Abstract
The rapid advancement of micro/nano-electromechanical systems (MEMS/NEMS) and precision manufacturing has fundamentally challenged traditional friction theories at the nanoscale. Classical continuum models fail to capture energy dissipation mechanisms at the atomic level, which are influenced by interfacial phenomena such as electron transfer, charge redistribution, and energy level realignment. Density functional theory (DFT), renowned for its accurate description of ground-state properties in many-electron systems, has emerged as a key tool for uncovering quantized friction mechanisms. By quantifying potential energy surface (PES) fluctuations, the evolution of interfacial charge density, and dynamic electronic band structures, DFT establishes a universal correlation between frictional dissipation and electronic behavior, transcending the limitations of conventional models in explaining stick–slip motion, superlubricity, and non-Amonton effects. Research breakthroughs in the application of DFT include characterizing frictional chemical potentials, designing heterojunction-based superlubricity, elucidating strain/load modulation mechanisms, and resolving electronic energy dissipation pathways. However, these advances remain scattered across interdisciplinary studies. This article systematically summarizes methodological innovations and cutting-edge applications of DFT in computational tribology, with the aim of constructing a unified framework for carrying out the “electronic structure–energy dissipation–frictional response” predictions. It provides a state of the art of using DFT to help design high-performance lubricants and actively control interfacial friction.
1. Introduction
Tribology, as a fundamental discipline investigating the interactions between surfaces in contact and in relative motion, has evolved alongside humanity’s exploration into the nature of energy dissipation and material failure. Since Leonardo da Vinci [1] first proposed the mechanical concept of friction in the 15th century and Amontons established the classical laws of friction in 1699, macroscopic friction theories dominated the understanding of interfacial behavior for over three centuries. The friction law Amonstons, widely adopted in a large number of situations, describes friction force as a linear function of normal load () and emphasizes the principle of area independence, providing an initial framework for engineering lubrication design. However, with the rapid advancement of micro/nano-electromechanical systems and precision manufacturing technologies, the limitations of traditional friction theories have become increasingly evident at nanoscale. Accounting for quantum effects on interfacial behavior at atomic scale poses fundamental challenges to classical friction and wear models.
The emergence of nanotribology brings new challenges. As the contact scale approaches atomic dimensions, the corresponding interfacial behavior exhibits unusual tribological characteristics. Specifically, the proportional relationship between friction force and real contact area breaks down. Instead, friction becomes highly dependent on nano- and micro-parameters, including lattice commensurability [2,3] and sliding velocity [4,5]. Non-Amontons behaviors, including negative friction coefficients [4,6] and pressure-induced superlubricity [7], frequently occur. The core contradiction lies in the fundamental conflict between the quantized nature of energy dissipation at micro-interfaces and the continuum assumption inherent in classical friction models. For instance, traditional models such as the Prandtl [8]–Tomlinson [9] (P-T) spring–mass system can qualitatively explain stick–slip motion but fail to quantify the modulation effect of electron redistribution on the potential energy surface. Although the Frenkel–Kontorova (F-K) model [10] incorporates periodic substrate potentials, it still treats atoms as classical particles and neglects the dynamic evolution of electronic states. In this context, density functional theory (DFT), with its capability for accurate description of material electronic structures, has emerged as a key tool for deciphering the physical essence of nanofriction.
By solving the Kohn–Sham equations for many-electron systems, DFT enables interfacial energy calculations from first principles. Its core value in tribological research is manifested through three major breakthroughs: (i) Quantification of frictional energy evolution: quasi-dynamic models are constructed to calculate relevant energies, directly correlating interfacial electronic structures with sliding energy barriers to reveal variations in friction force. (ii) Clarification of friction origin: a universal relationship between charge density fluctuations and frictional dissipation is established, providing a unified explanation for frictional phenomena such as structural superlubricity and load-induced superlubricity. (iii) Friction regulation design: the elaboration of interfacial electronic structures is achieved through strain engineering, heterojunction design, and electric field modulation, guiding the rational design of ultralow friction materials.
This article is a review focusing on the recent theoretical breakthroughs and practical applications of DFT in modeling and simulating some important tribological phenomena, as illustrated in Figure 1. The article is organized as follows. Section 2 describes the core methodologies of DFT in carrying out friction analysis (binding energy, sliding energy barrier, and charge density evolution). Section 3 is concerned with three ultralow friction mechanisms revealed by DFT: the electronic-scale nature of structural superlubricity, strain engineering strategies for continuous sliding modulation, and the critical pressure theory of load-induced superlubricity. Section 4 explores DFT-based design principles for two-dimensional material interfaces, covering intrinsic property screening, metal substrate modulation, and heterojunction interface design. Section 5 reviews the application of DFT in active friction control, including electric field modulation, chemical passivation, doping design, and defect engineering. Section 6 discusses the integration of DFT with machine learning in friction coating design and nanogenerators, highlighting some relevant future developments and applications in tribology research. This review aims to present theoretical breakthroughs and technological innovations of DFT in understanding tribological phenomena, to reveal electronic-scale friction mechanisms, and to provide scientific insights and paths for developing high-performance lubricants and low-dissipation motion systems.
Figure 1.
Schematic diagram of this review: computational methods, mechanisms of superlubricity (Image source [7]), material and surface design (Image source [11]), active friction control (Image source [12]), developments and applications (Image source [13]). The images are solely for content demonstration. All the images shown here will be presented again later, accompanied by detailed descriptions.
2. DFT-Based Methods for Analyzing Friction
Density Functional Theory (DFT) provides a powerful tool for the atomic-scale physical study of friction mechanisms by quantitatively calculating the electronic structure and energy evolution at material interfaces. Its core methodologies include analyzing interfacial binding energy, sliding energy barriers, and charge density, as illustrated in Figure 2. Together, the computation and analysis of these quantities allow us to elucidate the physical origins and regulatory mechanisms of friction.
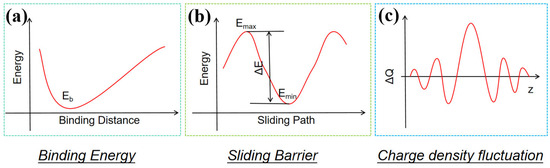
Figure 2.
Three approaches for analyzing friction via Density Functional Theory (DFT): (a) computation of binding energy, (b) determination of sliding energy barrier, and (c) analysis of charge density.
2.1. Binding Energy
DFT simulations provide a quantitative way for understanding the physical origin of static friction by calculating the binding energy of material interfaces at equilibrium. In fact, the binding energy reflects the strength of interactions between interfacial atoms [14], and its magnitude is directly related to the work required to separate the interfaces. Within the DFT framework, the binding energy is obtained by comparing the total energy difference between isolated systems and the contact system after optimizing the atomic configuration at a fixed interfacial distance. The binding energy is typically calculated by using the following formula:
where represents the total energy of a sliding system, and and denote the energies of the isolated fixed body and sliding body , respectively. For interfacial interactions of different natures, appropriate exchange–correlation functionals must be selected. For instance, van der Waals-corrected generalized gradient approximation (e.g., GGA-PBE functional) is commonly used to describe weak interfacial interactions in layered materials such as graphene, while the local density approximation (LDA) is more suitable for strongly bonded interfaces [15].
2.2. Sliding Energy Barrier
The energy dissipation mechanism during sliding can be evaluated using the Potential Energy Surface (PES) constructed via DFT. The PES depicts the relative energy variation in the interface along a specific sliding path, and the height of the energy barrier along this path is directly related to the minimum work required to overcome frictional resistance, i.e., the sliding energy barrier . This quantity is calculated as follows:
where and represent the maximum and minimum energies along the sliding path (Figure 2b). Two typical approaches exist for quantifying the sliding energy barrier [7,16,17,18,19,20]: one involves taking the maximum energy difference between adjacent extreme points on the PES, reflecting the theoretical maximum friction under extreme dissipation scenarios; the other calculates the barrier along the path of minimal energy gradient, which may more closely approximate the continuous energy dissipation in actual sliding processes. Both methods are important in the design of lubricating materials and the study of interface modifications; yet, a systematic comparison of their applicability and computational accuracy remains an area for further exploration.
In DFT simulations of friction, the normal load is computed by differentiating the interfacial binding energy with respect to the vertical separation distance z [7,19]: . The average lateral friction force is derived from the energy difference between the maximum and minimum energy points along the sliding path: where is the displacement. Dividing the normal load and the friction force by the contact area (i.e., the unit area of atomic contact) yields the normal stress and the frictional shear stress .
2.3. Charge Density
The continuously changing “contact” state between surfaces in relative motion leads to the dynamic variation in interfacial interactions, which fundamentally gives rise to friction. In other words, friction originates from the interactions between electrons and atomic nuclei at the interface of moving surfaces. Due to the significantly larger mass of atomic nuclei compared to electrons, the velocity response of nuclear positions during a relative motion is much slower than the redistribution of electrons. Therefore, the evolution of charge density at sliding interfaces serves as a key physical quantity for deciphering the microscopic mechanisms of friction [21,22]. Its evolutionary behavior can be precisely quantified through differential charge density analysis, providing a visual representation of the contribution of electronic interactions to friction during the sliding process.
As illustrated in Figure 3, Restuccia et al. [23] visualized charge distribution to elucidate the friction-reduction mechanism of graphene on steel surfaces (Figure 3a); Righi et al. [24,25] evaluated the stacking stability of graphene/graphene and the structural response of MoS2 under compression via interlayer charge redistribution (Figure 3b); Ciraci et al. [26] attributed the superlubricity of graphene on nickel substrates to a significant reduction in interfacial charge density (Figure 3c). These studies collectively established the correlation between charge density and friction. In 2018, Righi et al. [27] pioneered a semi-quantitative mapping between frictional properties and charge fluctuations across covalent, metallic, and van der Waals interfaces (Figure 3d–f), fitting the numerical relationship between charge redistribution and interfacial separation work. Sun et al. [28] demonstrated that the energy barrier variation on the sliding potential energy surface (PES) exhibits a linear positive correlation with the degree of charge density fluctuation (Figure 3g–j). This universal relationship holds across diverse interfacial systems, including van der Waals interfaces, ionic crystals, and covalent materials. Although normal load enhances the charge density–PES correlation by modulating interfacial separation, the slope of the linear relationship remains invariant. These results confirm that interfacial charge redistribution during sliding constitutes the dominant underlying mechanism of frictional energy dissipation.
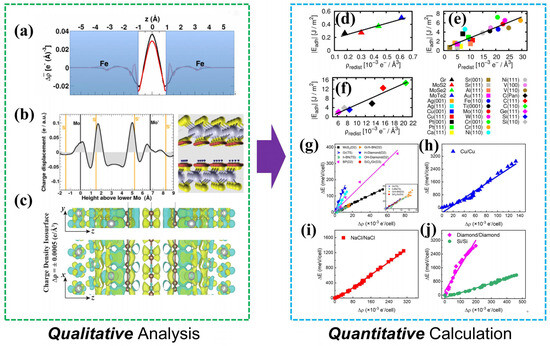
Figure 3.
Analysis of charge evolution in friction: (a) Average electron charge displacement at an iron interface [23] (the white central region corresponds to the optimized interfacial spacing); (b) Z-direction charge transfer and spatial distribution of charge transfer in MoS2/MoS2 [24] (red and blue indicate charge density accumulation or depletion, respectively); (c) Differential charge density map between top graphene and the underlying substrate [26] (yellow and blue represent charge density accumulation or depletion, respectively); Numerical fitting between the degree of charge redistribution and interfacial separation work [27]: (d) for van der Waals (vdW) bonded materials, (e) for simple metals and noble metals (squares) and transition metals of non-noble metals (circles), and (f) the fitting relationship between adhesion energy and interfacial charge redistribution parameter for covalently bonded materials. Correlation between charge density evolution during frictional sliding and potential energy surface (PES) fluctuations for (g) vdW, (h) metallic, (i) ionic, and (j) covalent bonded materials [28].
To quantify the evolution of charge redistribution during friction, the interfacial charge rearrangement induced by sliding contact, i.e., the differential charge density, is first be determined as follows [27,29]:
where represents the total charge density distribution of a sliding system consisting of bodies and , and denote the total charge density distributions of the isolated sliding bodies and .
By projecting the differential charge density along the interface normal direction [27,29], a one-dimensional distribution function is obtained:
where is the basal area of the supercell. Although intuitively reflects charge rearrangement at each sliding position, it cannot directly characterize the dynamic charge changes induced by sliding. Thus, the difference in distribution functions between the maximum and minimum energy points along the sliding path is calculated by
This equation directly reveals the difference in coupling interactions between two contact configurations at the charge distribution level. The interfacial charge rearrangement quantity is defined as:
where the integration interval represents the sliding interfacial region [29]. Finally, the charge fluctuation of the two-dimensional interface is quantitatively characterized as:
where and represent the interfacial charge distributions at the highest and lowest charge density points of a sliding system, respectively; describes the change in interfacial charge density during sliding from the highest energy position to the lowest one and is defined as the interfacial charge density fluctuation.
