
Personal Network Inference Unveils Heterogeneous Immune Response Patterns to Viral Infection in Children with Acute Wheezing

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
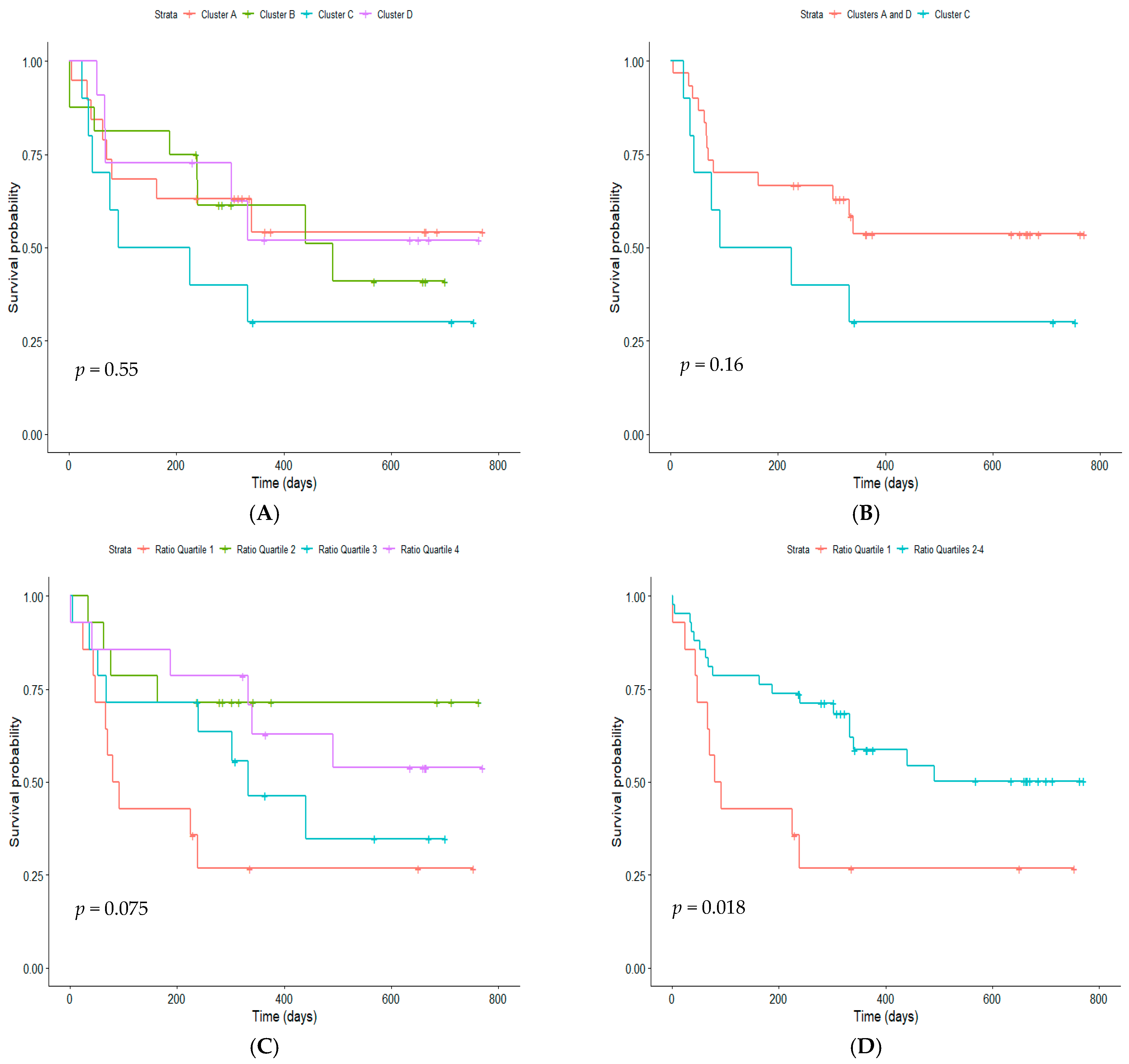
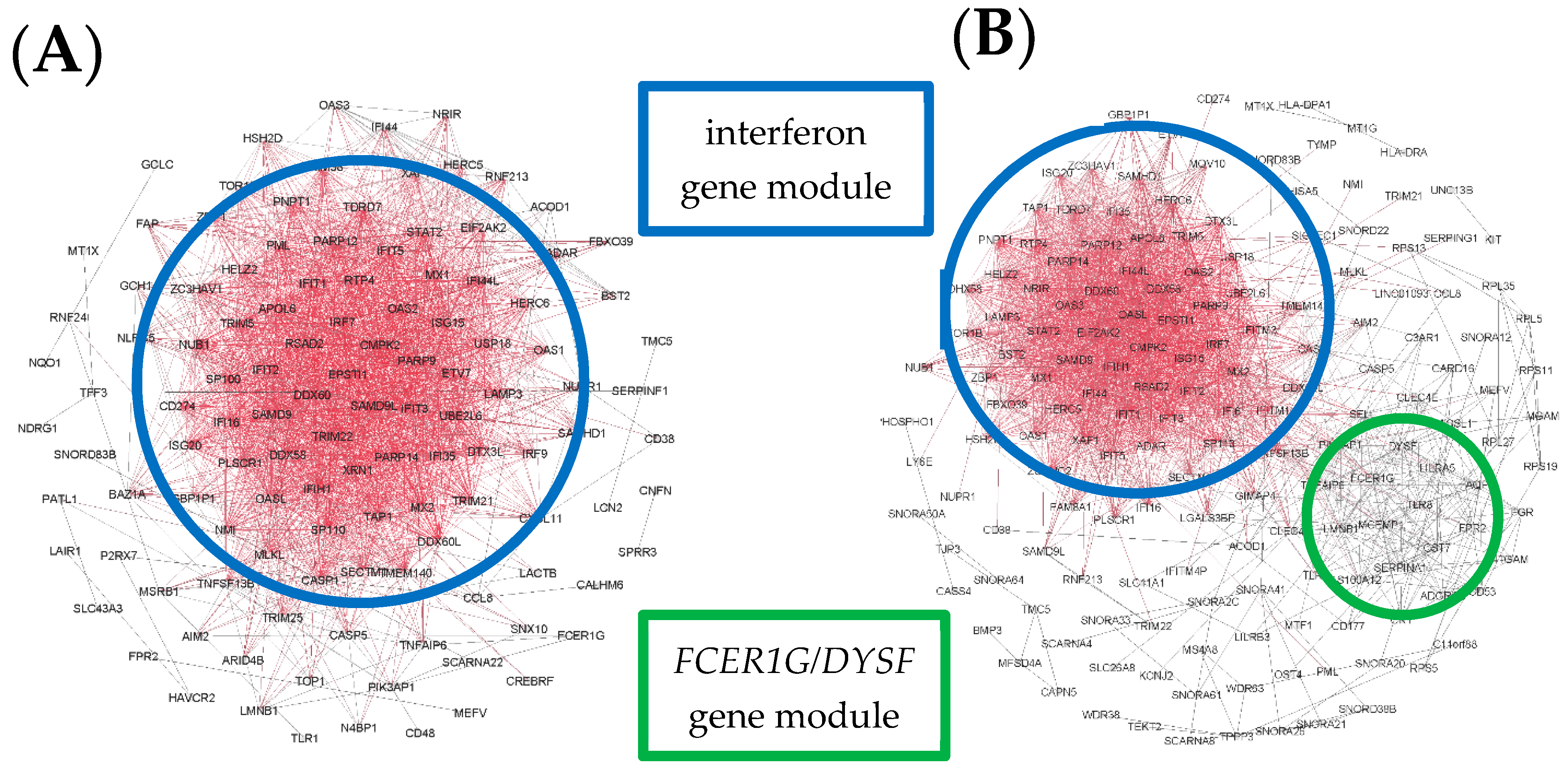
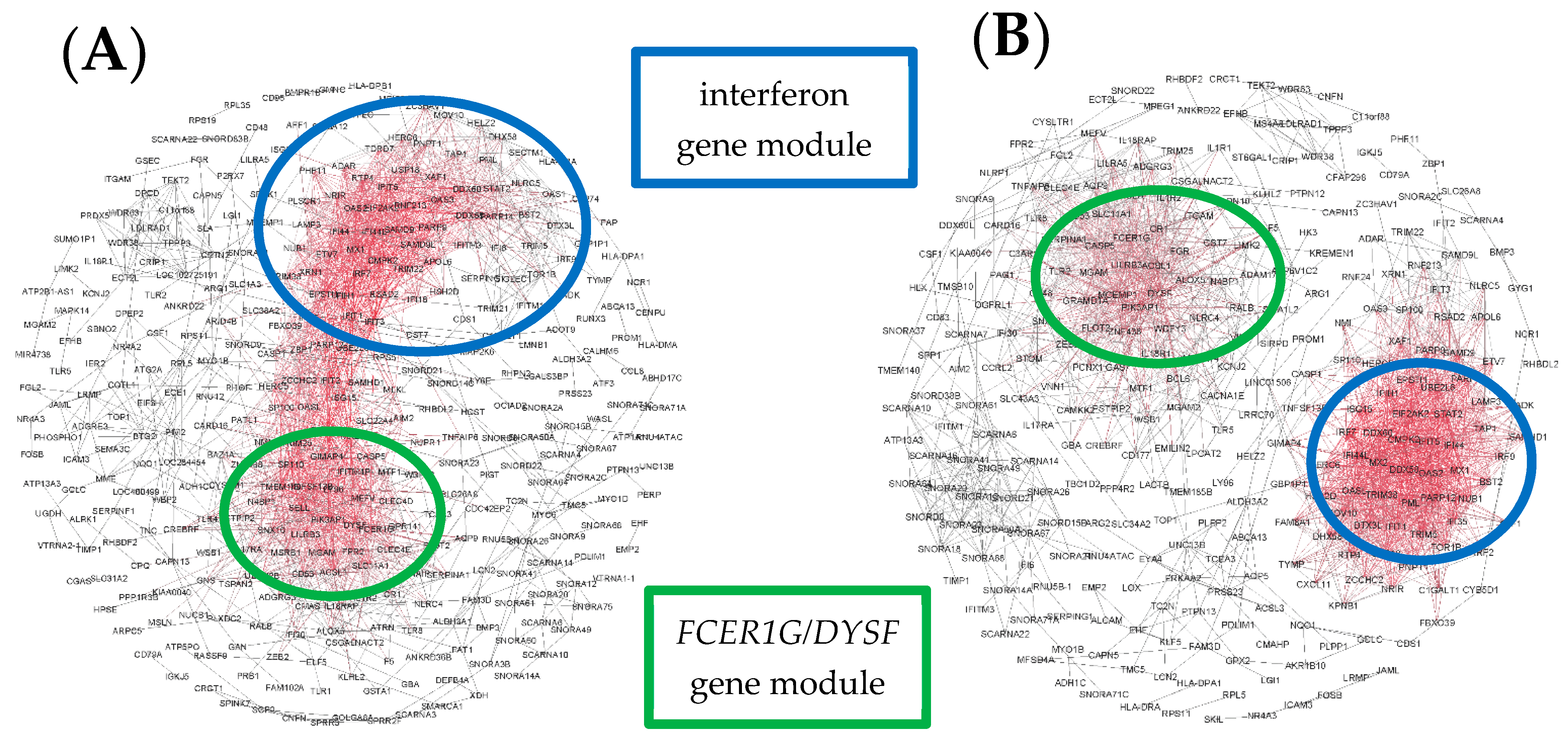
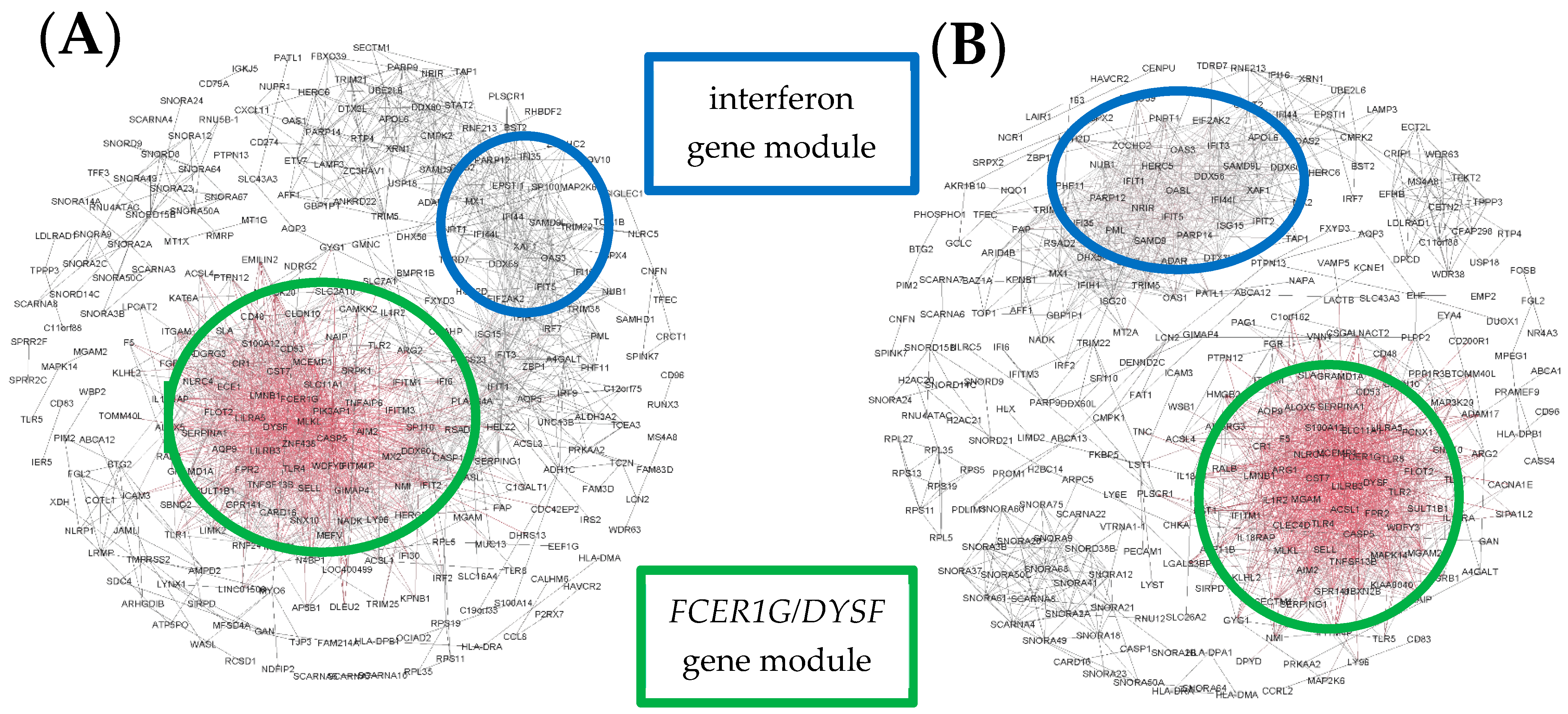
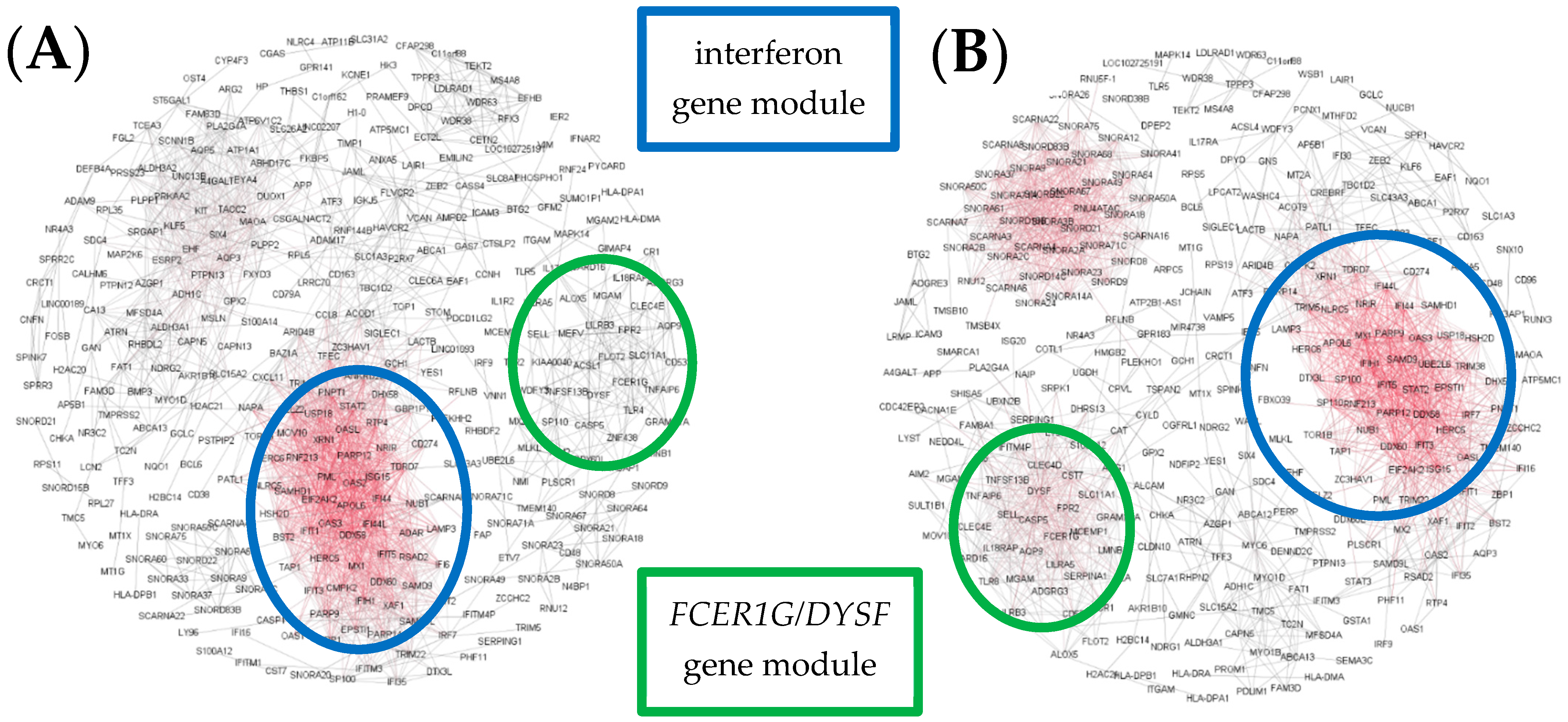
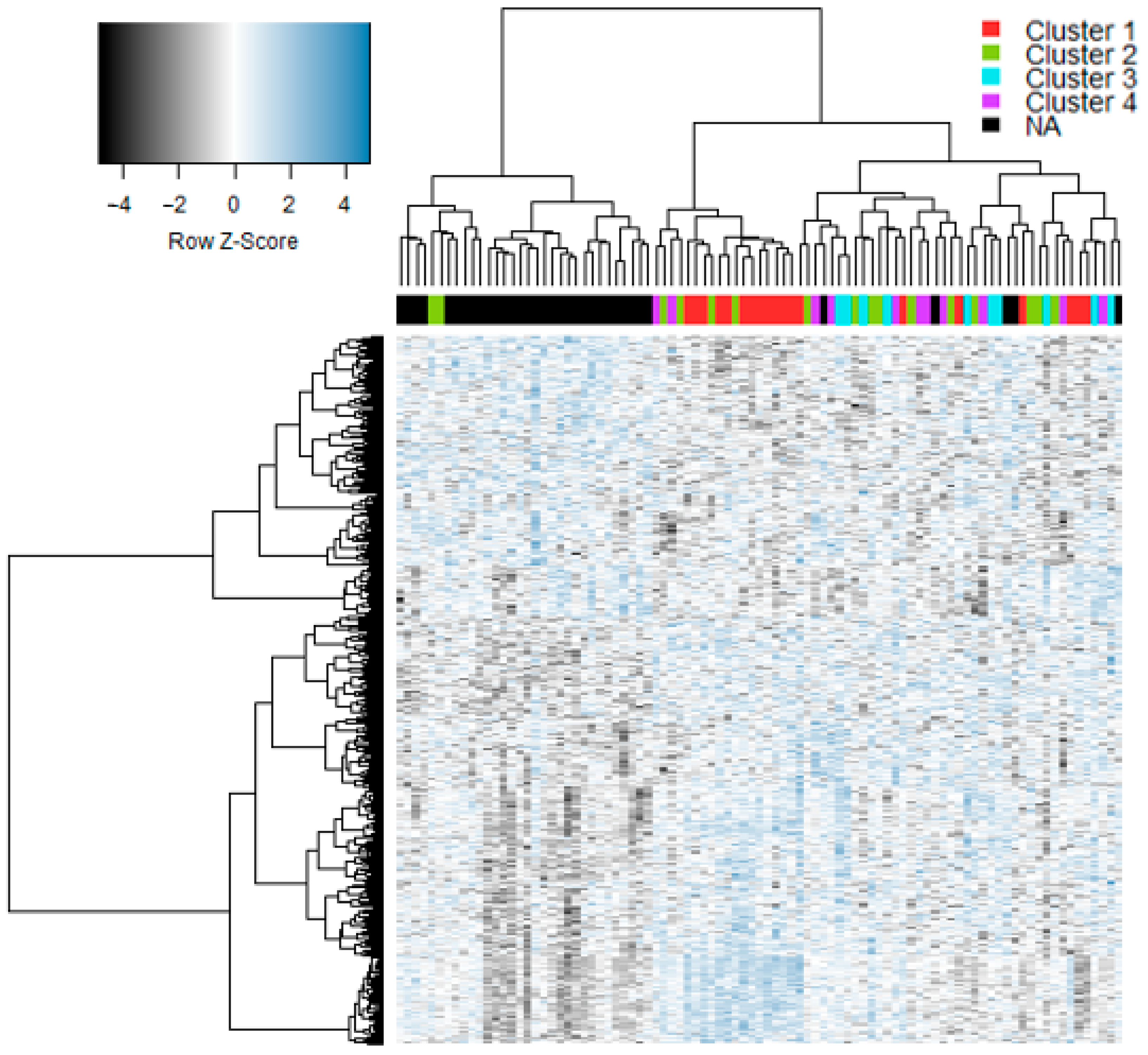
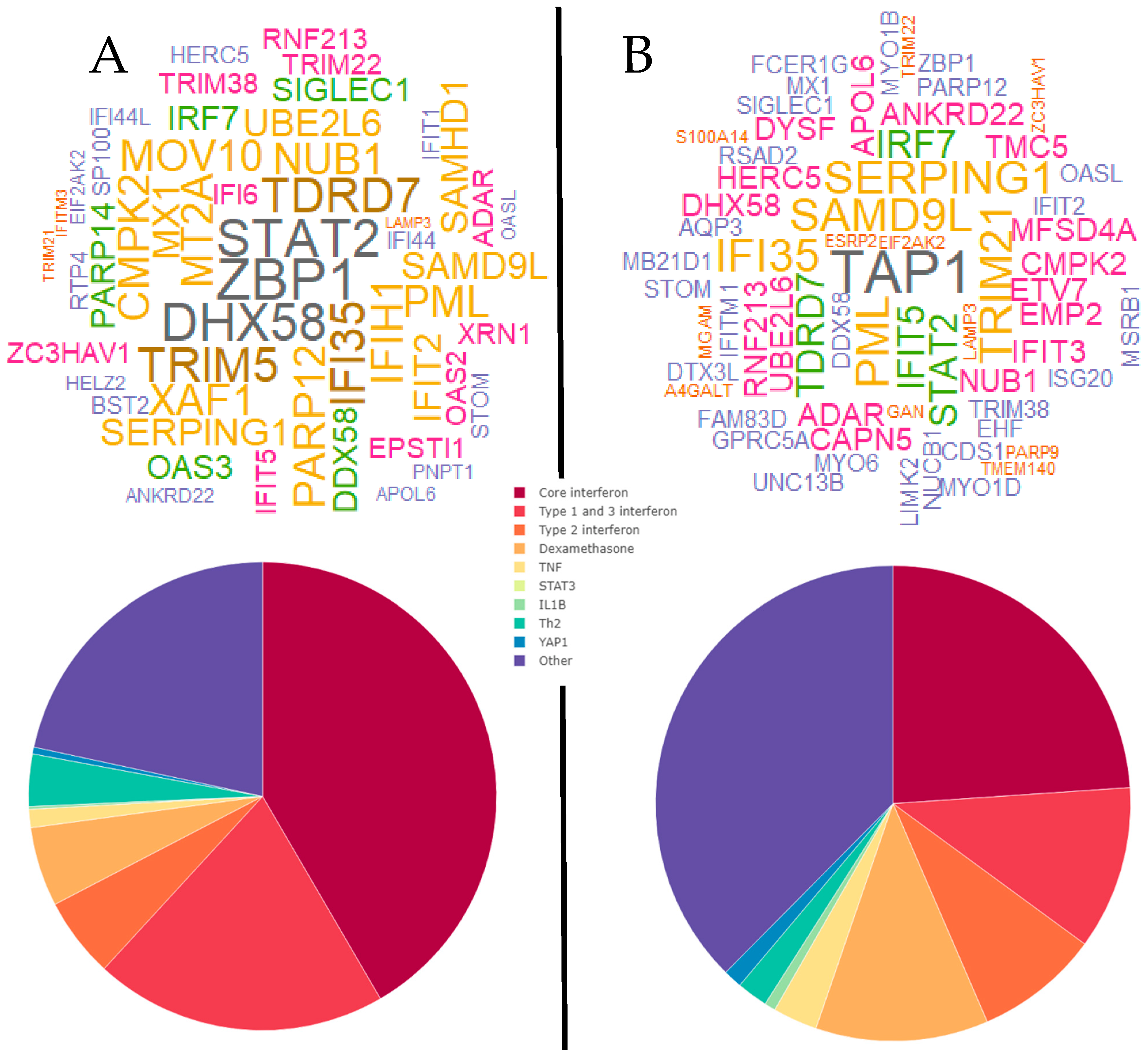
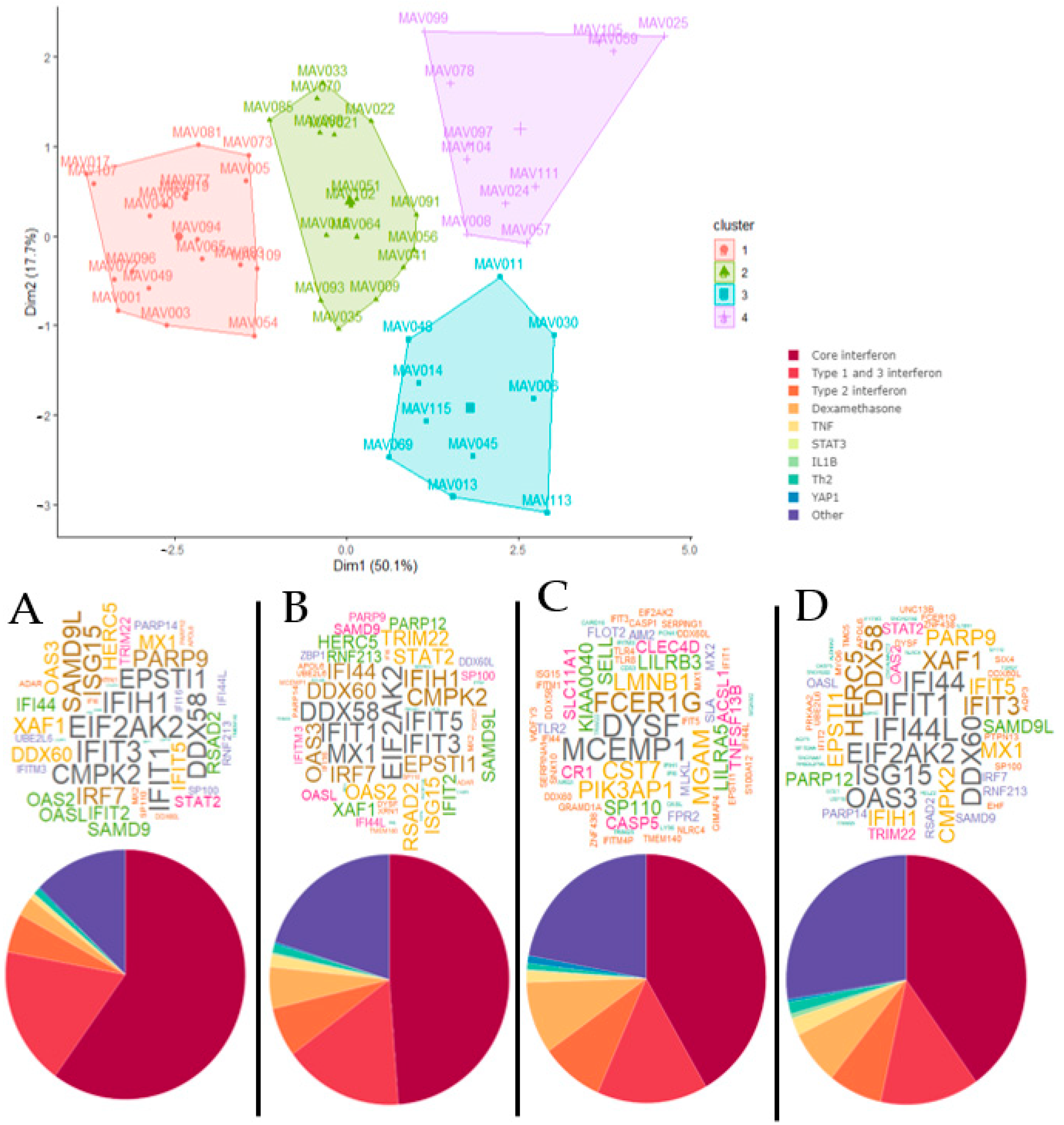
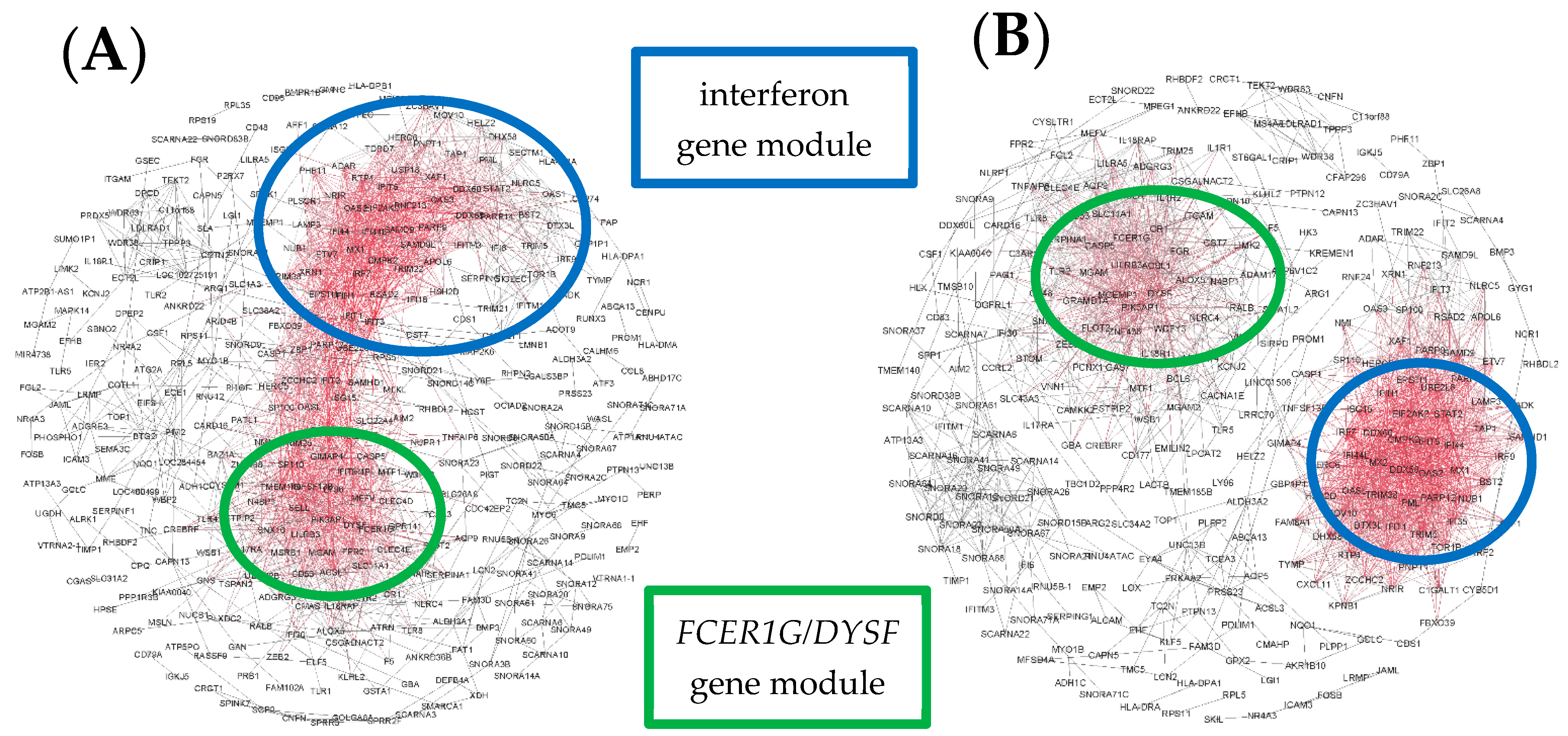
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Miller, E.K.; Hernandez, J.Z.; Wimmenauer, V.; Shepherd, B.E.; Hijano, D.; Libster, R.; Serra, M.E.; Bhat, N.; Batalle, J.P.; Mohamed, Y.; et al. A mechanistic role for type III IFN-l1 in asthma exacerbations mediated by human rhinoviruses. Am. J. Respir. Crit. Care Med. 2012, 185, 508–516. [Google Scholar] [CrossRef] [Green Version]
- Bizzintino, J.; Lee, W.-M.; Laing, I.A.; Vang, F.; Pappas, T.; Zhang, G.; Martin, A.C.; Geelhoed, G.C.; McMinn, P.C.; Goldblatt, J.; et al. Association between human rhinovirus C and severity of acute asthma in children. Eur. Respir. J. 2011, 37, 1037–1042. [Google Scholar] [CrossRef]
- Johnston, S.L.; Pattermore, P.K.; Sanderson, G.; Smith, S.; Lampe, F.; Josephs, L.; Symington, P.; O’Toole, S.; Myint, S.H.; Tyrrell, D.A.J.; et al. Community study of role of viral infections in exacerbations of asthma in 9–11 year old children. BMJ 1995, 310, 1225–1229. [Google Scholar] [CrossRef] [Green Version]
- Khetsuriani, N.; Kazerouni, N.N.; Erdman, D.D.; Lu, X.; Redd, S.C.; Anderson, L.J.; Teague, W.G. Prevalence of viral respiratory tract infections in children with asthma. J. Allergy Clin. Immunol. 2007, 119, 314–321. [Google Scholar] [CrossRef]
