
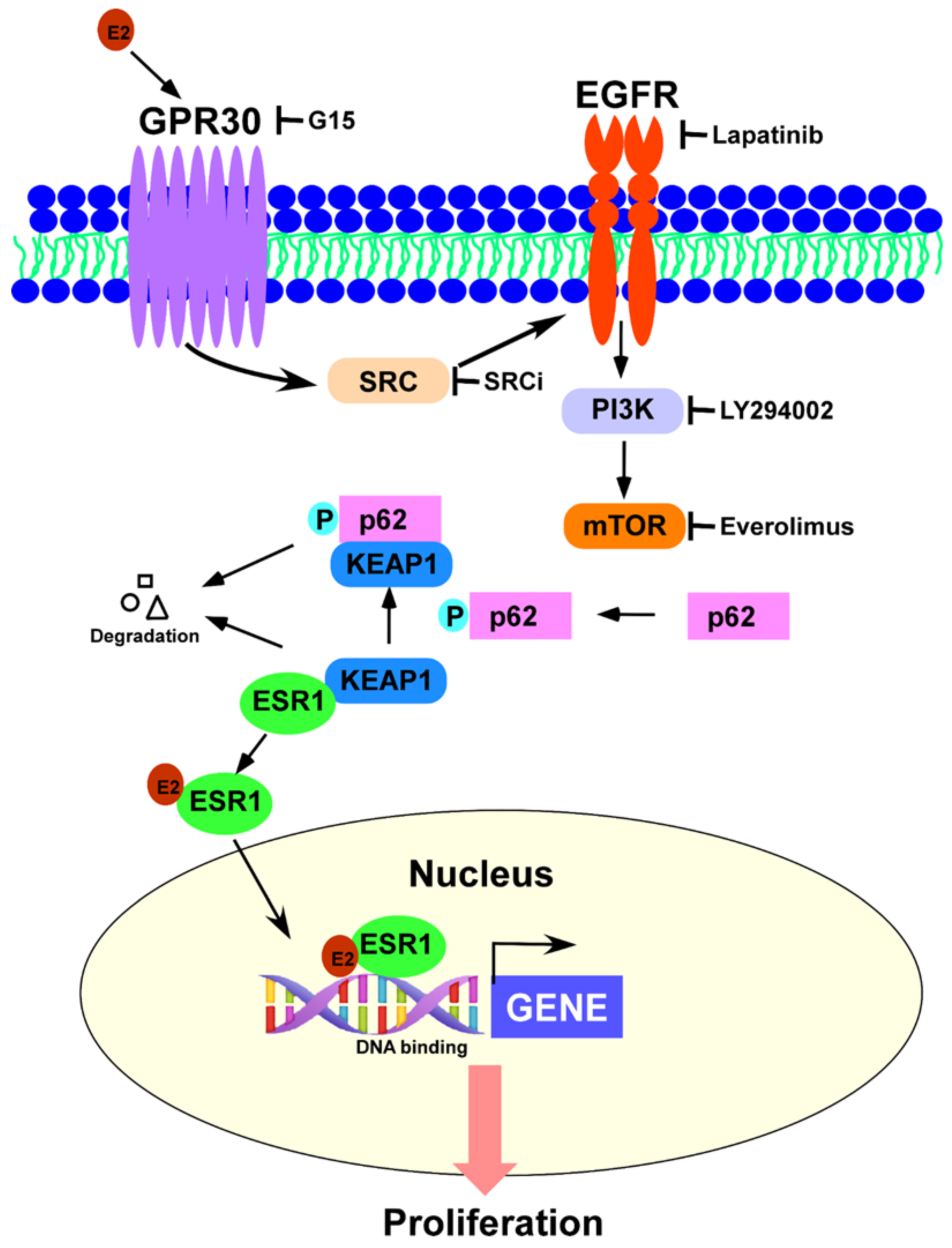
GPR30 Activation by 17β-Estradiol Promotes p62 Phosphorylation and Increases Estrogen Receptor α Protein Expression by Inducing Its Release from a Complex Formed with KEAP1

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatment of Cell Lines
2.2. Isolation of Primary Endometrial Stroma Cells
2.3. DNA Construction
2.4. DNA and siRNA Transfection
2.5. Cell Proliferation Assay
2.6. Western Blot
2.7. Immunoprecipitation
2.8. Confocal Microscopy
2.9. Proximity Ligation Assay
2.10. Animal Experiments and Immunohistochemistry
3. Results
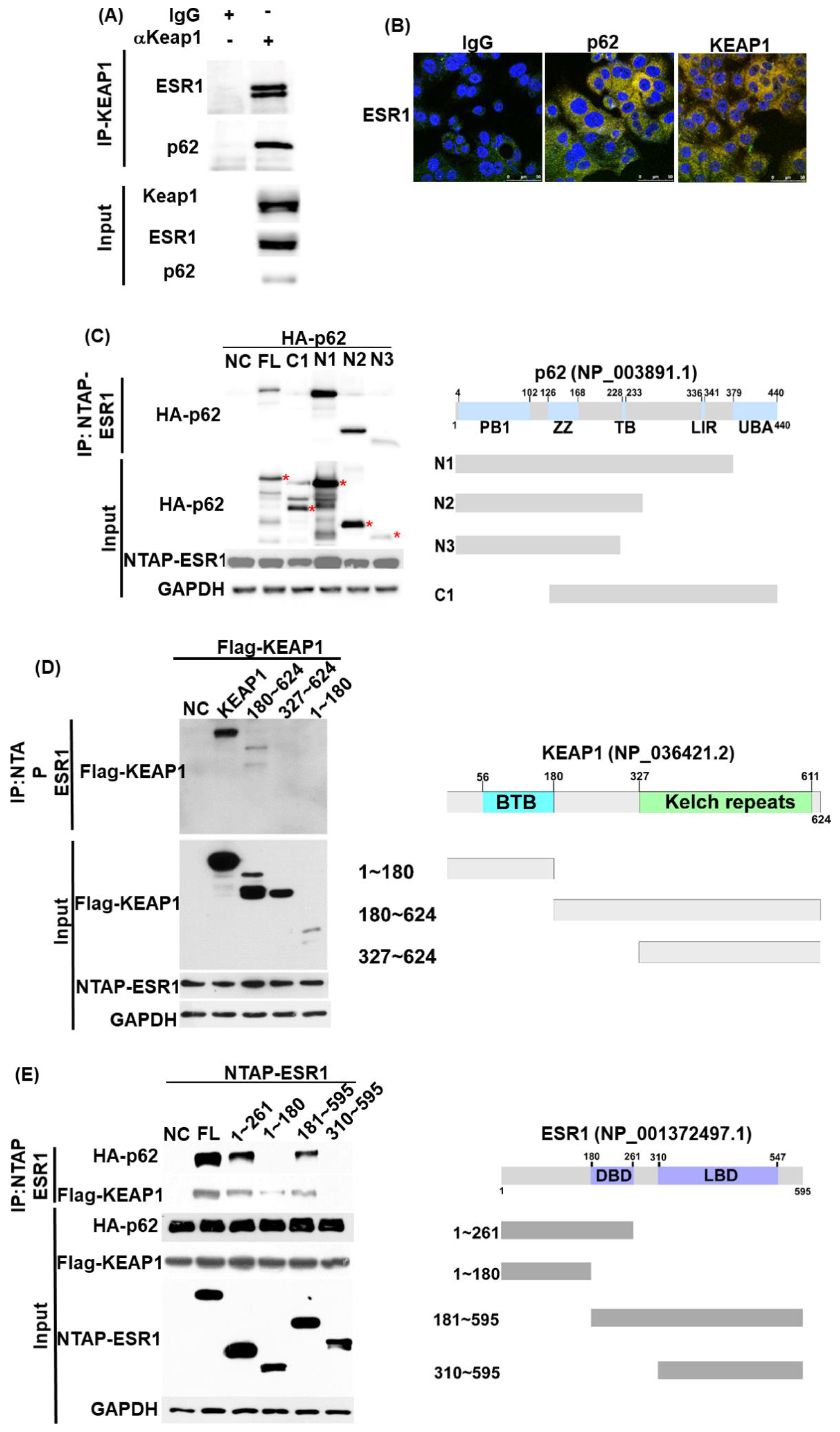
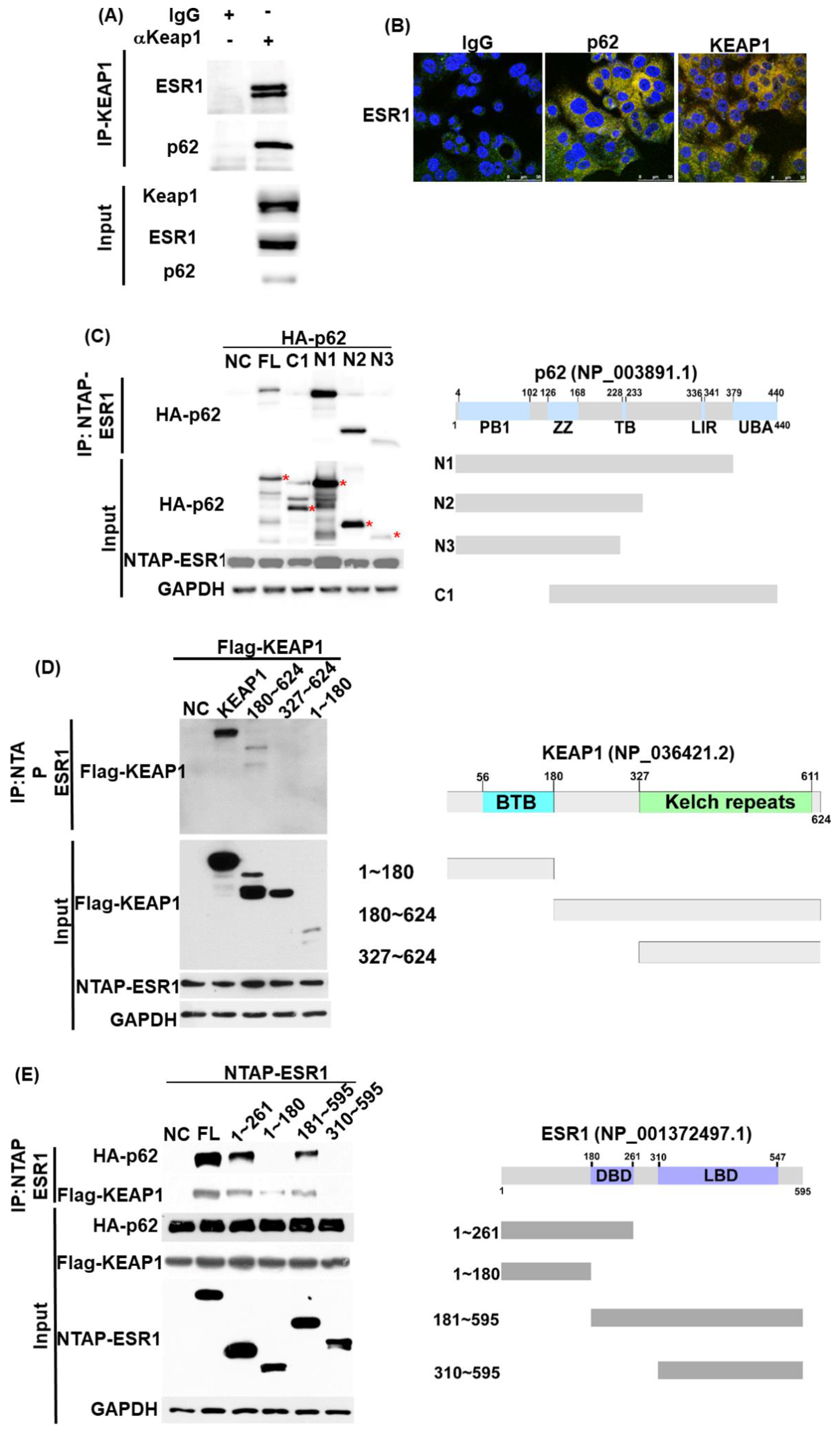
3.1. ESR1, KEAP1, and p62 Interact with Each Other and Are Capable of Forming a Complex
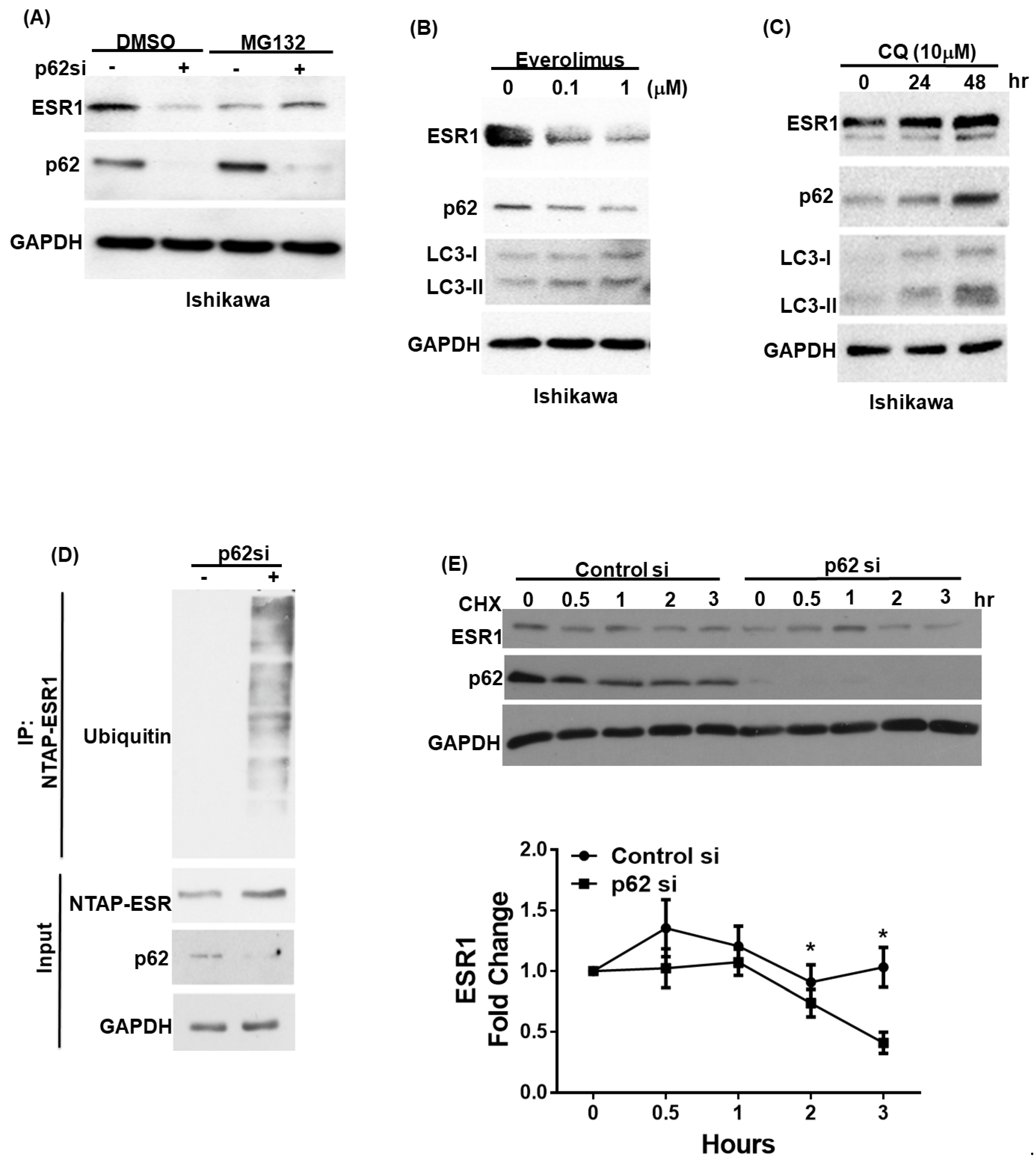
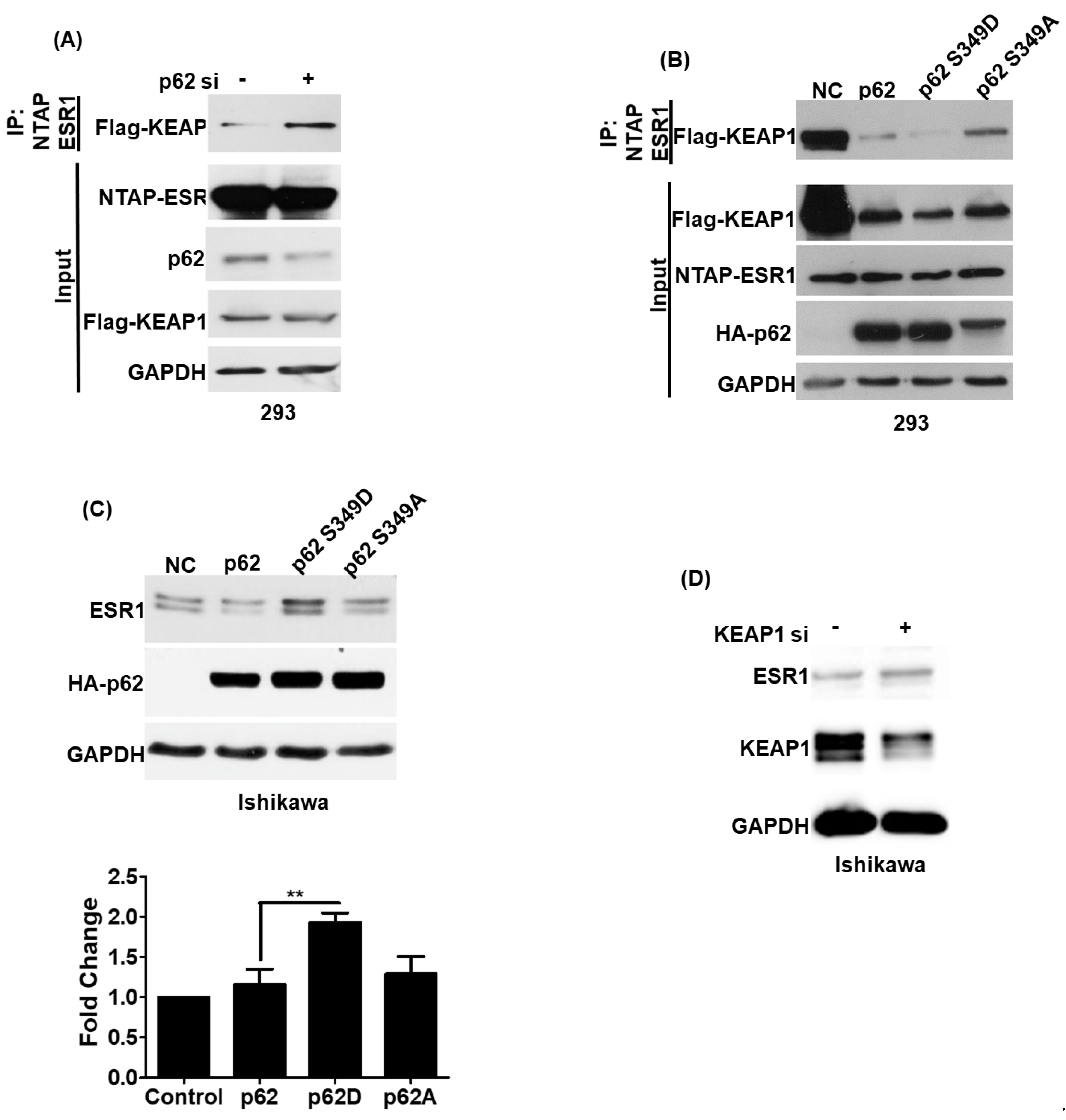
3.2. p62 Increases ESR1 Protein Stability
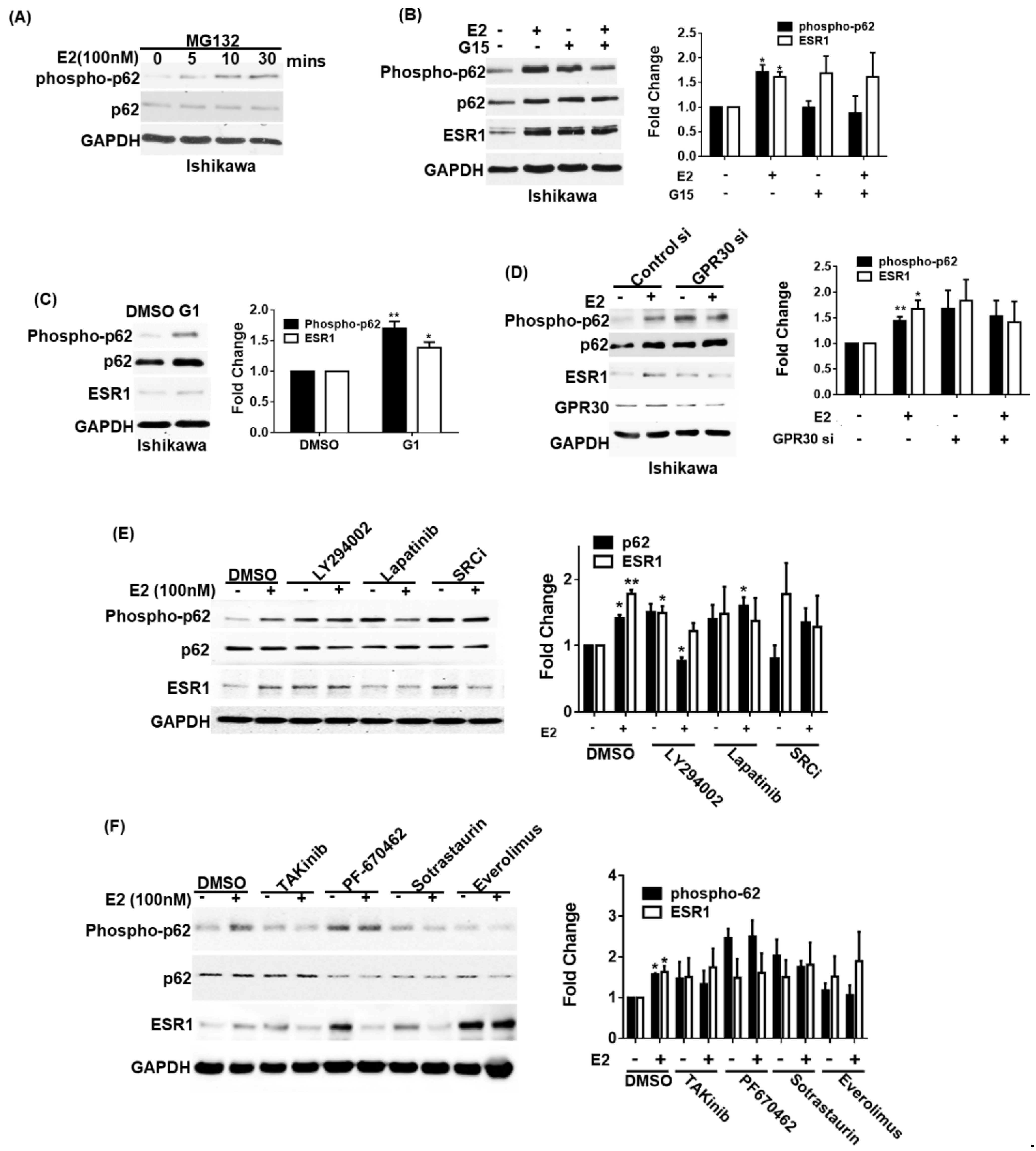
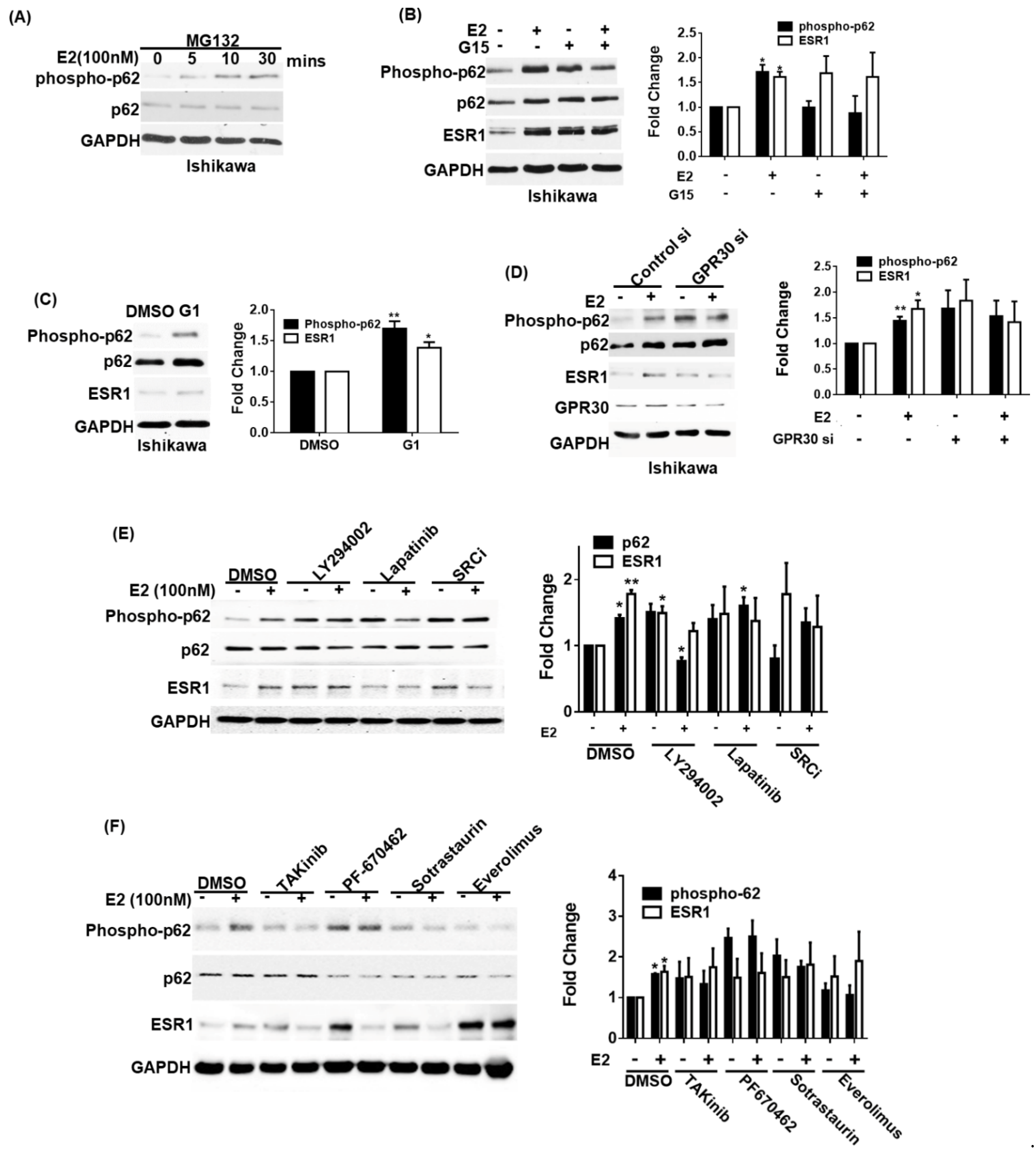
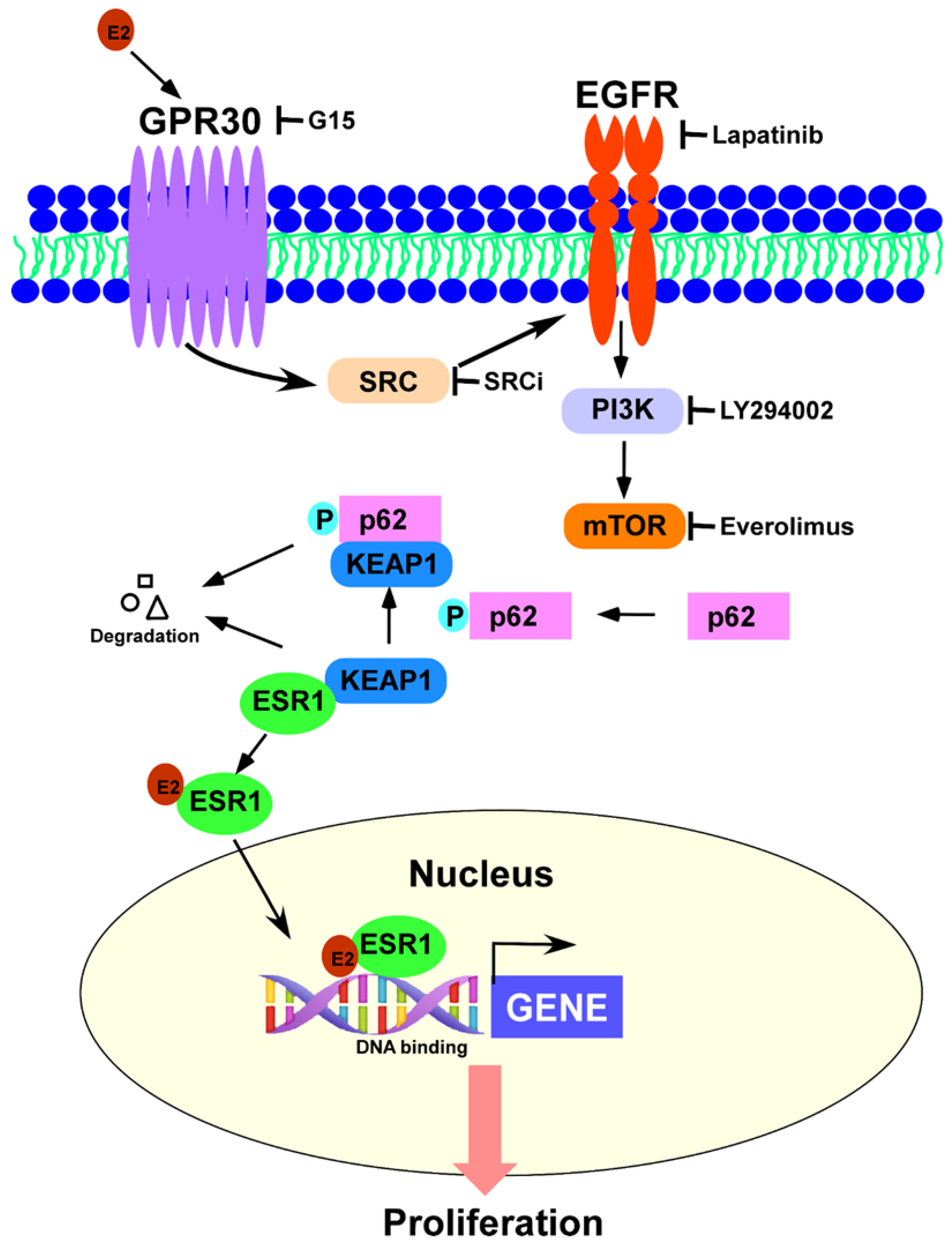
3.3. GPR30 Activation by 17β-Estradiol Promotes p62 Phosphorylation and ESR1 Expression
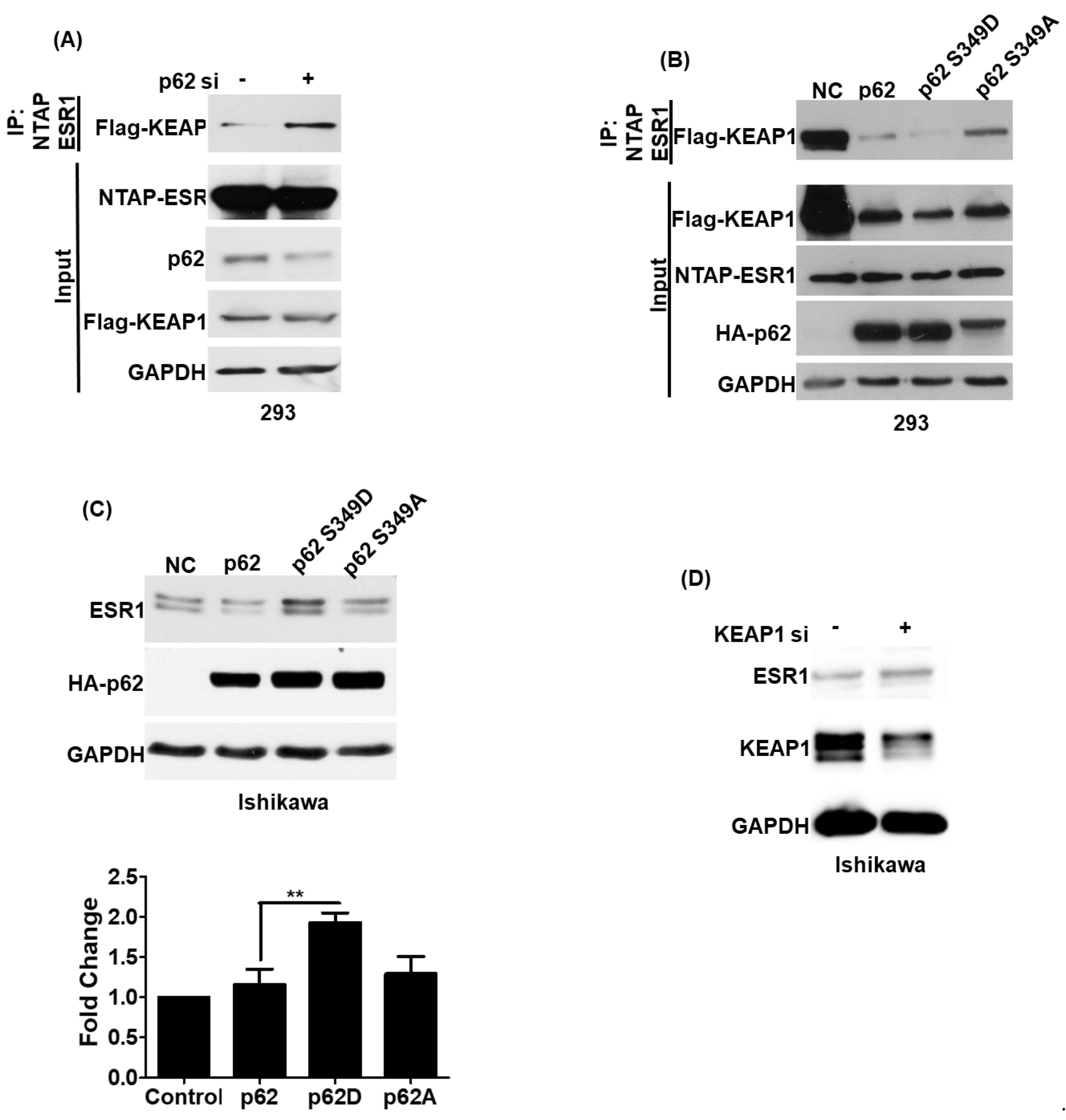
3.4. Phosphorylated p62 Binds to KEAP1 and Promotes the Release of ESR1 from a Protein Complex to Increase Its Expression
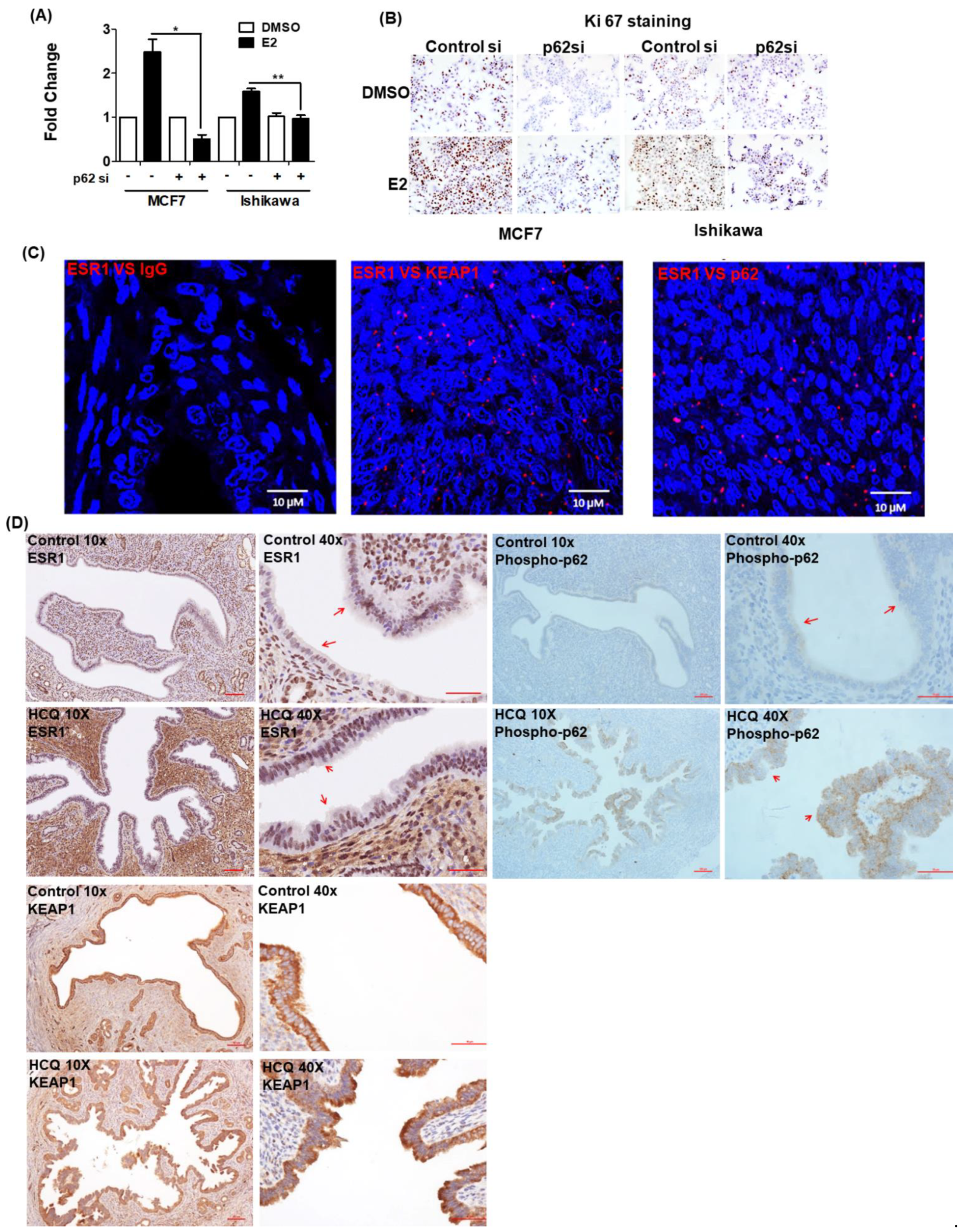
