
The Importance of Epigenetics in Diagnostics and Treatment of Major Depressive Disorder

 , ,
, ,
Abstract
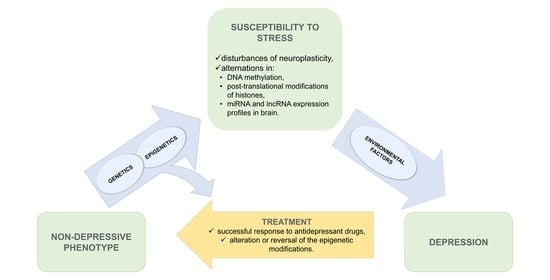
1. Introduction
2. DNA Methylation
2.1. DNA Methylation in Depression

{kind=link}
{kind=link}
| Gene | Material | Model/Study Description | Antidepressant | Results | Study |
|---|---|---|---|---|---|
| BDNF | Buccal swab | French patients (age > 65) with depression | - | Significantly higher methylation of gene promoter. | [22] |
| PBMC | Korean patients with MDD, before and after 12-week long antidepressant treatment | SSRIs, SNRIs, TCAs, | Better treatment response in patients with lower methylation of BDNF promoter. | [23] | |
| PBMC | Patients with MDD after antidepressant treatment | SSRIs, SNRIs | Decreased BDNF methylation after antidepressant treatment. | [24] | |
| PBMC | MDD patients before treatment with antidepressants and assessed outcomes at the study endpoint | - | Significantly lower methylation of BDNF in non-responders to antidepressant treatment. | [25] | |
| Buccal swab | Fifty-seven women with depressive symptoms during pregnancy and their infants | - | Prenatal depressive symptoms significantly decreased BDNF methylation in both male and female infants. | [26] | |
| SLC6A4 | PMBC and umbilical cord leukocytes (infants) | Women with depressive symptoms during pregnancy and their infants | - | Increased maternal depressed mood symptoms in the 2nd trimester were associated with the lower maternal and infant SLC6A4 promoter methylation status. | [27] |
| Peripheral blood | Patients with MDD and childhood trauma, during antidepressant treatment with SSRIs | SSRIs | Increased methylation of promoter after antidepressant treatment. | [28] | |
| Peripheral blood | Korean patients with MDD, before and after 12 week long antidepressant treatment | amitriptyline, bupropion, escitalopram, fluoxetine, imipramine, mirtazapine, paroxetine, sertraline, venlafaxine | Higher methylation of SLC6A4 was associated with worst treatment response and worst depressive symptoms remission. | [29] | |
| Peripheral blood | 84 monozygotic twin pairs with or without depressive symptoms | - | Intrapair DNA methylation variance was significantly correlated with intrapair differences in depressive symptoms. | [30] | |
| Peripheral blood | Caucasian MDD patients before and after antidepressant treatment with escitalopram | escitalopram | Hypomethylation of the promoter was associated with diminished response rate to escitalopram. | [31] | |
| Peripheral blood | Japanese patients with MDD before and after antidepressant treatment | paroxetine, fluvoxamine | Hypermethylation of the SLC6A4 promoter was associated with better treatment response for paroxetine and fluvoxamine. | [32] | |
| Peripheral blood | Caucasian patients with MDD receiving antidepressant drugs | SSRIs, SNRIs, TCAs | Hypomethylation of SLC6A4 predicted impaired antidepressant response. | [33] | |
| NR3C1 | Peripheral blood | MDD patients with childhood maltreatment and sexual abuse | - | Childhood sexual abuse, and maltreatment positively correlated with higher NR3C1 methylation. | [34] |
| Buccal swab | Fifty-seven women with depressive symptoms during pregnancy and their infants | - | Prenatal depressive symptoms significantly increased NR3C1 methylation in male infants. | [26] | |
| ZNF575 | Cord blood | Women with depressive symptoms during pregnancy, undergoing antidepressant treatment and their infants | SSRIs | Lower DNA methylation of ZNF575 in newborns exposed to antidepressants in utero. | [35] |
| PPFIA4 | Peripheral blood | Japanese patients with MDD receiving antidepressant drugs | paroxetine | Difference in promoter methylation was significantly correlated with treatment response to paroxetine. | [36] |
| HS3ST1 | Peripheral blood | Japanese patients with MDD receiving antidepressant drugs | paroxetine | Difference in promoter methylation was significantly correlated with treatment response to paroxetine. | [36] |
| IL-11 | Peripheral blood | Caucasian European patients with MDD receiving escitalopram or nortriptyline | escitalopram, nortriptyline | Lower methylation was associated with better treatment response; higher methylation was associated with better response to escitalopram, but with worse response to nortriptyline. | [37] |
| TPH2 | Peripheral blood | Han Chinese patients with MDD | SSRIs, SNRIs, SARIs, antipsychotic drugs | Hypomethylation was associated with treatment response rate. | [38] |
2.1.1. Brain-Derived Neurotrophic Factor (BDNF) Gene Methylation in Depression
| Gene | Material | Study Description | Antidepressant | Results | Study |
|---|---|---|---|---|---|
| BDNF | Prefrontal cortex and hippocampus | Rat model of infant maltreatment by a caregiver | - | Increased methylation of BDNF in the prefrontal cortex | [25] |
| Hippocampus | Mouse model of post-stroke depression | Fluoxetine | Increased methylation of BDNF promoter in hippocampus as well as reduction in depressive behavior | [44] | |
| NR3C1 | hippocampus | Rat model: pups with increased frequency of licking, grooming and arched-back nursing by mother versus neglected rats | - | Offspring neglected by a mother showed changes in GR methylation levels | [23] |
| THP1 | PBMC | Rats subjected to chronic mild stress procedure and administrated with venlafaxine | venlafaxine | Increased methylation status after antidepressant therapy | [45] |
| GPX4 | PBMC | Rats subjected to chronic mild stress procedure and administrated with agomelatine | agomelatine | Decreased methylation status after antidepressant therapy | [46] |
| NOS1 | Brain tissues | Rats subjected to chronic mild stress procedure and administrated with agomelatine | agomelatine | Decreased methylation status of gene promoter in amygdala and basal ganglia after antidepressant therapy | [46] |
| GPX1 | Brain tissues | Rats subjected to chronic mild stress procedure and administrated with agomelatine | agomelatine | Decreased methylation status of gene promoter in hippocampus and hypothalamus after antidepressant therapy | [46] |
| CAT | Brain tissues | Rats subjected to chronic mild stress procedure and administrated with agomelatine | agomelatine | Decreased methylation status of gene promoter in cerebral cortex after antidepressant therapy | [46] |
2.1.2. SLC6A4 Gene Methylation in Depression
2.1.3. NR3C1 Gene Methylation in Depression
2.2. DNA Methylation and Antidepressant Treatment
2.2.1. Human Research
2.2.2. Animal Models of Depression
3. Histone Modifications
3.1. Histone Modifications in the Diagnostics of Depression
3.1.1. Animal Models of Depression
3.1.2. Clinical Trials in Depression
3.2. Histone Modifications in the Treatment of Depression
3.2.1. Animal Models of Depression
3.2.2. Clinical Trials in Depression
4. miRNA
4.1. miRNAs and the Brain—A Link between Neuroplasticity and Depression
4.2. Circulating miRNAs as Biomarkers of Depression
4.2.1. Blood, Plasma and Serum miRNAs as Biomarkers of Depression
4.2.2. Cerebrospinal Fluid miRNA as a Biomarker of Depression
4.3. miRNAs and Depression Therapy
| Treatment | Model/Patients | Type of Biological Material | Results/Findings | Paper |
|---|---|---|---|---|
| Selective serotonin reuptake inhibitors (SSRIs) | ||||
| Fluoxetine | Mice infused with antidepressant into raphe nuclei | Raphe nuclei | Fluoxetine infusion caused overexpression of miR-16, which resulted in reduction of SERT expression. | [154] |
| MS in Sprague Dawley rat | Hippocampi | MS reduced expression of miR-451, while fluoxetine treatment reversed this change. | [166] | |
| SK-N-SH and SH-SY5Y neuroblastoma cell lines | Cell lysates | Fluoxetine increased expression of miR-572 and miR-663a, which are involved in fundamental neurodevelopmental processes. | [167] | |
| Genetically modified mouse models, expressing higher or lower levels of miR135; chronic social defeat model | Raphe nuclei | Mice overexpressing miR-135 were resilient to social defeat, while knockout induced anxiety-like behaviors and decreased the response to antidepressants. | [168] | |
| PTSD in mice | Prefrontal cortices | Reduction of miR-1971 expression after administration of fluoxetine. | [169] | |
| Paroxetine | 1C11 neuroectodermal cell line | Cell lysates | miR-16 overexpression resulted in reduction of SERT translation and [3H]-paroxetine binding sides. | [154] |
| U87 human glioblastoma–astrocytoma cell line | Cell lysates | Treatment with paroxetine induced expression of BDNF and was limited by upregulation of miR-30a-5p. | [170] | |
| 80 human LCLs form healthy adult females | Cell lysates | Paroxetine-sensitive LCLs overexpressed miR-151-3p. | [171] | |
| 84 depressed patients and 78 control volunteers | Serum | Eight-week treatment resulted in increased miRNA-451a levels, decreased miRNA-34a-5p and miRNA-221-3p levels and reduced HAM-D scores. | [172] | |
| Citalopram | 32 depressed patients, classified into remitters and non–responders, based on changes in HAM-D scores, and 18 controls | Blood | Downregulation of miR-1202 at baseline was found in remitters, when compared to non-responders and controls; eight-week citalopram administration upregulated miR-1202 only in remitters. | [173] |