- Kwon, J.-M.; Shim, J.W.; Kim, D.S.; Jung, H.L.; Park, M.S.; Shim, J.Y. Prevalence of respiratory viral infection in children hospitalized for acute lower respiratory tract diseases, and association of rhinovirus and influenza virus with asthma exacerbations. Korean J. Pediatr. 2014, 57, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Knowles, N.J.; Hovi, T.; Hyypiä, T.; King, A.M.Q.; Lindberg, A.M.; Pallansch, M.A.; Palmenberg, A.C.; Simmonds, P.; Skern, T.; Stanway, G.; et al. Family-Picornaviridae. In Virus Taxonomy; Elsevier: San Diego, CA, USA, 2012; pp. 855–880. [Google Scholar]
- Reed, S.E. The aetiology and epidemiology of common colds, and the possibilities of prevention. Clin. Otolaryngol. 1981, 6, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Parker, L.C.; Stokes, C.A.; Sabroe, I. Rhinoviral infection and asthma: The detection and management of rhinoviruses by airway epithelial cells. Clin. Exp. Allergy 2014, 44, 20–28. [Google Scholar] [CrossRef]
- Cox, D.W.; Bizzintino, J.; Ferrari, G.; Khoo, S.K.; Zhang, G.; Whelan, S.; Lee, W.M.; Bochkov, Y.A.; Geelhoed, G.C.; Goldblatt, J.; et al. Human rhinovirus species C infection in young children with acute wheeze is associated with increased acute respiratory hospital admissions. Am. J. Respir. Crit. Care Med. 2013, 188, 1358–1364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oo, S.W.C.; Khoo, S.K.; Cox, D.W.; Chidlow, G.; Franks, K.; Prastanti, F.; Bochkov, Y.A.; Borland, M.L.; Zhang, G.; Gern, J.E.; et al. Defining age-specific relationships of respiratory syncytial virus and rhinovirus species in hospitalized children with acute wheeze. Pediatric Infect. Dis. J. 2021, 40, 873–879. [Google Scholar] [CrossRef]