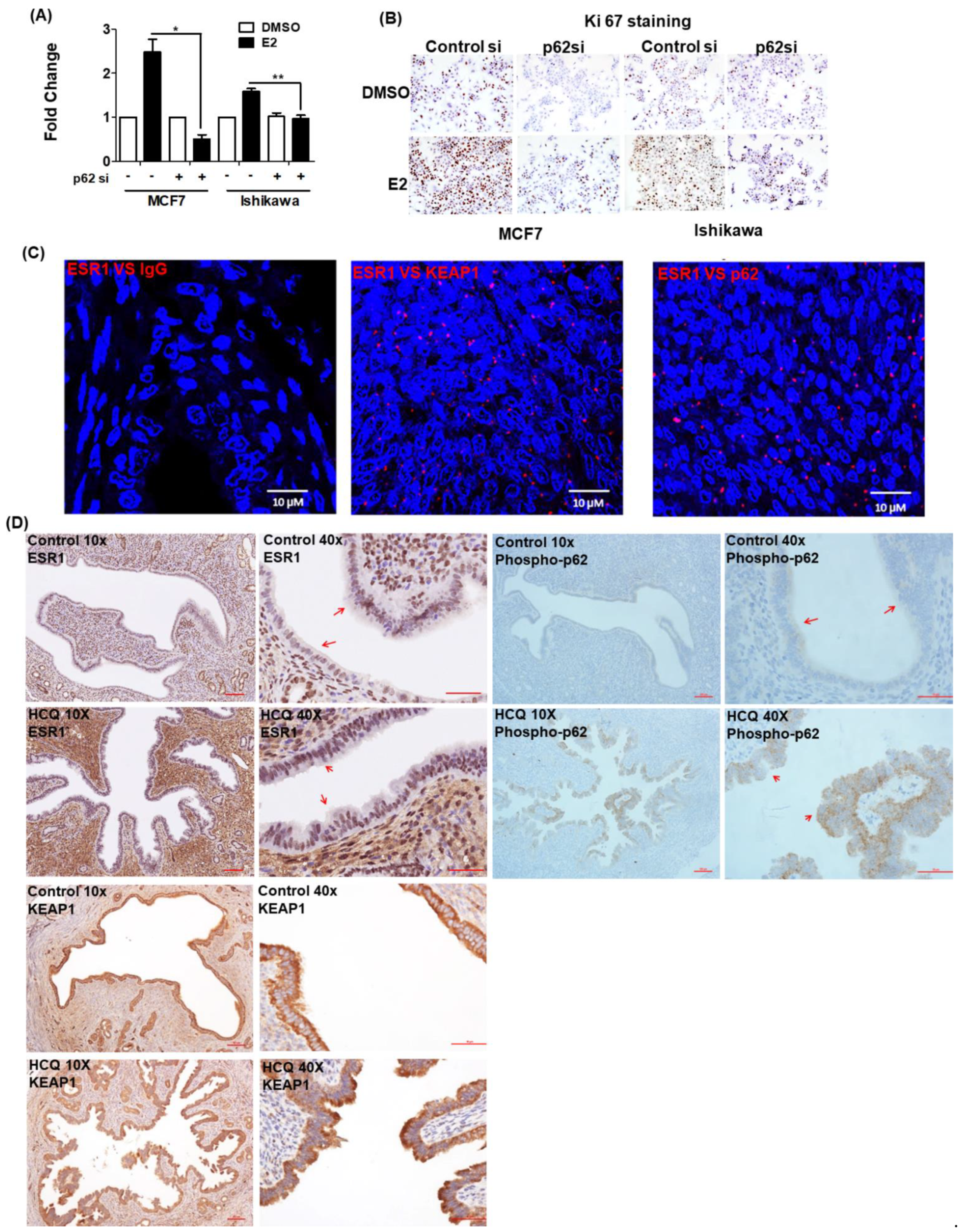
3.5. 17β-Estradiol-Induced Cell Proliferation Is Inhibited by p62 Silencing
3.6. ESR1 Interacts with Both KEAP1 and p62 In Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tang, Z.R.; Zhang, R.; Lian, Z.X.; Deng, S.L.; Yu, K. Estrogen-Receptor Expression and Function in Female Reproductive Disease. Cells 2019, 8, 1123. [Google Scholar] [CrossRef] [Green Version]
- Hewitt, S.C.; Winuthayanon, W.; Korach, K.S. What’s new in estrogen receptor action in the female reproductive tract. J. Mol. Endocrinol. 2016, 56, R55–R71. [Google Scholar] [CrossRef] [Green Version]
- Langer, G.; Bader, B.; Meoli, L.; Isensee, J.; Delbeck, M.; Noppinger, P.R.; Otto, C. A critical review of fundamental controversies in the field of GPR30 research. Steroids 2010, 75, 603–610. [Google Scholar] [CrossRef]
- Prossnitz, E.R.; Arterburn, J.B.; Smith, H.O.; Oprea, T.I.; Sklar, L.A.; Hathaway, H.J. Estrogen signaling through the transmembrane G protein-coupled receptor GPR30. Ann. Rev. Physiol. 2008, 70, 165–190. [Google Scholar] [CrossRef] [PubMed]
- Maggiolini, M.; Vivacqua, A.; Fasanella, G.; Recchia, A.G.; Sisci, D.; Pezzi, V.; Montanaro, D.; Musti, A.M.; Picard, D.; Ando, S. The G protein-coupled receptor GPR30 mediates c-fos up-regulation by 17beta-estradiol and