DFT simulations of sliding friction are typically performed in two modes: constant distance mode (CDM) and constant load mode (CLM). The static friction results relative to these two modes are, however, consistent when accounting for the work done. An exact equivalent conversion relationship exists between the corresponding loads [30]: the load at a specific interface height should be the arithmetic mean of the loads at the maximum and minimum energy points. Traditional constant height mode calculations may introduce systematic errors by ignoring pressure differences between positions. For a more detailed discussion, refer to the work by Lu et al. [30].
It is noteworthy that DFT-based calculations present some limitations in describing friction phenomena, which can be summarized as follows. First, their cost is high. This means that their applications are limited to the nanoscale or to micro-scale. For systems containing a large number of atoms, or for friction processes occurring at mesoscopic or even macroscopic scales, direct DFT simulations remain exceedingly challenging and require immense computational resources. Second, DFT-based computations are limited at the time-scale level. Indeed, DFT is essentially a method for determining ground-state electronic structures, and DFT-based dynamic simulations typically rely on quasi-static approximations. As a result, it is difficult to accurately capture time-dependent frictional dynamics, particularly those involving long-time-scale processes such as wear and fatigue, which lie well beyond the current capabilities of DFT-based approaches. Third, the material properties determined by DFT-based simulations are strongly dependent on the choice of the exchange–correlation functional chosen. However, physical quantities such as sliding energy barriers and charge density distributions are often of interest in tribological studies mainly for their relative variations. Although the selection of a functional may affect the absolute values of these quantities, it generally does not alter their relative variations. For example, consistent conclusions were reached for the same systems with different software packages and exchange–correlation functionals used (see Cheng et al. [31,32] and Sun et al. [28]).
3. Superlubricity
DFT simulations of sliding friction by precisely characterizing interlayer van der Waals forces and the dynamic evolution of electron density have established a universal linear relationship between frictional dissipation energy and charge density evolution. This theoretical breakthrough lays the foundation for understanding physical phenomena such as structural superlubricity, continuous sliding-induced superlubricity, and load-induced superlubricity.
3.1. Structural Superlubricity via Incommensurate Contact
Structural superlubricity (SSL), a groundbreaking phenomenon in tribology, has attracted widespread attention since its proposal [33,34]. The conceptual precursor can be traced back to the seminal studies by Dowson [35] and Israelachvili [36], which were based on contact models involving spherical rough surfaces and pebble geometries. In 1990, Hirano et al. [37] first theoretically demonstrated the possibility of achieving ultralow friction through incommensurate contact. The term “structural superlubricity” was formally coined by Müser et al. [38,39] in 2004. In the same year, Dienwiebel et al. [2] experimentally observed microscopic SSL behavior for the first time by rotating nanographic flakes (Figure 4a), marking a major advancement in the field of two-dimensional materials. By employing strategies such as graphene-coated microspheres, the microscopic SSL effect can be extended to the macroscopic scale, making related material design strategies a recent research focus.
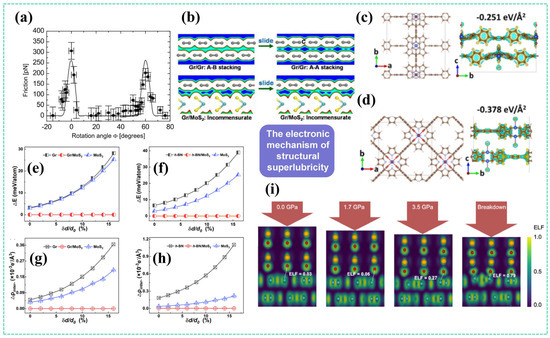
Figure 4.
DFT studies in structural superlubricity: (a) Experimental observation of rotation angle-dependent superlubricity between graphite layers [2]; (b) Interface charge density maps of Gr/Gr and Gr/MoS2 systems under low-energy (left) and high-energy (right) states of the potential energy surface [18] (yellow, blue, and gray spheres represent S, Mo, and C atoms, respectively); Sliding models (left) and differential charge density maps of minimum-energy configurations (right) for (c) bilayer graphene and (d) graphene/MoS2 heterojunctions [40]; (e) Sliding energy barriers and (g) charge fluctuations versus normal load for Gr/MoS2 heterojunctions, Gr/Gr homojunctions, and MoS2/MoS2 homojunctions [41]; (f) Sliding energy barriers and (h) charge fluctuations versus normal load for h-BN/MoS2 heterojunctions, h-BN/h-BN homojunctions, and MoS2/MoS2 homojunctions [41]; (i) Electron localization function (ELF) at the WO3/graphene interface, with dashed lines indicating atomic pairs with the strongest interactions [42].
In 2016, Wang et al. [43], for the first time, revealed the origin of structural superlubricity at the electronic scale: under incommensurate conditions, weak interlayer electron interactions dominate the frictional behavior, resulting in an energy barrier only one-tenth of that in the commensurate state. In commensurate contact, however, van der Waals forces and electronic interactions exhibit a synergistic effect, with van der Waals forces dominating friction in the low-pressure regime, while electronic interactions become significantly enhanced under high pressure.
In 2017, Luo et al. [18] observed a sharp drop in the friction coefficient in graphene/MoS2 heterostructures. Their DFT calculations uncovered the core mechanism (Figure 4b): minimal charge density fluctuations (≈0.0002 e/Å3) at the sliding interface govern the formation of an ultralow energy barrier (0.046 meV/atom), which is 195 times lower than that in MoS2 homojunctions. This demonstrates that dynamic electron distribution more accurately reflects the essence of friction than static binding energy. Similarly, Liu et al. [40] demonstrated extremely weak charge density fluctuations at the heterojunction interface during sliding (Figure 4c,d), leading to a maximum energy fluctuation significantly reduced to 6 × 10−5 eV/Å2—two orders of magnitude lower than that in their homojunction counterparts.
Sun et al. [42] conducted an in-depth study on the failure mechanism and electronic coupling dynamics of structural superlubricity under extreme pressure (Figure 4i). Using DFT, they systematically quantified the pressure-induced evolution of the electron localization function (ELF). When the pressure was below 1.7 GPa, the ELF value of interfacial oxygen–carbon atom pairs was less than 0.1, indicating purely van der Waals interactions. However, when the pressure reached a critical value of 3.50 GPa, the ELF value surged above 0.7, triggering the sudden formation of interfacial O–C covalent bonds. This abrupt change in electron density directly caused the shear strength to jump from 0.14 GPa in the superlubricity state to 3.65 GPa, accompanied by shear-induced tearing wear of the graphite layers.
Cheng et al. [41] investigated three types of interface systems—lattice-mismatched heterojunctions, rotation-mismatched homojunctions, and curvature-mismatched nanotubes—elucidating the universal origin of SSL at the electronic scale (Figure 4e–h). They concluded that interfacial electron density fluctuation is the key physical quantity governing superlubricity. This theory ultimately establishes a unified framework of “geometric mismatch → electron redistribution → energy fluctuation,” providing quantitative criteria for designing structural superlubricity. It is noteworthy that the patterns of electron density fluctuation revealed by DFT simulations are driving a fundamental paradigm shift in tribology—from traditional phenomenological models toward predictive frameworks at the quantum scale.
3.2. Strain-Modulated Continuous Sliding
In the field of solid lubrication, achieving the transition from intermittent stick–slip motion to continuous superlubricity represents a fundamental scientific challenge. The single-asperity contact model, known as the Prandtl–Tomlinson (P-T) model, provides a foundational theoretical framework for understanding this phenomenon (Figure 5a). This model attributes frictional dissipation to the dynamic competition between the tip–sample interaction potential (manifested as the sliding energy barrier to drastically increase from ) and the connecting spring stiffness . The critical criterion established by the model, formulated as , clearly delineates the system’s evolutionary path. In this formula, denotes the amplitude of the interaction potential between the tip and the periodic sliding surface atoms, and a represents the lattice period of the surface atoms. When , the system is confined to a high-dissipation stick–slip state, whereas a transition to continuous superlubricity occurs when . This classical model profoundly reveals that the essence of friction suppression lies in reducing energy fluctuations or/and enhancing interfacial stiffness. The underlying concept of “energy barrier vs. stiffness interplay” embodied in the model has become a crucial paradigm in cross-scale friction research [44,45].
Unlike traditional SSL that relies on incommensurate contact configurations, strain engineering opens a new pathway for regulating interfacial intrinsic properties. In 2014, Wang et al. [46] systematically investigated via DFT the modulation of frictional behavior in bilayer MoS2 under strain: they found that compressive strain ( to ) significantly reduces friction compared to tensile strain ( to ), with the effect persisting over a wide load range. The microscopic mechanism was revealed to be a dual synergistic effect: compressive strain promotes charge transfer from the Mo layer to the interfacial S layer, enhancing interlayer Coulomb repulsion; meanwhile, it induces band broadening and a more dispersed electronic density of states, weakening electron localization effects. Subsequent studies further expanded the dimensions of strain engineering: simulations by Wang et al. [47] on highly compressed MoS2 nanosheets indicated that strain reduces sliding resistance and enhances equivalent stiffness by altering atomic density and long-range potential overlap; whereas research by Wu et al. [48] on graphene showed that biaxial tensile strain induces lattice mismatch to form Moiré patterns, completely disrupting commensurate contact and achieving structural superlubricity. However, the common essence of these studies [48,49,50] remains the use of strain to break commensurate contact and suppress potential barrier fluctuations—reflecting that conventional strain strategies still operate within the paradigm of incommensurate contact.

Figure 5.
Strain-mediated continuous sliding theory: (a) Dynamics analysis of the transition from stick–slip to continuous sliding for a probe on a surface based on the P-T model [45]; (b) Process of in-plane strain-tuned commensurate contact in Gr/Gr interlayer sliding transitioning from stick–slip, to single-slip, to continuous sliding, and ultimately to superlubricity [32].
Figure 5.
Strain-mediated continuous sliding theory: (a) Dynamics analysis of the transition from stick–slip to continuous sliding for a probe on a surface based on the P-T model [45]; (b) Process of in-plane strain-tuned commensurate contact in Gr/Gr interlayer sliding transitioning from stick–slip, to single-slip, to continuous sliding, and ultimately to superlubricity [32].

Cheng et al. [51] systematically analyzed, via first-principles calculations, the mechanism by which biaxial strain influences interlayer friction in bilayer graphene (Figure 5b). Their work reveals the decisive role of strain direction in friction evolution. The study demonstrates that compressive strain () significantly reduces the interlayer sliding energy barrier, while tensile strain () enhances frictional dissipation. The microscopic mechanism stems from biaxial strain-driven interlayer electron redistribution: under compression, a decrease in the relative fluctuation of charge density smoothens the potential energy surface (PES) (energy barrier reduction > 40%), accompanied by increased interlayer charge density strengthening coupling strength; under tension, amplified charge density fluctuations (up to 60%) exacerbate energy barrier variations. Based on these insights, a biaxial strain-driven strategy was further proposed [32], achieving intrinsic superlubricity for the first time in a commensurate-contact graphene/graphene (Gr/Gr) system without relying on incommensurate configurations. DFT calculations show that at a biaxial compressive strain of 35%, the PES of the system becomes completely flat (); even at 15% strain, partial superlubricity emerges along specific sliding paths. Microscopic analysis reveals that strain eliminates differences in coupling strength between sites by mitigating fluctuations in interlayer charge density variations across different stacking configurations. More critically, quantitative analysis indicates that strain simultaneously enhances in-plane stiffness and reduces effective potential stiffness . As a result, to drastically increase from ≈0 to ≈104. This discovery fundamentally reshapes the understanding of strain regulation—demonstrating that a zero-friction state in commensurate systems can be achieved through precise modulation of electron density distribution, without relying on incommensurate contact.
3.3. Critical Pressure Theory for Load-Induced Superlubricity
Load-Induced Superlubricity, as a new paradigm that transcends conventional understanding of friction, has its theoretical foundation traced back to the pioneering work by Tománek et al. [19] in 1990 (Figure 6a–c). This study, employing first-principles calculations, revealed that the sliding energy barrier of a Pd atomic layer on a graphite surface exhibits significant load dependence: the friction coefficient μ demonstrates non-monotonic behavior with increasing external load, reaching a distinct minimum under a specific load. This finding suggested that compressing the interfacial separation (equivalent to applying a normal load) can reverse the corrugation of the potential energy surface (PES).
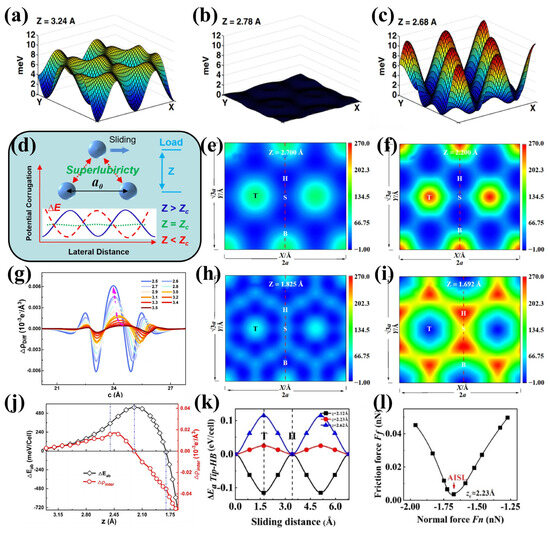
Figure 6.