| 45 untreated depressed patients, 32 treated with citalopram and 32 healthy volunteers | Plasma | miR-132 was upregulated in untreated depressed patients, when compared to healthy controls and citalopram-treated patients; and its expression decreased after two-month citalopram administration, while at the same time miR-124 expression increased. | [174] | |
| Escitalopram | MS and CUS in Sprague Dawley rats | NAc and striatum | Escitalopram normalized increased expression of miR-326 after MS and CUS. | [175] |
| 10 depressed patients | Blood | Treatment significantly altered expression of 30 miRNAs: 28 were upregulated, while two were downregulated. | [176] | |
| 158 depressed patients | Blood | Downregulation of miR-146a-5p, miR-146b-5p and miR-24-3p after eight-week treatment with escitalopram | [177] | |
| Serotonin–norepinephrine reuptake inhibitors (SNRIs) | ||||
| Duloxetine | Kunming mice in CUMS | Hippocampi and frontal lobes | Three-week administration of duloxetine caused increased expression of miR-18a, and miR-132 in hippocampus, while miR-134 and miR-124a were downregulated; only miR-18 was upregulated in frontal lobe. | [178] |
| 258 depressed patients | Blood | 16 miRNAs changed specifically according to duloxetine treatment, while miR-146a-5p, miR-146b-5p, miR-24-3p, miR-425-3p and miR-3074-5p were all downregulated and strongly correlated. | [177] | |
| Desvenlafaxine | 20 depressed patients | Blood | Using MRI miR-1202 levels were correlated with changes in brain activity and connectivity occurring during an eight-week antidepressant treatment. | [179] |
| Miscellaneous | ||||
| Imipramine (TCA) | Mice in CSDS | Blood | Administration of imipramine downregulated miR-146b-5p, miR-24-3p and miR-425, while no differences were detected in the non-responders or saline control groups. | [177] |
| Ketamine | MS in Sprague Dawley rats | Hippocampi | Upregulation of miR-598-5p after treatment. | [166] |
| Sprague Dawley rats | Hippocampi | Three injections of ketamine once per day caused downregulation of miR-206, while increase of BDNF level. | [180] | |
| ECT | MS in Sprague Dawley rats | Hippocampi | Upregulation of miR-598-5p after treatment. | [166] |
| ECS in Sprague Dawley rats | Blood and brains | Chronic and acute ECS upregulated miR-212 in dentate gyrus, while only chronic ECS increased level of this molecule in blood. | [181] | |
| 37 depressed patients and 34 healthy controls | Blood | Elevated levels of miR-126-3p and miR-106a-5p were normalized after ECT. | [182] | |
| 24 TRD depressed patients and 20 healthy controls | Blood | miR-let-7b and miR-let-7c were downregulated after treatment as compared to the controls. | [183] | |
| CBT | 24 depressed patients | Blood | Significantly higher levels of miR-135a after 12 weeks of CTB, when compared to the same patients before therapy and with population treated with SSRIs. | [168] |
| Different antidepressants | 68 patients treated with escitalopram (SSRI) or nortriptyline (TCA) | Blood | Downregulation of miR-146a-5p, miR-146b-5p, miR-24-3p and miR-425-3p after treatment. | [177] |
| 32 depressed patients | PBMCs | Significantly increased level of miR-124 in depressed patients as compared to the healthy controls being decreased after eight-week antidepressant treatment. | [184] | |
4.3.1. SSRI
4.3.2. SNRIs and TCA
5. lncRNA in Depression
5.1. lncRNA Description and Functions
5.2. lncRNA in the Nervous System
5.3. lncRNAs as Depression Biomarkers
5.4. Future Research
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Kennis, M.; Gerritsen, L.; Van Dalen, M.; Williams, A.; Cuijpers, P.; Bockting, C. Prospective biomarkers of major depressive disorder: A systematic review and meta-analysis. Mol. Psychiatry 2020, 25, 321–338. [Google Scholar] [CrossRef]
- Pitsillou, E.; Bresnehan, S.M.; Kagarakis, E.A.; Wijoyo, S.J.; Liang, J.; Hung, A.; Karagiannis, T.C. The cellular and molecular basis of major depressive disorder: Towards a unified model for understanding clinical depression. Mol. Biol. Rep. 2020, 47, 753–770. [Google Scholar] [CrossRef] [PubMed]
- Wray, N.R.; Ripke, S.; Mattheisen, M.; Trzaskowski, M.; Byrne, E.M.; Abdellaoui, A.; Adams, M.J.; Agerbo, E.; Air, T.M.; Andlauer, T.M.F.; et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat. Genet. 2018, 50, 668–681. [Google Scholar] [CrossRef]
- Sullivan, P.F.; Neale, M.C.; Kendler, K.S. Genetic Epidemiology of Major Depression: Review and Meta-Analysis. Am. J. Psychiatry 2000, 157, 1552–1562. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Kennedy, P.J.; Nestler, E.J. Epigenetics of the Depressed Brain: Role of Histone Acetylation and Methylation. Neuropsychopharmacology 2013, 38, 124–137. [Google Scholar] [CrossRef]
- Li, M.; D’Arcy, C.; Li, X.; Zhang, T.; Joober, R.; Meng, X. What do DNA methylation studies tell us about depression? A systematic review. Transl. Psychiatry 2019, 9, 1–14. [Google Scholar] [CrossRef]
- Uchida, S.; Yamagata, H.; Seki, T.; Watanabe, Y. Epigenetic mechanisms of major depression: Targeting neuronal plasticity. Psychiatry Clin. Neurosci. 2018, 72, 212–227. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Meng, L.; Pei, F.; Zheng, Y.; Leng, J. A review of DNA methylation in depression. J. Clin. Neurosci. 2017, 43, 39–46. [Google Scholar] [CrossRef]
- Kriaucionis, S.; Heintz, N. The Nuclear DNA Base 5-Hydroxymethylcytosine Is Present in Purkinje Neurons and the Brain. Science 2009, 324, 929–930. [Google Scholar] [CrossRef]
- Catalanotto, C.; Cogoni, C.; Zardo, G. MicroRNA in Control of Gene Expression: An Overview of Nuclear Functions. Int. J. Mol. Sci. 2016, 17, 1712. [Google Scholar] [CrossRef]
- Morlando, M.; Fatica, A. Alteration of Epigenetic Regulation by Long Noncoding RNAs in Cancer. Int. J. Mol. Sci. 2018, 19, 570. [Google Scholar] [CrossRef]
- Hulshoff, M.S.; Xu, X.; Krenning, G.; Zeisberg, E.M. Epigenetic Regulation of Endothelial-to-Mesenchymal Transition in Chronic Heart Disease. Arter. Thromb. Vasc. Biol. 2018, 38, 1986–1996. [Google Scholar] [CrossRef] [PubMed]
- Ma, N.; Tie, C.; Yu, B.; Zhang, W.; Wan, J. Identifying lncRNA-miRNA-mRNA networks to investigate Alzheimer’s disease pathogenesis and therapy strategy. Aging 2020, 12, 2897–2920. [Google Scholar] [CrossRef] [PubMed]
- Nafee, T.M.; Farrell, W.E.; Carroll, W.D.; Fryer, A.A.; Ismail, K.M.K. Epigenetic control of fetal gene expression. Int. J. Obstet. Gynaecol. 2007, 115, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Kondo, Y. Epigenetic Cross-Talk between DNA Methylation and Histone Modifications in Human Cancers. Yonsei Med. J. 2009, 50, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Vialou, V.; Feng, J.; Robison, A.J.; Nestler, E.J. Epigenetic Mechanisms of Depression and Antidepressant Action. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 59–87. [Google Scholar] [CrossRef]
- Murgatroyd, C.; Patchev, A.V.; Wu, Y.; Micale, V.; Bockmühl, Y.; Fischer, D.; Holsboer, F.; Wotjak, C.T.; Almeida, O.F.X.; Spengler, D. Dynamic DNA methylation programs persistent adverse effects of early-life stress. Nat. Neurosci. 2009, 12, 1559–1566. [Google Scholar] [CrossRef]
- Lin, E.; Tsai, S.-J. Epigenetics and Depression: An Update. Psychiatry Investig. 2019, 16, 654–661. [Google Scholar] [CrossRef]
- Volkow, N.D.; Koob, G.; Baler, R. Biomarkers in Substance Use Disorders. ACS Chem. Neurosci. 2015, 6, 522–525. [Google Scholar] [CrossRef] [PubMed]
- Saavedra, K.; Molina-Márquez, A.M.; Saavedra, N.; Zambrano, T.; Salazar, L.A. Epigenetic Modifications of Major Depressive Disorder. Int. J. Mol. Sci. 2016, 17, 1279. [Google Scholar] [CrossRef]
- Córdova-Palomera, A.; Fatjó-Vilas, M.; Gastó, C.; Navarro, V.; Krebs, M.-O.; Fañanás, L. Genome-wide methylation study on depression: Differential methylation and variable methylation in monozygotic twins. Transl. Psychiatry 2015, 5, e557. [Google Scholar] [CrossRef] [PubMed]
- Januar, V.; Ancelin, M.-L.; Ritchie, K.; Saffery, R.; Ryan, J. BDNF promoter methylation and genetic variation in late-life depression. Transl. Psychiatry 2015, 5, e619. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.-J.; Kim, J.-M.; Kim, S.-Y.; Kim, S.-W.; Shin, I.-S.; Kim, H.-R.; Park, M.-H.; Shin, M.-G.; Yoon, J.-H.; Yoon, J.-S. A Longitudinal Study of BDNF Promoter Methylation and Depression in Breast Cancer. Psychiatry Investig. 2015, 12, 523–531. [Google Scholar] [CrossRef]
- D’Addario, C.; Dell’Osso, B.; Galimberti, D.; Palazzo, M.C.; Benatti, B.; Di Francesco, A.; Scarpini, E.; Altamura, A.C.; Maccarrone, M. Epigenetic modulation of BDNF gene in patients with major depressive disorder. Biol. Psychiatry 2013, 73, e6–e7. [Google Scholar] [CrossRef]
- Tadić, A.; Müller-Engling, L.; Schlicht, K.F.; Kotsiari, A.; Dreimüller, N.; Kleimann, A.; Bleich, S.; Lieb, K.; Frieling, H. Methylation of the promoter of brain-derived neurotrophic factor exon IV and antidepressant response in major depression. Mol. Psychiatry 2014, 19, 281–283. [Google Scholar] [CrossRef]
- Braithwaite, E.C.; Kundakovic, M.; Ramchandani, P.G.; Murphy, S.E.; Champagne, F.A. Maternal prenatal depressive symptoms predict infant NR3C1 1F and BDNF IV DNA methylation. Epigenetics 2015, 10, 408–417. [Google Scholar] [CrossRef] [PubMed]
