
The Potential of Gamma Secretase as a Therapeutic Target for Cardiac Diseases




Abstract
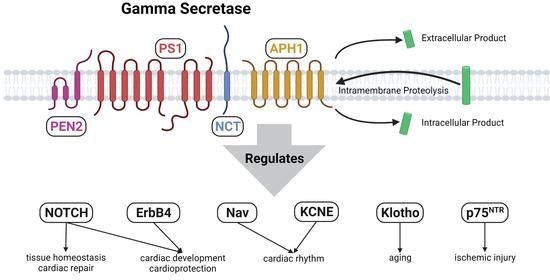
1. Introduction
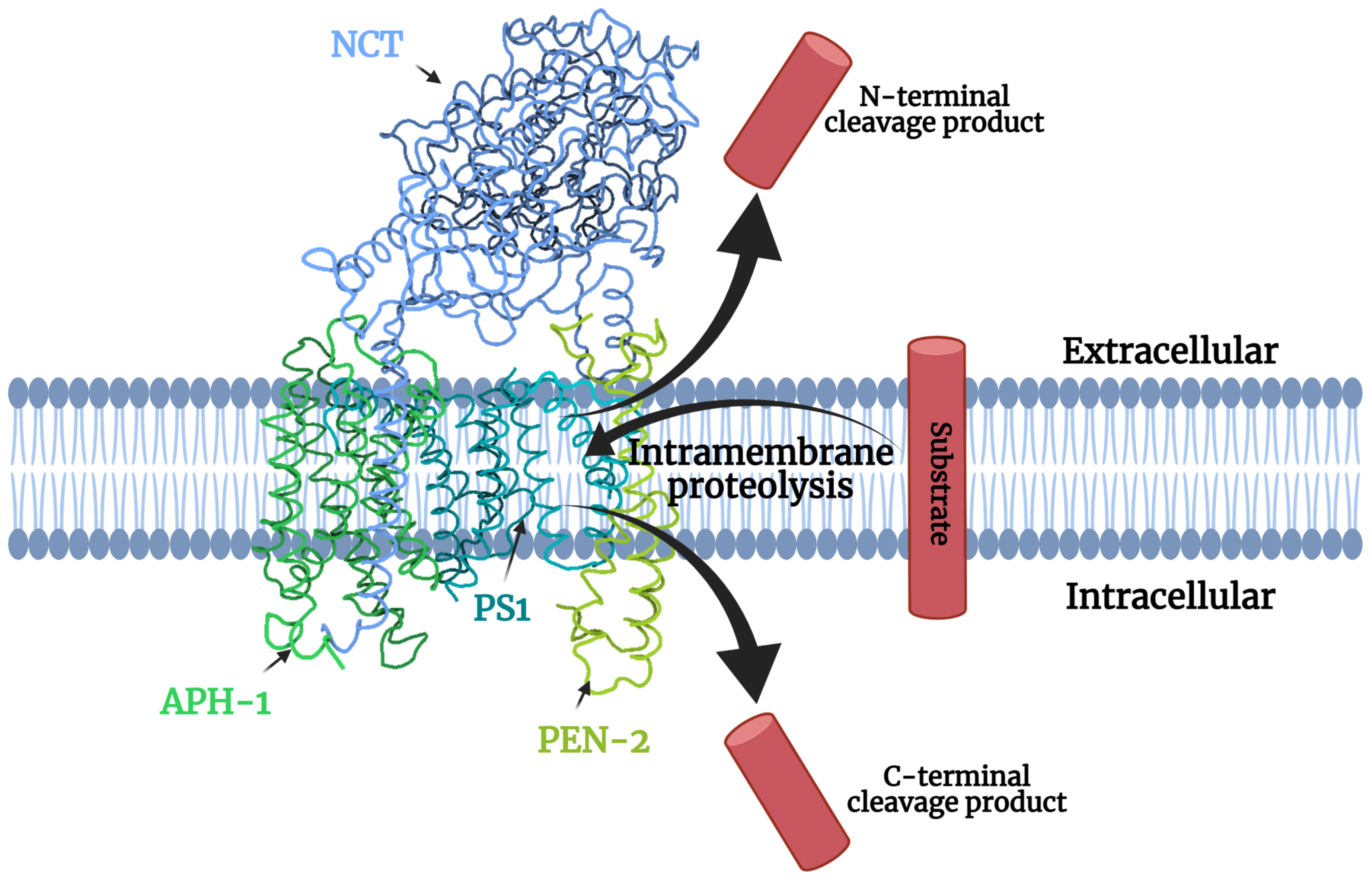
1.1. Gamma-Secretase Structure


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Pathways | Role in the Heart | Types of Cell Involved | Diseases Due to Dysregulation | Publications | Findings Suggesting Beneficial Effects in Adult Cardiac Disease |
|---|---|---|---|---|---|
| Beneficial Gamma-Secretase Substrates | |||||
| Notch | Heart development | Endocardial and endothelial cells | Cardiac fibrosis, heart failure, atherosclerosis, I/R injury, calcific aorta disease | [8] | LOF mutations lead to developmental defects |
| [9] | LOF mutations in endothelium lead to cardiac abnormalities | ||||
| [10] | LOF mutations lead to vascular defects and embryonic lethality | ||||
| Cardiomyocytes | [11] | Overexpression and selective silencing has been correlated to developmental defects and lethality | |||
| Angiogenesis and vasculature maintenance | Endothelial cells | [12] | Endothelial-specific Jagged1 LOF mutants showed decrease in angiogenesis Endothelial-specific Jagged 1 GOF mutants showed increased angiogenesis Curative approach | ||
| [13] | Notch-1 mutants and Notch-1/ Notch-4 double mutants have defective angiogenic vascular development | ||||
| [14] | LOF mutations in the NICD decrease angiogenesis. GOF mutations increase angiogenesis post I/R injury Curative approach | ||||
| Regulation of survival and regeneration in I/R injury | Cardiomyocytes | [15] | GOF ameliorated the increase apoptosis in cardiomyocytes seen in I/R injury and conferred cardioprotection Curative approach | ||
| [14] | LOF and GOF mutations in the Notch intracellular domain showed decrease and increase in angiogenesis respectively in human umbilical cord cells post I/R injury as well as mice models post MI Curative approach | ||||
| Regulation of cardiac fibrosis | Cardiac fibroblasts | [16] | Silencing of Notch-3 aggravates cardiac fibrosis in mice with MI as opposed to overexpression Preventive approach | ||
| Cardiomyocytes | [11] | Cardiomyocyte specific upregulation of Notch-1 reduced fibrosis in post-MI Curative approach | |||
| ErbB4 | Ventricular trabecular formation | Endocardial cells cardiomyocytes | Myocardial ischemia, systolic and diastolic heart failure | [17,18,19] | LOF causes embryonic lethality |
| Regulates ventricular wall development | Cardiomyocytes | [17,18] | LOF causes developmental disorders | ||
| Proliferation of cardiomyocytes | [20] | Inactivation of ErbB4 disrupted the normal proliferation of cardiomyocytes in postnatal mice | |||
| Regulation of cardiac fibrosis | Cardiac fibroblasts | [21,22,23] | GGF2(recombinant NRG1) treatment post MI increased protection against fibrosis Curative approach | ||
| Adaptation to changing heart demands | Cardiomyocytes | [20,24] | Prior Inhibition leads to decreased adaptation to changing heart demands during MI and pregnancy | ||
| Prevents systolic/diastolic heart failure | [25] | NRG1 attenuated the remodeling of ventricular wall and partially improved heart function in volume-overload HF mice Curative approach | |||
| [26] | Decreased expression correlates with terminal heart failure | ||||
| [27] | LOF induces DCM | ||||
| KCNE1-4 | Maintenance of cardiac rhythm | Cardiomyocytes | Ventricular fibrillation, atrial fibrillation, long QT syndrome | [28,29] | KCNE Null and GOF Mutations associated with atrial-fibrillation |
| [30] | Missense Mutations associated with long QT syndrome | ||||
| NAV1-4 | Maintenance of cardiac rhythm | Cardiomyocytes | Ventricular fibrillation, atrial fibrillation, long QT syndrome | [31] | Loss of function mutations lead to sudden cardiac death |
| Klotho | Attenuates ROS | Cardiomyocytes | Stroke, kidney disease-associated cardiovascular disease, cardiac hypertrophy | [32] | Overexpression improves cardiac function in aging, in endotoxemia, and reduces cardiomyocyte apoptosis in doxorubicin-induced injury Curative approach |
| Maintains ion homeostasis | [33] | ||||
| Regulation of cardiac hypertrophy | [34] | ||||
| [35] | Deficiency shows worsening of CV disease. Protective as shown to alleviate left ventricular hypertrophy. LOF studies show that Klotho null mice developed greater Left ventricular hypertrophy upon induction by indole-sulphate Preventive approach | ||||
| Detrimental Gamma-Secretase Substrates | |||||
| p75NTR | Regulation of sympathetic innervation | Neurons | Reduced heart rate, Sudden cardiac death | [36,37,38] | LOF leads to reduced sympathetic innervation and synaptic transmission, downregulated basal heart rate |
| Development of microvascular injury | Cardiac pericytes Microvascular endothelial cells | Microvascular injury and cardiomyopathy | [39] | Expression enables microvascular injury. LOF helps to rescues cardiomyopathy Preventive approach | |