phytoestrogens in breast cancer cells. J. Biol. Chem. 2004, 279, 27008–27016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, C.L.; Wu, H.M.; Lin, C.Y.; Lin, Y.J.; Chao, A.; Wang, T.H.; Hsueh, S.; Lai, C.H.; Wang, H.S. Estradiol and tamoxifen induce cell migration through GPR30 and activation of focal adhesion kinase (FAK) in endometrial cancers with low or without nuclear estrogen receptor alpha (ERalpha). PLoS ONE 2013, 8, e72999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, H.O.; Arias-Pulido, H.; Kuo, D.Y.; Howard, T.; Qualls, C.R.; Lee, S.J.; Verschraegen, C.F.; Hathaway, H.J.; Joste, N.E.; Prossnitz, E.R. GPR30 predicts poor survival for ovarian cancer. Gynecol. Oncol. 2009, 114, 465–471. [Google Scholar] [CrossRef] [Green Version]
- Smith, H.O.; Leslie, K.K.; Singh, M.; Qualls, C.R.; Revankar, C.M.; Joste, N.E.; Prossnitz, E.R. GPR30: A novel indicator of poor survival for endometrial carcinoma. Am. J. Obstet Gynecol. 2007, 196, 386.e1-9; discussion 386.e9-11. [Google Scholar] [CrossRef]
- Wang, X.S.; Yue, J.; Hu, L.N.; Tian, Z.; Zhang, K.; Yang, L.; Zhang, H.N.; Guo, Y.Y.; Feng, B.; Liu, H.Y.; et al. Activation of G protein-coupled receptor 30 protects neurons by regulating autophagy in astrocytes. Glia 2020, 68, 27–43. [Google Scholar] [CrossRef] [Green Version]