- Devlin, A.M.; Brain, U.; Austin, J.; Oberlander, T.F. Prenatal exposure to maternal depressed mood and the MTHFR C677T variant affect SLC6A4 methylation in infants at birth. PLoS ONE 2010, 5, e12201. [Google Scholar] [CrossRef]
- Booij, L.; Szyf, M.; Carballedo, A.; Frey, E.-M.; Morris, D.; Dymov, S.; Vaisheva, F.; Ly, V.; Fahey, C.; Meaney, J.; et al. DNA methylation of the serotonin transporter gene in peripheral cells and stress-related changes in hippocampal volume: A study in depressed patients and healthy controls. PLoS ONE 2015, 10, e0119061. [Google Scholar] [CrossRef]
- Kang, H.-J.; Kim, J.-M.; Stewart, R.; Kim, S.-Y.; Bae, K.-Y.; Kim, S.-W.; Shin, I.-S.; Shin, M.-G.; Yoon, J.-S. Association of SLC6A4 methylation with early adversity, characteristics and outcomes in depression. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2013, 44, 23–28. [Google Scholar] [CrossRef]
- Zhao, J.; Goldberg, J.; Bremner, J.D.; Vaccarino, V. Association between promoter methylation of serotonin transporter gene and depressive symptoms: A monozygotic twin study. Psychosom. Med. 2013, 75, 523–529. [Google Scholar] [CrossRef]
- Domschke, K.; Tidow, N.; Schwarte, K.; Deckert, J.; Lesch, K.-P.; Arolt, V.; Zwanzger, P.; Baune, B.T. Serotonin transporter gene hypomethylation predicts impaired antidepressant treatment response. Int. J. Neuropsychopharmacol. 2014, 17, 1167–1176. [Google Scholar] [CrossRef] [PubMed]
- Okada, S.; Morinobu, S.; Fuchikami, M.; Segawa, M.; Yokomaku, K.; Kataoka, T.; Okamoto, Y.; Yamawaki, S.; Inoue, T.; Kusumi, I.; et al. The potential of SLC6A4 gene methylation analysis for the diagnosis and treatment of major depression. J. Psychiatr. Res. 2014, 53, 47–53. [Google Scholar] [CrossRef]
- Schiele, M.A.; Zwanzger, P.; Schwarte, K.; Arolt, V.; Baune, B.T.; Domschke, K. Serotonin transporter gene promoter hypomethylation as a predictor of antidepressant treatment response in major depression—A replication study. Int. J. Neuropsychopharmacol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Perroud, N.A.; Paolonigiacobino, A.; Prada, P.B.; Olié, E.; Salzmann, A.; Nicastro, R.; Guillaume, S.; Mouthon-Mermoud, D.; Stouder, C.; Dieben, K.; et al. Increased methylation of glucocorticoid receptor gene (NR3C1) in adults with a history of childhood maltreatment: A link with the severity and type of trauma. Transl. Psychiatry 2011, 1, e59. [Google Scholar] [CrossRef]
- Cardenas, A.; Faleschini, S.; Hidalgo, A.C.; Rifas-Shiman, S.L.; Baccarelli, A.A.; DeMeo, D.L.; Litonjua, A.A.; Neumann, A.; Felix, J.F.; Jaddoe, V.W.V.; et al. Prenatal maternal antidepressants, anxiety, and depression and offspring DNA methylation: Epigenome-wide associations at birth and persistence into early childhood. Clin. Epigenet. 2019, 11, 56. [Google Scholar] [CrossRef]
- Takeuchi, N.; Nonen, S.; Kato, M.; Wakeno, M.; Takekita, Y.; Kinoshita, T.; Kugawa, F. Therapeutic Response to Paroxetine in Major Depressive Disorder Predicted by DNA Methylation. Neuropsychobiology 2018, 75, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Powell, T.R.; Smith, R.G.; Hackinger, S.; Schalkwyk, L.C.; Uher, R.; McGuffin, P.; Mill, J.; E Tansey, K. DNA methylation in interleukin-11 predicts clinical response to antidepressants in GENDEP. Transl. Psychiatry 2013, 3, e300. [Google Scholar] [CrossRef]
- Shen, T.; Li, X.; Chen, L.; Chen, Z.; Tan, T.; Hua, T.; Chen, B.; Yuan, Y.; Zhang, Z.; Kuney, L.; et al. The relationship of tryptophan hydroxylase-2 methylation to early-life stress and its impact on short-term antidepressant treatment response. J. Affect. Disord. 2020, 276, 850–858. [Google Scholar] [CrossRef] [PubMed]
- Sabunciyan, S.; Aryee, M.J.; Irizarry, R.A.; Rongione, M.; Webster, M.J.; Kaufman, W.E.; Murakami, P.; Lessard, A.; Yolken, R.H.; Feinberg, A.P.; et al. Genome-wide DNA methylation scan in major depressive disorder. PLoS ONE 2012, 7, e34451. [Google Scholar] [CrossRef]
- Fuchikami, M.; Morinobu, S.; Segawa, M.; Okamoto, Y.; Yamawaki, S.; Ozaki, N.; Inoue, T.; Kusumi, I.; Koyama, T.; Tsuchiyama, K.; et al. DNA methylation profiles of the brain-derived neurotrophic factor (BDNF) gene as a potent diagnostic biomarker in major depression. PLoS ONE 2011, 6, e23881. [Google Scholar] [CrossRef]
- Martinowich, K.; Hattori, D.; Wu, H.; Fouse, S.; He, F.; Hu, Y.; Fan, G.; Sun, Y.E. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science 2003, 302, 890–893. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.-J.; Kim, J.-M.; Lee, J.-Y.; Kim, S.-Y.; Bae, K.-Y.; Kim, S.-W.; Shin, I.-S.; Kim, H.-R.; Shin, M.-G.; Yoon, J.-S. BDNF promoter methylation and suicidal behavior in depressive patients. J. Affect. Disord. 2013, 151, 679–685. [Google Scholar] [CrossRef]
- Roth, T.L.; Lubin, F.D.; Funk, A.J.; Sweatt, J.D. Lasting epigenetic influence of early-life adversity on the BDNF gene. Biol. Psychiatry 2009, 65, 760–769. [Google Scholar] [CrossRef]
- Jin, H.-J.; Pei, L.; Li, Y.-N.; Zheng, H.; Yang, S.; Wan, Y.; Mao, L.; Xia, Y.-P.; He, Q.-W.; Li, M.; et al. Alleviative effects of fluoxetine on depressive-like behaviors by epigenetic regulation of BDNF gene transcription in mouse model of post-stroke depression. Sci. Rep. 2017, 7, 1–15. [Google Scholar] [CrossRef]
- Wigner, P.; Synowiec, E.; Jóźwiak, P.; Czarny, P.; Bijak, M.; Białek, K.; Szemraj, J.; Gruca, P.; Papp, M.; Śliwiński, T. The Effect of Chronic Mild Stress and Venlafaxine on the Expression and Methylation Levels of Genes Involved in the Tryptophan Catabolites Pathway in the Blood and Brain Structures of Rats. J. Mol. Neurosci. 2020, 70, 1425–1436. [Google Scholar] [CrossRef]
- Wigner, P.; Synowiec, E.; Jóźwiak, P.; Czarny, P.; Bijak, M.; Barszczewska, G.; Białek, K.; Szemraj, J.; Gruca, P.; Papp, M.; et al. The Changes of Expression and Methylation of Genes Involved in Oxidative Stress in Course of Chronic Mild Stress and Antidepressant Therapy with Agomelatine. Genes 2020, 11, 644. [Google Scholar] [CrossRef]
- Booij, L.; Szyf, M.; Carballedo, A.; Frey, E.-M.; Morris, D.; Ly, V.; Fahey, C.; Meaney, J.; Gill, M.; Frodl, T. The role of SLC6A4 DNA methylation in stress-related changes in hippocampal volume: A study in depressed patients and healthy controls. In Proceedings of the 29th CINP World Congress of Neuropsychopharmacology, Vancouver, BC, Canada, 22–26 June 2014. [Google Scholar]
- Chiarella, J.; Schumann, L.; Pomares, F.B.; Frodl, T.; Tozzi, L.; Nemoda, Z.; Yu, P.; Szyf, M.; Khalid-Khan, S.; Booij, L. DNA methylation differences in stress-related genes, functional connectivity and gray matter volume in depressed and healthy adolescents. J. Affect. Disord. 2020, 271, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Weaver, I.C.G.; Cervoni, N.; Champagne, F.A.; D’Alessio, A.C.; Sharma, S.; Seckl, J.R.; Dymov, S.; Szyf, M.; Meaney, M.J. Epigenetic programming by maternal behavior. Nat. Neurosci. 2004, 7, 847–854. [Google Scholar] [CrossRef]
- Fuchikami, M.; Morinobu, S.; Kurata, A.; Yamamoto, S.; Yamawaki, S. Single immobilization stress differentially alters the expression profile of transcripts of the brain-derived neurotrophic factor (BDNF) gene and histone acetylation at its promoters in the rat hippocampus. Int. J. Neuropsychopharmacol. 2009, 12, 73–82. [Google Scholar] [CrossRef]
- Gassen, N.C.; Fries, G.R.; Zannas, A.S.; Hartmann, J.; Zschocke, J.; Hafner, K.; Carrillo-Roa, T.; Steinbacher, J.; Preißinger, S.N.; Hoeijmakers, L.; et al. Chaperoning epigenetics: FKBP51 decreases the activity of DNMT1 and mediates epigenetic effects of the antidepressant paroxetine. Sci. Signal. 2015, 8, ra119. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, N.; Zschocke, J.; Perisic, T.; Yu, S.; Holsboer, F.; Rein, T. Antidepressants inhibit DNA methyltransferase 1 through reducing G9a levels. Biochem. J. 2012, 448, 93–102. [Google Scholar] [CrossRef]
- Le François, B.; Soo, J.; Millar, A.M.; Daigle, M.; Le Guisquet, A.-M.; Leman, S.; Minier, F.; Belzung, C.; Albert, P.R. Chronic mild stress and antidepressant treatment alter 5-HT1A receptor expression by modifying DNA methylation of a conserved Sp4 site. Neurobiol. Dis. 2015, 82, 332–341. [Google Scholar] [CrossRef] [PubMed]
- Elliott, E.; Ezra-Nevo, G.; Regev, L.; Neufeld-Cohen, A.; Chen, A. Resilience to social stress coincides with functional DNA methylation of the Crf gene in adult mice. Nat. Neurosci. 2010, 13, 1351–1353. [Google Scholar] [CrossRef]
- Sales, A.J.; Joca, S.R.L. Antidepressant administration modulates stress-induced DNA methylation and DNA methyltransferase expression in rat prefrontal cortex and hippocampus. Behav. Brain Res. 2018, 343, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Sales, A.J.; Biojone, C.; Terceti, M.S.; Guimarães, F.S.; Gomes, M.V.M.; Joca, S.R.L. Antidepressant-like effect induced by systemic and intra-hippocampal administration of DNA methylation inhibitors. Br. J. Pharmacol. 2011, 164, 1711–1721. [Google Scholar] [CrossRef]
- Sales, A.J.; Joca, S.R.L. Effects of DNA methylation inhibitors and conventional antidepressants on mice behaviour and brain DNA methylation levels. Acta Neuropsychiatr. 2015, 28, 11–22. [Google Scholar] [CrossRef]
- Nestler, E.J.; Peña, C.J.; Kundakovic, M.; Mitchell, A.; Akbarian, S. Epigenetic Basis of Mental Illness. Neuroscientist 2016, 22, 447–463. [Google Scholar] [CrossRef]