| Downstream Interactions | |||||
| AMPK (activated by Notch) | Regulation of cardiomyocyte growth and differentiation | Cardiomyocytes | Cardiac fibrosis, cardiac hypertrophy, heart failure | [40,41] | Silencing and mutations leads to increased cardiomyocyte hypertrophy, increasing expression attenuates cardiomyocyte hypertrophy Preventive approach |
| Regulation of angiogenesis | Endothelial cells | [42] | LOF inhibits angiogenesis and vascularization | ||
| Management of ROS-induced damage | Cardiomyocytes Endothelial cells | [43] | LOF increased ROS-mediated fibrosis in response to isoproterenol Preventive approach | ||
| Regulation of cardiac hypertrophy | [44,45,46] | Activation attenuates cardiac hypertrophy and improves survival and limits infarct size Curative approach | |||
| Regulation of post-MI injury | [47] | ||||
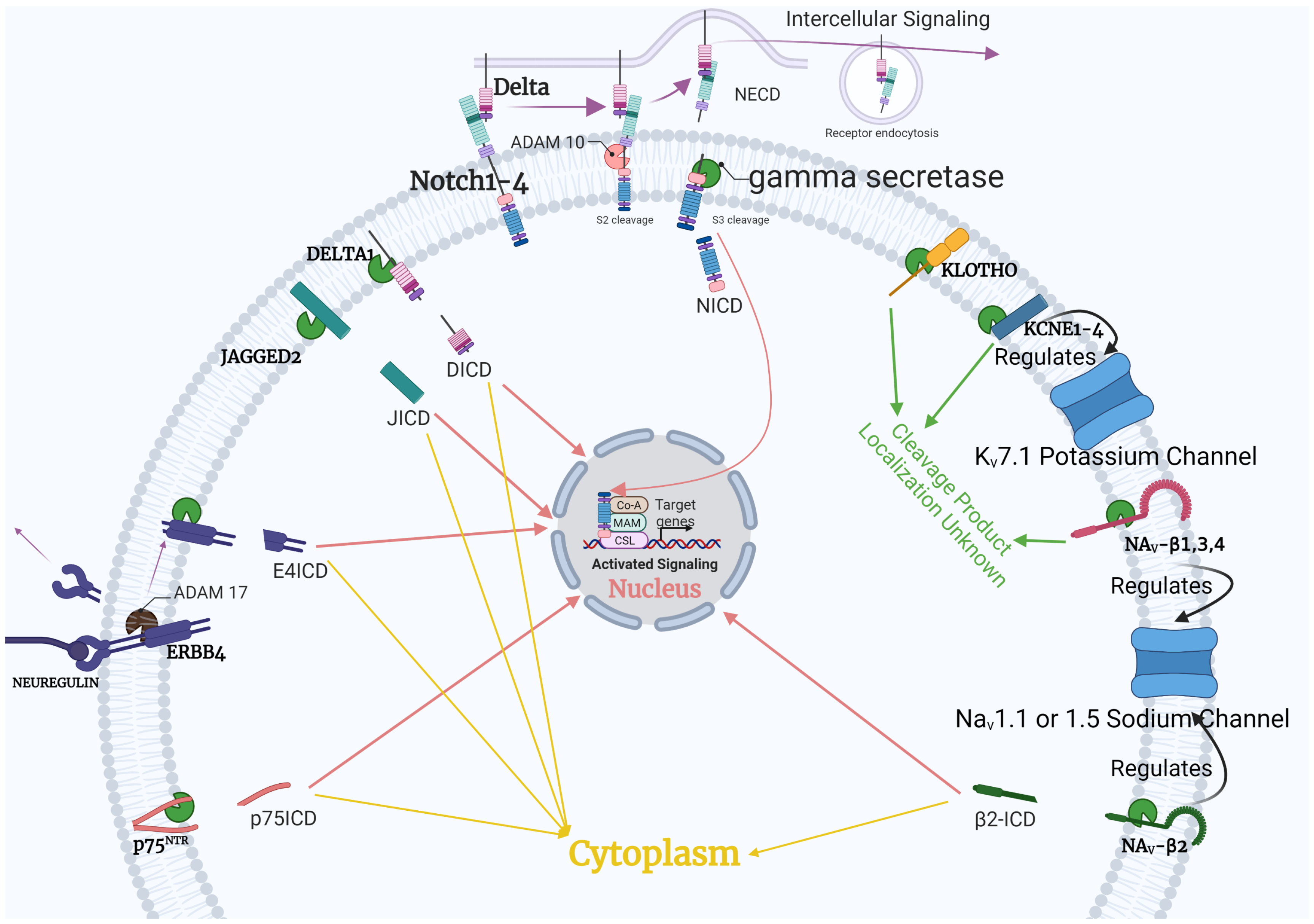
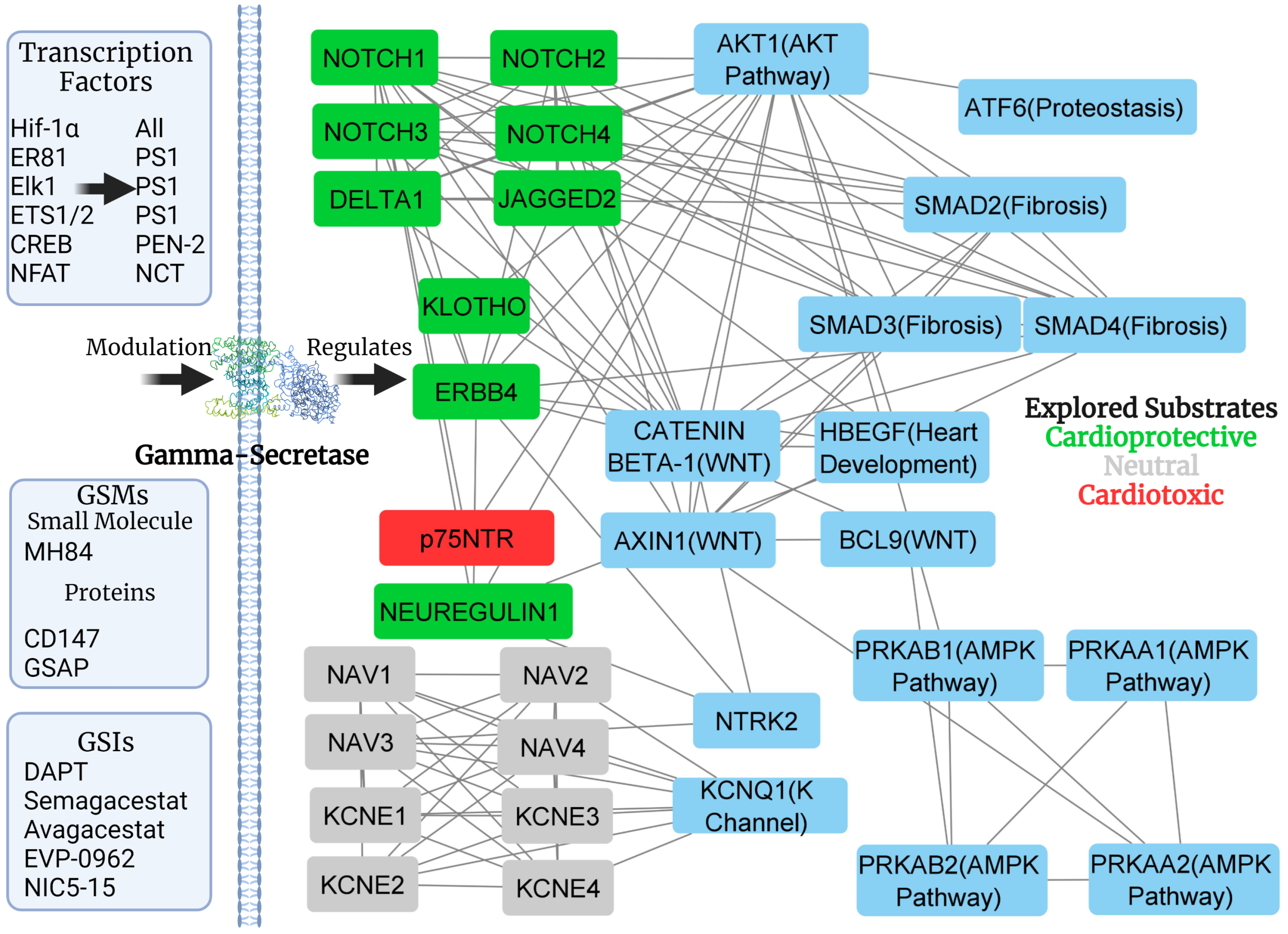
1.2. Gamma-Secretase Regulation
1.3. Gamma-Secretase Function
2. Gamma-Secretase Animal Models
3. Presenilin Mutations in Cardiomyopathy
4. Substrates of Gamma-Secretase—Implications for Cardiac Disease
4.1. Notch Pathway
4.2. ErbB4
4.3. Voltage-Gated Channels
4.4. Klotho
4.5. p75NTR
5. Downstream Interaction: Notch, AKT, AMPK
Downstream Complexity
6. Difficulty in Drug Development
7. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Virani, S.S.; Alonso, A.; Aparicio, H.J.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics—2021 Update. Circulation 2021, 143, e254–e743. [Google Scholar] [CrossRef] [PubMed]
- Jurisch-Yaksi, N.; Sannerud, R.; Annaert, W. A fast growing spectrum of biological functions of γ-secretase in development and disease. Biochim. Biophys. Acta-(BBA)-Biomembr. 2013, 1828, 2815–2827. [Google Scholar] [CrossRef]
- de la Pompa, J.L.; Epstein, J.A. Coordinating Tissue Interactions: Notch Signaling in Cardiac Development and Disease. Dev. Cell 2012, 22, 244–254. [Google Scholar] [CrossRef]
- Gude, N.; Sussman, M. Notch signaling and cardiac repair. J. Mol. Cell. Cardiol. 2012, 52, 1226–1232. [Google Scholar] [CrossRef] [PubMed]
- Gertsik, N.; Chiu, D.; Li, Y.M. Complex regulation of γ-secretase: From obligatory to modulatory subunits. Front. Aging Neurosci. 2015, 6, 342. [Google Scholar] [CrossRef]
- Zhou, S.; Zhou, H.; Walian, P.J.; Jap, B.K. The discovery and role of CD147 as a subunit of gamma-secretase complex. Drug News Perspect. 2006, 19, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.C.; Yan, C.; Yang, G.; Lu, P.; Ma, D.; Sun, L.; Zhou, R.; Scheres, S.H.W.; Shi, Y. An atomic structure of human γ-secretase. Nature 2015, 525, 212–217. [Google Scholar] [CrossRef]
- Grego-Bessa, J.; Luna-Zurita, L.; del Monte, G.; Bolós, V.; Melgar, P.; Arandilla, A.; Garratt, A.N.; Zang, H.; Mukouyama, Y.S.; Chen, H.; et al. Notch Signaling Is Essential for Ventricular Chamber Development. Dev. Cell 2007, 12, 415–429. [Google Scholar] [CrossRef]
- Hofmann, J.J.; Briot, A.; Enciso, J.; Zovein, A.C.; Ren, S.; Zhang, Z.W.; Radtke, F.; Simons, M.; Wang, Y.; Iruela-Arispe, M.L. Endothelial deletion of murine Jag1 leads to valve calcification and congenital heart defects associated with Alagille syndrome. Development 2012, 139, 4449–4460. [Google Scholar] [CrossRef]
- Limbourg, F.P.; Takeshita, K.; Radtke, F.; Bronson, R.T.; Chin, M.T.; Liao, J.K. Essential Role of Endothelial Notch1 in Angiogenesis. Circulation 2005, 111, 1826–1832. [Google Scholar] [CrossRef]
- Kratsios, P.; Catela, C.; Salimova, E.; Huth, M.; Berno, V.; Rosenthal, N.; Mourkioti, F. Distinct Roles for Cell-Autonomous Notch Signaling in Cardiomyocytes of the Embryonic and Adult Heart. Circ. Res. 2010, 106, 559–572. [Google Scholar] [CrossRef]
- Benedito, R.; Roca, C.; Sörensen, I.; Adams, S.; Gossler, A.; Fruttiger, M.; Adams, R.H. The notch ligands Dll4 and Jagged1 have opposing effects on angiogenesis. Cell 2009, 137, 1124–1135. [Google Scholar] [CrossRef]
- Krebs, L.T.; Xue, Y.; Norton, C.R.; Shutter, J.R.; Maguire, M.; Sundberg, J.P.; Gallahan, D.; Closson, V.; Kitajewski, J.; Callahan, R.; et al. Notch signaling is essential for vascular morphogenesis in mice. Genes Dev. 2000, 14, 1343–1352. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.L.; Zhu, R.R.; Liu, S.; Xu, H.; Xu, X.; Wu, Q.C.; Liu, J.C. Notch signaling promotes angiogenesis and improves cardiac function after myocardial infarction. J. Cell. Biochem. 2018, 119, 7105–7112. [Google Scholar] [CrossRef]
- Zhou, X.; Wan, L.; Xu, Q.; Zhao, Y.; Liu, J. Notch Signaling Activation Contributes to Cardioprotection Provided by Ischemic Preconditioning and Postconditioning. J. Transl. Med. 2013, 11, 251. [Google Scholar] [CrossRef]