- Katsuragi, Y.; Ichimura, Y.; Komatsu, M. p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J. 2015, 282, 4672–4678. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Martin, P.; Saito, T.; Komatsu, M. p62/SQSTM1: ‘Jack of all trades’ in health and cancer. FEBS J. 2019, 286, 8–23. [Google Scholar] [CrossRef] [Green Version]
- Ichimura, Y.; Waguri, S.; Sou, Y.S.; Kageyama, S.; Hasegawa, J.; Ishimura, R.; Saito, T.; Yang, Y.; Kouno, T.; Fukutomi, T.; et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol. Cell 2013, 51, 618–631. [Google Scholar] [CrossRef] [Green Version]
- Ichimura, Y.; Komatsu, M. Activation of p62/SQSTM1-Keap1-Nuclear Factor Erythroid 2-Related Factor 2 Pathway in Cancer. Front. Oncol. 2018, 8, 210. [Google Scholar] [CrossRef]
- Nishida, M. The Ishikawa cells from birth to the present. Hum. Cell 2002, 15, 104–117. [Google Scholar] [CrossRef]
- Yen, C.F.; Kim, S.H.; Liao, S.K.; Atabekoglu, C.; Uckac, S.; Arici, A.; Arlier, S.; Lee, C.L.; Wang, H.S.; Kayisli, U.A. Increased expression of integrin-linked kinase during decidualization regulates the morphological transformation of endometrial stromal cells. Fertil. Steril. 2017, 107, 803–812. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.H.; Kang, H.J.; Na, H.; Lee, M.O. Trichostatin A enhances acetylation as well as protein stability of ERalpha through induction of p300 protein. Breast Cancer Res. 2010, 12, R22. [Google Scholar] [CrossRef] [Green Version]