DFT simulations of load-induced superlubricity: The “frictional collapse” of rare gases on metal surfaces [52], showing the continuous evolution of the sliding potential energy surface V(x, y, z) from (a) positive corrugation to (b) flat and finally to (c) negative corrugation; (d) Inversion of sliding energy barrier curvature under load leading to superlubricity, where the applied normal load drives the transition of the potential energy surface from positive corrugation (solid line: ) to negative corrugation (dashed line: ), with flattening of the energy landscape (dotted line: ) and disappearance of sliding friction dissipation at a critical pressure [7]; Evolution of PES corrugation in graphene/graphene interlayer sliding with decreasing interlayer spacing [7]: (e) slight corrugation, (f) severe corrugation, (h) flattened state, and (i) negative corrugation; (g) Evolution of interlayer charge density fluctuations in bilayer Gr/Gr under normal load and (j) its correlation with interlayer sliding energy barriers; (k) Inversion evolution of energy barriers along the sliding path at different heights for a Ag probe sliding on borophene supported by a Ti substrate, and (l) the corresponding friction force as a function of normal load [53].
In 2008, Righi et al. [52] confirmed via DFT that pressure induces a “frictional collapse” phenomenon: rare gases (Xe/Kr/Ar) exhibit a unique “anti-corrugation” effect on Cu(111) and Ag(111) surfaces. When the adsorption distance is compressed to a critical value, the potential energy surface becomes completely flattened. This overturns the classical rule that increased load always enhances friction. Instead, it reveals that normal pressure can achieve superlubricity by reconstructing the geometric morphology of the potential energy surface. Using van der Waals-corrected DFT calculations, Lu et al. [54] discovered that within the attraction-dominated region in the Xe/Cu(111) system, at a critical pressure of 0.75 GPa, the corrugation of the potential energy surface vanishes. The essence of its near-zero friction stems from the energy-level crossing effect induced by the van der Waals attraction far from the surface. They demonstrated that this law holds for a wide class of two-dimensional materials [7]: the graphene/graphene system exhibits a continuous evolution of the potential energy surface from a corrugated state → flattening → anti-corrugated state under ultrahigh pressure of ~280 GPa (Figure 6e,f,h,i), accompanied by a collapse of the frictional shear strength to nearly zero.
Similarly, pressure-induced reconstruction of the potential energy surface was observed in multicomponent systems such as Xe/Cu and Pd/graphene. Meanwhile, this pressure-induced superlubricity can be mutually verified with atomic force microscopy (AFM) theory [55]: simulations of a Cu tip scanning on a graphene surface revealed that the transition of the potential energy surface corrugation state strictly corresponds to the contrast inversion in AFM frequency shift signals. This correlation originates from the simultaneous crossing of the potential energy surface, interaction forces, and their second derivatives at the critical height. This mutual verification mechanism bridges superlubric states with experimental characterization for the first time, opening a new pathway for detecting zero-friction states.
Lin et al. [56] reported that compressing the interlayer spacing of silicon-supported graphene to 2.65 Å resulted in an anomalous reduction in the friction coefficient to the range of 0.0006–0.0011. This phenomenon was attributed to the smoothing of the potential energy landscape, induced by weak charge transfer (with a maximum of approximately 0.0093 eV) from the silicon substrate. He et al. [31] further elucidated that when the fluctuation in interfacial charge density reaches a critical threshold, the energy barrier becomes extremely low, enabling the achievement of superlubricity under experimentally feasible pressures. This decouples the conventional positive correlation between pressure and friction enhancement. Moreover, the authors of [53] observed, for the first time in a non-rare-gas system, attraction-induced superlubricity (AISL) (Figure 6k,l). This phenomenon arises from Coulomb-dominated electron redistribution regulation—at a critical height, the difference in charge density approaches zero, which can be identified through contrast reversal in AFM images. This provides a theoretical pathway for the experimental direct detection of AISL.
4. Design of Two-Dimensional Lubrication Interfaces
The friction mechanisms revealed by DFT can be directly used for the design of sliding interfaces with two-dimensional materials. Indeed, in light of the friction mechanisms identified, it is possible to modulate intrinsic material properties, leverage substrate effects and optimize heterojunction interfaces so as to obtain the desired tribological properties.
4.1. Screening of Intrinsic Material Properties
In designing lubricating interfaces based on two-dimensional materials, screening intrinsic material properties is a fundamental step toward constructing high-performance systems. Intrinsic properties not only determine the ultimate lubrication potential of materials, but also directly influence friction dissipation mechanisms through key parameters such as electronic structure, interlayer interactions, and lattice dynamics. DFT simulations, with their capability to precisely elucidate electronic behavior from first principles, provide an efficient theoretical framework for uncovering the intrinsic regulatory mechanisms of the lubricating properties of two-dimensional materials.
In 2015, Righi et al. [57] revealed key differences between graphene and transition metal dichalcogenides (TMDs) in terms of interlayer adhesion, the characteristics of the sliding potential energy surface (PES), and ideal shear strength through DFT analysis (Figure 7a). They found that although TMDs exhibit a higher PES energy barrier () and stronger interlayer adhesion () due to their larger interfacial atomic size, graphene possesses a higher density of energy barrier points on its PES owing to its smaller lattice constant. More importantly, they proposed and quantified the ideal interfacial shear strength as a direct and highly comparable intrinsic parameter for evaluating the inherent sliding capability of two-dimensional materials. Based on this insight, the authors of [58] subsequently discovered ultralow friction in a black phosphorus system that is even lower than that of graphene.
In 2021, Luo et al. [59] investigated five types of two-dimensional metal–organic frameworks (MOFs) with square grid structures. Using DFT calculations, they found that weak interactions between metal sites on the MOF surface and hydroxyl groups on a silica probe dominate frictional energy dissipation. The strength of this interaction is directly related to coordination stability. In the same year, Serles et al. [60] studied the low-friction characteristics of magnetene, a non-van der Waals two-dimensional material (Figure 7b). Using DFT analysis, they precisely quantified the low energy corrugation on its cleavage plane and revealed the mechanism whereby two-dimensional confinement-induced reduction in valence state significantly decreases the density of surface adsorbates.
Liu et al. [61] systematically investigated the interlayer frictional behavior of the two-dimensional magnetic material CrI3, observing near-zero and negative differential friction coefficients in van der Waals homostructures. Their DFT simulations showed a synergistic regulation mechanism involving magnetic phase transitions and electronic orbital reconstruction (Figure 7c): when an interlayer ferromagnetic-to-antiferromagnetic phase transition occurs, the sliding energy barrier is significantly reduced. Meanwhile, a pressure-induced intralayer transition of electronic orbital dominance from to leads to a non-monotonic evolution of the friction coefficient with normal load through triple coupling of van der Waals interactions, electrostatic potential, and lattice deformation.
Luo et al. [62] analyzed the intrinsic frictional behavior of covalent organic frameworks (COFs) (Figure 7d) and metal–organic frameworks (MOFs) (Figure 7e), observing that the inorganic–organic hybrid structure of MOFs exhibits superior lubrication advantages. Recently, He et al. [63] systematically evaluated the intrinsic frictional properties of nine types of hexagonal two-dimensional materials and identified germanene, phumphonene, and arsenene as promising candidates with lubricating potential. These three materials demonstrate unique advantages in strain response, far exceeding the minor deformation capability of graphene. Such DFT simulations enable preliminary exploration of the frictional properties of materials, thereby providing valuable guidance for subsequent experimental studies.
Remark that the low-friction properties exhibited by black phosphorus [64], magnetene [60], and MOFs [65] have been experimentally verified. Therefore, DFT-based calculations can be regarded as an effective screening approach for exploring new lubricating materials. However, it should be pointed out that such simulations are generally based on idealized crystal structures (e.g., perfect lattice, defect-free conditions), whereas the tribological performance of real materials is susceptible to various non-ideal factors such as defects, chemical doping, surface adsorption, and interfacial environments. Consequently, discrepancies may still arise between DFT predictions and experimental measurements, and caution must be taken when extrapolating theoretical results to practical applications.

Figure 7.
DFT analysis of intrinsic frictional properties of materials: (a) Variation in separation energy during sliding in different TMD systems [57]; (b) Changes in surface potential energy during sliding for hematene, magnetene, chromiteene, graphene, and MoS2 [60]; (c) Weight of the and orbitals at the highest-energy () and lowest-energy () sites for a Cr3I homojunction [61]; Structural diagrams (left) and sliding energy barriers (right) for (d) COF and (e) MOF homojunctions [62].
Figure 7.
DFT analysis of intrinsic frictional properties of materials: (a) Variation in separation energy during sliding in different TMD systems [57]; (b) Changes in surface potential energy during sliding for hematene, magnetene, chromiteene, graphene, and MoS2 [60]; (c) Weight of the and orbitals at the highest-energy () and lowest-energy () sites for a Cr3I homojunction [61]; Structural diagrams (left) and sliding energy barriers (right) for (d) COF and (e) MOF homojunctions [62].

4.2. Metallic Substrate Modulation Mechanisms
Due to the atomic-scale thickness of two-dimensional lubricating materials, when deposited onto a substrate as solid lubricants, charge transfer between the substrate and the material inevitably influences their surface frictional properties. Therefore, the modulation of the frictional behavior of two-dimensional lubricants by the supporting substrate cannot be overlooked [66,67,68,69]. Typically, when materials such as graphene and molybdenum disulfide are posed on a substrate, their electronic structure, interfacial coupling, and deformation characteristics are significantly modulated, thereby altering the friction dissipation mechanisms. Early experimental studies have confirmed the wide existence of this effect: Munzir et al. [66] observed a two-fold difference in the frictional force of graphene on different substrates; Qi et al. [67] found that edge friction is governed by both the passivation state of dangling bonds and edge pinning effects. Zeng [68] and Shi [69] demonstrated that enhancing the material–substrate binding energy can suppress wrinkling in graphene and structural instability in molybdenum disulfide, respectively, offering pathways for achieving ultralow friction. These experimental findings require interpretation through microscopic theory, and DFT is well-suited to elucidate the interaction mechanisms at the substrate–lubricant interface.
The DFT study by Kelly et al. [70] was the first one to systematically investigate the regulatory role of the substrate, confirming that substrates alter the electronic properties of the material supported. In 2017, Qiao et al. [11] focused on strongly bonded substrate systems. Through DFT simulations of the sliding behavior of an Ir/Au tip on a graphene/Ni(111) surface (Figure 8b,c), they uncovered a unique electronic orbital competition mechanism: significant hybridization between the tip and graphene orbitals induces strong charge transfer, leading to an inversion of the adsorption energy sequence. This study broke through conventional contact mechanics understanding by demonstrating that strongly bonded substrates enable active design of frictional behavior through modulation of orbital hybridization states.
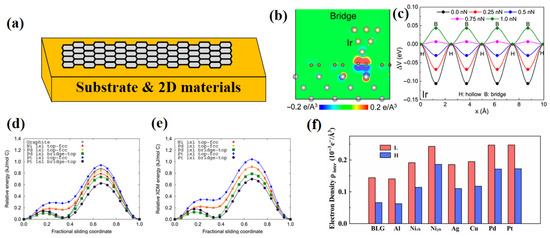
Figure 8.
DFT investigations of substrate effects on friction: (a) Schematic of a two-dimensional material on a substrate [11]; (b) Charge transfer at the Bridge site and (c) sliding energy barrier for an Ir probe sliding on graphene supported by a Ni substrate; (d) Sliding energy barrier and (e) dispersion-contributed sliding energy barrier for a monolayer graphene sliding on another metal-supported graphene layer [71]; (f) Interlayer charge density at the L and H sites (where H and L denote the highest and lowest energy sites, respectively) in a bilayer graphene system supported by a metal substrate [72].
Johnson et al. [71] extended the research to bilayer graphene (BLG) systems (Figure 8d,e), proposing a dual regulatory model involving substrate binding strength and lattice orientation. Physically adsorbed substrates (e.g., Al, Cu, Ag, Au) influence interlayer sliding only through slight strain, whereas chemically adsorbed substrates (e.g., Ni, Pd, Pt) significantly reduce the interlayer spacing of BLG (). The enhanced dispersion forces stabilize high-energy transition states (such as AA stacking) in the upper graphene layer, diminishing the sliding energy barrier by up to 30%. In 2023, He et al. [72], using both point and surface model systems, elucidated universal substrate modulation mechanisms from a charge density perspective (Figure 8f): substrates alter the friction on the graphene surface by modulating fluctuations in the interlayer charge density. This implies that energy barrier suppression can be achieved by reducing interfacial charge density fluctuations, providing a design road towards optimizing the lubricating performance of two-dimensional materials.
4.3. Heterointerface Design Strategies
Heterostructure interface design serves as a crucial strategy for achieving structural superlubricity (SSL) (Figure 9a). Unlike strain or rotation modulation, heterostructures gives inherently rise to incommensurate contact interfaces by directly combining different two-dimensional materials [21,73]. However, whether a heterostructure achieves superlubricity depends not only on geometric matching but also on the complex interactions between the materials. Theoretical guidance through computational studies can effectively prevent wasteful trial-and-error experimentation.