- Lee, W.-M.; Lemanske, R.F., Jr.; Evans, M.D.; Vang, F.; Pappas, T.; Gangnon, R.; Jackson, D.J.; Gern, J.E. Human rhinovirus species and season of infection determine illness severity. Am. J. Respir. Crit. Care Med. 2012, 186, 886–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linder, J.E.; Kraft, D.C.; Mohamed, Y.; Lu, Z.; Heil, L.; Tollefson, S.; Saville, B.R.; Wright, P.F.; Williams, J.V.; Miller, E.K. Human rhinovirus C: Age, season, and lower respiratory illness over the past 3 decades. J. Allergy Clin. Immunol. 2013, 131, 69–77. [Google Scholar] [CrossRef]
- Xiao, Q.; Zheng, S.; Zhou, L.; Ren, L.; Xie, X.; Deng, Y.; Tian, D.; Zhao, Y.; Fu, Z.; Li, T.; et al. Impact of human rhinovirus types and viral load on the severity of illness in hospitalized children with lower respiratory tract infections. Pediatric Infect. Dis. J. 2015, 34, 1187–1192. [Google Scholar] [CrossRef]
- Bochkov, Y.A.; Watters, K.; Ashraf, S.; Griggs, T.F.; Devries, M.K.; Jackson, D.J.; Palmenberg, A.C.; Gern, J.E. Cadherin-related family member 3, a childhood asthma susceptibility gene product, mediates rhinovirus C binding and replication. Proc. Natl. Acad. Sci. USA 2015, 112, 5485–5490. [Google Scholar] [CrossRef] [Green Version]
- Everman, J.L.; Sajuthi, S.; Saef, B.; Rios, C.; Stoner, A.M.; Numata, M.; Hu, D.; Eng, C.; Oh, S.; Rodriguez-Santana, J.; et al. Functional genomics of CDHR3 confirms its role in HRV-C infection and childhood asthma exacerbations. J. Allergy Clin. Immunol. 2019, 144, 962–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bønnelykke, K.; Sleiman, P.; Nielsen, K.; Kreiner-Møller, E.; Mercader, J.M.; Belgrave, D.; den Dekker, H.T.; Husby, A.; Sevelsted, A.; Faura-Tellez, G.; et al. A genome-wide association study identifies CDHR3 as a susceptibility locus for early childhood asthma with severe exacerbations. Nat. Genet. 