- Belzeaux, R.; Lin, R.; Ju, C.; Chay, M.-A.; Fiori, L.M.; Lutz, P.-E.; Turecki, G. Transcriptomic and epigenomic biomarkers of antidepressant response. J. Affect. Disord. 2018, 233, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, M.; Fan, M.; Song, Y.; Yu, H.; Zhi, X.; Xiao, K.; Lai, S.; Zhang, J.; Jin, X.; et al. Chromodomain Y-like Protein-Mediated Histone Crotonylation Regulates Stress-Induced Depressive Behaviors. Biol. Psychiatry 2019, 85, 635–649. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.C.; Maze, I. Histone Crotonylation Makes Its Mark in Depression Research. Biol. Psychiatry 2019, 85, 616–618. [Google Scholar] [CrossRef] [PubMed]
- Renthal, W.; Maze, I.; Krishnan, V.; Covington, H.E., 3rd; Xiao, G.; Kumar, A.; Russo, S.J.; Graham, A.; Tsankova, N.; Kippin, T.E. Histone deacetylase 5 epigenetically controls behavioral adaptations to chronic emotional stimuli. Neuron 2007, 56, 517–529. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Fan, W.; Zhang, X.; Dong, E. Gestational stress induces depressive-like and anxiety-like phenotypes through epigenetic regulation of BDNF expression in offspring hippocampus. Epigenetics 2016, 11, 150–162. [Google Scholar] [CrossRef]
- Covington, H.E., 3rd; Maze, I.; LaPlant, Q.C.; Vialou, V.F.; Ohnishi, Y.N.; Berton, O.; Fass, D.M.; Renthal, W.; Rush, A.J., 3rd; Wu, E.Y. Antidepressant actions of histone deacetylase inhibitors. J. Neurosci. 2009, 29, 11451–11460. [Google Scholar] [CrossRef]
- Covington, H.E., 3rd; Maze, I.; Sun, H.; Bomze, H.M.; DeMaio, K.D.; Wu, E.Y.; Dietz, D.M.; Lobo, M.K.; Ghose, S.; Mouzon, E. A role for repressive histone methylation in cocaine-induced vulnerability to stress. Neuron 2011, 71, 656–670. [Google Scholar] [CrossRef] [PubMed]
- Romero, S.M.; Montesinos, J.; Pascual, M.; Aguilar, M.; Roger-Sanchez, C.; Guerri, C.; Miñarro, J.; Rodríguez-Arias, M. Up-regulation of histone acetylation induced by social defeat mediates the conditioned rewarding effects of cocaine. Prog. Neuropsychopharmacol. Biol. Psychiatry 2016, 70, 39–48. [Google Scholar] [CrossRef]
- Han, A.; Sung, Y.-B.; Chung, S.-Y.; Kwon, M.-S. Possible additional antidepressant-like mechanism of sodium butyrate: Targeting the hippocampus. Neuropharmacology 2014, 81, 292–302. [Google Scholar] [CrossRef]
- Maury, E.; Hashizume, R. Epigenetic modification in chromatin machinery and its deregulation in pediatric brain tumors: Insight into epigenetic therapies. Epigenetic 2017, 12, 353–369. [Google Scholar] [CrossRef] [PubMed]
- Tsankova, N.M.; Berton, O.; Renthal, W.; Kumar, A.; Neve, R.L.; Nestler, E.J. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat. Neurosci. 2006, 9, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Wang, J.; Zhang, K.; Zhao, M.; Ellenbroek, B.; Shao, F.; Wang, W. Effects of adolescent social stress and antidepressant treatment on cognitive inflexibility and Bdnf epigenetic modifications in the mPFC of adult mice. Psychoneuroendocrinology 2018, 88, 92–101. [Google Scholar] [CrossRef]
- Chen, L.; Miao, Z.; Xu, X. β-hydroxybutyrate alleviates depressive behaviors in mice possibly by increasing the histone3-lysine9-β-hydroxybutyrylation. Biochem. Biophys. Res. Commun. 2017, 490, 117–122. [Google Scholar] [CrossRef]
- Francis, D.D.; Szegda, K.; Campbell, G.; Martin, W.D.; Insel, T.R. Epigenetic sources of behavioral differences in mice. Nat. Neurosci. 2003, 6, 445–446. [Google Scholar] [CrossRef] [PubMed]
- Tarantino, L.M.; Sullivan, P.F.; Meltzer-Brody, S. Using animal models to disentangle the role of genetic, epigenetic, and environmental influences on behavioral outcomes associated with maternal anxiety and depression. Front. Psychiatry 2011, 2, 44. [Google Scholar] [CrossRef]
- Uchida, S.; Hara, K.; Kobayashi, A.; Otsuki, K.; Yamagata, H.; Hobara, T.; Suzuki, T.; Miyata, N.; Watanabe, Y. Epigenetic status of Gdnf in the ventral striatum determines susceptibility and adaptation to daily stressful events. Neuron 2011, 69, 359–372. [Google Scholar] [CrossRef]
- Köhler, J.C.; Gröger, N.; Lesse, A.; Ciurana, S.G.; Rether, K.; Fegert, J.; Bock, J.; Braun, K. Early-Life Adversity Induces Epigenetically Regulated Changes in Hippocampal Dopaminergic Molecular Pathways. Mol. Neurobiol. 2019, 56, 3616–3625. [Google Scholar] [CrossRef] [PubMed]
- Seo, M.K.; Kim, S.-G.; Seog, D.-H.; Bahk, W.-M.; Kim, S.-H.; Park, S.W.; Lee, J.G. Effects of Early Life Stress on Epigenetic Changes of the Glucocorticoid Receptor 1(7) Promoter during Adulthood. Int. J. Mol. Sci. 2020, 21, 6331. [Google Scholar] [CrossRef] [PubMed]
- Kenworthy, C.; Sengupta, A.; Luz, S.; Hoeve, E.V.; Meda, K.; Bhatnagar, S.; Abel, T. Social defeat induces changes in histone acetylation and expression of histone modifying enzymes in the ventral hippocampus, prefrontal cortex, and dorsal raphe nucleus. Neuroscience 2014, 264, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Qiu, H.-M.; Fei, H.-Z.; Hu, X.-Y.; Xia, H.-J.; Wang, L.-J.; Qin, L.-J.; Jiang, X.-H.; Zhou, Q.-X. Histone acetylation and expression of mono-aminergic transmitters synthetases involved in CUS-induced depressive rats. Exp. Biol. Med. 2014, 239, 330–336. [Google Scholar] [CrossRef]
- Li, H.-Y.; Jiang, Q.-S.; Fu, X.-Y.; Jiang, X.-H.; Zhou, Q.-X.; Qiu, H.-M. Abnormal modification of histone acetylation involved in depression-like behaviors of rats induced by chronically unpredicted stress. Neuroreport 2017, 28, 1054–1060. [Google Scholar] [CrossRef] [PubMed]
- Wan, Q.; Gao, K.; Rong, H.; Wu, M.; Wang, H.; Wang, X.; Wang, G.; Liu, Z. Histone modifications of the Crhr1 gene in a rat model of depression following chronic stress. Behav. Brain Res. 2014, 271, 1–6. [Google Scholar] [CrossRef]
- Hunter, R.G.; McCarthy, K.J.; Milne, T.A.; Pfaff, D.W.; McEwen, B.S. Regulation of hippocampal H3 histone methylation by acute and chronic stress. Proc. Natl. Acad. Sci. USA 2009, 106, 20912–20917. [Google Scholar] [CrossRef]
- Taufique, S.K.T.; Prabhat, A.; Kumar, V. Illuminated night alters hippocampal gene expressions and induces depressive-like responses in diurnal corvids. Eur. J. Neurosci. 2018, 48, 3005–3018. [Google Scholar] [CrossRef]
- Hobara, T.; Uchida, S.; Otsuki, K.; Matsubara, T.; Funato, H.; Matsuo, K.; Suetsugi, M.; Watanabe, Y. Altered gene expression of histone deacetylases in mood disorder patients. J. Psychiatr. Res. 2010, 44, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Iga, J.-I.; Ueno, S.-I.; Yamauchi, K.; Numata, S.; Kinouchi, S.; Tayoshi-Shibuya, S.; Song, H.; Ohmori, T. Altered HDAC5 and CREB mRNA expressions in the peripheral leukocytes of major depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 2007, 31, 628–632. [Google Scholar] [CrossRef] [PubMed]
- Nagy, C.; Torres-Platas, S.G.; Mechawar, N.; Turecki, G. Repression of Astrocytic Connexins in Cortical and Subcortical Brain Regions and Prefrontal Enrichment of H3K9me3 in Depression and Suicide. Int. J. Neuropsychopharmacol. 2017, 20, 50–57. [Google Scholar] [CrossRef]
- Cruceanu, C.; Alda, M.; Nagy, C.; Freemantle, E.; Rouleau, G.A.; Turecki, G. H3K4 tri-methylation in synapsin genes leads to different expression patterns in bipolar disorder and major depression. Int. J. Neuropsychopharmacol. 2013, 16, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Fass, D.M.; Reis, S.A.; Ghosh, B.; Hennig, K.M.; Joseph, N.F.; Zhao, W.-N.; Nieland, T.J.; Guan, J.-S.; Kuhnle, C.E.G.; Tang, W.; et al. Crebinostat: A novel cognitive enhancer that inhibits histone deacetylase activity and modulates chromatin-mediated neuroplasticity. Neuropharmacology 2013, 64, 81–96. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, B.; Zhao, W.-N.; Reis, S.A.; Patnaik, D.; Fass, D.M.; Tsai, L.-H.; Mazitschek, R.; Haggarty, S.J. Dissecting structure-activity-relationships of crebinostat: Brain penetrant HDAC inhibitors for neuroepigenetic regulation. Bioorg. Med. Chem. Lett. 2016, 26, 1265–1271. [Google Scholar] [CrossRef]
- Gurbani, S.S.; Yoon, Y.; Weinberg, B.D.; Salgado, E.; Press, R.H.; Cordova, J.S.; Ramesh, K.K.; Liang, Z.; Vega, J.V.; Voloschin, A.; et al. Assessing Treatment Response of Glioblastoma to an HDAC Inhibitor Using Whole-Brain Spectroscopic MRI. Tomography 2019, 5, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Yamawaki, Y.; Yoshioka, N.; Nozaki, K.; Ito, H.; Oda, K.; Harada, K.; Shirawachi, S.; Asano, S.; Aizawa, H.; Yamawaki, S.; et al. Sodium butyrate abolishes lipopolysaccharide-induced depression-like behaviors and hippocampal microglial activation in mice. Brain Res. 2018, 1680, 13–38. [Google Scholar] [CrossRef]
- Gundersen, B.B.; Blendy, J.A. Effects of the histone deacetylase inhibitor sodium butyrate in models of depression and anxiety. Neuropharmacology 2009, 57, 67–74. [Google Scholar] [CrossRef]
- Schroeder, F.A.; Lin, C.L.; Crusio, W.E.; Akbarian, S. Antidepressant-like effects of the histone deacetylase inhibitor, sodium butyrate, in the mouse. Biol. Psychiatry 2007, 62, 55–64. [Google Scholar] [CrossRef]
- Covington, H.E., 3rd; Vialou, V.F.; LaPlant, Q.; Ohnishi, Y.N.; Nestler, E.J. Hippocampal-dependent antidepressant-like activity of histone deacetylase inhibition. Neurosci. Lett. 2011, 493, 122–126. [Google Scholar] [CrossRef]