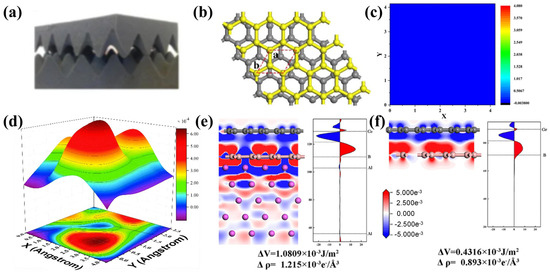
Figure 9.
DFT studies on heterostructure design: (a) Schematic of an incommensurate model [73]; (b) Heterostructure model and (c) corresponding potential energy surface (PES) plot for graphene/borophene assemblies [74], where a and b represent the lattice parameters inside the plane; (d) PES plot for a heterostructure composed of graphene and tellurene [75]; Differential charge density (left) and quantitative profile of the corresponding differential charge density (right) for (e) graphene on Al-supported borophene and (f) graphene on freestanding borophene [76].
Lu et al. [74] used DFT calculations to predict the ultralow friction mechanism in a graphene/hexagonal borophene heterostructure (Figure 9b,c). The sliding energy corrugation in this system is only 0.052 meV/atom—two orders of magnitude lower than that of a graphene homostructure. Analysis of the microscopic mechanism indicates that the electron-deficient structure of borophene promotes slight charge transfer from graphene, augmenting the electron density in the boron layer. Simultaneously, a 4.65% lattice mismatch weakens the atom-to-atom registry effect. These two factors synergistically suppress the energy barrier.
It is noteworthy that intrinsic borophene requires substrate support due to its structural instability [76]. An Al(111) substrate can stabilize the honeycomb structure of borophene and reduce the activity of dangling bonds, enabling the heterostructure to maintain low charge density fluctuations (Figure 9e,f) and an ultralow energy corrugation of 0.052 meV/atom even when supported. Zhang et al. [75] designed a Graphene/Antimonene heterostructure and demonstrated that structural superlubricity can be achieved between graphene and an antimonene monolayer under misaligned contact conditions. This phenomenon is primarily attributed to a reduced work of separation, which helps maintain an ultralow friction coefficient. Recently, He et al. [63] observed ultralow friction in heterojunctions assembled from germanene, phosphorene, and arsenene with graphene, compared to their homojunction counterparts. An extremely low sliding energy barrier between black phosphorus and graphene was also a key finding by Xu et al. [77].
However, not all heterostructures satisfying the incommensurate condition can achieve superlubricity. Through a systematic DFT study of 208 types of two-dimensional heterostructures, Liu et al. [78] proposed a triple-criterion constraint (Figure 10a–c): the interfacial binding energy must be below a critical value of 0.5 J/m2; the in-plane stiffness must suppress lattice deformation; and a dimensionless parameter ζ, representing the ratio of the two, must exceed a material-dependent threshold. A notable counterexample is the C2/Sc3C2 heterostructure: although it satisfies the geometric condition of incommensurability, its binding energy reaches 1.29 J/m2—far exceeding the 0.5 J/m2 threshold—and its ζ value is lower than that of a MoS2 homostructure, resulting in an average frictional stress as high as 271.78 MPa. Analysis of the underlying mechanism reveals that high binding energy induces significant in-plane deformation, promoting the formation of locally commensurate contact regions. When the binding energy exceeds 1.0 J/m2, covalent bonds may even form (as in the Si2/B5 system), completely eliminating sliding degrees of freedom. This work demonstrates that achieving structural superlubricity in heterostructures requires coordinated control of both interfacial binding strength and mechanical properties of the materials.
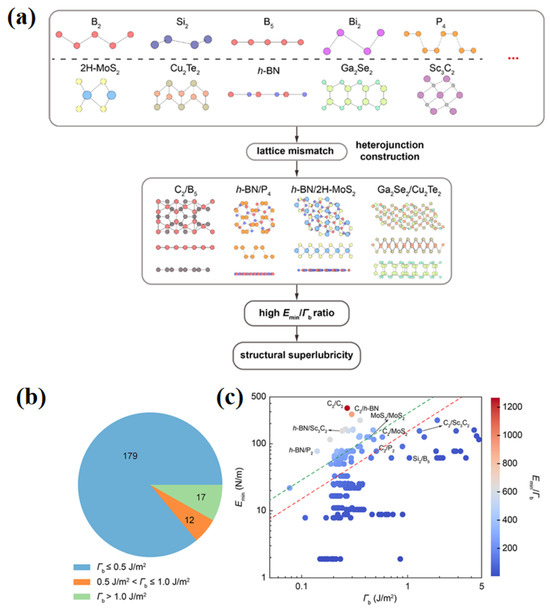
Figure 10.
Screening of heterostructural superlubricity materials via DFT [78]: (a) Screening workflow for identifying superlubricative heterointerfaces from 2D material candidates; (b) Distribution of binding energies (Γb) across the 208 heterostructures screened in (a); (c) In-plane stiffness versus binding energy (Γb) for the 208 heterostructures, compared with C2/C2 (graphite) and MoS2/MoS2 homojunctions.
5. Active Control of Nanofriction
5.1. Electric Field Modulation of Interfacial Charge
Electric field regulation is a core non-contact method for friction control, enabling active manipulation of tribological behavior by altering interfacial charge distribution or molecular arrangement. In wet tribological systems, electric fields can induce the formation of short-range ordered structures in ionic solutions [79], thereby modifying boundary lubrication characteristics. In dry friction systems, applied bias voltages influence charge density distribution by modulating surface potential, as exemplified by the precise manipulation of sample friction via tip bias in atomic force microscopy [80,81,82]. This control strategy has attracted significant attention due to its non-contact nature. DFT simulations can effectively elucidate their underlying microscopic mechanisms.
Early DFT studies focused primarily on the influence of electric fields on the intrinsic electronic properties of materials. In 2015, Wang et al. [83], using van der Waals-corrected DFT calculations, identified a non-monotonic modulation mechanism of a vertical electric field on the interlayer friction of MoS2: when the field strength is below 0.25 V/Å, charge accumulation between adjacent sulfur planes enhances chemical coupling, leading to a slight increase in the frictional energy barrier and lateral force; once the field strength exceeds the critical value of 0.25 V/Å, charge dissipation weakens interfacial bonding, resulting in a significant reduction in friction. This study confirmed at the electronic scale that electric fields can modulate interfacial bonding states by reconstructing interlayer charge distribution. One year later, the same team reported a new discovery in graphene systems [84]: a vertical electric field induces asymmetric charge redistribution by altering the electronic structure difference between AA and AB stacking, enabling continuous modulation of the friction coefficient.
In 2024, Ouyang et al. [85] designed a graphene/h-BN/graphene/h-BN heterostructure device and achieved decoupled regulation of electronic friction and adhesion force in atomic force microscopy experiments. By precisely controlling the carrier density in the top graphene layer, they observed a 28% monotonic increase in friction with carrier density and revealed the underlying mechanism using DFT (Figure 11d,g): the electric field-induced interfacial charge redistribution exhibits a linear correlation with the sliding energy barrier.
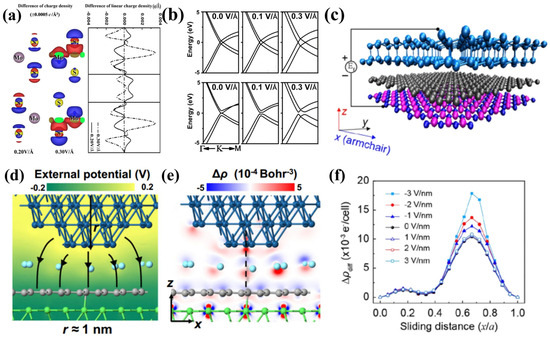
Figure 11.
DFT analysis of material friction under external electric fields: (a) Differential charge density maps of MoS2 under different electric fields (left) and their quantitative profiles along the z-axis (right) [83]; (b) Band structures of bilayer graphene under varying electric fields [84]; (c) Simulation model of a Si tip sliding on a graphene/h-BN heterostructure under an external electric field and (f) quantitative profile of differential charge density along the sliding path [85]; (d) Electrostatic potential and (e) differential charge density between an Ir probe and Ni-supported graphene [12].
Ma et al. [12] utilized low-amplitude alternating current (AC) to achieve an ultralow friction state in a Gr/Ni system: the friction force was reduced to 25% of its original value and maintained continuous sliding for over 70,000 s (nearly 20 h) under a high contact pressure of 9.1 GPa. Scanning electron microscopy confirmed the absence of wear at the interface. Through combining DFT and non-equilibrium Green’s function (NEGF) calculations, they elucidated the core mechanism of AC (Figure 11e,f): a dynamic bias voltage drives periodic fluctuations in interfacial charge density, inducing lateral force perturbations on the tip atoms. Using an extended Prandtl–Tomlinson model with thermal activation (PTT-E), the same team quantitatively explained the physical essence whereby electron density fluctuations reduce the effective energy barrier, leading to “early slip”. This mechanism has also proven to be effective in systems such as Ir/Gr/Cu and Ir/h-BN/Au, with its modulation efficiency exhibiting a positive correlation with interfacial conductivity. In contrast to conventional methods, such as static electric fields, which rely on bonding strength modulation, or mechanical vibrations that require precise resonance matching, the AC strategy offers distinctive advantages including resonance-free tuning, low driving voltage, zero macroscopic vibration, and high durability. This approach provides a promising pathway to overcome the high-pressure friction and wear bottlenecks in nano-electromechanical systems (NEMS).
5.2. Chemical Terminal Passivation
Chemical passivation constitutes an effective approach to modulating the frictional behavior of two-dimensional materials. Through treatments such as fluorination and oxidation, it alters the surface chemical state, thereby influencing electronic structure and interfacial interactions. Such modifications are often inevitable in practical applications, and their mechanisms can also be precisely identified using DFT calculations.
In 2012, Ma et al. [86] revealed the fundamental mechanism of friction modulation in graphene oxide (Figure 12a,b). The oxidation process introduces epoxy (-O-) and hydroxyl (-OH) functional groups, leading to three major effects: localized wrinkling and sp2-to-sp3 hybridization transition; charge redistribution; and significant expansion of interlayer spacing. This study demonstrated that oxidation reconstructs interfacial bonding properties via functional groups, providing a microscopic model for understanding friction hysteresis induced by environmental humidity.
Jia et al. [87] systematically compared the frictional behavior of hydrogenated and fluorinated diamond films, uncovering the microscopic regulation mechanism of chemical terminal passivation (Figure 12d,e,h,i). The fluorinated surface, due to the high electronegativity of fluorine atoms, causes electron accumulation from carbon toward the fluorine layer, forming a dense electron shielding layer that effectively reduces interfacial energy corrugation. In contrast, the hydrogenated surface, dominated by covalent C–H bonds, only induces localized charge redistribution with a weaker electron shielding effect. In 2013, Wang et al. [88] analyzed the differences in frictional behavior between fluorinated graphene (FG) and graphane (HG). They revealed that fluorine atoms, with higher electronegativity, induce significant charge transfer, causing electron accumulation in the fluorine layer and forming a strong electrostatic repulsion interface. This results in an interlayer spacing expansion to 5.76 Å (much larger than HG’s 4.4 Å) and significantly suppresses interlayer spacing fluctuations during sliding. It was confirmed that fluorination reduces the friction coefficient by 47% compared to hydrogenation by weakening interfacial energy corrugation.
Wang et al. [89] combined fluorination with heterostructure design to achieve superlubricity in a fluorinated graphene/MoS2 (FG/MoS2) system. DFT calculations (Figure 12f,j) showed that a 4.8% lattice mismatch generates a Moiré pattern, reducing the interlayer energy barrier to 0.0096 meV/atom—only 3% of that in FG/FG homostructure and 0.4% of MoS2/MoS2. The mechanism lies in the mutual cancelation of localized energy fluctuations within the Moiré superlattice, resulting in an ultra-flat potential energy surface. This synergistic effect yields a shear strength as low as 2.9 MPa, one to two orders of magnitude lower than in homogeneous systems. This work demonstrates that combining chemical modification with geometric mismatch can exceed the performance limits of a single modulation strategy.
Sun et al. [90], through experiments and DFT calculations on a hydrogenated DLC system, evidenced the core role of interfacial electronic interactions in achieving ultralow friction (Figure 12c,g). The study showed that high hydrogen content (34.56%) DLC can achieve a friction coefficient as low as 0.005 ± 0.001 in a vacuum environment, nearly an order of magnitude lower than that of low hydrogen content samples (5.4%). Hydrogen atoms significantly suppress interfacial charge interactions by saturating surface dangling bonds: at 100% hydrogen coverage, the interfacial adhesion energy decreases to 16.77 meV/Å2 and charge redistribution is only 0.63 × 10−3 e−/Å2, representing reductions of 98% and 99.8%, respectively, compared to a hydrogen-free system. More importantly, charge evolution during sliding exhibits a linear relationship with frictional dissipation barriers, and static adhesion energy also correlates linearly with charge redistribution. The consistency of these two linear relationships indicates that chemical passivation collectively weakens interfacial electronic coupling by inhibiting charge flow, fundamentally reducing energy dissipation.