2014, 46, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Basta, H.A.; Ashraf, S.; Sgro, J.Y.; Bochkov, Y.A.; Gern, J.E.; Palmenberg, A.C. Modeling of the human rhinovirus C capsid suggests possible causes for antiviral drug resistance. Virology 2014, 448, 82–90. [Google Scholar] [CrossRef] [Green Version]
- McErlean, P.; Shackelton, L.A.; Andrews, E.; Webster, D.R.; Lambert, S.B.; Nissen, M.D.; Sloots, T.P.; Mackay, I.M. Distinguishing molecular features and clinical characteristics of a putative new rhinovirus species, human rhinovirus C (HRV C). PLoS ONE 2008, 3, e1847. [Google Scholar] [CrossRef]
- Basta, H.A.; Sgro, J.Y.; Palmenberg, A.C. Modeling of the human rhinovirus C capsid suggests a novel topography with insights on receptor preference and immunogenicity. Virology 2014, 448, 176–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suhrbier, A.; Linn, M.L. Suppression of antiviral responses by antibody-dependent enhancement of macrophage infection. Trends Immunol. 2003, 24, 165–168. [Google Scholar] [CrossRef]
- Iwasaki, J.; Smith, W.A.; Khoo, S.K.; Bizzintino, J.; Zhang, G.; Cox, D.W.; Laing, I.A.; Le Souëf, P.N.; Thomas, W.R.; Hales, B.J. Comparison of rhinovirus antibody titers in children with asthma exacerbations and species-specific rhinovirus infection. J. Allergy Clin. Immunol. 2014, 134, 25–32. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, K.; Linnemann, R.W.; Avadhanula, V.; Mansbach, J.M.; Piedra, P.A.; Gern, J.E.; Camargo, C.A., Jr. Detection of respiratory syncytial virus and rhinovirus in healthy infants. BMC Res. Notes 2015, 8, 718. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, A.; Carraro, E.; Kamikawa, J.; Leal, E.; Granato, C.; Bellei, N. Rhinovirus species and their clinical presentation among different risk groups of non-hospitalized patients. J. Med. Virol. 