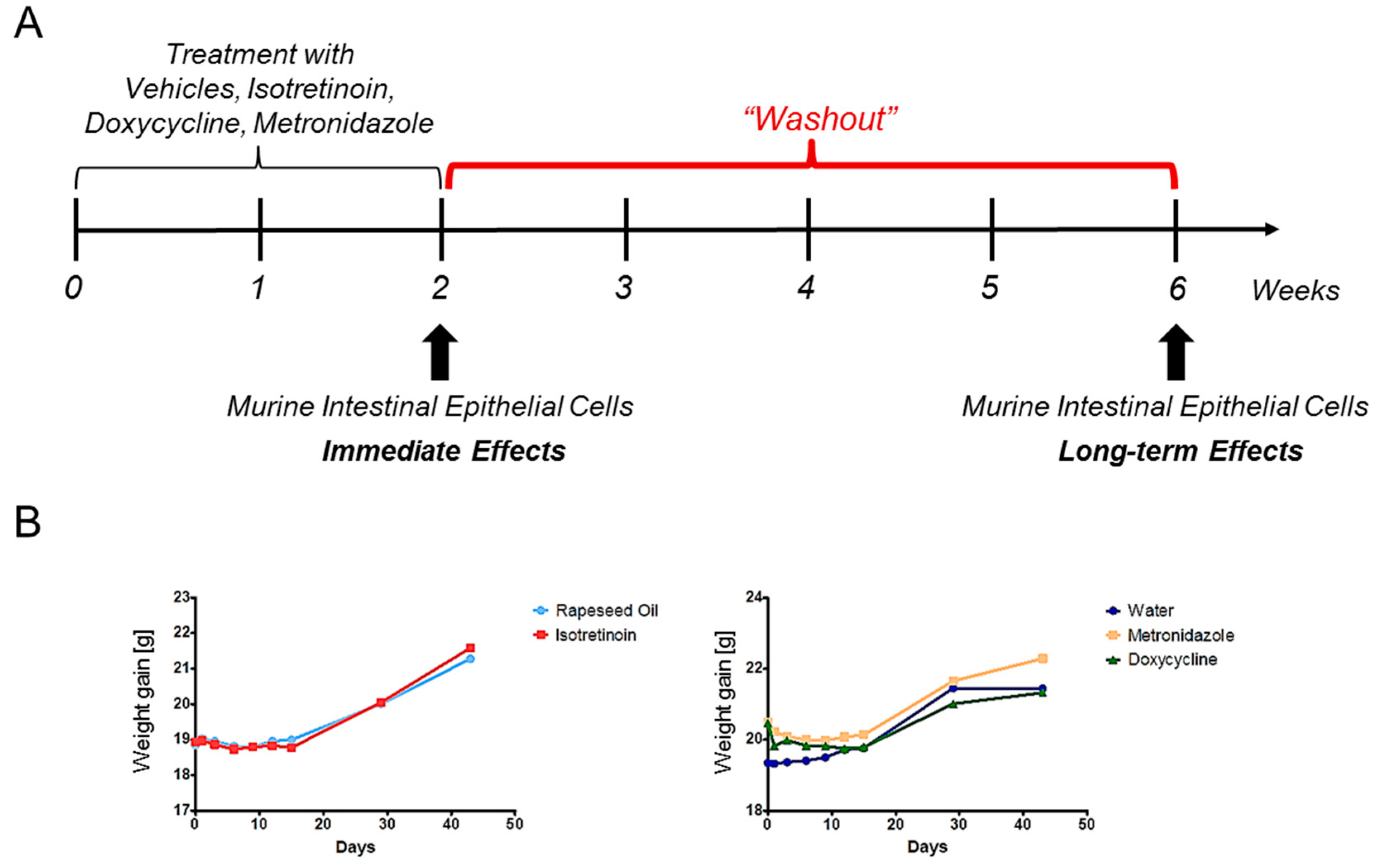
4.3. Study Design
4.4. Isolation of Murine Intestinal Epithelial Cells from the Colon
4.5. Genomic DNA and Total RNA Procedures
4.6. Sequencing Procedures
4.6.1. Library Preparations
4.6.2. Clustering and Sequencing
4.7. Bioinformatics
DNA Methylation-Sequencing
4.8. Statistics
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Molodecky, N.A.; Soon, I.S.; Rabi, D.M.; Ghali, W.A.; Ferris, M.; Chernoff, G.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Barkema, H.W.; et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology 2012, 142, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Khor, B.; Gardet, A.; Xavier, R.J. Genetics and pathogenesis of inflammatory bowel disease. Nature 2011, 474, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Iborra, M.; Bernuzzi, F.; Invernizzi, P.; Danese, S. MicroRNAs in autoimmunity and inflammatory bowel disease: Crucial regulators in immune response. Autoimmun. Rev. 2012, 11, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Pekow, J.R.; Kwon, J.H. MicroRNAs in inflammatory bowel disease. Inflamm. Bowel Dis. 2012, 18, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Kalla, R.; Ventham, N.T.; Kennedy, N.A.; Quintana, J.F.; Nimmo, E.R.; Buck, A.H.; Satsangi, J. MicroRNAs: New players in IBD. Gut 2015, 64, 504–517. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Zhou, J.; Zhong, Y.; Jiang, L.; Mu, P.; Li, Y.; Singh, N.; Nagarkatti, M.; Nagarkatti, P. Expression, regulation and function of microRNAs in multiple sclerosis. Int. J. Med. Sci. 2014, 11, 810–818. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.Y.; Li, L.; Wang, M.J.; Chen, X.M.; Huang, Q.C.; Lu, C.J. An Exploration of the Role of MicroRNAs in Psoriasis: A Systematic Review of the Literature. Medicine (Baltimore) 2015, 94, e2030. [Google Scholar] [CrossRef] [PubMed]
- Amarilyo, G.; La Cava, A. miRNA in systemic lupus erythematosus. Clin. Immunol. 2012, 144, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Sedivy, J.M.; Banumathy, G.; Adams, P.D. Aging by epigenetics—A consequence of chromatin damage? Exp. Cell Res. 2008, 314, 1909–1917. [Google Scholar] [CrossRef] [PubMed]
- Deaton, A.M.; Bird, A. CpG islands and the regulation of transcription. Genes Dev. 2011, 25, 1010–1022. [Google Scholar] [CrossRef] [PubMed]
- Santos-Rosa, H.; Schneider, R.; Bannister, A.J.; Sherriff, J.; Bernstein, B.E.; Emre, N.C.; Schreiber, S.L.; Mellor, J.; Kouzarides, T. Active genes are tri-methylated at K4 of histone H3. Nature 2002, 419, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.E.; Kamal, M.; Lindblad-Toh, K.; Bekiranov, S.; Bailey, D.K.; Huebert, D.J.; McMahon, S.; Karlsson, E.K.; Kulbokas, E.J., 3rd; Gingeras, T.R.; et al. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell 2005, 120, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Rana, T.M. Therapeutic targeting of microRNAs: Current status and future challenges. Nat. Rev. Drug Discov. 2014, 13, 622–638. [Google Scholar] [CrossRef] [PubMed]