- Zhang, M.; Pan, X.; Zou, Q.; Xia, Y.; Chen, J.; Hao, Q.; Wang, H.; Sun, D. Notch3 Ameliorates Cardiac Fibrosis After Myocardial Infarction by Inhibiting the TGF-β1/Smad3 Pathway. Cardiovasc. Toxicol. 2016, 16, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Gassmann, M.; Casagranda, F.; Orioli, D.; Simon, H.; Lai, C.; Klein, R.; Lemke, G. Aberrant neural and cardiac development in mice lacking the ErbB4 neuregulin receptor. Nature 1995, 378, 390–394. [Google Scholar] [CrossRef]
- Lee, K.F.; Simon, H.; Chen, H.; Bates, B.; Hung, M.C.; Hauser, C. Requirement for neuregulin receptor erbB2 in neural and cardiac development. Nature 1995, 378, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Bressan, M.; Hassel, D.; Huisken, J.; Staudt, D.; Kikuchi, K.; Poss, K.D.; Mikawa, T.; Stainier, D.Y.R. A dual role for ErbB2 signaling in cardiac trabeculation. Development 2010, 137, 3867–3875. [Google Scholar] [CrossRef] [PubMed]
- Bersell, K.; Arab, S.; Haring, B.; Kühn, B. Neuregulin1/ErbB4 signaling induces cardiomyocyte proliferation and repair of heart injury. Cell 2009, 138, 257–270. [Google Scholar] [CrossRef]
- Geissler, A.; Ryzhov, S.; Sawyer, D.B. Neuregulins: Protective and reparative growth factors in multiple forms of cardiovascular disease. Clin. Sci. 2020, 134, 2623–2643. [Google Scholar] [CrossRef] [PubMed]
- Dugaucquier, L.; Feyen, E.; Mateiu, L.; Bruyns, T.A.M.; De Keulenaer, G.W.; Segers, V.F.M. The role of endothelial autocrine NRG1/ERBB4 signaling in cardiac remodeling. Am. J.-Physiol.-Heart Circ. Physiol. 2020, 319, H443–H455. [Google Scholar] [CrossRef]
- Galindo, C.L.; Kasasbeh, E.; Murphy, A.; Ryzhov, S.; Lenihan, S.; Ahmad, F.A.; Williams, P.; Nunnally, A.; Adcock, J.; Song, Y.; et al. Anti-Remodeling and Anti-Fibrotic Effects of the Neuregulin-1β Glial Growth Factor 2 in a Large Animal Model of Heart Failure. J. Am. Heart Assoc. 2014, 3, e000773. [Google Scholar] [CrossRef] [PubMed]
- Lemmens, K.; Doggen, K.; De Keulenaer, G.W. Activation of the neuregulin/ErbB system during physiological ventricular remodeling in pregnancy. Am. J.-Physiol.-Heart Circ. Physiol. 2011, 300, H931–H942. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhuo, X.; Gao, J.; Liu, H.; Lin, F.; Ma, A. Neuregulin-1β Partially Improves Cardiac Function in Volume-Overload Heart Failure Through Regulation of Abnormal Calcium Handling. Front. Pharmacol. 2019, 10, 616. [Google Scholar] [CrossRef]
- Rohrbach, S.; Niemann, B.; Silber, R.E.; Holtz, J. Neuregulin receptors erbB2 and erbB4 in failing human myocardium: Depressed expression and attenuated activation. Basic Res. Cardiol. 2005, 100, 240–249. [Google Scholar] [CrossRef]
- García Rivello, H.; Taranda, J.; Said, M.; Cabeza-Meckert, P.; Vila-Petroff, M.; Scaglione, J.; Ghio, S.; Chen, J.; Lai, C.; Laguens, R.; et al. Dilated cardiomyopathy in Erb-b4-deficient ventricular muscle. Am. J.-Physiol.-Heart Circ. Physiol. 2005, 289, H1153–H1160. [Google Scholar] [CrossRef]
- Temple, J.; Frias, P.; Rottman, J.; Yang, T.; Wu, Y.; Verheijck, E.E.; Zhang, W.; Siprachanh, C.; Kanki, H.; Atkinson, J.B.; et al. Atrial Fibrillation in KCNE1-Null Mice. Circ. Res. 2005, 97, 62–69. [Google Scholar] [CrossRef]
- Olesen, M.S.; Bentzen, B.H.; Nielsen, J.B.; Steffensen, A.B.; David, J.P.; Jabbari, J.; Jensen, H.K.; Haunsø, S.; Svendsen, J.H.; Schmitt, N. Mutations in the potassium channel subunit KCNE1 are associated with early-onset familial atrial fibrillation. BMC Med. Genet. 2012, 13, 24. [Google Scholar] [CrossRef]
- Splawski, I.; Tristani-Firouzi, M.; Lehmann, M.H.; Sanguinetti, M.C.; Keating, M.T. Mutations in the hminK gene cause long QT syndrome and suppress lKs function. Nat. Genet. 1997, 17, 338–340. [Google Scholar] [CrossRef]
- Yuan, L.; Koivumäki, J.T.; Liang, B.; Lorentzen, L.G.; Tang, C.; Andersen, M.N.; Svendsen, J.H.; Tfelt-Hansen, J.; Maleckar, M.; Schmitt, N.; et al. Investigations of the Navβ1b sodium channel subunit in human ventricle; functional characterization of the H162P Brugada syndrome mutant. Am. J.-Physiol.-Heart Circ. Physiol. 2014, 306, H1204–H1212. [Google Scholar] [CrossRef]
- Zhu, H.; Gao, Y.; Zhu, S.; Cui, Q.; Du, J. Klotho Improves Cardiac Function by Suppressing Reactive Oxygen Species (ROS) Mediated Apoptosis by Modulating Mapks/Nrf2 Signaling in Doxorubicin-Induced Cardiotoxicity. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2017, 23, 5283–5293. [Google Scholar] [CrossRef] [PubMed]
- Olejnik, A.; Franczak, A.; Krzywonos-Zawadzka, A.; Kałużna-Oleksy, M.; Bil-Lula, I. The Biological Role of Klotho Protein in the Development of Cardiovascular Diseases. BioMed Res. Int. 2018, 2018, 5171945. [Google Scholar] [CrossRef] [PubMed]
- Hui, H.; Zhai, Y.; Ao, L.; Cleveland, J.C., Jr.; Liu, H.; Fullerton, D.A.; Meng, X. Klotho suppresses the inflammatory responses and ameliorates cardiac dysfunction in aging endotoxemic mice. Oncotarget 2017, 8, 15663–15676. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Wang, C.; Nie, L.; Zhao, X.; Gu, J.; Guan, X.; Wang, S.; Xiao, T.; Xu, X.; He, T.; et al. Klotho Protects Against Indoxyl Sulphate-Induced Myocardial Hypertrophy. J. Am. Soc. Nephrol. 2015, 26, 2434–2446. [Google Scholar] [CrossRef]