2010, 82, 2110–2115. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.J.; Arnold, J.C.; Fairchok, M.P.; Danaher, P.J.; McDonough, E.A.; Blair, P.J.; Garcia, J.; Halsey, E.S.; Schofield, C.; Ottolini, M.; et al. Epidemiologic, clinical, and virologic characteristics of human rhinovirus infection among otherwise healthy children and adults: Rhinovirus among adults and children. J. Clin. Virol. Off. Publ. Pan Am. Soc. Clin. Virol. 2015, 64, 74–82. [Google Scholar] [CrossRef]
- Granados, A.; Goodall, E.C.; Luinstra, K.; Smieja, M.; Mahony, J. Comparison of asymptomatic and symptomatic rhinovirus infections in university students: Incidence, species diversity, and viral load. Diagn. Microbiol. Infect. Dis. 2015, 82, 292–296. [Google Scholar] [CrossRef]
- Martin, E.T.; Kuypers, J.; Chu, H.Y.; Foote, S.; Hashikawa, A.; Fairchok, M.P.; Englund, J.A. Heterotypic infection and spread of rhinovirus A, B, and C among childcare attendees. J. Infect. Dis. 2018, 218, 848–855. [Google Scholar] [CrossRef]
- Savolainen-Kopra, C.; Blomqvist, S.; Kaijalainen, S.; Jounio, U.; Juvonen, R.; Peitso, A.; Saukkoriipi, A.; Vainio, O.; Hovi, T.; Roivainen, M. All known human rhinovirus species are present in sputum specimens of military recruits during respiratory infection. Viruses 2009, 1, 1178–1189. [Google Scholar] [CrossRef]
- Nakagome, K.; Bochkov, Y.A.; Ashraf, S.; Brockman-Schneider, R.A.; Evans, M.D.; Pasic, T.R.; Gern, J.E. Effects of rhinovirus species on viral replication and cytokine production. J. Allergy Clin. Immunol. 2014, 134, 332–341. [Google Scholar] [CrossRef] [Green Version]
- Aoki, T.; Matsumoto, Y.; Hirata, K.; Ochiai, K.; Okada, M.; Ichikawa, K.; Shibasaki, M.; Arinami, T.; Sumazaki, R.; Noguchi, E. Expression profiling of genes related to asthma exacerbations. Clin. Exp. Allergy 2009, 39, 213–221. [Google Scholar] [CrossRef] [Green Version]