Figure 12.
DFT analysis of chemical terminal passivation: (a) Structural diagram and (b) variation in sliding energy (top) and lateral force (bottom) along a specified path for the C8OH system [86]; (c) Schematic of passivated DLC structure (where dC and dH represent distinct equilibrium distances) and (g) consistency between charge redistribution and electronic mechanisms underlying ultralow friction fluctuations in DLC [90]; (d,e) Structural diagrams and (h,i) charge density maps for (d,h) hydrogenated and (e,i) fluorinated DLC [87]; (f) Structural diagram and (j) sliding energy along a specified path for a heterostructure assembled from fluorinated graphene and MoS2 [89].
Figure 12.
DFT analysis of chemical terminal passivation: (a) Structural diagram and (b) variation in sliding energy (top) and lateral force (bottom) along a specified path for the C8OH system [86]; (c) Schematic of passivated DLC structure (where dC and dH represent distinct equilibrium distances) and (g) consistency between charge redistribution and electronic mechanisms underlying ultralow friction fluctuations in DLC [90]; (d,e) Structural diagrams and (h,i) charge density maps for (d,h) hydrogenated and (e,i) fluorinated DLC [87]; (f) Structural diagram and (j) sliding energy along a specified path for a heterostructure assembled from fluorinated graphene and MoS2 [89].

5.3. Doping Design and Defect Engineering
Doping design and defect engineering act as atomic-scale strategies for modulating electronic structure, enabling the targeted optimization of the tribological properties of two-dimensional materials through precise control of dopant elements and defect concentrations. DFT calculations play a central role in elucidating the structure–property relationships between doping and frictional performance.
Lu et al. [91] compared nitrogen/boron-doped graphene systems (Figure 13a,b) and elucidated the key role of electronegativity gradients: nitrogen atoms (electronegativity 3.04) donate electrons, forming interlayer electrostatic repulsion. At a doping concentration of 12.5%, the energy barrier on the potential energy surface decreased to 0.630 meV/atom—a 51% reduction compared to the intrinsic system—resulting in a 30% decrease in the friction coefficient under high load (>1.13 nN/atom). In contrast, boron atoms (electronegativity 2.04) enhance interlayer attraction due to hole doping, increasing the friction coefficient by 55%. This work established a quantitative correlation between the electronegativity sequence “N > C > B” and frictional behavior, confirming the advantageous role of electron-donor characteristics in improving lubrication.
Jia et al. [92] conducted an atomic-scale study of phosphorus-doped nanocrystalline diamond films (Figure 13d,e,h) and found that phosphorus doping induces interfacial charge redistribution and significantly enhances the C–H dipole repulsion between hydrogenated diamond films, reducing the friction coefficient to 12.5% of that of the undoped system (under a 0.5 nN load). This effect is highly dependent on the interfacial chemical environment: a 40% friction reduction was achieved on hydrogenated surfaces, while on fluorinated surfaces it diminished to 15% due to electron shielding, confirming the sensitivity of doping effects to the chemical terminal.
In 2022, Du et al. [93] used DFT calculations to identify the regulation mechanism of Mo doping on the tribological properties of Cr2TiAlC2-based MAX materials. After Mo atoms substituted Cr sites, the adsorption energy of oxygen atoms on the (010) crystal plane significantly decreased (Figure 13c). For example, the adsorption energy at the O3 site dropped from 5.878 eV in Cr2TiAlC2 to a lower value in the doped system, indicating enhanced oxidation resistance. Meanwhile, Mo doping increased the interlayer binding energy, with Cr1.8Ti0.8Mo0.4AlC2 reaching −274 meV/Å2—a 3.8% augmentation compared to the undoped system (−264 meV/Å2)—attributed to Mo-induced charge redistribution and interlayer space charge accumulation. The study formed a closed loop of theoretical calculations and experimental observations, confirming the non-linear regulation of tribological behavior by doping concentration.

Figure 13.
DFT Analysis of Friction Doping Design and Defect Engineering: (a) Structural diagram and (b) variation in potential energy along a specified path for boron- and nitrogen-doped bilayer graphene [91]; (c) Adsorption energy of oxygen on undoped and Mo-doped Cr2TiAlC2 [93]; (d) Energy versus interlayer distance relationship and (e) coefficient of friction (COF) as a function of pressure for undoped and phosphorus-doped DLC; (f) Structural diagram and (g) variation in interaction energy along a specified path for defective bilayer graphene [94].
Figure 13.
DFT Analysis of Friction Doping Design and Defect Engineering: (a) Structural diagram and (b) variation in potential energy along a specified path for boron- and nitrogen-doped bilayer graphene [91]; (c) Adsorption energy of oxygen on undoped and Mo-doped Cr2TiAlC2 [93]; (d) Energy versus interlayer distance relationship and (e) coefficient of friction (COF) as a function of pressure for undoped and phosphorus-doped DLC; (f) Structural diagram and (g) variation in interaction energy along a specified path for defective bilayer graphene [94].

Minkin et al. [94] investigated the influence of defect engineering on the superlubricity state (Figure 13f,g,i,j), noting that excessive doping (e.g., Mo at 0.4 atomic ratio) induces lattice distortion and electronic structure perturbation, leading to abnormally enhanced interlayer binding energy (−274 meV/Å2), which hinders the self-adaptive reconstruction of the friction film during shear. Experiments confirmed that under these conditions, the friction coefficient sharply increases to 0.49 at 600 °C, and the wear rate (6.0 × 10−7 mm3N−1m−1) rises nearly tenfold compared to the optimized composition (0.2 atomic ratio Mo), revealing a critical inverse relationship between defect concentration and the superlubricity state. In a study on defect engineering, Lu et al. [91] quantified the negative impact of vacancy defects on the frictional properties of graphene. They found that under high pressure, vacancy defects significantly exacerbate friction, increasing the friction coefficient by approximately 55% compared to the intrinsic system—even higher than that of the boron-doped system.
6. Development and Applications of DFT
6.1. DFT + Machine Learning
In simulating friction by DFT, a quasi-static approach is often employed to approximate dynamic processes. For simple systems, such as bilayer graphene, the high symmetry allows for improved computational efficiency through sparse sampling of points. However, for complex interfacial systems, such as multi-atom heterostructures, the need for dense sampling on a high-dimensional potential energy surface (PES) leads to prohibitive computational costs. Recently, the implementation of DFT with machine learning (ML) strategies has effectively opened new perspectives.
Lu et al. [95] innovatively constructed a descriptor database encompassing geometric structure, mechanical properties, and electronic states. Using a random forest algorithm, they achieved high-precision prediction of the maximum sliding energy barrier () for two-dimensional materials, and identified a strong correlation between the initial interfacial charge transfer amount and frictional performance—model accuracy improved significantly when this parameter was used as a descriptor. The automated platform developed in this study (LICP-FPHTC-Platform) enhanced the efficiency of PES generation by an order of magnitude, offering a new approach for high-throughput screening of low-friction materials.
In 2024, Guo et al. [96] developed a deep neural network (DNN) trained on DFT data, which can predict charge density in real time using only atomic positions as input, successfully enabling nanosecond-scale simulation of water–graphene interfaces. The study established an inverse relationship between slip length and charge density, explaining the mechanism behind the reduced friction coefficient in defective systems. Also in the same year, Woods et al. [97], based on a global dataset of 760 bilayer materials (BMDS), used a gradient-boosted regression model to achieve cross-material prediction of adhesion energy, van der Waals energy, and corrugation energy. This model established a quantitative structure–property relationship linking atomic attributes to macroscopic interfacial behavior.
Wang et al. [98] recently conducted high-throughput DFT screening of 6351 types of monolayer materials and identified 25 high-performance sliding ferroelectrics. Big data analysis revealed that the effective van der Waals radius (quantifying interlayer interactions) is a key hidden variable governing the energy barrier. Based on this, an equivariant machine learning model was developed, enabling high-accuracy prediction of out-of-plane polarization.
6.2. Analysis of Friction Coatings
DFT can provide unique insights into addressing the tribological challenges of modern industrial equipment under extreme operating conditions by revealing the microscopic mechanisms of solid lubricant coatings.
Liu et al. [99] used DFT calculations to elucidate the unique plastic deformation behavior of the layered oxide Na2ZrO3 (Figure 14a,b,d). A key finding was that the interlayer sliding energy barrier on the (001) plane is significantly lower than the cleavage energy, highlighting the solid lubricating potential of layered oxides. Geng et al. [100] combined DFT calculations with high-temperature experiments to systematically explain the mechanism underlying a 30% reduction in the friction coefficient (to 0.30) and an 83% decrease in wear rate in a NiCrAlY-MoO3/CoO composite coating at 800 °C. DFT simulations revealed that the core mechanism involves the evolution of metal–oxygen bond strengths in the layered ternary oxides CoMoO4 and NiMoO4 formed in situ on the worn surface (Figure 14c): the Co–O bond energy (−1.05 eV) and Ni–O bond energy (−0.61 eV) are significantly lower than those in the corresponding binary oxides CoO (−1.99 eV) and NiO (−1.44 eV). This weakened bonding character makes the layered lattice more prone to preferential fracture under high-temperature shear, promoting interlayer sliding as the dominant friction mechanism and leading to a breakthrough in high-temperature lubrication.
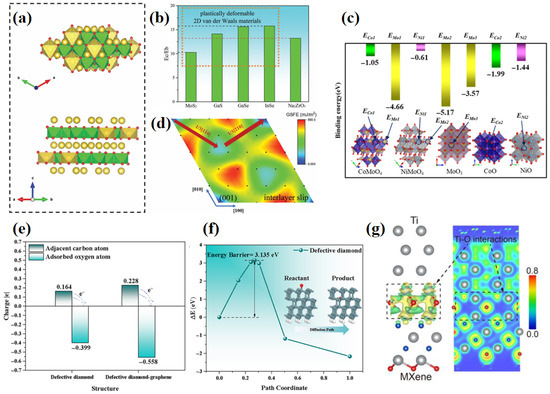
Figure 14.
DFT Analysis of Friction Coatings: (a) Crystal structure of Na2ZrO3; (b) Cleavage energy ()/binding energy () for the (001) planes of MoS2, GaS, GaSe, InSe, and Na2ZrO3; (d) Generalized stacking fault energy (GSFE) surface for the (001) plane of Na2ZrO3 [99]; (c) Binding energies of Co-O, Mo-O, and Ni-O bonds in CoMoO4, NiMoO4, MoO3, CoO, and NiO [101]; (e) Adsorption energy for defective DLC and defective DLC-graphene interfaces, and (f) the diffusion energy barrier for an oxygen atom migrating from the reactant to product state in defective DLC [98]; (g) Schematic of interfacial interactions between metallic Ti and Ti3C2Tx MXene [100].
Wang et al. [98] employed DFT to clarify the mechanism responsible for a 35% reduction in the friction coefficient and for significantly improved wear resistance in a diamond-graphene heterostructure coating under high-temperature friction conditions (Figure 14e,f). DFT results showed that the adsorption energy of oxygen atoms on the defective graphene layer (−0.2702 eV) increased by 123% compared to that on a pure diamond surface (−0.1211 eV), while the diffusion energy barrier for oxygen atoms rose sharply to 13.029 eV—a 316% increase compared to the value without a graphene coating (3.135 eV). This strong carbon–oxygen interaction, induced by defect sites in graphene, forms a high-energy diffusion barrier that effectively suppresses oxygen penetration into the diamond substrate. Coupled with the formation of epoxy groups (C–O–C) on the coating surface, this mechanism promotes preferential self-sacrificial fracture in the flexible graphene layer, delaying brittle failure of the diamond matrix and resulting in a breakthrough enhancement of high-temperature lubrication and anti-wear performance.
Geng et al. [100] used DFT to study the interlayer interactions at the interface between metallic Ti and Ti3C2Tx MXene. Charge density difference analysis revealed significant interfacial charge transfer (0.35e) (Figure 14g), forming a strong chemisorption structure. This unique bonding behavior reduces the energy barrier for tribochemical protection (with an experimentally measured activation energy of only 1.00 ± 0.24 eV), enabling the nanofilm to achieve a near-superlubricious state with a friction coefficient below 0.01 under a contact pressure of 10 GPa and temperature of 473 K, while the wear rate approaches zero (<10−9 mm3/N·m).
6.3. Mechanisms of Nanoscale Triboelectric Generators
DFT has also turned out to be efficient in elucidating the microscopic energy conversion mechanisms of triboelectric nanogenerators (TENGs). By precisely analyzing key parameters such as surface electronic structure, charge transfer barriers, and interfacial binding energy, this theory provides an atomic-scale foundation for the functional modification of triboelectric materials, facilitating the transition of micro–nano energy harvesting technology from basic research to engineering applications.