- Habecker, B.A.; Bilimoria, P.; Linick, C.; Gritman, K.; Lorentz, C.U.; Woodward, W.; Birren, S.J. Regulation of cardiac innervation and function via the p75 neurotrophin receptor. Auton. Neurosci. 2008, 140, 40–48. [Google Scholar] [CrossRef]
- Lorentz, C.U.; Woodward, W.R.; Tharp, K.; Habecker, B.A. Altered norepinephrine content and ventricular function in p75NTR-/- mice after myocardial infarction. Auton. Neurosci. 2011, 164, 13–19. [Google Scholar] [CrossRef]
- Lorentz, C.U.; Parrish, D.C.; Alston, E.N.; Pellegrino, M.J.; Woodward, W.R.; Hempstead, B.L.; Habecker, B.A. Sympathetic denervation of peri-infarct myocardium requires the p75 neurotrophin receptor. Exp. Neurol. 2013, 249, 111–119. [Google Scholar] [CrossRef]
- Siao, C.J.; Lorentz, C.U.; Kermani, P.; Marinic, T.; Carter, J.; McGrath, K.; Padow, V.A.; Mark, W.; Falcone, D.J.; Cohen-Gould, L.; et al. ProNGF, a cytokine induced after myocardial infarction in humans, targets pericytes to promote microvascular damage and activation. J. Exp. Med. 2012, 209, 2291–2305. [Google Scholar] [CrossRef]
- Takano, A.P.C.; Diniz, G.P.; Barreto-Chaves, M.L.M. AMPK signaling pathway is rapidly activated by T3 and regulates the cardiomyocyte growth. Mol. Cell. Endocrinol. 2013, 376, 43–50. [Google Scholar] [CrossRef]
- Davies, J.K.; Wells, D.J.; Liu, K.; Whitrow, H.R.; Daniel, T.D.; Grignani, R.; Lygate, C.A.; Schneider, J.E.; Noël, G.; Watkins, H.; et al. Characterization of the role of γ2 R531G mutation in AMP-activated protein kinase in cardiac hypertrophy and Wolff-Parkinson-White syndrome. Am. J.-Physiol.-Heart Circ. Physiol. 2006, 290, H1942–H1951. [Google Scholar] [CrossRef]
- Nagata, D.; Mogi, M.; Walsh, K. AMP-activated protein kinase (AMPK) signaling in endothelial cells is essential for angiogenesis in response to hypoxic stress. J. Biol. Chem. 2003, 278, 31000–31006. [Google Scholar] [CrossRef]
- Ma, X.; Fu, Y.; Xiao, H.; Song, Y.; Chen, R.; Shen, J.; An, X.; Shen, Q.; Li, Z.; Zhang, Y. Cardiac Fibrosis Alleviated by Exercise Training Is AMPK-Dependent. PLoS ONE 2015, 10, e0129971. [Google Scholar] [CrossRef] [PubMed]
- Gélinas, R.; Mailleux, F.; Dontaine, J.; Bultot, L.; Demeulder, B.; Ginion, A.; Daskalopoulos, E.P.; Esfahani, H.; Dubois-Deruy, E.; Lauzier, B.; et al. AMPK activation counteracts cardiac hypertrophy by reducing O-GlcNAcylation. Nat. Commun. 2018, 9, 374. [Google Scholar] [CrossRef] [PubMed]
- Gundewar, S.; Calvert, J.W.; Jha, S.; Toedt-Pingel, I.; Yong Ji, S.; Nunez, D.; Ramachandran, A.; Anaya-Cisneros, M.; Tian, R.; Lefer, D.J. Activation of AMP-Activated Protein Kinase by Metformin Improves Left Ventricular Function and Survival in Heart Failure. Am. Heart Assoc. 2009, 104, 403–411. [Google Scholar] [CrossRef]
- Timm, K.N.; Tyler, D.J. The Role of AMPK Activation for Cardioprotection in Doxorubicin-Induced Cardiotoxicity. Cardiovasc. Drugs Ther. 2020, 34, 255–269. [Google Scholar] [CrossRef]
- Cieslik, K.A.; Taffet, G.E.; Crawford, J.R.; Trial, J.; Mejia Osuna, P.; Entman, M.L. AICAR-dependent AMPK activation improves scar formation in the aged heart in a murine model of reperfused myocardial infarction. J. Mol. Cell. Cardiol. 2013, 63, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, Y.; Xu, H.; Zhang, Y.W. The γ-secretase complex: From structure to function. Front. Cell. Neurosci. 2014, 8, 427. [Google Scholar] [CrossRef]
- Wolfe, M.S. Structure and Function of the γ-Secretase Complex. Biochemistry 2019, 58, 2953–2966. [Google Scholar] [CrossRef]
- Wong, E.; Liao, G.P.; Chang, J.C.; Xu, P.; Li, Y.M.; Greengard, P. GSAP modulates γ-secretase specificity by inducing conformational change in PS1. Proc. Natl. Acad. Sci. USA 2019, 116, 6385–6390. [Google Scholar] [CrossRef]
- Villa, J.C.; Chiu, D.; Brandes, A.H.; Escorcia, F.E.; Villa, C.H.; Maguire, W.F.; Hu, C.J.; de Stanchina, E.; Simon, M.C.; Sisodia, S.S.; et al. Nontranscriptional Role of Hif-1α in Activation of γ-Secretase and Notch Signaling in Breast Cancer. Cell Rep. 2014, 8, 1077–1092. [Google Scholar] [CrossRef]
- Kim, D.Y.; Carey, B.W.; Wang, H.; Ingano, L.A.M.; Binshtok, A.M.; Wertz, M.H.; Pettingell, W.H.; He, P.; Lee, V.M.Y.; Woolf, C.J.; et al. BACE1 regulates voltage-gated sodium channels and neuronal activity. Nat. Cell Biol. 2007, 9, 755–764. [Google Scholar] [CrossRef]
- Jo, E.H.; Ahn, J.S.; Mo, J.S.; Yoon, J.H.; Ann, E.J.; Baek, H.J.; Lee, H.J.; Kim, S.H.; Kim, M.Y.; Park, H.S. Akt1 phosphorylates Nicastrin to regulate its protein stability and activity. J. Neurochem. 2015, 134, 799–810. [Google Scholar] [CrossRef] [PubMed]
- Kopan, R.; Ilagan, M.X.G. γ-Secretase: Proteasome of the membrane? Nat. Rev. Mol. Cell Biol. 2004, 5, 499–504. [Google Scholar] [CrossRef]