- Wilkinson, M.B.; Xiao, G.; Kumar, A.; LaPlant, Q.; Renthal, W.; Sikder, D.; Kodadek, T.J.; Nestler, E.J. Imipramine treatment and resiliency exhibit similar chromatin regulation in the mouse nucleus accumbens in depression models. J. Neurosci. 2009, 29, 7820–7832. [Google Scholar] [CrossRef]
- Schmauss, C. An HDAC-dependent epigenetic mechanism that enhances the efficacy of the antidepressant drug fluoxetine. Sci. Rep. 2015, 5, 8171. [Google Scholar] [CrossRef]
- Ferland, C.L.; Schrader, L.A. Regulation of histone acetylation in the hippocampus of chronically stressed rats: A potential role of sirtuins. Neuroscience 2011, 174, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Yamawaki, Y.; Fuchikami, M.; Morinobu, S.; Segawa, M.; Matsumoto, T.; Yamawaki, S. Antidepressant-like effect of sodium butyrate (HDAC inhibitor) and its molecular mechanism of action in the rat hippocampus. World J. Biol. Psychiatry 2012, 13, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Geng, X.; Dang, W.; Wu, B.; Dai, Z.; Li, Y.; Yang, Y.; Zhang, H.; Shi, J. Molecular mechanisms associated with the antidepressant effects of the class I histone deacetylase inhibitor MS-275 in the rat ventrolateral orbital cortex. Brain Res. 2012, 1447, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-Y.; Zhang, H.; Gatta, E.; Glover, E.J.; Pandey, S.C.; Lasek, A.W. The histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA) alleviates depression-like behavior and normalizes epigenetic changes in the hippocampus during ethanol withdrawal. Alcohol 2019, 78, 79–87. [Google Scholar] [CrossRef]
- Park, S.W.; Seo, M.K.; Lee, J.G.; Hien, L.T.; Kim, Y.H. Effects of maternal separation and antidepressant drug on epigenetic regulation of the brain-derived neurotrophic factor exon I promoter in the adult rat hippocampus. Psychiatry Clin. Neurosci. 2018, 72, 255–265. [Google Scholar] [CrossRef]
- Qiao, M.; Jiang, Q.-S.; Liu, Y.-J.; Hu, X.-Y.; Wang, L.-J.; Zhou, Q.-X.; Qiu, H.-M. Antidepressant mechanisms of venlafaxine involving increasing histone acetylation and modulating tyrosine hydroxylase and tryptophan hydroxylase expression in hippocampus of depressive rats. Neuroreport 2019, 30, 255–261. [Google Scholar] [CrossRef]
- Jiang, H.; Zhang, X.; Lu, J.; Meng, H.; Sun, Y.; Yang, X.; Zhao, B.; Bao, T. Antidepressant-Like Effects of Acupuncture-Insights From DNA Methylation and Histone Modifications of Brain-Derived Neurotrophic Factor. Front. Psychiatry 2018, 9, 102. [Google Scholar] [CrossRef] [PubMed]
- Tsankova, N.M.; Kumar, A.; Nestler, E.J. Histone modifications at gene promoter regions in rat hippocampus after acute and chronic electroconvulsive seizures. J. Neurosci. 2004, 24, 5603–5610. [Google Scholar] [CrossRef] [PubMed]
- Lopez, J.P.; Mamdani, F.; LaBonte, B.; Beaulieu, M.-M.; Yang, J.P.; Berlim, M.T.; Ernst, C.; Turecki, G. Epigenetic regulation of BDNF expression according to antidepressant response. Mol. Psychiatry 2013, 18, 398–399. [Google Scholar] [CrossRef]
- Targum, S.D.; Cameron, B.R.; Ferreira, L.; MacDonald, I.D. An augmentation study of MSI-195 (S-adenosylmethionine) in Major Depressive Disorder. J. Psychiatr. Res. 2018, 107, 86–96. [Google Scholar] [CrossRef]
- Gebert, L.F.R.; MacRae, I.J. Regulation of microRNA function in animals. Nat. Rev. Mol. Cell Biol. 2019, 20, 21–37. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef]
- Lytle, J.R.; Yario, T.A.; Steitz, J.A. Target mRNAs are repressed as efficiently by microRNA-binding sites in the 5′ UTR as in the 3′ UTR. Proc. Natl. Acad. Sci. USA 2007, 104, 9667–9672. [Google Scholar] [CrossRef]
- Lee, I.; Ajay, S.S.; Yook, J.I.; Kim, H.S.; Hong, S.H.; Kim, N.H.; Dhanasekaran, S.M.; Chinnaiyan, A.M.; Athey, B.D. New class of microRNA targets containing simultaneous 5′-UTR and 3′-UTR interaction sites. Genome Res. 2009, 19, 1175–1183. [Google Scholar] [CrossRef] [PubMed]
- Forman, J.J.; Coller, H.A. The code within the code: MicroRNAs target coding regions. Cell Cycle 2010, 9, 1533–1541. [Google Scholar] [CrossRef]
- Chi, S.W.; Hannon, G.J.; Darnell, R.B. An alternative mode of microRNA target recognition. Nat. Struct. Mol. Biol. 2012, 19, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Seok, H.; Ham, J.; Jang, E.S.; Chi, S.W. MicroRNA target recognition: Insights from transcriptome-wide non-canonical interactions. Mol. Cells 2016, 39, 375–381. [Google Scholar]
- Liu, H.; Lei, C.; He, Q.; Pan, Z.; Xiao, D.; Tao, Y. Nuclear functions of mammalian MicroRNAs in gene regulation, immunity and cancer. Mol. Cancer 2018, 17, 1–14. [Google Scholar] [CrossRef]
- Li, L.-C.; Okino, S.T.; Zhao, H.; Pookot, D.; Place, R.F.; Urakami, S.; Enokida, H.; Dahiya, R. Small dsRNAs induce transcriptional activation in human cells. Proc. Nat. Acad. Sci. USA 2006, 103, 17337–17342. [Google Scholar] [CrossRef] [PubMed]
- Huang, V.; Place, R.F.; Portnoy, V.; Wang, J.; Qi, Z.; Jia, Z.; Yu, A.; Shuman, M.; Yu, J.; Li, L.-C. Upregulation of Cyclin B1 by miRNA and its implications in cancer. Nucleic Acids Res. 2012, 40, 1695–1707. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, J.C.; Younger, S.T.; Nguyen, N.-B.; Hardy, D.B.; Monia, B.P.; Corey, D.R.; A Janowski, B. Antisense transcripts are targets for activating small RNAs. Nat. Struct. Mol. Biol. 2008, 15, 842–848. [Google Scholar] [CrossRef] [PubMed]
- Matsui, M.; Chu, Y.; Zhang, H.; Gagnon, K.T.; Shaikh, S.; Kuchimanchi, S.; Manoharan, M.; Corey, D.R.; Janowski, B.A. Promoter RNA links transcriptional regulation of inflammatory pathway genes. Nucleic Acids Res. 2013, 41, 10086–10109. [Google Scholar] [CrossRef]
- Pisignano, G.; Napoli, S.; Magistri, M.; Mapelli, S.N.; Pastori, C.; Di Marco, S.; Civenni, G.; Albino, D.; Enriquez, C.; Allegrini, S.; et al. A promoter-proximal transcript targeted by genetic polymorphism controls E-cadherin silencing in human cancers. Nat. Commun. 2017, 8, 15622. [Google Scholar] [CrossRef]
- Meng, X.; Jiang, Q.; Chang, N.; Wang, X.; Liu, C.; Xiong, J.; Cao, H.; Liang, Z. Small activating RNA binds to the genomic target site in a seed-region-dependent manner. Nucleic Acids Res. 2016, 44, 2274–2282. [Google Scholar] [CrossRef]
- Zheng, L.; Chen, Y.; Ye, L.; Jiao, W.; Qiangsong, T.; Mei, H.; Li, D.; Yang, F.; Li, H.; Huang, K.; et al. miRNA-584-3p inhibits gastric cancer progression by repressing Yin Yang 1-facilitated MMP-14 expression. Sci. Rep. 2017, 7, 8967. [Google Scholar] [CrossRef]
- Zhang, Y.; Fan, M.; Zhang, X.; Huang, F.; Wu, K.; Zhang, J.; Liu, J.; Huang, Z.; Luo, H.; Tao, L.; et al. Cellular microRNAs up-regulate transcription via interaction with promoter TATA-box motifs. RNA 2014, 20, 1878–1889. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Lan, X.; Han, R.; Wang, J.; Huang, Y.; Sun, J.; Guo, W.; Chen, H. Erratum: miR-2478 inhibits TGFβ1 expression by targeting the transcriptional activation region downstream of the TGFβ1 promoter in dairy goats. Sci. Rep. 2018, 8, 46966. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, H. RNAa Induced by TATA Box-Targeting MicroRNAs. Adv. Exp. Med. Biol. 2017, 983, 91–111. [Google Scholar]
- Paugh, S.W.; Coss, D.R.; Bao, J.; Laudermilk, L.T.; Grace, C.R.; Ferreira, A.M.; Waddell, M.B.; Ridout, G.; Naeve, D.; Leuze, M.; et al. MicroRNAs Form Triplexes with Double Stranded DNA at Sequence-Specific Binding Sites; a Eukaryotic Mechanism via which microRNAs Could Directly Alter Gene Expression. PLoS Comput. Biol. 2016, 12, e1004744. [Google Scholar] [CrossRef]
- Toscano-Garibay, J.D.; Aquino-Jarquin, G. Transcriptional regulation mechanism mediated by miRNA-DNA•DNA triplex structure stabilized by Argonaute. Biochim. Biophys. Acta 2014, 1839, 1079–1083. [Google Scholar] [CrossRef] [PubMed]
- Włodarski, A.; Strycharz, J.; Wróblewski, A.; Kasznicki, J.; Drzewoski, J.; Śliwińska, A. The Role of microRNAs in Metabolic Syndrome-Related Oxidative Stress. Int. J. Mol. Sci. 2020, 21, 6902. [Google Scholar] [CrossRef]
- Roy, B.; Yoshino, Y.; Allen, L.; Prall, K.; Schell, G.; Dwivedi, Y. Exploiting Circulating MicroRNAs as Biomarkers in Psychiatric Disorders. Mol. Diagn. 2020, 24, 279–298. [Google Scholar] [CrossRef]
- Iorio, M.V.; Croce, C.M. MicroRNA dysregulation in cancer: Diagnostics, monitoring and therapeutics. A comprehensive review. EMBO Mol. Med. 2012, 4, 143–159. [Google Scholar] [CrossRef] [PubMed]
- Singh, T.; Yadav, S. Role of microRNAs in neurodegeneration induced by environmental neurotoxicants and aging. Ageing Res. Rev. 2020, 60, 101068. [Google Scholar] [CrossRef]
- Yuan, H.; Mischoulon, D.; Fava, M.; Otto, M.W. Circulating microRNAs as biomarkers for depression: Many candidates, few finalists. J. Affect. Disord. 2018, 233, 68–78. [Google Scholar] [CrossRef]
- Dwivedi, Y. Pathogenetic and therapeutic applications of microRNAs in major depressive disorder. Prog. Neuropsychopharmacol. Biol. Psychiatry 2016, 64, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Lennox, A.L.; Mao, H.; Silver, D.L. RNA on the brain: Emerging layers of post-transcriptional regulation in cerebral cortex development. Wiley Interdiscip. Rev. Dev. Biol. 2018, 7, e290. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Wang, Z.Z.; Zhao, M.; Zhang, Y.; Chen, N.H. Role of non-coding RNA in the pathogenesis of depression. Gene 2020, 735, 144276. [Google Scholar] [CrossRef]