- Bosco, A.; Ehteshami, S.; Panyala, S.; Martinez, F.D. IRF7 is a major hub connecting interferon-mediated responses in virus-induced asthma exacerbations in vivo. J. Allergy Clin. Immunol. 2012, 129, 88–94. [Google Scholar] [CrossRef] [Green Version]
- Bosco, A.; Ehteshami, S.; Stern, D.A.; Martinez, F.D. Decreased activation of inflammatory networks during acute asthma exacerbations is associated with chronic airflow obstruction. Mucosal Immunol. 2010, 3, 399–409. [Google Scholar] [CrossRef] [Green Version]
- Subrata, L.S.; Bizzintino, J.; Mamessier, E.; Bosco, A.; McKenna, K.L.; Wikstrom, M.E.; Goldblatt, J.; Sly, P.D.; Hales, B.J.; Thomas, W.R.; et al. Interactions between innate antiviral and atopic immunoinflammatory pathways precipitate and sustain asthma exacerbations in children. J. Immunol. 2009, 183, 2793–2800. [Google Scholar] [CrossRef]
- Khoo, S.K.; Read, J.; Franks, K.; Zhang, G.; Bizzintino, J.; Coleman, L.; McCrae, C.; Oberg, L.; Troy, N.M.; Prastanti, F.; et al. Upper airway cell transcriptomics identify a major new immunological phenotype with strong clinical correlates in young children with acute wheezing. J. Immunol. 2019, 202, 1845–1858. [Google Scholar] [CrossRef] [Green Version]
- de la Fuente, A. From ‘differential expression’ to ‘differential networking’-identification of dysfunctional regulatory networks in diseases. Trends Genet. 2010, 26, 326–333. [Google Scholar] [CrossRef]
- Kuijjer, M.L.; Tung, M.G.; Yuan, G.; Quackenbush, J.; Glass, K. Estimating sample-specific regulatory networks. iScience 2019, 14, 226–240. [Google Scholar] [CrossRef] [Green Version]