- Doedens, J.R.; Mahimkar, R.M.; Black, R.A. TACE/ADAM-17 enzymatic activity is increased in response to cellular stimulation. Biochem. Biophys. Res. Commun. 2003, 308, 331–338. [Google Scholar] [CrossRef]
- Haapasalo, A.; Kovacs, D.M. The Many Substrates of Presenilin/γ-Secretase. J. Alzheimer’s Dis. 2011, 25, 3–28. [Google Scholar] [CrossRef]
- Boulton, M.E.; Cai, J.; Grant, M.B. gamma-Secretase: A multifaceted regulator of angiogenesis. J. Cell. Mol. Med. 2008, 12, 781–795. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, M.; Moriizumi, E.; Koseki, H.; Shirasawa, T. Presenilin 1 is essential for cardiac morphogenesis. Dev. Dyn. 2004, 230, 795–799. [Google Scholar] [CrossRef]
- High, F.A.; Epstein, J.A. The multifaceted role of Notch in cardiac development and disease. Nat. Rev. Genet. 2008, 9, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Bovo, E.; Nikolaienko, R.; Kahn, D.; Cho, E.; Robia, S.L.; Zima, A.V. Presenilin 1 is a direct regulator of the cardiac sarco/endoplasmic reticulum calcium pump. Cell Calcium 2021, 99, 102468. [Google Scholar] [CrossRef]
- Takeda, T.; Asahi, M.; Yamaguchi, O.; Hikoso, S.; Nakayama, H.; Kusakari, Y.; Kawai, M.; Hongo, K.; Higuchi, Y.; Kashiwase, K.; et al. Presenilin 2 regulates the systolic function of heart by modulating Ca2+ signaling. FASEB J. 2005, 19, 2069–2071. [Google Scholar] [CrossRef]
- Serneels, L.; Dejaegere, T.; Craessaerts, K.; Horre, K.; Jorissen, E.; Tousseyn, T.; Hebert, S.; Coolen, M.; Martens, G.; Zwijsen, A.; et al. Differential contribution of the three Aph1 genes to -secretase activity in vivo. Proc. Natl. Acad. Sci. USA 2005, 102, 1719–1724. [Google Scholar] [CrossRef]
- Nguyen, V.; Hawkins, C.; Bergeron, C.; Supala, A.; Huang, J.; Westaway, D.; St George-Hyslop, P.; Rozmahel, R. Loss of nicastrin elicits an apoptotic phenotype in mouse embryos. Brain Res. 2006, 1086, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Bammens, L.; Chávez-Gutiérrez, L.; Tolia, A.; Zwijsen, A.; Strooper, B.D. Functional and Topological Analysis of Pen-2, the Fourth Subunit of the γ-Secretase Complex. J. Biol. Chem. 2011, 286, 12271–12282. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Yang, Y. The roles of CyPA and CD147 in cardiac remodelling. Exp. Mol. Pathol. 2018, 104, 222–226. [Google Scholar] [CrossRef]
- Wang, L.; Tang, R.; Zhang, Y.; Chen, S.; Guo, Y.; Wang, X.; Liu, Z.; Liu, H.; Zhang, X.; Liu, B.C. PTH-induced EndMT via miR-29a-5p/GSAP/Notch1 pathway contributed to valvular calcification in rats with CKD. Cell Prolif. 2021, 54, e13018. [Google Scholar] [CrossRef]
- Liu, Y.; Li, P.; Liu, K.; He, Q.; Han, S.; Sun, X.; Li, T.; Shen, L. Timely Inhibition of Notch Signaling by DAPT Promotes Cardiac Differentiation of Murine Pluripotent Stem Cells. PLoS ONE 2014, 9, e109588. [Google Scholar] [CrossRef]
- Li, D.; Parks, S.B.; Kushner, J.D.; Nauman, D.; Burgess, D.; Ludwigsen, S.; Partain, J.; Nixon, R.R.; Allen, C.N.; Irwin, R.P.; et al. Mutations of Presenilin Genes in Dilated Cardiomyopathy and Heart Failure. Am. J. Hum. Genet. 2006, 79, 1030–1039. [Google Scholar] [CrossRef]
- Morales, A.; Painter, T.; Li, R.; Siegfried, J.D.; Li, D.; Norton, N.; Hershberger, R.E. Rare Variant Mutations in Pregnancy-Associated or Peripartum Cardiomyopathy. Circulation 2010, 121, 2176–2182. [Google Scholar] [CrossRef] [PubMed]
- Kelleher, R.J.; Shen, J. Presenilin-1 mutations and Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, 629–631. [Google Scholar] [CrossRef]
- Gianni, D.; Li, A.; Tesco, G.; McKay, K.M.; Moore, J.; Raygor, K.; Rota, M.; Gwathmey, J.K.; Dec, G.W.; Aretz, T.; et al. Protein Aggregates and Novel Presenilin Gene Variants in Idiopathic Dilated Cardiomyopathy. Circulation 2010, 121, 1216–1226. [Google Scholar] [CrossRef]
- Xia, W. γ-Secretase and its modulators: Twenty years and beyond. Neurosci. Lett. 2019, 701, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Ikeuchi, T.; Sisodia, S.S. The Notch ligands, Delta1 and Jagged2, are substrates for presenilin-dependent “gamma-secretase” cleavage. J. Biol. Chem. 2003, 278, 7751–7754. [Google Scholar] [CrossRef] [PubMed]
- Stupnikov, M.R.; Yang, Y.; Mori, M.; Lu, J.; Cardoso, W.V. Jagged and Delta-like ligands control distinct events during airway progenitor cell differentiation. Elife 2019, 8, e50487. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, E.B.; Conner, S.D. γ-Secretase-Dependent Cleavage Initiates Notch Signaling from the Plasma Membrane. Traffic 2010, 11, 1234–1245. [Google Scholar] [CrossRef] [PubMed]
- Luxán, G.; D’Amato, G.; MacGrogan, D.; de la Pompa, J.L. Endocardial Notch Signaling in Cardiac Development and Disease. Circ. Res. 2016, 118, e1–e18. [Google Scholar] [CrossRef]
- [PDF] Notch Pathway and Its Role in Cardiovascular System: Review|Semantic Scholar. Available online: https://www.semanticscholar.org/paper/Notch-Pathway-and-its-Role-in-Cardiovascular-Review-Badr-Mahran/a42580d709cc813d711eea759e05baf3ff868460 (accessed on 30 October 2021).