- Chao, A.; Lin, C.Y.; Chao, A.N.; Tsai, C.L.; Chen, M.Y.; Lee, L.Y.; Chang, T.C.; Wang, T.H.; Lai, C.H.; Wang, H.S. Lysine-specific demethylase 1 (LSD1) destabilizes p62 and inhibits autophagy in gynecologic malignancies. Oncotarget 2017, 8, 74434–74450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.H.; Chao, A.; Tsai, C.L.; Chang, C.L.; Chen, S.H.; Lee, Y.S.; Chen, J.K.; Lin, Y.J.; Chang, P.Y.; Wang, C.J.; et al. Stress-induced phosphoprotein 1 as a secreted biomarker for human ovarian cancer promotes cancer cell proliferation. Mol. Cell Proteomics 2010, 9, 1873–1884. [Google Scholar] [CrossRef] [Green Version]
- Tsai, C.L.; Chao, A.; Jung, S.M.; Tsai, C.N.; Lin, C.Y.; Chen, S.H.; Sue, S.C.; Wang, T.H.; Wang, H.S.; Lai, C.H. Stress-induced phosphoprotein-1 maintains the stability of JAK2 in cancer cells. Oncotarget 2016, 7, 50548–50563. [Google Scholar] [CrossRef]
- Jiang, X.; Bao, Y.; Liu, H.; Kou, X.; Zhang, Z.; Sun, F.; Qian, Z.; Lin, Z.; Li, X.; Liu, X.; et al. VPS34 stimulation of p62 phosphorylation for cancer progression. Oncogene 2017, 36, 6850–6862. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, Y.; Tsujimura, A.; Taguchi, K.; Tanaka, M. HSF1 stress response pathway regulates autophagy receptor SQSTM1/p62-associated proteostasis. Autophagy 2017, 13, 133–148. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, K.; Simmons, A.N.; Kajino-Sakamoto, R.; Tsuji, Y.; Ninomiya-Tsuji, J. TAK1 Regulates the Nrf2 Antioxidant System Through Modulating p62/SQSTM1. Antioxid Redox Signal. 2016, 25, 953–964. [Google Scholar] [CrossRef] [Green Version]
- McChesney, E.W. Animal toxicity and pharmacokinetics of hydroxychloroquine sulfate. Am. J. Med. 1983, 75, 11–18. [Google Scholar] [CrossRef]
- Plante, B.J.; Lessey, B.A.; Taylor, R.N.; Wang, W.; Bagchi, M.K.; Yuan, L.; Scotchie, J.; Fritz, M.A.; Young, S.L. G protein-coupled estrogen receptor (GPER) expression in normal and abnormal endometrium. Reprod. Sci. 2012, 19, 684–693. [Google Scholar] [CrossRef] [Green Version]
- Tong, K.I.; Kobayashi, A.; Katsuoka, F.; Yamamoto, M. Two-site substrate recognition model for the Keap1-Nrf2 system: A hinge and latch mechanism. Biol. Chem. 2006, 387, 1311–1320. [Google Scholar] [CrossRef]
- Xiang, J.; Liu, X.; Ren, J.; Chen, K.; Wang, H.L.; Miao, Y.Y.; Qi, M.M. How does estrogen work on autophagy? Autophagy 2019, 15, 197–211. [Google Scholar] [CrossRef] [Green Version]