- Chidlow, G.R.; Harnett, G.B.; Shellam, G.R.; Smith, D.W. An economical tandem multiplex real-time PCR technique for the detection of a comprehensive range of respiratory pathogens. Viruses 2009, 1, 42–56. [Google Scholar] [CrossRef] [Green Version]
- Bochkov, Y.A.; Grindle, K.; Vang, F.; Evans, M.D.; Gern, J.E. Improved molecular typing assay for rhinovirus species A, B, and C. J. Clin. Microbiol. 2014, 52, 2461–2471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, M.; Wang, P.; Boyd, A.D.; Kostov, G.; Athey, B.; Jones, E.G.; Bunney, W.E.; Myers, R.M.; Speed, T.P.; Akil, H.; et al. Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res. 2005, 33, e175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Smyth, G.K. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004, 3, 3. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Leek, J.T.; Storey, J.D. Capturing heterogeneity in gene expression studies by “surrogate variable analysis”. PLoS Genet. 2007, 3, e161. [Google Scholar] [CrossRef] [Green Version]
- Jaffe, A.E.; Hyde, T.; Kleinman, J.; Weinbergern, D.R.; Chenoweth, J.G.; McKay, R.D.; Leek, J.T.; Colantuoni, C. Practical impacts of genomic data “cleaning” on biological discovery using surrogate variable analysis. BMC Bioinform. 2015, 16, 372. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Kerns, R.T.; Peddada, S.D.; Bushel, P.R. Principal component analysis-based filtering improves detection for Affymetrix gene expression arrays. Nucleic Acids Res. 2011, 39, e86. [Google Scholar] [CrossRef] [Green Version]
- Kuijjer, M.L.; Quackenbush, J.; Glass, K. lionessR: Single-sample network reconstruction in R. bioRxiv 2019, 582098. [Google Scholar] [CrossRef]
- Freytag, S. RUVcorr: Removal of Unwanted Variation for Gene-Gene Correlations and Related Analysis. Available online: https://www.bioconductor.org/packages/release/bioc/html/RUVcorr.html (accessed on 5 March 2020).