Li et al. [13] fabricated a triboelectric nanogenerator (TENG) based on reduced graphene oxide film (rGOF), introducing varying proportions of oxygen and carbon defects by controlling annealing temperatures (from 500 °C to 3000 °C). DFT analysis (Figure 15a–c) revealed that oxygen defects form outward C–O dipoles, increasing the out-of-plane component of electron density and raising the work function from 4.48 eV to 5.40 eV. In contrast, Stone–Wales carbon defects shorten the average C–C bond length (from 1.42 Å to 1.41 Å), significantly reducing the work function to 4.35 eV. This atomic-scale mechanism explains the minimal work function (4.49 eV) of the rGOF-2000 sample. As a positive triboelectric material, it maximizes electron transfer efficiency, enabling the TENG to achieve an output voltage of 190 V and a power density of 5.04 W/m2—a nine-fold increase compared to the pristine GOF. Additionally, in samples annealed above 2500 °C, high crystallinity and enhanced interlayer van der Waals interactions improve tensile strength to 209.8 kPa, while reduced interfacial wear is achieved due to significantly improved mechanical stability.
Zhang et al. [102] developed a hydrogenated diamond-like carbon (DLC)-based superlubricious triboelectric generator (SL-TENG) that, under a contact stress of 1.37 GPa, achieves a friction coefficient below 0.01 and an ultralow wear rate (2.6 × 10−7 mm3/N·m)—an 87.5% reduction compared to graphite-like carbon (GLC) film. Using a sliding model with hydrogen-terminated diamond substrate and a bilayer graphene interface, DFT calculations indicated that the hydrogen-terminated surface suppresses the sliding energy barrier to as low as 17.8 meV/cell (one-fifth of that of a dangling-bond surface). By inhibiting interfacial mechanical interlocking and adhesion, this study reveals the physical essence of decoupling electron transfer and mechanical dissipation in the superlubricious state.

Figure 15.
DFT Analysis of Nanotriboelectric Generators: (a,b) Work function of graphene with (a) oxygen vacancies and (b) carbon vacancies; (c) Charge density of pristine graphene and with 1, 5, and 10 oxygen adatoms [13]; (d,e) DFT simulation diagrams of porous cellulose (LIPC) under (d) stress-free conditions and (e) 0.1 MPa stress, showing (i) side view of the optimized structure, (ii) top view and (iii) side view of the electrostatic surface potential [103]; (f,g) Work function calculation plots corresponding to (d) and (e), respectively [103]; (h,i) Total charge density maps for cellulose under (h) stress-free conditions and (i) 0.1 MPa stress [104].
Figure 15.
DFT Analysis of Nanotriboelectric Generators: (a,b) Work function of graphene with (a) oxygen vacancies and (b) carbon vacancies; (c) Charge density of pristine graphene and with 1, 5, and 10 oxygen adatoms [13]; (d,e) DFT simulation diagrams of porous cellulose (LIPC) under (d) stress-free conditions and (e) 0.1 MPa stress, showing (i) side view of the optimized structure, (ii) top view and (iii) side view of the electrostatic surface potential [103]; (f,g) Work function calculation plots corresponding to (d) and (e), respectively [103]; (h,i) Total charge density maps for cellulose under (h) stress-free conditions and (i) 0.1 MPa stress [104].

Nguyen et al. [103] developed a Tribo–Hygro–Electric Generator (THEG), an innovative device that leverages the synergistic effect between liquid-infused porous cellulose (LIPC) and metal contacts (such as aluminum and copper) to enable combined mechanical and hygroscopic energy harvesting. Using DFT, they conducted an in-depth analysis of the microscopic energy conversion mechanism of LIPC under pressure (Figure 15d–g): simulations of the geometrically optimized structure and electrostatic potential distribution of cellulose chains under stress evidenced a transition from insulating to semiconductor-like behavior under 0.01 MPa stress—the band gap energy decreased significantly from 11.03 eV to 2.65 eV, while the work function dropped from 6.44 eV to 4.53 eV. This atomic-scale insight clarifies the formation principle of the Schottky contact at the Al/LIPC interface and the unidirectional charge transfer process driven by the internal electric field, providing a theoretical basis for THEG’s high current density output and multi-functional sensing capabilities. On the basis of this work, the same team [104] further developed a Metal–Organic Polymer-based Heterojunction Triboelectric Generator (MOPH-TEG). This device integrates a heterostructure composed of water-infused porous cellulose (WIPC), carbon fiber sheets (CF), and aluminum/copper electrodes to achieve efficient conversion of mechanical and moisture energy. Simulations showed that under 0.1 MPa compressive stress, the geometrically optimized structure of cellulose undergoes molecular rearrangement (Figure 15h,i), leading to a notable change in electrostatic potential distribution. The work function decreased from 6.44 eV (stress-free) to 4.302 eV, while the band gap narrowed from 2.65 eV to 2.17 eV, confirming the material’s transition from an insulating state to p-type semiconductor behavior. Charge density difference mapping further revealed a positive charge region at the cellulose/CF interface (facilitating hole transport) and the formation of a built-in electric field at the Al/cellulose interface (driving directional electron transfer), giving atomic-scale evidence for the charge separation and low-barrier transport mechanism in the Schottky contact.
7. Conclusions
In the field of tribology, different approaches have been proposed. The classical theoretical, experimental and numerical methods based on continuum mechanics and continuum physics are suitable for investigating the tribological behaviors of systems at the macroscopic level [105,106]. The approach using molecular dynamics (MD) allows studying the tribological behaviors of systems at the microscopic scale and also bridging the microscopic and macroscopic scales [107]. The approach base on Density Functional Theory (DFT) has been proved to be a powerful and versatile tool for tribological research. DFT has not only uncovered the quantum mechanical nature of frictional dissipation but also established a universal paradigm of “geometric structure–electronic redistribution–energy dissipation.” This article aims mainly to systematically review the contributions of DFT to investigating structural superlubricity, continuous sliding regulation and pressure-induced superlubricity. The essence of structural superlubricity lies in the minimization of interfacial charge density fluctuations, offering quantitative criteria for the design of incommensurate contacts. The strain-coordination strategy has achieved a frictionless state in commensurate systems for the first time, challenging the conventional understanding that relies solely on incommensurability. The critical pressure model for pressure-induced superlubricity demonstrates that the pressure required for the occurrence of superlubricity can be reduced from theoretically high levels to engineering-feasible ranges, in addition, while the concordance of the results from the potential energy surface flattening and the atomic force microscopy image inversion bridges theoretical predictions with experimental investigations. These advancements contribute significantly to the development of tribology at the micro-scale.
In the field of two-dimensional material interface engineering, the DFT-guided three-dimensional design pathway demonstrates its robust potential: intrinsic property screening identifies low-dissipation lubricating materials such as phosphorene and black phosphorus; metallic substrates dynamically optimize interface states through charge transfer modulation; heterojunction design, characterized by “incommensurability–energy barrier–electron” coupling, provides a universal blueprint for structural superlubricity. This multi-parameter synergistic design philosophy advances friction control from empirical adjustments to rational designs. Cutting-edge strategies for active friction regulation have been revitalized under DFT analysis. Electric field modulation achieves continuous superlubricity over 70,000 s at 9.1 GPa pressure in the Gr/Ni system by inducing periodic charge fluctuations through alternating current; chemical terminal engineering triggers Moiré superlattice effects in fluorinated graphene/MoS2 heterostructures, leading to a sharp drop in shear strength; nitrogen-doped graphene reduces the sliding energy barrier by altering electronic properties; the passivation of dangling bonds with hydrogen molecules produces an exponential decay in friction, etc. These breakthroughs demonstrate the deterministic transition of DFT from passive analysis to active design.
The current research on tribology using DFT-based computations still faces three major bottlenecks. First, DFT methods are constrained by both spatial and temporal scales. Based on first principles for electronic structures, DFT itself is resource-intensive and costly, making it difficult to directly simulate large-scale systems or long-term dynamic processes such as wear and fretting fatigue. Although some cross-scale methods invoking molecular dynamics (such as AIMD [108,109] and QM/MM [110,111]) have been developed, a systematic and universally applicable multi-scale modeling framework is still lacking. Second, existing DFT-based studies of friction primarily focus on solid dry interfaces, so that explanation of the mechanisms in liquid lubrication environments remains insufficient. Indeed, lubricated media involve dynamic electron transfer and rearrangement processes, the applicability and predictive accuracy of the current DFT theoretical framework in wet friction systems require further developments. Finally, the understanding of the behavior of magnetic materials during friction processes remains insufficient. In particular, the mechanisms underlying friction-induced changes in magnetic states, spin-dependent electronic excitations, and their influence on energy dissipation pathways are still not well understood, which makes the application of DFT in studying the effect of magnetism on tribology largely open.
As DFT continues to deepen our understanding of tribological mechanisms, research is developing by combining phenomenon observation and mechanism-driven investigation, advancing humanity toward the ultimate goal of “quasi-zero dissipation moving interfaces.” With further developments in DFT-based simulations, the research of tribology is expected to achieve a transition from “passive analysis” to “active design” at the atomic scale, advancing the manufacture of micro/nano energy systems.
Author Contributions
H.F.: methodology, data curation, formal analysis, writing—original draft and editing; Z.C.: supervision, editing and review; Z.L.: supervision; Q.H.: editing, supervision, funding acquisition, and review. All authors have read and agreed to the published version of the manuscript.
Funding
This work was financially supported by the National Natural Science Foundation of China (Grant Nos. 11890673, 11972344) and the Chengdu Aeronautic Polytechnic University Foundation (Grant Nos. ZZX0624164).