- Reddy, D.; Siegel, C.A.; Sands, B.E.; Kane, S. Possible association between isotretinoin and inflammatory bowel disease. Am. J. Gastroenterol. 2006, 101, 1569–1573. [Google Scholar] [CrossRef] [PubMed]
- Shale, M.; Kaplan, G.G.; Panaccione, R.; Ghosh, S. Isotretinoin and intestinal inflammation: What gastroenterologists need to know. Gut 2009, 58, 737–741. [Google Scholar] [CrossRef] [PubMed]
- Crockett, S.D.; Porter, C.Q.; Martin, C.F.; Sandler, R.S.; Kappelman, M.D. Isotretinoin use and the risk of inflammatory bowel disease: A case-control study. Am. J. Gastroenterol. 2010, 105, 1986–1993. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, C.N.; Nugent, Z.; Longobardi, T.; Blanchard, J.F. Isotretinoin is not associated with inflammatory bowel disease: A population-based case-control study. Am. J. Gastroenterol. 2009, 104, 2774–2778. [Google Scholar] [CrossRef] [PubMed]
- Popescu, C.M.; Popescu, R. Isotretinoin therapy and inflammatory bowel disease. Arch. Dermatol. 2011, 147, 724–729. [Google Scholar] [CrossRef] [PubMed]
- Thakrar, B.T.; Robinson, N.J. Isotretinoin use and the risk of inflammatory bowel disease. Am. J. Gastroenterol. 2011, 106, 1000–1002. [Google Scholar] [CrossRef] [PubMed]
- Frey-Wagner, I.; Fischbeck, A.; Cee, A.; Leonardi, I.; Gruber, S.; Becker, E.; Atrott, K.; Lang, S.; Rogler, G. Effects of retinoids in mouse models of colitis: Benefit or danger to the gastrointestinal tract? Inflamm. Bowel Dis. 2013, 19, 2356–2365. [Google Scholar] [CrossRef] [PubMed]
- Wojtal, K.A.; Wolfram, L.; Frey-Wagner, I.; Lang, S.; Scharl, M.; Vavricka, S.R.; Rogler, G. The effects of vitamin A on cells of innate immunity in vitro. Toxicol. In Vitro 2013, 27, 1525–1532. [Google Scholar] [CrossRef] [PubMed]
- Margolis, D.J.; Fanelli, M.; Hoffstad, O.; Lewis, J.D. Potential association between the oral tetracycline class of antimicrobials used to treat acne and inflammatory bowel disease. Am. J. Gastroenterol. 2010, 105, 2610–2616. [Google Scholar] [CrossRef] [PubMed]
- Williams, H.C.; Dellavalle, R.P.; Garner, S. Acne vulgaris. Lancet 2012, 379, 361–372. [Google Scholar] [CrossRef]
- Shaw, S.Y.; Blanchard, J.F.; Bernstein, C.N. Association between the use of antibiotics in the first year of life and pediatric inflammatory bowel disease. Am. J. Gastroenterol. 2010, 105, 2687–2692. [Google Scholar] [CrossRef] [PubMed]