- MacGrogan, D.; Münch, J.; de la Pompa, J.L. Notch and interacting signalling pathways in cardiac development, disease, and regeneration. Nat. Rev. Cardiol. 2018, 15, 685–704. [Google Scholar] [CrossRef]
- Siekmann, A.F.; Lawson, N.D. Notch signalling limits angiogenic cell behaviour in developing zebrafish arteries. Nature 2007, 445, 781–784. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, B.; Chamberlain, A.A.; Lui, W.; Koirala, P.; Susztak, K.; Klein, D.; Taylor, V.; Zhou, B. Endocardial to Myocardial Notch-Wnt-Bmp Axis Regulates Early Heart Valve Development. PLoS ONE 2013, 8, e60244. [Google Scholar] [CrossRef]
- Zhou, X.L.; Liu, J.C. Role of Notch Signaling in the Mammalian Heart. Braz. J. Med Biol. Res. 2014, 47, 1–10. [Google Scholar] [CrossRef]
- Travisano, S.I.; Oliveira, V.L.; Prados, B.; Grego-Bessa, J.; Piñeiro-Sabarís, R.; Bou, V.; Gómez, M.J.; Sánchez-Cabo, F.; MacGrogan, D.; de la Pompa, J.L. Coronary arterial development is regulated by a Dll4-Jag1-EphrinB2 signaling cascade. Elife 2019, 8, e49977. [Google Scholar] [CrossRef]
- Lu, P.; Wang, Y.; Liu, Y.; Wang, Y.; Wu, B.; Zheng, D.; Harvey, R.P.; Zhou, B. Perinatal angiogenesis from pre-existing coronary vessels via DLL4–NOTCH1 signalling. Nat. Cell Biol. 2021, 23, 967–977. [Google Scholar] [CrossRef] [PubMed]
- Nistri, S.; Sassoli, C.; Bani, D. Notch Signaling in Ischemic Damage and Fibrosis: Evidence and Clues from the Heart. Front. Pharmacol. 2017, 8, 187. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Yu, L.; Liu, D.; Yang, X.; Zheng, Y.; Gui, Y.; Wang, H. MIB1 mutations reduce Notch signaling activation and contribute to congenital heart disease. Clin. Sci. 2018, 132, 2483–2491. [Google Scholar] [CrossRef]
- Zhao, Z.Q.; Corvera, J.S.; Halkos, M.E.; Kerendi, F.; Wang, N.P.; Guyton, R.A.; Vinten-Johansen, J. Inhibition of myocardial injury by ischemic postconditioning during reperfusion: Comparison with ischemic preconditioning. Am. J.-Physiol.-Heart Circ. Physiol. 2003, 285, H579–H588. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Suk, J.; Park, J.; Kim, S.B.; Kwak, S.S.; Kim, J.W.; Lee, C.H.; Byun, B.; Ahn, J.K.; Joe, C.O. Notch Signal Activates Hypoxia Pathway through HES1-Dependent SRC/Signal Transducers and Activators of Transcription 3 Pathway. Mol. Cancer Res. 2009, 7, 1663–1671. [Google Scholar] [CrossRef] [PubMed]
- Nemir, M.; Metrich, M.; Plaisance, I.; Lepore, M.; Cruchet, S.; Berthonneche, C.; Sarre, A.; Radtke, F.; Pedrazzini, T. The Notch pathway controls fibrotic and regenerative repair in the adult heart. Eur. Heart J. 2014, 35, 2174–2185. [Google Scholar] [CrossRef]
- Fan, Z.; Guan, J. Antifibrotic therapies to control cardiac fibrosis. Biomater. Res. 2016, 20, 13. [Google Scholar] [CrossRef]
- Hara, H.; Takeda, N.; Komuro, I. Pathophysiology and therapeutic potential of cardiac fibrosis. Inflamm. Regen. 2017, 37, 13. [Google Scholar] [CrossRef]
- Fan, Y.H.; Dong, H.; Pan, Q.; Cao, Y.J.; Li, H.; Wang, H.C. Notch Signaling May Negatively Regulate Neonatal Rat Cardiac Fibroblast-Myofibroblast Transformation. Physiol. Res. 2011, 60, 739–748. [Google Scholar] [CrossRef]
- Bolduc, D.M.; Montagna, D.R.; Seghers, M.C.; Wolfe, M.S.; Selkoe, D.J. The amyloid-beta forming tripeptide cleavage mechanism of γ-secretase. Elife 2016, 5, e17578. [Google Scholar] [CrossRef]
- Funamoto, S.; Sasaki, T.; Ishihara, S.; Nobuhara, M.; Nakano, M.; Watanabe-Takahashi, M.; Saito, T.; Kakuda, N.; Miyasaka, T.; Nishikawa, K.; et al. Substrate ectodomain is critical for substrate preference and inhibition of γ-secretase. Nat. Commun. 2013, 4, 2529. [Google Scholar] [CrossRef]
- Meckler, X.; Checler, F. Presenilin 1 and Presenilin 2 Target γ-Secretase Complexes to Distinct Cellular Compartments. J. Biol. Chem. 2016, 291, 12821–12837. [Google Scholar] [CrossRef]
- Merilahti, J.A.M.; Elenius, K. Gamma-secretase-dependent signaling of receptor tyrosine kinases. Oncogene 2019, 38, 151–163. [Google Scholar] [CrossRef] [PubMed]
- Ni, C.Y.; Murphy, M.P.; Golde, T.E.; Carpenter, G. γ-Secretase Cleavage and Nuclear Localization of ErbB-4 Receptor Tyrosine Kinase. Science 2001, 294, 2179–2181. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Jung, K.M.; Huang, Y.Z.; Bennett, L.B.; Lee, J.S.; Mei, L.; Kim, T.W. Presenilin-dependent γ-Secretase-like Intramembrane Cleavage of ErbB4. J. Biol. Chem. 2002, 277, 6318–6323. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, R.; Mine, N.; Mizushima, H.; Mekada, E. ErbB1 and ErbB4 generate opposing signals regulating mesenchymal cell proliferation during valvulogenesis. J. Cell Sci. 2017, 130, 1321–1332. [Google Scholar] [CrossRef]
- Odiete, O.; Hill, M.F.; Sawyer, D.B. Neuregulin in cardiovascular development and disease. Circ. Res. 2012, 111, 1376–1385. [Google Scholar] [CrossRef]
- Wadugu, B.; Kühn, B. The role of neuregulin/ErbB2/ErbB4 signaling in the heart with special focus on effects on cardiomyocyte proliferation. Am. J.-Physiol.-Heart Circ. Physiol. 2012, 302, H2139–H2147. [Google Scholar] [CrossRef]
- Brown, D.; Samsa, L.A.; Ito, C.; Ma, H.; Batres, K.; Arnaout, R.; Qian, L.; Liu, J. Neuregulin-1 is essential for nerve plexus formation during cardiac maturation. J. Cell. Mol. Med. 2018, 22, 2007–2017. [Google Scholar] [CrossRef]
- Boyette, L.C.; Manna, B. Physiology, Myocardial Oxygen Demand; StatPearls Publishing: Treasure Island, FL, USA, 2021.