- Gureev, A.P.; Popov, V.N.; Starkov, A.A. Crosstalk between the mTOR and Nrf2/ARE signaling pathways as a target in the improvement of long-term potentiation. Exp. Neurol. 2020, 328, 113285. [Google Scholar] [CrossRef]
- Tang, Z.; Hu, B.; Zang, F.; Wang, J.; Zhang, X.; Chen, H. Nrf2 drives oxidative stress-induced autophagy in nucleus pulposus cells via a Keap1/Nrf2/p62 feedback loop to protect intervertebral disc from degeneration. Cell Death Dis. 2019, 10, 510. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Jiang, D.; Wu, K. p62 promotes bladder cancer cell growth by activating KEAP1/NRF2-dependent antioxidative response. Cancer Sci. 2020, 111, 1156–1164. [Google Scholar] [CrossRef]
- Gong, M.; Li, Y.; Ye, X.; Zhang, L.; Wang, Z.; Xu, X.; Shen, Y.; Zheng, C. Loss-of-function mutations in KEAP1 drive lung cancer progression via KEAP1/NRF2 pathway activation. Cell Commun. Signal. 2020, 18, 98. [Google Scholar] [CrossRef]
- Lui, A.; New, J.; Ogony, J.; Thomas, S.; Lewis-Wambi, J. Everolimus downregulates estrogen receptor and induces autophagy in aromatase inhibitor-resistant breast cancer cells. BMC Cancer 2016, 16, 487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsai, C.-L.; Lin, C.-Y.; Chao, A.; Lee, Y.-S.; Wu, R.-C.; Tsai, C.-N.; Yen, C.-F.; Chao, A.-S. GPR30 Activation by 17β-Estradiol Promotes p62 Phosphorylation and Increases Estrogen Receptor α Protein Expression by Inducing Its Release from a Complex Formed with KEAP1. J. Pers. Med. 2021, 11, 906. https://doi.org/10.3390/jpm11090906