- Hviid, A.; Svanstrom, H.; Frisch, M. Antibiotic use and inflammatory bowel diseases in childhood. Gut 2011, 60, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Shaw, S.Y.; Blanchard, J.F.; Bernstein, C.N. Association between the use of antibiotics and new diagnoses of Crohn’s disease and ulcerative colitis. Am. J. Gastroenterol. 2011, 106, 2133–2142. [Google Scholar] [CrossRef] [PubMed]
- Ungaro, R.; Bernstein, C.N.; Gearry, R.; Hviid, A.; Kolho, K.L.; Kronman, M.P.; Shaw, S.; Van Kruiningen, H.; Colombel, J.F.; Atreja, A. Antibiotics associated with increased risk of new-onset Crohn’s disease but not ulcerative colitis: A meta-analysis. Am. J. Gastroenterol. 2014, 109, 1728–1738. [Google Scholar] [CrossRef] [PubMed]
- Becker, E.; Bengs, S.; Aluri, S.; Opitz, L.; Atrott, K.; Stanzel, C.; Castro, P.A.; Rogler, G.; Frey-Wagner, I. Doxycycline, metronidazole and isotretinoin: Do they modify microRNA/mRNA expression profiles and function in murine T-cells? Sci. Rep. 2016, 6, 37082. [Google Scholar] [CrossRef] [PubMed]
- Shin, C.; Nam, J.W.; Farh, K.K.; Chiang, H.R.; Shkumatava, A.; Bartel, D.P. Expanding the microRNA targeting code: functional sites with centered pairing. Mol. Cell 2010, 38, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Shivdasani, R.A. MicroRNAs: Regulators of gene expression and cell differentiation. Blood 2006, 108, 3646–3653. [Google Scholar] [CrossRef] [PubMed]
- Smith, Z.D.; Meissner, A. DNA methylation: Roles in mammalian development. Nat. Rev. Genet. 2013, 14, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Ventham, N.T.; Kennedy, N.A.; Nimmo, E.R.; Satsangi, J. Beyond gene discovery in inflammatory bowel disease: The emerging role of epigenetics. Gastroenterology 2013, 145, 293–308. [Google Scholar] [CrossRef] [PubMed]
- Karatzas, P.S.; Gazouli, M.; Safioleas, M.; Mantzaris, G.J. DNA methylation changes in inflammatory bowel disease. Ann. Gastroenterol. 2014, 27, 125–132. [Google Scholar] [PubMed]
- Tschurtschenthaler, M.; Kachroo, P.; Heinsen, F.A.; Adolph, T.E.; Ruhlemann, M.C.; Klughammer, J.; Offner, F.A.; Ammerpohl, O.; Krueger, F.; Smallwood, S.; et al. Paternal chronic colitis causes epigenetic inheritance of susceptibility to colitis. Sci. Rep. 2016, 6, 31640. [Google Scholar] [CrossRef] [PubMed]
- Low, D.; Mizoguchi, A.; Mizoguchi, E. DNA methylation in inflammatory bowel disease and beyond. World J. Gastroenterol. 2013, 19, 5238–5249. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.A.; Wang, T.; Coarfa, C.; Nagarajan, R.P.; Hong, C.; Downey, S.L.; Johnson, B.E.; Fouse, S.D.; Delaney, A.; Zhao, Y.; et al. Comparison of sequencing-based methods to profile DNA methylation and identification of monoallelic epigenetic modifications. Nat. Biotechnol. 2010, 28, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Aberg, K.A.; Xie, L.; Chan, R.F.; Zhao, M.; Pandey, A.K.; Kumar, G.; Clark, S.L.; van den Oord, E.J. Evaluation of Methyl-Binding Domain Based Enrichment Approaches Revisited. PLoS ONE 2015, 10, e0132205. [Google Scholar] [CrossRef] [PubMed]