- Russell, K.F.; Stern, D.F.; Polverini, P.J.; Bender, J.R. Neuregulin activation of ErbB receptors in vascular endothelium leads to angiogenesis. Am. J. Physiol. 1999, 277, H2205–H2211. [Google Scholar] [CrossRef]
- Abbott, G.W. KCNE genetics and pharmacogenomics in cardiac arrhythmias: Much ado about nothing? Expert Rev. Clin. Pharmacol. 2013, 6, 49–60. [Google Scholar] [CrossRef][Green Version]
- Sachse, C.C.; Kim, Y.H.; Agsten, M.; Huth, T.; Alzheimer, C.; Kovacs, D.M.; Kim, D.Y. BACE1 and presenilin/γ-secretase regulate proteolytic processing of KCNE1 and 2, auxiliary subunits of voltage-gated potassium channels. FASEB J. 2013, 27, 2458–2467. [Google Scholar] [CrossRef]
- Abbott, G.W. KCNE1 and KCNE3: The yin and yang of voltage-gated K(+) channel regulation. Gene 2016, 576, 1–13. [Google Scholar] [CrossRef]
- Abbott, G.W. KCNE4 and KCNE5: K+ channel regulation and cardiac arrhythmogenesis. Gene 2016, 593, 249–260. [Google Scholar] [CrossRef]
- Bouza, A.A.; Isom, L.L. Chapter 14: Voltage-gated sodium channel β subunits and their related diseases. Volt.-Gated Sodium Channels Struct. Funct. Channelopathies 2018, 246, 423–450. [Google Scholar] [CrossRef]
- Kim, D.Y.; Gersbacher, M.T.; Inquimbert, P.; Kovacs, D.M. Reduced sodium channel Na(v)1.1 levels in BACE1-null mice. J. Biol. Chem. 2011, 286, 8106–8116. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Hwang, K.H.; Park, K.S.; Kong, I.D.; Cha, S.K. Biological Role of Anti-aging Protein Klotho. J. Lifestyle Med. 2015, 5, 1–6. [Google Scholar] [CrossRef]
- Bloch, L.; Sineshchekova, O.; Reichenbach, D.; Reiss, K.; Saftig, P.; Kuro-o, M.; Kaether, C. Klotho is a substrate for α-, β- and γ-secretase. FEBS Lett. 2009, 583, 3221–3224. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.P.; Mendes, F.; Carias, E.; Gonçalves, R.B.; Fragoso, A.; Dias, C.; Tavares, N.; Café, H.M.; Santos, N.; Rato, F.; et al. Plasmatic Klotho and FGF23 Levels as Biomarkers of CKD-Associated Cardiac Disease in Type 2 Diabetic Patients. Int. J. Mol. Sci. 2019, 20, 1536. [Google Scholar] [CrossRef]
- Ding, J.; Tang, Q.; Luo, B.; Zhang, L.; Lin, L.; Han, L.; Hao, M.; Li, M.; Yu, L.; Li, M. Klotho inhibits angiotensin II-induced cardiac hypertrophy, fibrosis, and dysfunction in mice through suppression of transforming growth factor-β1 signaling pathway. Eur. J. Pharmacol. 2019, 859, 172549. [Google Scholar] [CrossRef]
- Saar-Kovrov, V.; Donners, M.M.P.C.; van der Vorst, E.P.C. Shedding of Klotho: Functional Implications in Chronic Kidney Disease and Associated Vascular Disease. Front. Cardiovasc. Med. 2021, 7, 407. [Google Scholar] [CrossRef] [PubMed]
- Bibel, M. Neurotrophins: Key regulators of cell fate and cell shape in the vertebrate nervous system. Genes Dev. 2000, 14, 2919–2937. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Rahn, J.J.; Lun, X.; Sun, B.; Kelly, J.J.P.; Weiss, S.; Robbins, S.M.; Forsyth, P.A.; Senger, D.L. Gamma-Secretase Represents a Therapeutic Target for the Treatment of Invasive Glioma Mediated by the p75 Neurotrophin Receptor. PLoS Biol. 2008, 6, e289. [Google Scholar] [CrossRef] [PubMed]
- Kreitman, M.; Noronha, A.; Yarden, Y. Irreversible modifications of receptor tyrosine kinases. FEBS Lett. 2018, 592, 2199–2212. [Google Scholar] [CrossRef]
- Abid, M.R.; Guo, S.; Minami, T.; Spokes, K.C.; Ueki, K.; Skurk, C.; Walsh, K.; Aird, W.C. Vascular endothelial growth factor activates PI3K/Akt/forkhead signaling in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 294–300. [Google Scholar] [CrossRef]
- Kontos, C.D.; Cha, E.H.; York, J.D.; Peters, K.G. The endothelial receptor tyrosine kinase Tie1 activates phosphatidylinositol 3-kinase and Akt to inhibit apoptosis. Mol. Cell. Biol. 2002, 22, 1704–1713. [Google Scholar] [CrossRef]
- Aoyagi, T.; Matsui, T. Phosphoinositide-3 kinase signaling in cardiac hypertrophy and heart failure. Curr. Pharm. Des. 2011, 17, 1818–1824. [Google Scholar] [CrossRef]
- Shioi, T.; McMullen, J.R.; Kang, P.M.; Douglas, P.S.; Obata, T.; Franke, T.F.; Cantley, L.C.; Izumo, S. Akt/Protein Kinase B Promotes Organ Growth in Transgenic Mice. Mol. Cell. Biol. 2002, 22, 2799–2809. [Google Scholar] [CrossRef]
- Haq, S.; Choukroun, G.; Kang, Z.B.; Ranu, H.; Matsui, T.; Rosenzweig, A.; Molkentin, J.D.; Alessandrini, A.; Woodgett, J.; Hajjar, R.; et al. Glycogen Synthase Kinase-3β Is a Negative Regulator of Cardiomyocyte Hypertrophy. J. Cell Biol. 2000, 151, 117–130. [Google Scholar] [CrossRef]
- Chaanine, A.H.; Hajjar, R.J. AKT signalling in the failing heart. Eur. J. Heart Fail. 2011, 13, 825–829. [Google Scholar] [CrossRef]
- Walsh, K. Akt Signaling and Growth of the Heart. Circulation 2006, 113, 2032–2034. [Google Scholar] [CrossRef] [PubMed]
- Takeshita, K.; Satoh, M.; Ii, M.; Silver, M.; Limbourg, F.P.; Mukai, Y.; Rikitake, Y.; Radtke, F.; Gridley, T.; Losordo, D.W.; et al. Critical Role of Endothelial Notch1 Signaling in Postnatal Angiogenesis. Circ. Res. 2007, 100, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Ge, W.; Ren, J. mTOR-STAT3-notch signalling contributes to ALDH2-induced protection against cardiac contractile dysfunction and autophagy under alcoholism. J. Cell. Mol. Med. 2012, 16, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Long, Y.C.; Zierath, J.R. AMP-activated protein kinase signaling in metabolic regulation. Diabetes-Am. Diabetes Assoc. 2006, 116, 1776–1783. [Google Scholar] [CrossRef]
- Yang, H.; Sun, W.; Quan, N.; Wang, L.; Chu, D.; Cates, C.; Liu, Q.; Zheng, Y.; Li, J. Cardioprotective actions of Notch1 against myocardial infarction via LKB1-dependent AMPK signaling pathway. Biochem. Pharmacol. 2016, 108, 47–57. [Google Scholar] [CrossRef]
- Dyck, J.R.B.; Lopaschuk, G.D. AMPK alterations in cardiac physiology and pathology: Enemy or ally? J. Physiol. 2006, 574, 95–112. [Google Scholar] [CrossRef]
- Su, Z.; Liu, Y.; Zhang, H. Adaptive Cardiac Metabolism Under Chronic Hypoxia: Mechanism and Clinical Implications. Front. Cell Dev. Biol. 2021, 9, 91. [Google Scholar] [CrossRef]
- Zhang, Y.; Lee, T.S.; Kolb, E.M.; Sun, K.; Lu, X.; Sladek, F.M.; Kassab, G.S.; Garland, T.; Shyy, J.Y.J. AMP-activated protein kinase is involved in endothelial NO synthase activation in response to shear stress. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1281–1287. [Google Scholar] [CrossRef]
- Kim, J.E.; Kim, Y.W.; Lee, I.K.; Kim, J.Y.; Kang, Y.J.; Park, S.Y. AMP-activated protein kinase activation by 5-aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside (AICAR) inhibits palmitate-induced endothelial cell apoptosis through reactive oxygen species suppression. J. Pharmacol. Sci. 2008, 106, 394–403. [Google Scholar] [CrossRef]