- Rajasethupathy, P.; Fiumara, F.; Sheridan, R.; Betel, D.; Puthanveettil, S.V.; Russo, J.J.; Sander, C.; Tuschl, T.; Kandel, E. Characterization of small RNAs in Aplysia reveals a role for miR-124 in constraining synaptic plasticity through CREB. Neuron 2009, 63, 803–817. [Google Scholar] [CrossRef] [PubMed]
- Schratt, G.M.; Tuebing, F.; Nigh, E.A.; Kane, C.G.; Sabatini, M.E.; Kiebler, M.; Greenberg, M.E. A brain-specific microRNA regulates dendritic spine development. Nature 2006, 439, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Chakrapani, S.; Eskander, N.; De Los Santos, L.A.; Omisore, B.A.; Mostafa, J.A. Neuroplasticity and the Biological Role of Brain Derived Neurotrophic Factor in the Pathophysiology and Management of Depression. Cureus 2020, 12, e11396. [Google Scholar] [CrossRef]
- Drevets, W.C.; Price, J.L.; Furey, M.L. Brain structural and functional abnormalities in mood disorders: Implications for neurocircuitry models of depression. Brain Struct. Funct. 2008, 213, 93–118. [Google Scholar] [CrossRef]
- MacQueen, G.M.; Yucel, K.; Taylor, V.H.; Macdonald, K.; Joffe, R. Posterior hippocampal volumes are associated with remission rates in patients with major depressive disorder. Biol. Psychiatry 2008, 64, 880–883. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Liu, R.-J.; Dwyer, J.M.; Banasr, M.; Lee, B.; Son, H.; Li, X.-Y.; Aghajanian, G.; Duman, R.S. Glutamate N-methyl-D-aspartate receptor antagonists rapidly reverse behavioral and synaptic deficits caused by chronic stress exposure. Biol. Psychiatry 2011, 69, 754–761. [Google Scholar] [CrossRef]
- Watanabe, Y.; Gould, E.; McEwen, B.S. Stress induces atrophy of apical dendrites of hippocampal CA3 pyramidal neurons. Brain Res. 1992, 588, 341–345. [Google Scholar] [CrossRef]
- Surget, A.; Saxe, M.; Leman, S.; Ibarguen-Vargas, Y.; Chalon, S.; Griebel, G.; Hen, R.; Belzung, C. Drug-dependent requirement of hippocampal neurogenesis in a model of depression and of antidepressant reversal. Biol. Psychiatry 2008, 64, 293–301. [Google Scholar] [CrossRef]
- Santarelli, L.; Saxe, M.; Gross, C.; Surget, A.; Battaglia, F.; Dulawa, S.; Weisstaub, N.; Lee, J.; Duman, R.; Arancio, O.; et al. Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science 2003, 301, 805–809. [Google Scholar] [CrossRef]
- Boldrini, M.; Santiago, A.N.; Hen, R.; Dwork, A.J.; Rosoklija, G.B.; Tamir, H.; Arango, V.; Mann, J.J. Hippocampal granule neuron number and dentate gyrus volume in antidepressant-treated and untreated major depression. Neuropsychopharmacology 2013, 38, 1068–1077. [Google Scholar] [CrossRef]
- Boldrini, M.; Underwood, M.D.; Hen, R.; Rosoklija, G.B.; Dwork, A.J.; Mann, J.J.; Arango, V. Antidepressants increase neural progenitor cells in the human hippocampus. Neuropsychopharmacology 2009, 34, 2376–2389. [Google Scholar] [CrossRef]
- Boldrini, M.; Hen, R.; Underwood, M.D.; Rosoklija, G.B.; Dwork, A.J.; Mann, J.J.; Arango, V. Hippocampal angiogenesis and progenitor cell proliferation are increased with antidepressant use in major depression. Biol. Psychiatry 2012, 72, 562–571. [Google Scholar] [CrossRef]
- Duman, R.S.; Aghajanian, G.K. Synaptic dysfunction in depression: Potential therapeutic targets. Science 2012, 338, 68–72. [Google Scholar] [CrossRef]
- Davis, T.H.; Cuellar, T.L.; Koch, S.M.; Barker, A.J.; Harfe, B.D.; McManus, M.T.; Ullian, E.M. Conditional loss of Dicer disrupts cellular and tissue morphogenesis in the cortex and hippocampus. J. Neurosci. 2008, 28, 4322–4330. [Google Scholar] [CrossRef]
- Schaefer, A.; O’Carroll, D.; Tan, C.L.; Hillman, D.; Sugimori, M.; Llinas, R.; Greengard, P. Cerebellar neurodegeneration in the absence of microRNAs. J. Exp. Med. 2007, 204, 1553–1558. [Google Scholar] [CrossRef]
- Stark, K.L.; Xu, B.; Bagchi, A.; Lai, W.-S.; Liu, H.; Hsu, R.; Wan, X.; Pavlidis, P.; A Mills, A.; Karayiorgou, M.; et al. Altered brain microRNA biogenesis contributes to phenotypic deficits in a 22q11-deletion mouse model. Nat. Genet. 2008, 40, 751–760. [Google Scholar] [CrossRef]
- Carniel, B.P.; da Rocha, N.S. Brain-derived neurotrophic factor (BDNF) and inflammatory markers: Perspectives for the management of depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 2020, 110151. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Liu, Y.; Wang, X.; Wu, J.; Liu, K.; Zhou, J.; Liu, L.; Zhang, C. Identification of differential microRNAs in cerebrospinal fluid and serum of patients with major depressive disorder. PLoS ONE 2015, 10, e0121975. [Google Scholar] [CrossRef]
- Fan, H.-M.; Sun, X.-Y.; Guo, W.; Zhong, A.-F.; Niu, W.; Zhao, L.; Dai, Y.-H.; Guo, Z.-M.; Zhang, L.-Y.; Lu, J. Differential expression of microRNA in peripheral blood mononuclear cells as specific biomarker for major depressive disorder patients. J. Psychiatr. Res. 2014, 59, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Smalheiser, N.R. The RNA-centred view of the synapse: Non-coding RNAs and synaptic plasticity. Philos. Trans. R. Soc. Lond B Biol. Sci. 2014, 369. [Google Scholar] [CrossRef] [PubMed]
- Baudry, A.; Mouillet-Richard, S.; Schneider, B.; Launay, J.-M.; Kellermann, O. miR-16 targets the serotonin transporter: A new facet for adaptive responses to antidepressants. Science 2010, 329, 1537–1541. [Google Scholar] [CrossRef] [PubMed]
- Launay, J.M.; Mouillet-Richard, S.; Baudry, A.; Pietri, M.; Kellermann, O. Raphe-mediated signals control the hippocampal response to SRI antidepressants via miR-16. Transl. Psychiatry 2011, 1, e56. [Google Scholar] [CrossRef] [PubMed]
- Song, M.-F.; Dong, J.-Z.; Wang, Y.-W.; He, J.; Ju, X.; Zhang, L.; Zhang, Y.-H.; Shi, J.-F.; Lv, Y.-Y. CSF miR-16 is decreased in major depression patients and its neutralization in rats induces depression-like behaviors via a serotonin transmitter system. J. Affect. Disord. 2015, 178, 25–31. [Google Scholar] [CrossRef]
- Zurawek, D.; Kusmider, M.; Faron-Gorecka, A.; Gruca, P.; Pabian, P.; Kolasa, M.; Solich, J.; Szafran-Pilch, K.; Papp, M.; Dziedzicka-Wasylewska, M. Time-dependent miR-16 serum fluctuations together with reciprocal changes in the expression level of miR-16 in mesocortical circuit contribute to stress resilient phenotype in chronic mild stress—An animal model of depression. Eur. Neuropsychopharmacol. 2016, 26, 23–36. [Google Scholar] [CrossRef]
- Gallego, J.A.; Gordon, M.L.; Claycomb, K.; Bhatt, M.; Lencz, T.; Malhotra, A.K. In vivo microRNA detection and quantitation in cerebrospinal fluid. J. Mol. Neurosci. 2012, 47, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.Y.; Xia, Q.H.; Xia, Q.R.; Zhang, X.L.; Liang, J. Microrna-based biomarkers in the diagnosis and monitoring of therapeutic response in patients with depression. Neuropsychiatr. Dis. Treat. 2019, 15, 3583–3597. [Google Scholar] [CrossRef]
- Van den Berg, M.M.J.; Krauskopf, J.; Ramaekers, J.G.; Kleinjans, J.C.S.; Prickaerts, J.; Briedé, J.J. Circulating microRNAs as potential biomarkers for psychiatric and neurodegenerative disorders. Prog. Neurobiol. 2020, 185, 101732. [Google Scholar] [CrossRef]
- Stoicea, N.; Du, A.; Lakis, D.C.; Tipton, C.; Arias-Morales, C.E.; Bergese, S.D. The MiRNA Journey from Theory to Practice as a CNS Biomarker. Front. Genet. 2016, 7, 11. [Google Scholar] [CrossRef]
- Wei, D.; Wan, Q.; Li, L.; Jin, H.; Liu, Y.; Wang, Y.; Zhang, G. MicroRNAs as Potential Biomarkers for Diagnosing Cancers of Central Nervous System: A Meta-analysis. Mol. Neurobiol. 2015, 51, 1452–1461. [Google Scholar] [CrossRef] [PubMed]
- Iwata, M.; Ota, K.T.; Duman, R.S. The inflammasome: Pathways linking psychological stress, depression, and systemic illnesses. Brain Behav. Immun. 2013, 31, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Moschos, S.A.; Williams, A.E.; Perry, M.M.; Birrell, M.A.; Belvisi, M.G.; Lindsay, M.A. Expression profiling in vivo demonstrates rapid changes in lung microRNA levels following lipopolysaccharide-induced inflammation but not in the anti-inflammatory action of glucocorticoids. BMC Genom. 2007, 8, 240. [Google Scholar] [CrossRef]
- Belzeaux, R.; Lin, R.; Turecki, G. Potential Use of MicroRNA for Monitoring Therapeutic Response to Antidepressants. CNS Drugs 2017, 31, 253–262. [Google Scholar] [CrossRef]
- O’Connor, R.M.; Grenham, S.; Dinan, T.G.; Cryan, J.F. microRNAs as novel antidepressant targets: Converging effects of ketamine and electroconvulsive shock therapy in the rat hippocampus. Int. J. Neuropsychopharmacol. 2013, 16, 1885–1892. [Google Scholar] [CrossRef]
- Vasu, M.M.; Anitha, A.; Takahashi, T.; Thanseem, I.; Iwata, K.; Asakawa, T.; Suzuki, K. Fluoxetine Increases the Expression of miR-572 and miR-663a in Human Neuroblastoma Cell Lines. PLoS ONE 2016, 11, e0164425. [Google Scholar]
- Issler, O.; Haramati, S.; Paul, E.D.; Maeno, H.; Navon, I.; Zwang, R.; Gil, S.; Mayberg, H.S.; Dunlop, B.W.; Menke, A.; et al. MicroRNA 135 is essential for chronic stress resiliency, antidepressant efficacy, and intact serotonergic activity. Neuron 2014, 83, 344–360. [Google Scholar] [CrossRef]
- Schmidt, U.; Herrmann, L.; Hagl, K.; Novak, B.; Huber, C.; Holsboer, F.; Wotjak, C.T.; Buell, D.R. Therapeutic Action of Fluoxetine is Associated with a Reduction in Prefrontal Cortical miR-1971 Expression Levels in a Mouse Model of Posttraumatic Stress Disorder. Front. Psychiatry 2013, 4, 66. [Google Scholar] [CrossRef] [PubMed]