- Pasquinelli, A.E. MicroRNAs and their targets: Recognition, regulation and an emerging reciprocal relationship. Nat. Rev. Genet. 2012, 13, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Pedrotti, L.P.; Barrios, B.E.; Maccio-Maretto, L.; Bento, A.F.; Sena, A.A.; Rodriguez-Galan, M.C.; Calixto, J.B.; Correa, S.G. Systemic IL-12 burst expands intestinal T-lymphocyte subsets bearing the α4β7 integrin in mice. Eur. J. Immunol. 2016, 46, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Henriques, C.M.; Rino, J.; Nibbs, R.J.; Graham, G.J.; Barata, J.T. IL-7 induces rapid clathrin-mediated internalization and JAK3-dependent degradation of IL-7Rα in T cells. Blood 2010, 115, 3269–3277. [Google Scholar] [CrossRef] [PubMed]
- Faller, E.M.; Ghazawi, F.M.; Cavar, M.; MacPherson, P.A. IL-7 induces clathrin-mediated endocytosis of CD127 and subsequent degradation by the proteasome in primary human CD8 T cells. Immunol. Cell Biol. 2016, 94, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Hegarty, J.P.; Yu, W.; Cappel, J.A.; Chen, X.; Faber, P.W.; Wang, Y.; Poritz, L.S.; Fan, J.B.; Koltun, W.A. Identification of disease-associated DNA methylation in B cells from Crohn’s disease and ulcerative colitis patients. Dig. Dis. Sci. 2012, 57, 3145–3153. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.Z.; van Sommeren, S.; Huang, H.; Ng, S.C.; Alberts, R.; Takahashi, A.; Ripke, S.; Lee, J.C.; Jostins, L.; Shah, T.; et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet. 2015, 47, 979–986. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, K.; Tsuzuki, Y.; Matsunaga, H.; Inoue, T.; Miyazaki, J.; Hokari, R.; Okada, Y.; Kawaguchi, A.; Nagao, S.; Itoh, K.; et al. In vivo demonstration of T lymphocyte migration and amelioration of ileitis in intestinal mucosa of SAMP1/Yit mice by the inhibition of MAdCAM-1. Clin. Exp. Immunol. 2005, 140, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Soler, D.; Chapman, T.; Yang, L.L.; Wyant, T.; Egan, R.; Fedyk, E.R. The binding specificity and selective antagonism of vedolizumab, an anti-α4β7 integrin therapeutic antibody in development for inflammatory bowel diseases. J. Pharmacol. Exp. Ther. 2009, 330, 864–875. [Google Scholar] [CrossRef] [PubMed]