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Pitenis, A.A.; Dowson, D.; Gregory Sawyer, W. Leonardo da Vinci’s friction experiments: An old story acknowledged and repeated. Tribol. Lett. 2014, 56, 509–515. [Google Scholar] [CrossRef]
- Dienwiebel, M.; Verhoeven, G.S.; Pradeep, N.; Frenken, J.W.; Heimberg, J.A.; Zandbergen, H.W. Superlubricity of graphite. Phys. Rev. Lett. 2004, 92, 126101. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Ning, Z.; Zhang, Y.; Zheng, Q.; Chen, Q.; Xie, H.; Zhang, Q.; Qian, W.; Wei, F. Superlubricity in centimetres-long double-walled carbon nanotubes under ambient conditions. Nat. Nanotechnol. 2013, 8, 912–916. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Smolyanitsky, A.; Li, Q.; Feng, X.-Q.; Cannara, R.J. Adhesion-dependent negative friction coefficient on chemically modified graphite at the nanoscale. Nat. Mater. 2012, 11, 1032–1037. [Google Scholar] [CrossRef] [PubMed]
- Gnecco, E.; Bennewitz, R.; Gyalog, T.; Loppacher, C.; Bammerlin, M.; Meyer, E.; Güntherodt, H.-J. Velocity dependence of atomic friction. Phys. Rev. Lett. 2000, 84, 1172. [Google Scholar] [CrossRef]
- Liu, B.; Wang, J.; Zhao, S.; Qu, C.; Liu, Y.; Ma, L.; Zhang, Z.; Liu, K.; Zheng, Q.; Ma, M. Negative friction coefficient in microscale graphite/mica layered heterojunctions. Sci. Adv. 2020, 6, eaaz6787. [Google Scholar] [CrossRef]
- Sun, J.; Zhang, Y.; Lu, Z.; Li, Q.; Xue, Q.; Du, S.; Pu, J.; Wang, L. Superlubricity enabled by pressure-induced friction collapse. J. Phys. Chem. Lett. 2018, 9, 2554–2559. [Google Scholar] [CrossRef]
- Prandtl, L. Ein Gedankenmodell zur kinetischen Theorie der festen Körper. ZAMM-J. Appl. Math. Mech./Z. Für Angew. Math. Und Mech. 1928, 8, 85–106. [Google Scholar] [CrossRef]
- Tomlinson, G.A. CVI. A molecular theory of friction. Lond. Edinb. Dublin Philos. Mag. J. Sci. 1929, 7, 905–939. [Google Scholar] [CrossRef]
- Aubry, S.; Le Daeron, P.-Y. The discrete Frenkel-Kontorova model and its extensions: I. Exact results for the ground-states. Phys. D Nonlinear Phenom. 1983, 8, 381–422. [Google Scholar] [CrossRef]
- Gao, L.; Ma, Y.; Liu, Y.; Song, A.; Ma, T.; Hu, Y.; Su, Y.; Qiao, L. Anomalous frictional behaviors of Ir and Au tips sliding on graphene/Ni (111) substrate: Density functional theory calculations. J. Phys. Chem. C 2017, 121, 21397–21404. [Google Scholar] [CrossRef]
- Song, A.; Zhao, J.-X.; Tang, X.; Wu, H.-J.; Xu, Z.; Cao, J.; Liu, X.; Wang, H.; Li, Q.; Hu, Y.-Z.; et al. Tuning friction force and reducing wear by applying alternating electric current in conductive AFM experiments. Nat. Commun. 2025, 16, 4704. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhang, J.; Deng, Y.; Zhao, H.; Zhang, P.; Fu, S.; Xu, X.; Li, H. Defect-mediated work function regulation in graphene film for high-performing triboelectric nanogenerators. Nano Energy 2022, 99, 107411. [Google Scholar] [CrossRef]
- Luo, C.; Jiang, Y.; Liu, Y.; Wang, Y.; Sun, J.; Qian, L.; Chen, L. Role of interfacial bonding in tribochemical wear. Front. Chem. 2022, 10, 852371. [Google Scholar] [CrossRef] [PubMed]
- Siegel, D.J.; Hamilton, J. First-principles study of the solubility, diffusion, and clustering of C in Ni. Phys. Rev. B 2003, 68, 094105. [Google Scholar] [CrossRef]
- Baksi, M.; Toffoli, D.; Gulseren, O.; Ustunel, H. Nanotribological properties of the h-BN/Au (111) interface: A DFT study. J. Phys. Chem. C 2019, 123, 28411–28418. [Google Scholar] [CrossRef]
- De Barros Bouchet, M.-I.; Zilibotti, G.; Matta, C.; Righi, M.C.; Vandenbulcke, L.; Vacher, B.; Martin, J.-M. Friction of diamond in the presence of water vapor and hydrogen gas. Coupling gas-phase lubrication and first-principles studies. J. Phys. Chem. C 2012, 116, 6966–6972. [Google Scholar] [CrossRef]
- Wang, L.; Zhou, X.; Ma, T.; Liu, D.; Gao, L.; Li, X.; Zhang, J.; Hu, Y.; Wang, H.; Dai, Y.; et al. Superlubricity of a graphene/MoS2 heterostructure: A combined experimental and DFT study. Nanoscale 2017, 9, 10846–10853. [Google Scholar] [CrossRef]
- Zhong, W.; Tománek, D. First-principles theory of atomic-scale friction. Phys. Rev. Lett. 1990, 64, 3054–3057. [Google Scholar] [CrossRef]
- Li, H.; Shi, W.; Guo, Y.; Guo, W. Nonmonotonic interfacial friction with normal force in two-dimensional crystals. Phys. Rev. B 2020, 102, 085427. [Google Scholar] [CrossRef]
- Leven, I.; Krepel, D.; Shemesh, O.; Hod, O. Robust superlubricity in graphene/h-BN heterojunctions. J. Phys. Chem. Lett. 2013, 4, 115–120. [Google Scholar] [CrossRef]
- Dong, Y.; Vadakkepatt, A.; Martini, A. Analytical models for atomic friction. Tribol. Lett. 2011, 44, 367–386. [Google Scholar] [CrossRef]
- Restuccia, P.; Righi, M.C. Tribochemistry of graphene on iron and its possible role in lubrication of steel. Carbon 2016, 106, 118–124. [Google Scholar] [CrossRef]
- Levita, G.; Cavaleiro, A.; Molinari, E.; Polcar, T.; Righi, M.C. Sliding properties of MoS2 layers: Load and interlayer orientation effects. J. Phys. Chem. C 2014, 118, 13809–13816. [Google Scholar] [CrossRef]
- Reguzzoni, M.; Fasolino, A.; Molinari, E.; Righi, M.C. Potential energy surface for graphene on graphene: Ab initioderivation, analytical description, and microscopic interpretation. Phys. Rev. B 2012, 86, 245434. [Google Scholar] [CrossRef]
- Cahangirov, S.; Ciraci, S.; Özçelik, V.O. Superlubricity through graphene multilayers between Ni(111) surfaces. Phys. Rev. B 2013, 87, 205428. [Google Scholar] [CrossRef]
- Wolloch, M.; Levita, G.; Restuccia, P.; Righi, M.C. Interfacial charge density and its connection to adhesion and frictional forces. Phys. Rev. Lett. 2018, 121, 026804. [Google Scholar] [CrossRef]
- Sun, J.; Zhang, X.; Du, S.; Pu, J.; Wang, Y.; Yuan, Y.; Qian, L.; Francisco, J.S. Charge density evolution governing interfacial friction. J. Am. Chem. Soc. 2023, 145, 5536–5544. [Google Scholar] [CrossRef]
- Cheng, Z.; Zhang, B.; An, X.; Lu, Z.; Zhang, G.; Ma, F.; Zhou, F. First-principles delimitation of the boundary between intralayer and interlayer in two-dimensional structures. J. Phys. Chem. C 2019, 123, 26912–26920. [Google Scholar] [CrossRef]
- Cao, Y.; Zhao, Z.; Lv, Q.; Dong, X.; Niu, Y.; He, W.; Wang, Y.; Lu, Z. Potential negative friction coefficient predicted by first-principles calculations: A possible consequence of inaccurate computational models. Phys. Rev. B 2025, 111, 054109. [Google Scholar] [CrossRef]
- Cheng, Z.; Sun, J.; Zhang, B.; Lu, Z.; Ma, F.; He, Q.C. Superlubricity enabled by load-driven redistribution of electrons. Adv. Mater. Interfaces 2022, 9, 2101589. [Google Scholar] [CrossRef]
- Cheng, Z.; Feng, H.; Sun, J.; Lu, Z.; He, Q.C. Strain-Driven superlubricity of graphene/graphene in commensurate contact. Adv. Mater. Interfaces 2023, 10, 2202062. [Google Scholar] [CrossRef]
- Zhang, S.; Ma, T.; Erdemir, A.; Li, Q. Tribology of two-dimensional materials: From mechanisms to modulating strategies. Mater. Today 2019, 26, 67–86. [Google Scholar] [CrossRef]
- Vanossi, A.; Bechinger, C.; Urbakh, M. Structural lubricity in soft and hard matter systems. Nat. Commun. 2020, 11, 4657. [Google Scholar] [CrossRef] [PubMed]
- Tabor, D. History of Tribology: D. Dowson; Elsevier: Amsterdam, The Netherlands, 1979. [Google Scholar]
- Israelachvili, J.N. Adhesion, friction and lubrication of molecularly smooth surfaces. In Fundamentals of Friction: Macroscopic and Microscopic Processes; Springer: Dordrecht, The Netherlands, 1992; pp. 351–385. [Google Scholar]
- Hirano, M.; Shinjo, K. Atomistic locking and friction. Phys. Rev. B 1990, 41, 11837. [Google Scholar] [CrossRef]
- Müser, M.H. Structural lubricity: Role of dimension and symmetry. Europhys. Lett. (EPL) 2004, 66, 97–103. [Google Scholar] [CrossRef]
- Müser, M.H. Theoretical studies of superlubricity. In Fundamentals of Friction and Wear on the Nanoscale; Springer: Dordrecht, The Netherlands, 2015; pp. 209–232. [Google Scholar]
- Liu, L.; Li, Y.; Wang, H.; Yang, Z.; Wang, K.; Luo, J.; Liu, Y. The correlation between molecular structure and superlubricity in homojunctions of 2D materials. Mater. Sci. Eng. R Rep. 2024, 161, 100868. [Google Scholar] [CrossRef]
- Cheng, Z.; Feng, H.; Lu, Y.; Lu, Z.; He, Q.-C. Electron-scale origin of structural superlubricity. Tribol. Int. 2025, 202, 110294. [Google Scholar] [CrossRef]
- Sun, T.; Gao, E.; Jia, X.; Bian, J.; Wang, Z.; Ma, M.; Zheng, Q.; Xu, Z. Robust structural superlubricity under gigapascal pressures. Nat. Commun. 2024, 15, 5952. [Google Scholar] [CrossRef]
- Wang, L.; Ma, T.; Hu, Y.; Wang, H. Understanding the atomic-scale friction in graphene: The distinction in behaviors of interlayer interactions during sliding. J. Appl. Phys. 2016, 120, 205302. [Google Scholar] [CrossRef]
- Socoliuc, A.; Bennewitz, R.; Gnecco, E.; Meyer, E. Transition from stick-slip to continuous sliding in atomic friction: Entering a new regime of ultralow friction. Phys. Rev. Lett. 2004, 92, 134301. [Google Scholar] [CrossRef]
- Cahangirov, S.; Ataca, C.; Topsakal, M.; Sahin, H.; Ciraci, S. Frictional figures of merit for single layered nanostructures. Phys. Rev. Lett. 2012, 108, 126103. [Google Scholar] [CrossRef]
- Wang, C.; Li, H.; Zhang, Y.; Sun, Q.; Jia, Y. Effect of strain on atomic-scale friction in layered MoS2. Tribol. Int. 2014, 77, 211–217. [Google Scholar] [CrossRef]
- Wang, K.; Ouyang, W.; Cao, W.; Ma, M.; Zheng, Q. Robust superlubricity by strain engineering. Nanoscale 2019, 11, 2186–2193. [Google Scholar] [CrossRef]
- Wu, S.; Meng, Z.; Tao, X.; Wang, Z. Superlubricity of molybdenum disulfide subjected to large compressive strains. Friction 2021, 10, 209–216. [Google Scholar] [CrossRef]
- Zhang, S.; Hou, Y.; Li, S.; Liu, L.; Zhang, Z.; Feng, X.Q.; Li, Q. Tuning friction to a superlubric state via in-plane straining. Proc. Natl. Acad. Sci. USA 2019, 117, 6951. [Google Scholar] [CrossRef] [PubMed]
- Androulidakis, C.; Koukaras, E.N.; Paterakis, G.; Trakakis, G.; Galiotis, C. Tunable macroscale structural superlubricity in two-layer graphene via strain engineering. Nat. Commun. 2020, 11, 1595. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Sun, J.; Zhang, B.; Lu, Z.; Ma, F.; Zhang, G.; Xue, Q. Strain effects of vertical separation and horizontal sliding in commensurate two-dimensional homojunctions. J. Phys. Chem. Lett. 2020, 11, 5815–5822. [Google Scholar] [CrossRef]
- Righi, M.C.; Ferrario, M. Pressure induced friction collapse of rare gas boundary layers sliding over metal surfaces. Phys. Rev. Lett. 2007, 99, 176101. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Cheng, Z.; Yang, T.; Lu, Z.; He, Q.-C. Attraction-Induced superlubricity and its detection. Appl. Surf. Sci. 2023, 640, 158423. [Google Scholar] [CrossRef]
- Sun, J.; Zhang, Y.; Lu, Z.; Xue, Q.; Wang, L. Attraction induced frictionless sliding of rare gas monolayer on metallic surfaces: An efficient strategy for superlubricity. Phys. Chem. Chem. Phys. 2017, 19, 11026–11031. [Google Scholar] [CrossRef]
- Sun, J.; Chang, K.; Mei, D.; Lu, Z.; Pu, J.; Xue, Q.; Huang, Q.; Wang, L.; Du, S. Mutual identification between the pressure-induced superlubricity and the image contrast inversion of carbon nanostructures from AFM technology. J. Phys. Chem. Lett. 2019, 10, 1498–1504. [Google Scholar] [CrossRef]
- Lin, J.; Zhang, C.; Liao, N.; Zhang, M. A DFT analysis on the superlubricity performance of structural superlubricity micro/nano-component on silicon-based surface. Tribol. Int. 2023, 189, 109005. [Google Scholar] [CrossRef]
- Levita, G.; Molinari, E.; Polcar, T.; Righi, M.C. First-principles comparative study on the interlayer adhesion and shear strength of transition-metal dichalcogenides and graphene. Phys. Rev. B 2015, 92, 085434. [Google Scholar] [CrossRef]