- Angelucci, F.; Croce, N.; Spalletta, G.; DiNallo, V.; Gravina, P.; Bossù, P.; Federici, G.; Caltagirone, C.; Bernardini, S. Paroxetine rapidly modulates the expression of brain-derived neurotrophic factor mRNA and protein in a human glioblastoma-astrocytoma cell line. Pharmacology 2011, 87, 5–10. [Google Scholar] [CrossRef]
- Oved, K.; Morag, A.; Pasmanik-Chor, M.; Oron-Karni, V.; Shomron, N.; Rehavi, M.; Stingl, J.C.; Gurwitz, D. Genome-wide miRNA expression profiling of human lymphoblastoid cell lines identifies tentative SSRI antidepressant response biomarkers. Pharmacogenomics 2012, 13, 1129–1139. [Google Scholar] [CrossRef]
- Kuang, W.-H.; Dong, Z.-Q.; Tian, L.-T.; Li, J. MicroRNA-451a, microRNA-34a-5p, and microRNA-221-3p as predictors of response to antidepressant treatment. Braz. J. Med. Biol. Res. 2018, 51, e7212. [Google Scholar] [CrossRef] [PubMed]
- Lopez, J.P.; Lim, R.; Cruceanu, C.; Crapper, L.; Fasano, C.; LaBonte, B.; Maussion, G.; Yang, J.P.; Yerko, V.; Vigneault, E.; et al. miR-1202 is a primate-specific and brain-enriched microRNA involved in major depression and antidepressant treatment. Nat. Med. 2014, 20, 764–768. [Google Scholar] [CrossRef]
- Fang, Y.; Qiu, Q.; Zhang, S.; Sun, L.; Li, G.; Xiao, S.; Li, X. Changes in miRNA-132 and miR-124 levels in non-treated and citalopram-treated patients with depression. J. Affect. Disord. 2018, 227, 745–751. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Y.; Wang, L.; Bai, M.; Zhang, X.; Zhu, X. Dopamine Receptor D2 and Associated microRNAs Are Involved in Stress Susceptibility and Resistance to Escitalopram Treatment. Int. J. Neuropsychopharmacol. 2015, 18. [Google Scholar] [CrossRef]
- Bocchio-Chiavetto, L.; Maffioletti, E.; Bettinsoli, P.; Giovannini, C.; Bignotti, S.; Tardito, D.; Corrada, D.; Milanesi, L.; Gennarelli, M. Blood microRNA changes in depressed patients during antidepressant treatment. Eur. Neuropsychopharmacol. 2013, 23, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Lopez, J.P.; Fiori, L.M.; Cruceanu, C.; Lin, R.; LaBonte, B.; Cates, H.M.; Heller, E.A.; Vialou, V.; Ku, S.M.; Gerald, C.; et al. MicroRNAs 146a/b-5 and 425-3p and 24-3p are markers of antidepressant response and regulate MAPK/Wnt-system genes. Nat. Commun. 2017, 8, 15497. [Google Scholar] [CrossRef]
- Pan, B.; Liu, Y. Effects of duloxetine on microRNA expression profile in frontal lobe and hippocampus in a mouse model of depression. Int. J. Clin. Exp. Pathol. 2015, 8, 15454–15461. [Google Scholar]
- Lopez, J.P.; Pereira, F.R.S.; Richard-Devantoy, S.; Berlim, M.; Chachamovich, E.; Fiori, L.M.; Niola, P.; Turecki, G.; Jollant, F. Co-Variation of Peripheral Levels of miR-1202 and Brain Activity and Connectivity During Antidepressant Treatment. Neuropsychopharmacology 2017, 42, 2043–2051. [Google Scholar] [CrossRef]
- Yang, X.; Yang, Q.; Wang, X.; Luo, C.; Wan, Y.; Li, J.; Liu, K.; Zhou, M.; Zhang, C. MicroRNA expression profile and functional analysis reveal that miR-206 is a critical novel gene for the expression of BDNF induced by ketamine. Neuromol. Med. 2014, 16, 594–605. [Google Scholar] [CrossRef] [PubMed]
- Ryan, K.M.; O’Donovan, S.M.; McLoughlin, D.M. Electroconvulsive stimulation alters levels of BDNF-associated microRNAs. Neurosci. Lett. 2013, 549, 125–129. [Google Scholar] [CrossRef]
- Kolshus, E.; Ryan, K.M.; Blackshields, G.; Smyth, P.; Sheils, O.; McLoughlin, D.M. Peripheral blood microRNA and VEGFA mRNA changes following electroconvulsive therapy: Implications for psychotic depression. Acta Psychiatr. Scand. 2017, 136, 594–606. [Google Scholar] [CrossRef] [PubMed]
- Gururajan, A.; E Naughton, M.; A Scott, K.; O’Connor, R.M.; Moloney, G.; Clarke, G.; Dowling, J.; Walsh, A.; Ismail, F.; Shorten, G.; et al. MicroRNAs as biomarkers for major depression: A role for let-7b and let-7c. Transl. Psychiatry 2016, 6, e862. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Liu, X.; Jiang, K.; Peng, D.; Hong, W.; Huafang, L.; Qian, Y.; Yu, S.; Li, H. Alterations of microRNA-124 expression in peripheral blood mononuclear cells in pre- and post-treatment patients with major depressive disorder. J. Psychiatr. Res. 2016, 78, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Belzeaux, R.; Bergon, A.; Jeanjean, V.; Loriod, B.; Formisano-Tréziny, C.; Verrier, L.; Loundou, A.; Baumstarck-Barrau, K.; Boyer, L.; Gall, V.; et al. Responder and nonresponder patients exhibit different peripheral transcriptional signatures during major depressive episode. Transl. Psychiatry 2012, 2, e185. [Google Scholar] [CrossRef]
- Schmitz, S.U.; Grote, P.; Herrmann, B.G. Mechanisms of long noncoding RNA function in development and disease. Cell. Mol. Life Sci. 2016, 73, 2491–2509. [Google Scholar] [CrossRef]
- Chen, X.; Sun, Y.; Cai, R.; Wang, G.; Shu, X.; Pang, W. Long noncoding RNA: Multiple players in gene expression. BMB Rep. 2018, 51, 280–289. [Google Scholar] [CrossRef]
- Tsagakis, I.; Douka, K.; Birds, I. Long non-coding RNAs in development and disease: Conservation to mechanisms. J. Pathol. 2020, 250, 480–495. [Google Scholar] [CrossRef]
- Huang, X.; Luo, Y.-L.; Mao, Y.-S.; Ji, J.-L. The link between long noncoding RNAs and depression. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2017, 73, 73–78. [Google Scholar] [CrossRef]
- Cui, X.; Sun, X.; Niu, W.; Kong, L.; He, M.; Zhong, A.; Chen, S.; Jiang, K.; Zhang, L.; Cheng, Z. Long non-coding RNA: Potential diagnostic and therapeutic biomarker for major depressive disorder. Med. Sci. Monit. 2016, 22, 5240–5248. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.-N.; Goodwin, J.E.; Liu, B.; Li, D.; Wen, R.; Yang, N.; Xia, J.; Zhou, H.; Zhang, T.; Song, W.-L.; et al. Characterization of Long Noncoding RNA and mRNA Profiles in Sepsis-Induced Myocardial Depression. Mol. Nucleic Acids 2019, 17, 852–866. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.L. Linking Long Noncoding RNA Localization and Function. Trends Biochem. Sci. 2016, 41, 761–772. [Google Scholar] [CrossRef]
- Awan, H.M.; Shah, A.; Rashid, F.; Shan, G. Primate-specific Long Non-coding RNAs and MicroRNAs. Genom. Proteom. Bioinf. 2017, 15, 187–195. [Google Scholar] [CrossRef]
- Ma, L.; Bajic, V.B.; Zhang, Z. On the classification of long non-coding RNAs. RNA Biol. 2013, 10, 924–933. [Google Scholar] [CrossRef]
- Wu, P.; Zuo, X.; Deng, H.; Liu, X.; Liu, L.; Ji, A. Roles of long noncoding RNAs in brain development, functional diversification and neurodegenerative diseases. Brain Res. Bull. 2013, 97, 69–80. [Google Scholar] [CrossRef]
- Guennewig, B.; Cooper, A.A. The central role of noncoding RNA in the brain. Int. Rev. Neurobiol. 2014, 116, 153–194. [Google Scholar]
- Torres-Berrío, A.; Issler, O.; Parise, E.M.; Nestler, E.J. Unraveling the epigenetic landscape of depression: Focus on early life stress. Dialogues Clin. Neurosci. 2019, 21, 341–357. [Google Scholar] [PubMed]
- Chen, S.; Zhu, X.; Niu, W.; Yao, G.; Kong, L.; He, M.; Chen, C.; Lu, Z.; Cui, X.; Zhang, L. Regulatory Role of lncRNA NONHSAT089447 in the Dopamine Signaling Pathway in Schizophrenic Patients. Med. Sci. Monit. 2019, 25, 4322–4332. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, E.; Bagheri-Hosseinabadi, Z.; De Toma, I.; Jafarisani, M.; Sadeghi, I. The importance of long non-coding RNAs in neuropsychiatric disorders. Mol. Asp. Med. 2019, 70, 127–140. [Google Scholar] [CrossRef]
- Li, C.; Cao, F.; Li, S.; Huang, S.; Li, W.; Abumaria, N. Profiling and co-expression network analysis of learned helplessness regulated mRNAs and lncRNAs in the mouse hippocampus. Front. Mol. Neurosci. 2018, 10, 1–13. [Google Scholar] [CrossRef]
- Liu, S.; Zhou, B.; Wang, L.; Hu, H.; Yao, C.; Cai, Z.; Cui, X. Therapeutic antidepressant potential of NONHSAG045500 in regulating serotonin transporter in major depressive disorder. Med. Sci. Monit. 2018, 24, 4465–4473. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Niu, W.; Kong, L.; He, M.; Jiang, K.; Chen, S.; Zhong, A.; Li, W.; Lu, J.; Zhang, L. Long noncoding RNA expression in peripheral blood mononuclear cells and suicide risk in Chinese patients with major depressive disorder. Brain Behav. 2017, 7, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xue, M.; Mei, Y.; Li, Z.; Ceng, Z.; Li, Y.; Zhang, Y.; Li, N.; Teng, H.; Sun, Z.S.; et al. Co-expression Network of mRNAs and lncRNAs Regulated by Stress-Linked Behavioral Assays. Psychopharmacology 2020, 237, 571–582. [Google Scholar] [CrossRef]
- Watson, C.N.; Belli, A.; Pietro, V.; Di Budini, M.F. Small Non-coding RNAs: New Class of Biomarkers and Potential Therapeutic Targets in Neurodegenerative Disease. Front. Genet. 2019, 10, 1–14. [Google Scholar] [CrossRef]
- Liu, Z.; Li, X.; Sun, N.; Xu, Y.; Meng, Y.; Yang, C.; Wang, Y.; Zhang, K. Microarray Profiling and Co-Expression Network Analysis of Circulating lncRNAs and mRNAs Associated with Major Depressive Disorder. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Niu, W.; Kong, L.; He, M.; Jiang, K.; Chen, S.; Zhong, A.; Zhang, Q.; Li, W.; Lu, J.; et al. Long noncoding RNA as an indicator differentiating schizophrenia from major depressive disorder and generalized anxiety disorder in nonpsychiatric hospital. Biomark. Med. 2017, 11, 221–228. [Google Scholar] [CrossRef]