- Coombes, J.L.; Siddiqui, K.R.; Arancibia-Carcamo, C.V.; Hall, J.; Sun, C.M.; Belkaid, Y.; Powrie, F. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-β and retinoic acid-dependent mechanism. J. Exp. Med. 2007, 204, 1757–1764. [Google Scholar] [CrossRef] [PubMed]
- Wittmann, A.; Bron, P.A.; van Svam, I.I.; Kleerebezem, M.; Adam, P.; Gronbach, K.; Menz, S.; Flade, I.; Bender, A.; Schafer, A.; et al. TLR signaling-induced CD103-expressing cells protect against intestinal inflammation. Inflamm. Bowel Dis. 2015, 21, 507–519. [Google Scholar] [CrossRef] [PubMed]
- D’Agostino, P.; Arcoleo, F.; Barbera, C.; Di Bella, G.; La Rosa, M.; Misiano, G.; Milano, S.; Brai, M.; Cammarata, G.; Feo, S.; et al. Tetracycline inhibits the nitric oxide synthase activity induced by endotoxin in cultured murine macrophages. Eur. J. Pharmacol. 1998, 346, 283–290. [Google Scholar] [CrossRef]
- Griffin, M.O.; Fricovsky, E.; Ceballos, G.; Villarreal, F. Tetracyclines: A pleitropic family of compounds with promising therapeutic properties. Review of the literature. Am. J. Physiol. Cell Physiol. 2010, 299, C539–C548. [Google Scholar] [CrossRef]
- Becker, E.; Schmidt, T.S.B.; Bengs, S.; Poveda, L.; Opitz, L.; Atrott, K.; Stanzel, C.; Biedermann, L.; Rehman, A.; Jonas, D.; et al. Effects of oral antibiotics and isotretinoin on the murine gut microbiota. Int. J. Antimicrob. Agents 2017, 50, 342–351. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, Y.; Sato, T.; Ohteki, T. Commensal Gram-positive bacteria initiates colitis by inducing monocyte/macrophage mobilization. Mucosal Immunol. 2015, 8, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.J.; Servant, N.; Toedling, J.; Sarazin, A.; Marchais, A.; Duvernois-Berthet, E.; Cognat, V.; Colot, V.; Voinnet, O.; Heard, E.; et al. ncPRO-seq: A tool for annotation and profiling of ncRNAs in sRNA-seq data. Bioinformatics 2012, 28, 3147–3149. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Caffo, B.; Jaffee, H.A.; Irizarry, R.A.; Feinberg, A.P. Redefining CpG islands using hidden Markov models. Biostatistics 2010, 11, 499–514. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A | |||||
|---|---|---|---|---|---|
| Treatment vs. Vehicles per Time Point | Regulation | microRNA | Log2FC | p-Value | In silico microRNA targets |
| Isotretinoin | Induction | mmu-miR-451a | 1.361 | 4.03 × 10−4 | 24↓ |
| 2 weeks | |||||
| Isotretinoin | Induction | mmu-miR-98-3p | 2.078 | 2.65 × 10−5 | 5326↓ |
| 6 weeks | mmu-miR-142b | 1.805 | 1.99 × 10−4 | 3830↓ | |
| Metronidazole | Induction | mmu-miR-19b-3p | 1.2 | 5.72 × 10−6 | 1075↓ |
| 2 weeks | mmu-miR-1983 | 1.719 | 1.05 × 10−5 | 4154↓ | |
| mmu-miR-219a-5p | 1.599 | 1.16 × 10−5 | 388↓ | ||
| mmu-miR-19a-3p | 1.295 | 3.25 × 10−5 | 1075↓ | ||
| mmu-miR-219c-3p | 1.387 | 7.97 × 10−5 | 4342↓ | ||