- Losi, G.; Restuccia, P.; Righi, M. Superlubricity in phosphorene identified by means of ab initio calculations. 2D Mater. 2020, 7, 025033. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, Y.; Qiao, Y.; Tan, S.; Feng, S.; Ma, J.; Liu, Y.; Luo, J. 2D metal-organic frameworks with square grid structure: A promising new-generation superlubricating material. Nano Today 2021, 40, 101262. [Google Scholar] [CrossRef]
- Serles, P.; Arif, T.; Puthirath, A.B.; Yadav, S.; Wang, G.; Cui, T.; Balan, A.P.; Yadav, T.P.; Thibeorchews, P.; Chakingal, N. Friction of magnetene, a non–van der Waals 2D material. Sci. Adv. 2021, 7, eabk2041. [Google Scholar] [CrossRef]
- Liu, K.; Cheng, J.; Zhao, X.; Zhu, Y.; Ren, X.; Shi, J.; Guo, Z.; Shan, C.; Liu, H.; Li, S. Negative differential friction coefficients of two-dimensional commensurate contacts dominated by electronic phase transition. Nano Res. 2022, 15, 5758–5766. [Google Scholar] [CrossRef]
- Zhang, L.; Tan, X.; Jiao, J.; Guo, D.; Luo, J. Interlayer friction behavior of molybdenum ditelluride with different structures. Nano Res. 2023, 16, 11375–11382. [Google Scholar] [CrossRef]
- Feng, H.; Cheng, Z.; Lu, Z.; He, Q.-C. Investigating lubrication capacities of novel 2D hexagonal materials by DFT simulations. Tribol. Int. 2025, 204, 110475. [Google Scholar] [CrossRef]
- Dong, S.; Shi, J.; Li, H.; Zhang, R.; Ma, X.; Yang, Y.; Cui, C.; Wang, W.; Li, J. Phytic acid-modified black phosphorus nanosheets achieve ultrahigh load bearing and rapid superlubrication on engineered steel surfaces. Adv. Funct. Mater. 2025, 35, 2500057. [Google Scholar] [CrossRef]
- Liu, L.; Wang, K.; Liu, Y. Interlayer superlubricity of layered Metal-organic frameworks and its heterojunctions enabled by highly oriented crystalline films. Chem. Eng. J. 2022, 450, 138249. [Google Scholar] [CrossRef]
- Munther, M.; Palma, T.; Beheshti, A.; Davami, K. Substrate-regulated nanoscale friction of graphene. Mater. Lett. 2018, 221, 54–56. [Google Scholar] [CrossRef]
- Qi, Y.; Liu, J.; Dong, Y.; Feng, X.-Q.; Li, Q. Impacts of environments on nanoscale wear behavior of graphene: Edge passivation vs. substrate pinning. Carbon 2018, 139, 59–66. [Google Scholar] [CrossRef]
- Zeng, X.; Peng, Y.; Lang, H. A novel approach to decrease friction of graphene. Carbon 2017, 118, 233–240. [Google Scholar] [CrossRef]
- Shi, B.; Gan, X.; Lang, H.; Zou, K.; Wang, L.; Sun, J.; Lu, Y.; Peng, Y. Ultra-low friction and patterning on atomically thin MoS 2 via electronic tight-binding. Nanoscale 2021, 13, 16860–16871. [Google Scholar] [CrossRef]
- Khomyakov, P.; Giovannetti, G.; Rusu, P.; Brocks, G.v.; Van den Brink, J.; Kelly, P.J. First-principles study of the interaction and charge transfer between graphene and metals. Phys. Rev. B—Condens. Matter Mater. Phys. 2009, 79, 195425. [Google Scholar] [CrossRef]
- Christian, M.S.; Johnson, E.R. Effect of a metal substrate on interlayer interactions in bilayer graphene. J. Phys. Chem. C 2018, 122, 8910–8918. [Google Scholar] [CrossRef]
- Feng, H.; Cheng, Z.; Long, D.; Yang, T.; Lu, Z.; He, Q. How Do Substrates Affect the Friction on Graphene at the Nanoscale? Lubricants 2023, 11, 465. [Google Scholar] [CrossRef]
- Hod, O.; Meyer, E.; Zheng, Q.; Urbakh, M. Structural superlubricity and ultralow friction across the length scales. Nature 2018, 563, 485–492. [Google Scholar] [CrossRef]
- Xu, Y.; Cheng, Z.; Zhu, X.; Lu, Z.; Zhang, G. Ultra-low friction of graphene/honeycomb borophene heterojunction. Tribol. Lett. 2021, 69, 44. [Google Scholar] [CrossRef]
- Jiang, X.; Lu, Z.; Zhang, R. The unusual tribological properties of graphene/antimonene heterojunctions: A first-principles investigation. Materials 2021, 14, 1201. [Google Scholar] [CrossRef]
- Xu, Y.; Zhu, X.; Cheng, Z.; Lu, Z.; He, W.; Zhang, G. A novel ultra-low friction heterostructure: Aluminum substrate-honeycomb borophene/graphene heterojunction. Comput. Mater. Sci. 2022, 205, 111236. [Google Scholar] [CrossRef]
- Sun, K.; Yu, S.; Wang, X.; Yao, L.; Xu, Y. Atomic-scale investigation of the tribological behaviors of black phosphorus/graphene heterostructures. Tribol. Int. 2025, 211, 110881. [Google Scholar] [CrossRef]
- Gao, E.; Wu, B.; Wang, Y.; Jia, X.; Ouyang, W.; Liu, Z. Computational prediction of superlubric layered heterojunctions. ACS Appl. Mater. Interfaces 2021, 13, 33600–33608. [Google Scholar] [CrossRef]
- Zhu, X.; Yuan, Q.; Zhao, Y.-P. Phase transitions of a water overlayer on charged graphene: From electromelting to electrofreezing. Nanoscale 2014, 6, 5432–5437. [Google Scholar] [CrossRef]
- Park, J.Y.; Ogletree, D.; Thiel, P.; Salmeron, M. Electronic control of friction in silicon pn junctions. Science 2006, 313, 186. [Google Scholar] [CrossRef]
- Lang, H.; Peng, Y.; Cao, X.a.; Zou, K. Atomic-scale friction characteristics of graphene under conductive AFM with applied voltages. ACS Appl. Mater. Interfaces 2020, 12, 25503–25511. [Google Scholar] [CrossRef]
- Lang, H.; Peng, Y.; Yu, K. Dynamic electron transfer for reducing nanofriction of graphene at electrified interfaces. Appl. Surf. Sci. 2020, 520, 146327. [Google Scholar] [CrossRef]
- Wang, C.; Chen, W.; Zhang, Y.; Sun, Q.; Jia, Y. Effects of vdW interaction and electric field on friction in MoS2. Tribol. Lett. 2015, 59, 7. [Google Scholar] [CrossRef]
- Wang, J.; Li, J.; Li, C.; Cai, X.; Zhu, W.; Jia, Y. Tuning the nanofriction between two graphene layers by external electric fields: A density functional theory study. Tribol. Lett. 2016, 61, 4. [Google Scholar] [CrossRef]
- Li, Y.; Wu, B.; Ouyang, W.; Liu, Z.; Wang, W. Experimental decoding and tuning electronic friction of Si nanotip sliding on graphene. Nano Lett. 2024, 24, 1130–1136. [Google Scholar] [CrossRef]
- Wang, L.-F.; Ma, T.-B.; Hu, Y.-Z.; Wang, H. Atomic-scale friction in graphene oxide: An interfacial interaction perspective from first-principles calculations. Phys. Rev. B—Condens. Matter Mater. Phys. 2012, 86, 125436. [Google Scholar] [CrossRef]
- Wang, J.; Wang, F.; Li, J.; Sun, Q.; Yuan, P.; Jia, Y. Comparative study of friction properties for hydrogen-and fluorine-modified diamond surfaces: A first-principles investigation. Surf. Sci. 2013, 608, 74–79. [Google Scholar] [CrossRef]
- Wang, L.-F.; Ma, T.-B.; Hu, Y.-Z.; Wang, H.; Shao, T.-M. Ab initio study of the friction mechanism of fluorographene and graphane. J. Phys. Chem. C 2013, 117, 12520–12525. [Google Scholar] [CrossRef]
- Wang, L.-F.; Ma, T.-B.; Hu, Y.-Z.; Zheng, Q.; Wang, H.; Luo, J. Superlubricity of two-dimensional fluorographene/MoS2 heterostructure: A first-principles study. Nanotechnology 2014, 25, 385701. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Xiao, C.; Sun, J.; Li, J.; Qian, L. Electronic insight into ultralow friction in hydrogenated diamond. Tribol. Int. 2024, 193, 109410. [Google Scholar] [CrossRef]
- Zhang, B.; Zhang, G.; Cheng, Z.; Ma, F.; Lu, Z. Atomic-scale friction adjustment enabled by doping-induced modification in graphene nanosheet. Appl. Surf. Sci. 2019, 483, 742–749. [Google Scholar] [CrossRef]
- Wang, J.; Li, M.; Zhang, X.; Cai, X.; Yang, L.; Li, J.; Jia, Y. An atomic scale study of ultralow friction between phosphorus-doped nanocrystalline diamond films. Tribol. Int. 2015, 86, 85–90. [Google Scholar] [CrossRef]
- Du, C.-F.; Xue, Y.; Zeng, Q.; Wang, J.; Zhao, X.; Wang, Z.; Wang, C.; Yu, H.; Liu, W. Mo-doped Cr-Ti-Mo ternary o-MAX with ultra-low wear at elevated temperatures. J. Eur. Ceram. Soc. 2022, 42, 7403–7413. [Google Scholar] [CrossRef]
- Minkin, A.S.; Lebedeva, I.V.; Popov, A.M.; Knizhnik, A.A. Atomic-scale defects restricting structural superlubricity: Ab initio study on the example of the twisted graphene bilayer. Phys. Rev. B 2021, 104, 075444. [Google Scholar] [CrossRef]
- Niu, Y.; Wang, Y.; He, M.; He, W.; Zhao, Z.; Lu, Z. Calculation and prediction of sliding energy barriers by first-principles combined with machine learning. Ceram. Int. 2023, 49, 24752–24761. [Google Scholar] [CrossRef]
- Li, H.; Guo, W.; Guo, Y. Impart of heterogeneous charge polarization and distribution on friction at water-graphene interfaces: A density-functional-theory based machine learning study. J. Phys. Chem. Lett. 2024, 15, 6585–6591. [Google Scholar] [CrossRef] [PubMed]
- Barik, R.K.; Woods, L.M. Frictional Properties of Two-Dimensional Materials: Data-Driven Machine Learning Predictive Modeling. ACS Appl. Mater. Interfaces 2024, 16, 40149–40159. [Google Scholar] [CrossRef]
- Wang, X.; Li, Y.; Zhang, Y.; Wang, P.; Zhao, Y.-M.; Zhou, J.; Yang, J.; Wang, X.; Shen, L. Machine learning assisted identification of homobilayer sliding ferroelectrics with large out-of-plane polarization and low sliding energy barriers. Phys. Rev. B 2025, 111, 094106. [Google Scholar] [CrossRef]
- Zhang, G.; Chen, Y.; Cao, Y.; Liu, B.; Huang, Q.; Liu, Y. Superior self-lubricating coatings with heterogeneous nanocomposite structures on Ti–Nb–Zr–Ta–Hf refractory multi-principal element alloy. Adv. Funct. Mater. 2024, 34, 2405657. [Google Scholar] [CrossRef]
- Geng, Y.; Chen, H.; Luo, S.; Teng, Y.; He, Q.; Zhang, L.; Zhang, Z.; Yang, Z.; Shi, Y.; Wang, Q. In-Situ formed Titanium-MXene nanomembrane as ultrathin van-der-Waals lubricant. Mater. Today 2025, 88, 328–337. [Google Scholar] [CrossRef]
- Guo, D.; Jia, J.; Zhang, Z.; Yang, J.; Wang, J.; Jiao, X.; Shi, Y.; Cai, L.; He, N. High-temperature tribological characteristics and lubrication mechanisms of NiCrAlY-MoO3/CoO composite coatings: An experimental and DFT computation study. Surf. Coat. Technol. 2025, 496, 131701. [Google Scholar] [CrossRef]
- Zhang, L.; Cai, H.; Xu, L.; Ji, L.; Wang, D.; Zheng, Y.; Feng, Y.; Sui, X.; Guo, Y.; Guo, W. Macro-superlubric triboelectric nanogenerator based on tribovoltaic effect. Matter 2022, 5, 1532–1546. [Google Scholar] [CrossRef]
- Nguyen, Q.T.; Vu, D.L.; Le, C.D.; Choi, W.M.; Ahn, K.-K. Tribo-hygro-electric generator: Harnessing mechanical energy with liquid-infused porous cellulose for multiple sensing and DC power generation. Nano Energy 2024, 132, 110353. [Google Scholar] [CrossRef]
- Nguyen, Q.T.; Van Tam, T.; Vu, D.L.; Ahn, J.H.; Choi, W.M.; Ahn, K.K. Ultrahigh current density DC triboelectric generator for energy harvesting and self-powered sensing. Nano Energy 2025, 143, 111291. [Google Scholar] [CrossRef]
- Hölscher, H.; Ebeling, D.; Schwarz, U.D. Friction at atomic-scale surface steps: Experiment and theory. Phys. Rev. Lett. 2008, 101, 246105. [Google Scholar] [CrossRef]
- Liu, L.; Zhou, M.; Jin, L.; Li, L.; Mo, Y.; Su, G.; Li, X.; Zhu, H.; Tian, Y. Recent advances in friction and lubrication of graphene and other 2D materials: Mechanisms and applications. Friction 2019, 7, 199–216. [Google Scholar] [CrossRef]
- He, J.; Tang, H.; Wang, C. Advances of molecular dynamics simulation in tribochemistry and lubrication investigations: A review. J. Ind. Eng. Chem. 2023, 126, 1–19. [Google Scholar] [CrossRef]
- Tran, N.V.; Tieu, A.K.; Zhu, H. First-principles molecular dynamics study on the surface chemistry and nanotribological properties of MgAl layered double hydroxides. Nanoscale 2021, 13, 5014–5025. [Google Scholar] [CrossRef] [PubMed]
- Leriche, C.; Pedretti, E.; Kang, D.; Righi, M.C.; Weber, B. Passivation species suppress atom-by-atom wear of micro-crystalline diamond. Available at SSRN 5181990. [CrossRef]
- Restuccia, P.; Ferrario, M.; Righi, M. Monitoring water and oxygen splitting at graphene edges and folds: Insights into the lubricity of graphitic materials. Carbon 2020, 156, 93–103. [Google Scholar] [CrossRef]
- Peeters, S.; Charrin, C.; Duron, I.; Loehlé, S.; Thiebaut, B.; Righi, M. Importance of the catalytic effect of the substrate in the functionality of lubricant additives: The case of molybdenum dithiocarbamates. Mater. Today Chem. 2021, 21, 100487. [Google Scholar] [CrossRef]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).