Tsai C-L, Lin C-Y, Chao A, Lee Y-S, Wu R-C, Tsai C-N, Yen C-F, Chao A-S. GPR30 Activation by 17β-Estradiol Promotes p62 Phosphorylation and Increases Estrogen Receptor α Protein Expression by Inducing Its Release from a Complex Formed with KEAP1. Journal of Personalized Medicine. 2021; 11(9):906. https://doi.org/10.3390/jpm11090906
Chicago/Turabian StyleTsai, Chia-Lung, Chiao-Yun Lin, Angel Chao, Yun-Shien Lee, Ren-Chin Wu, Chi-Neu Tsai, Chih-Feng Yen, and An-Shine Chao. 2021. "GPR30 Activation by 17β-Estradiol Promotes p62 Phosphorylation and Increases Estrogen Receptor α Protein Expression by Inducing Its Release from a Complex Formed with KEAP1" Journal of Personalized Medicine 11, no. 9: 906. https://doi.org/10.3390/jpm11090906
APA StyleTsai, C.-L., Lin, C.-Y., Chao, A., Lee, Y.-S., Wu, R.-C., Tsai, C.-N., Yen, C.-F., & Chao, A.-S. (2021). GPR30 Activation by 17β-Estradiol Promotes p62 Phosphorylation and Increases Estrogen Receptor α Protein Expression by Inducing Its Release from a Complex Formed with KEAP1. Journal of Personalized Medicine, 11(9), 906. https://doi.org/10.3390/jpm11090906