| Suppression | mmu-miR-1247-5p | −1.356 | 1.76 × 10−4 | 909↑ | |
| mmu-miR-409-5p | −1.141 | 3.90 × 10−4 | 120↑ | ||
| Metronidazole | - | - | - | - | - |
| 6 weeks | |||||
| Doxycycline | Induction | mmu-miR-6236 | 2.39 | 1.55 × 10−7 | 126↓ |
| 2 weeks | mmu-miR-5099 | 1.551 | 7.66 × 10−5 | 1404↓ | |
| mmu-miR-5121 | 1.337 | 5.85 × 10−5 | 4001↓ | ||
| mmu-miR-1940 | 2.27 | 1.36 × 10−4 | * | ||
| mmu-miR-1291 | 1.582 | 2.35 × 10−4 | 4934↓ | ||
| Suppression | mmu-miR-409-5p | −1.778 | 4.35 × 10−6 | 120↑ | |
| mmu-miR-6240** | −2.231 | 1.43 × 10−4 | 840↑ | ||
| Doxycycline | Suppression | mmu-miR-144-3p | −1.338 | 1.64 × 10−4 | 849↑ |
| 6 weeks | mmu-miR-144-5p | −1.271 | 1.32 × 10−4 | 2254↑ | |
| mmu-miR-31-5p | −1.009 | 4.10 × 10−4 | 414↑ |
| B | ||||
|---|---|---|---|---|
| 6 weeks vs. 2 weeks per Treatment | Regulation | microRNA | Log2FC | p-Value |
| Water | Suppression | mmu-miR-1940 | −2.198 | 3.43 × 10−5 |
| mmu-miR-877-3p | −1.648 | 3.43 × 10−5 | ||
| mmu-miR-409-5p | −1.213 | 1.85 × 10−4 | ||
| mmu-miR-485-3p | −1.472 | 1.60 × 10−4 | ||
| Seed oil | Suppression | mmu-miR-150-5p | −1.945 | 4.10 × 10−5 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Becker, E.; Bengs, S.; Aluri, S.; Opitz, L.; Atrott, K.; Rost, F.; Leonardi, I.; Stanzel, C.; Raselli, T.; Kasper, S.; et al. Large-Scale Integrative Analysis of Epigenetic Modifications Induced by Isotretinoin, Doxycycline and Metronidazole in Murine Colonic Intestinal Epithelial Cells. Epigenomes 2017, 1, 24. https://doi.org/10.3390/epigenomes1030024
Becker E, Bengs S, Aluri S, Opitz L, Atrott K, Rost F, Leonardi I, Stanzel C, Raselli T, Kasper S, et al. Large-Scale Integrative Analysis of Epigenetic Modifications Induced by Isotretinoin, Doxycycline and Metronidazole in Murine Colonic Intestinal Epithelial Cells. Epigenomes. 2017; 1(3):24. https://doi.org/10.3390/epigenomes1030024
Chicago/Turabian StyleBecker, Eugenia, Susan Bengs, Sirisha Aluri, Lennart Opitz, Kirstin Atrott, Felix Rost, Irina Leonardi, Claudia Stanzel, Tina Raselli, Stephanie Kasper, and et al. 2017. "Large-Scale Integrative Analysis of Epigenetic Modifications Induced by Isotretinoin, Doxycycline and Metronidazole in Murine Colonic Intestinal Epithelial Cells" Epigenomes 1, no. 3: 24. https://doi.org/10.3390/epigenomes1030024
APA StyleBecker, E., Bengs, S., Aluri, S., Opitz, L., Atrott, K., Rost, F., Leonardi, I., Stanzel, C., Raselli, T., Kasper, S., Ruiz, P. A., & Rogler, G. (2017). Large-Scale Integrative Analysis of Epigenetic Modifications Induced by Isotretinoin, Doxycycline and Metronidazole in Murine Colonic Intestinal Epithelial Cells. Epigenomes, 1(3), 24. https://doi.org/10.3390/epigenomes1030024