- Pastorcic, M.; Das, H.K. Ets transcription factors ER81 and Elk1 regulate the transcription of the human presenilin 1 gene promoter. Mol. Brain Res. 2003, 113, 57–66. [Google Scholar] [CrossRef]
- Wang, R.; Zhang, Y.w.; Sun, P.; Liu, R.; Zhang, X.; Zhang, X.; Xia, K.; Xia, J.; Xu, H.; Zhang, Z. Transcriptional Regulation of PEN-2, a Key Component of the γ-Secretase Complex, by CREB. Mol. Cell. Biol. 2006, 26, 1347–1354. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Zhou, H.; Walian, P.J.; Jap, B.K. CD147 is a regulatory subunit of the γ-secretase complex in Alzheimer’s disease amyloid β-peptide production. Proc. Natl. Acad. Sci. USA 2005, 102, 7499–7504. [Google Scholar] [CrossRef]
- Pohland, M.; Hagl, S.; Pellowska, M.; Wurglics, M.; Schubert-Zsilavecz, M.; Eckert, G.P. MH84: A Novel γ-Secretase Modulator/PPARγ Agonist—Improves Mitochondrial Dysfunction in a Cellular Model of Alzheimer’s Disease. Neurochem. Res. 2016, 41, 231–242. [Google Scholar] [CrossRef]
- Golde, T.E.; Koo, E.H.; Felsenstein, K.M.; Osborne, B.A.; Miele, L. γ-Secretase Inhibitors and Modulators. Biochim. Biophys. Acta-(BBA)-Biomembr. 2013, 1828, 2898–2907. [Google Scholar] [CrossRef]
- Takami, M.; Nagashima, Y.; Sano, Y.; Ishihara, S.; Morishima-Kawashima, M.; Funamoto, S.; Ihara, Y. gamma-Secretase: Successive tripeptide and tetrapeptide release from the transmembrane domain of beta-carboxyl terminal fragment. J. Neurosci. 2009, 29, 13042–13052. [Google Scholar] [CrossRef]
- Chávez-Gutiérrez, L.; Bammens, L.; Benilova, I.; Vandersteen, A.; Benurwar, M.; Borgers, M.; Lismont, S.; Zhou, L.; Van Cleynenbreugel, S.; Esselmann, H.; et al. The mechanism of γ-Secretase dysfunction in familial Alzheimer disease. EMBO J. 2012, 31, 2261–2274. [Google Scholar] [CrossRef]
- Shelton, C.C.; Zhu, L.; Chau, D.; Yang, L.; Wang, R.; Djaballah, H.; Zheng, H.; Li, Y.M. Modulation of -secretase specificity using small molecule allosteric inhibitors. Proc. Natl. Acad. Sci. USA 2009, 106, 20228–20233. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Yu, J.; Kang, J.K.; Morrow, J.P.; Pajvani, U.B. Liver-selective γ-secretase inhibition ameliorates diet-induced hepatic steatosis, dyslipidemia and atherosclerosis. Biochem. Biophys. Res. Commun. 2020, 527, 979–984. [Google Scholar] [CrossRef]
- McCaw, T.R.; Inga, E.; Chen, H.; Jaskula-Sztul, R.; Dudeja, V.; Bibb, J.A.; Ren, B.; Rose, J.B. Gamma Secretase Inhibitors in Cancer: A Current Perspective on Clinical Performance. Oncologist 2021, 26, e608–e621. [Google Scholar] [CrossRef]
- Imbimbo, B.P.; Del Giudice, E.; Cenacchi, V.; Volta, R.; Villetti, G.; Facchinetti, F.; Riccardi, B.; Puccini, P.; Moretto, N.; Grassi, F.; et al. In vitro and in vivo profiling of CHF5022 and CHF5074 Two beta-amyloid1-42 lowering agents. Pharmacol. Res. 2007, 55, 318–328. [Google Scholar] [CrossRef]
- Braune, E.B.; Lendahl, U. Notch—A goldilocks signaling pathway in disease and cancer therapy. Discov. Med. 2016, 21, 189–196. [Google Scholar]
- Ratni, H.; Alker, A.; Bartels, B.; Bissantz, C.; Chen, W.; Gerlach, I.; Limberg, A.; Lu, M.; Neidhart, W.; Pichereau, S.; et al. Discovery of RO7185876, a Highly Potent γ-Secretase Modulator (GSM) as a Potential Treatment for Alzheimer’s Disease. ACS Med. Chem. Lett. 2020, 11, 1257–1268. [Google Scholar] [CrossRef]
- Rivera-Torres, J.; Guzmán-Martínez, G.; Villa-Bellosta, R.; Orbe, J.; González-Gómez, C.; Serrano, M.; Díez, J.; Andrés, V.; Maraver, A. Targeting γ-secretases protect against angiotensin II-induced cardiac hypertrophy. J. Hypertens. 2015, 33, 843–850, discussion 850. [Google Scholar] [CrossRef]
- He, G.; Luo, W.; Li, P.; Remmers, C.; Netzer, W.J.; Hendrick, J.; Bettayeb, K.; Flajolet, M.; Gorelick, F.; Wennogle, L.P.; et al. Gamma-secretase activating protein is a therapeutic target for Alzheimer’s disease. Nature 2010, 467, 95–98. [Google Scholar] [CrossRef] [PubMed]
- May, P.; Reddy, Y.K.; Herz, J. Proteolytic Processing of Low Density Lipoprotein Receptor-related Protein Mediates Regulated Release of Its Intracellular Domain. J. Biol. Chem. 2002, 277, 18736–18743. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Tang, P.; Wang, P.; Boissy, R.E.; Zheng, H. Regulation of tyrosinase trafficking and processing by presenilins: Partial loss of function by familial Alzheimer’s disease mutation. Proc. Natl. Acad. Sci. USA 2005, 103, 353–358. [Google Scholar] [CrossRef] [PubMed]


| Potential Gamma-Secretase Alterations | Cardiac Disorders Observed | Publication |
|---|---|---|
| GAMMA-SECRETASE KNOCKOUT MODELS | ||
| Presenilin 1 | Cardiac outflow tract development defect, ventricular septal defects, Stenosis of the pulmonary artery | [59] [58] |
| Presenilin 2 | Increased cardiac contractility, abnormal calcium homeostasis | [61] |
| Nicastrin | Heart development abnormalities and increased apoptosis in abnormally developed regions Embryonic lethal | [63] |
| Aph-1 | Reduced angiogenesis Embryonic lethal | [62] |
| PEN-2 | Notch deficiency-like phenotype, poor embryonic development of heart Embryos have a large pericardial sac | [64] |
| GAMMA-SECRETASE MODULATORS (PROTEIN) | ||
| CD147 | Promotes cardiovascular inflammation, myocardial remodeling, and myocardial I/R injury | [65] |
| GSAP | Valvular calcification | [66] |
| GAMMA-SECRETASE MODULATORS (SMALL MOLECULES) | ||
| MH84 | No cardiac adverse effects have been reported in these agents yet | Not reported yet in literature |
| GAMMA-SECRETASE INHIBITORS | ||
| DAPT | Prevents differentiation in development | [67] |
| Semagecestat | No cardiac adverse effects have been reported in these agents yet | Not reported yet in literature |
| Avagacestat | ||
| EVP-0962 | ||
| NIC5-15 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sen, S.; Hallee, L.; Lam, C.K. The Potential of Gamma Secretase as a Therapeutic Target for Cardiac Diseases. J. Pers. Med. 2021, 11, 1294. https://doi.org/10.3390/jpm11121294
Sen S, Hallee L, Lam CK. The Potential of Gamma Secretase as a Therapeutic Target for Cardiac Diseases. Journal of Personalized Medicine. 2021; 11(12):1294. https://doi.org/10.3390/jpm11121294
Chicago/Turabian StyleSen, Sujoita, Logan Hallee, and Chi Keung Lam. 2021. "The Potential of Gamma Secretase as a Therapeutic Target for Cardiac Diseases" Journal of Personalized Medicine 11, no. 12: 1294. https://doi.org/10.3390/jpm11121294
APA StyleSen, S., Hallee, L., & Lam, C. K. (2021). The Potential of Gamma Secretase as a Therapeutic Target for Cardiac Diseases. Journal of Personalized Medicine, 11(12), 1294. https://doi.org/10.3390/jpm11121294