- Cui, X.; Niu, W.; Kong, L.; He, M.; Jiang, K.; Chen, S.; Zhong, A.; Li, W.; Lu, J.; Zhang, L. Can lncRNAs be indicators for the diagnosis of early onset or acute schizophrenia and distinguish major depressive disorder and generalized anxiety disorder?—A cross validation analysis. Am. J. Med. Genet. Part. B Neuropsychiatr. Genet. 2017, 174, 335–341. [Google Scholar] [CrossRef]
- Carter, G.; Miladinovic, B.; Patel, A.A.; Deland, L.; Mastorides, S.; Patel, N.A. Circulating long noncoding RNA GAS5 levels are correlated to prevalence of type 2 diabetes mellitus. BBA Clin. 2015, 4, 102–107. [Google Scholar] [CrossRef]
- Pandey, G.K.; Mitra, S.; Subhash, S.; Hertwig, F.; Kanduri, M.; Mishra, K.; Fransson, S.; Ganeshram, A.; Mondal, T.; Bandaru, S.; et al. The Risk-Associated Long Noncoding RNA NBAT-1 Controls Neuroblastoma Progression by Regulating Cell Proliferation and Neuronal Differentiation. Cancer Cell 2014, 26, 722–737. [Google Scholar] [CrossRef]
- Thin, K.Z. Clinica Chimica Acta. Clin. Chim. Acta 2019, 494, 38–47. [Google Scholar] [CrossRef]

| Histone Modification | Material | Model of Depression/Patients | Results | Paper |
|---|---|---|---|---|
| H3K12ac | Ventral hippocampus | Chronic social defeat stress in rats | Increased level of acetylation | [77] |
| H3K14ac | Nucleus accumbens | Social defeat stress in mice | Decreased level of acetylation | [64] |
| Hippocampus | Gestational stress in offspring of pregnant mice | Increased level of acetylation | [63] | |
| Prefrontal cortex | Chronic unpredicted stress in rats | Decreased level of acetylation | [79] | |
| Dorsal raphe | Chronic social defeat stress in rats | Increased level of acetylation | [77] | |
| Hippocampus | Chronic unpredicted stress in rats | Decreased level of acetylation | [79] | |
| H3K18ac | Medial prefrontal cortex | Chronic social defeat stress in rats | Increased level of acetylation | [77] |
| Ventral hippocampus | Chronic social defeat stress in rats | Increased level of acetylation | [77] | |
| H3K23ac | Prefrontal cortex | Chronic unpredicted stress in rats | Decreased level of acetylation | [79] |
| Hippocampus | Chronic unpredicted stress in rats | Decreased level of acetylation | [79] | |
| H3K27me3 | Ventral striatum | Chronic stress in C57BL/6 mice | Increased level of methylation | [74] |
| Hippocampus | Acute and restraint stress in rats | Decreased level of methylation | [81] | |
| Dentate gyrus | Acute and restraint stress in rats | Decreased level of methylation | [81] | |
| H3K4me3 | Hippocampus | Social defeat stress in mice | Decreased level of methylation | [66] |
| Ventral striatum | Chronic stress in BALB mice | Decreased level of methylation | [74] | |
| Hippocampus | Restraint stress in rats | Decreased level of methylation | [81] | |
| Prefrontal cortex | MDD suicide victims | Increased level of methylation | [86] | |
| H3K9ac | Hippocampus | Social defeat stress in mice | Decreased level of acetylation | [66] |
| Hippocampus | Chronic unpredicted stress in rats | Decreased level of acetylation | [78] | |
| Dorsal raphe | Chronic social defeat stress in rats | Increased level of acetylation | [77] | |
| H3K9me1 | Hippocampus | Acute stress in rats | Decreased level of methylation | [81] |
| Dentate gyrus | Acute stress in rats | Decreased level of methylation | [81] | |
| H3K9me2 | Medial prefrontal cortex | Adolescent social stress in mice | Decreased level of methylation | [70] |
| H3K9me3 | Hypothalamus | Chronic unpredictable mild stress in rats | Decreased level of methylation | [80] |
| Dentate gyrus | Chronic restraint stress in rats | Decreased level of methylation | [81] | |
| Prefrontal cortex | MDD suicide victims | Increased level of methylation | [85] | |
| H4K12ac | Hippocampus | Social defeat stress in mice | Increased level of acetylation | [66] |
| Ventral hippocampus | Chronic social defeat stress in rats | Decreased level of acetylation | [77] | |
| Hippocampus | Chronic unpredicted stress in rats | Decreased level of acetylation | [78] | |
| Dorsal raphe | Chronic social defeat stress in rats | Increased level of acetylation | [77] | |
| H4K16ac | Prefrontal cortex | Chronic unpredicted stress in rats | Decreased level of acetylation | [79] |
| Hippocampus | Chronic unpredicted stress in rats | Decreased level of acetylation | [79] | |
| Dorsal raphe | Chronic social defeat stress in rats | Increased level of acetylation | [77] | |
| H4K5ac | Dorsal raphe | Chronic social defeat stress in rats | Increased level of acetylation | [77] |
| H4K8ac | Ventral hippocampus | Chronic social defeat stress in rats | Decreased level of acetylation | [77] |
| Dorsal raphe | Chronic social defeat stress in rats | Increased level of acetylation | [77] |
| Treatment | Material | Model of Depression/Patients | Results | Paper |
|---|---|---|---|---|
| Histone deacetylase inhibitors | ||||
| Crebinostat | Primary neurons | Mice | Inhibition of the activity of HDAC1, 2, 3, 6 and 8 and elevation of H3ac and H4ac | [87] |
| Ms-275 | Ventrolateral orbital cortex | Social defeat stress in rats | Increase in H3ac | [98] |
| Neurinostat | Primary neurons | Mice | Inhibition of the activity of HDAC1, 2, 3 | [88] |
| Sodium butyrate | Hippocampus | Acute or chronic stress in mice | Increase in H4ac after acute administration, but decrease after chronic treatment | [91] |
| Hippocampus and frontal cortex | Chronic restraint stress in mice | Increase in H3ac and H4ac in the hippocampus and H3ac in the frontal cortex | [92] | |
| Hippocampus | Chronic variable stress in rats | Increase in H4K12ac | [96] | |
| Selective serotonin reuptake inhibitors | ||||
| Citalopram | Blood | MDD patients | Decrease in H3K27me3 | [104] |
| Escitalopram | Hippocampus | Maternal separation in rats | Increase in H3ac | [100] |
| Fluoxetine | Forebrain neocortex | Infant maternal separation in mice | Increase in H4K12ac | [95] |
| Dentate gyrus | Chronic restraint stress in rats | Increase in H3K9me3 | [81] | |
| Venlafaxine | Hippocampus | Chronic unpredicted stress in rats | Inhibition of HDAC5 expression, increase and decrease in H3K9ac | [101] |
| Tricyclic antidepressants | ||||
| Imipramine | Nucleus accumbens | Social defeat in mice | Reversed changes in H3me triggered by stress | [94] |
| Hippocampus | Chronic social defeat stress in mice | Decrease in H3K4me2, increase in histone acetylation at BDNF promoters and downregulation of HDAC5 | [69] | |
| lncRNA | Mechanism of Action | Function | Pathological State | References |
|---|---|---|---|---|
| BC200 | Inhibition of translation initiation in dendrites | Support of long-term synaptic plasticity | Alzheimer’s disease and Parkinson’s disease | [188,195] |
| UCHL1-AS | Enhancement of translation of UCHL1 mRNA | Involved in proper neurons development | Parkinson’s disease | [188] |
| HTT-AS | Negative regulation of HTT expression | Maintenance of proper nervous system functioning | Huntington’s disease | [188] |
| Camkk1 | Regulation of Ca2+/calmodulin-dependent protein kinase II | Long-term memory maintenance | Lack of evidence | [195,199] |
| Evx1as and Hox5b/6as are | Interaction with H3K4 histone and histone methyltransferase | Maintenance of proper nervous system functioning | Lack of evidence | [199] |
| Tuna | Regulation of gene expression in neural cells | Maintenance of proper nervous system functioning | Lack of evidence | [199] |
| Sox2OT | Overlapping with Sox2 gene | Involved in development of neural stem and progenitor cells | Alzhaimer’s disease and Parkinson’s disease | [195,199] |
| RMST | Regulation of neural cells differentiation | Involved in neurogenesis | Lack of evidence | [199] |
| Nkx2.2AS | Regulation of Nkx2.2 mRNA level | Maintenance of oligodendrocytes differentiation | Lack of evidence | [195,199] |
| HAR1F | Regulation of migration of neural cells and forebrain organization | Development of human embryonic neocortex | Huntington’s disease | [195,199] |
| Malat1 | Regulation of SR-family splicing factors | Involved in synaptogenesis | Lack of evidence | [199] |
| BDNF-AS | Reducing level of BDNF factor | Involved in neurons development | Schizophrenia | [195,199] |
| Gomafu | Binding to splicing factors | Involved in neural development | Schizophrenia | [195,199] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Czarny, P.; Białek, K.; Ziółkowska, S.; Strycharz, J.; Barszczewska, G.; Sliwinski, T. The Importance of Epigenetics in Diagnostics and Treatment of Major Depressive Disorder. J. Pers. Med. 2021, 11, 167. https://doi.org/10.3390/jpm11030167
Czarny P, Białek K, Ziółkowska S, Strycharz J, Barszczewska G, Sliwinski T. The Importance of Epigenetics in Diagnostics and Treatment of Major Depressive Disorder. Journal of Personalized Medicine. 2021; 11(3):167. https://doi.org/10.3390/jpm11030167
Chicago/Turabian StyleCzarny, Piotr, Katarzyna Białek, Sylwia Ziółkowska, Justyna Strycharz, Gabriela Barszczewska, and Tomasz Sliwinski. 2021. "The Importance of Epigenetics in Diagnostics and Treatment of Major Depressive Disorder" Journal of Personalized Medicine 11, no. 3: 167. https://doi.org/10.3390/jpm11030167
APA StyleCzarny, P., Białek, K., Ziółkowska, S., Strycharz, J., Barszczewska, G., & Sliwinski, T. (2021). The Importance of Epigenetics in Diagnostics and Treatment of Major Depressive Disorder. Journal of Personalized Medicine, 11(3), 167. https://doi.org/10.3390/jpm11030167