- Jacob, L.; Gagnon-Bartsch, J.A.; Speed, T.P. Correcting gene expression data when neither the unwanted variation nor the factor of interest are observed. Biostatistics 2016, 17, 16–28. [Google Scholar] [CrossRef] [Green Version]
- Bastian, M.; Heymann, S.; Jacomy, M. Gephi: An open source software for exploring and manipulating networks. In Proceedings of the Third International AAAI Conference on Weblogs and Social Media, San Jose, CA, USA, 17–20 May 2009; Adar, E., Hurst, M., Finin, T., Glance, N., Nicolov, N., Tseng, B., Eds.; AAAI Press: Menlo Park, CA, USA, 2009. [Google Scholar]
- Breuer, K.; Foroushani, A.K.; Laird, M.R.; Chen, C.; Sribnaia, A.; Lo, R.; Winsor, G.L.; Hancock, R.E.W.; Brinkman, F.S.L.; Lynn, D.J. InnateDB: Systems biology of innate immunity and beyond—Recent updates and continuing curation. Nucleic Acids Res. 2013, 41, D1228–D1233. [Google Scholar] [CrossRef] [PubMed]
- Lê, S.; Josse, J.; Husson, F. FactoMineR: An R package for multivariate analysis. J. Stat. Softw. 2008, 25, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Therneau, T.M. A Package for Survival Analysis in R. Available online: https://CRAN.R-project.org/package=survival (accessed on 7 January 2021).
- Kassambara, A.; Kosinski, M.; Biecek, P. Survminer: Drawing Survival Curves Using ‘Ggplot2’. Available online: https://CRAN.R-project.org/package=survminer (accessed on 10 September 2021).
- Law, C.W.; Chen, Y.; Shi, W.; Smyth, G.K. Voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014, 15, R29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altman, M.C.; Gill, M.A.; Whalen, E.; Babineau, D.C.; Shao, B.; Liu, A.H.; Jepson, B.; Gruchalla, R.S.; O’Connor, G.T.; Pongracic, J.A.; et al. Transcriptome networks identify mechanisms of viral and nonviral asthma exacerbations in children. Nat. Immunol. 2019, 20, 637–651. [Google Scholar] [CrossRef]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards suite: From gene data mining to disease genome sequence analyses. Curr. Protoc. Bioinform. 2016, 54, 1–30. [Google Scholar] [CrossRef]
- Busse, W.W.; Morgan, W.J.; Gergen, P.J.; Mitchell, H.E.; Gern, J.E.; Liu, A.H.; Gruchalla, R.S.; Kattan, M.; Teach, S.J.; Pongracic, J.A.; et al. Randomized trial of omalizumab (anti-IgE) for asthma in inner-city children. N. Engl. J. Med. 2011, 364, 1005–1015. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, A.L. Network pharmacology: The next paradigm in drug discovery. Nat. Chem. Biol. 2008, 4, 682–690. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RV-Negative Controls | RV-Negative Convalescence | RV-Negative Cases | RV-A Cases | RV-C Cases | |
|---|---|---|---|---|---|
| n | 21 | 14 | 19 | 11 | 26 |
| Age in years, mean (SD) | 6.62 (4.50) | 4.18 (1.67) | 3.47 (3.26) # | 6.42 (3.30) * | 4.11 (3.09) # |
| Males, n (%) | 8 (38) | 7 (50) | 10 (53) | 6 (55) | 14 (54) |
| Overall atopy, n (%) | 10/17 (59) | 7/11 (64) | 8 (42) | 10 (91) ** | 16 (62) |
| Atopy to aeroallergens only, n (%) | 10/17 (59) | 7/11 (64) | 8 (42) | 9 (82) * | 14 (54) |
| Children with virus detected, n (%) | 8 (38) | 3 (21) | 12 (63) | 11 (100) *###††† | 26 (100) ***###††† |
| Time between first symptoms and hospital presentation in days, mean (SD) | NA | NA | 5.21 (4.22) | 5.00 (4.56) | 3.62 (2.50) |
| Children diagnosed with acute asthma, bronchiolitis or wheeze, n (%) | NA | NA | 17 (89) | 11 (100) | 26 (100) |
| Exacerbation severity Z score, mean (SD) | NA | NA | 0.56 (0.79) | 0.44 (0.76) | 0.43 (0.88) |
| Acute use of corticosteroid, n (%) | NA | NA | 11/14 (79) | 8/8 (100) | 15/17 (88) |
| Cluster A | Cluster B | Cluster C | Cluster D | |
|---|---|---|---|---|
| n | 19 | 16 | 10 | 11 |
| Age in years, mean (SD) | 4.10 (2.67) | 4.21 (2.67) | 5.42 (5.24) | 3.99 (3.24) |
| Males, n (%) | 12 (63) | 10 (62) | 3 (30) | 5 (45) |
| Overall atopy, n (%) | 14 (74) | 11 (69) | 5 (50) | 4 (36) * |
| Atopy to aeroallergens only, n (%) | 13 (68) | 11 (69) | 4 (40) | 3 (27) *† |
| Children with virus detected, n (%) | 18 (95) | 12 (75) | 8 (80) | 11 (100) |
| RV species, RV-A n (%), RV-C n (%), RV-negative n (%) | 3 (16), 9 (47), 7 (37) | 3 (19), 7 (44), 6 (37) | 3 (30), 3 (30), 4 (40) | 2 (18), 7 (64), 2 (18) |
| Time between first symptoms and hospital presentation in days, mean (SD) | 3.63 (3.30) | 5.50 (4.05) | 5.20 (4.21) | 3.55 (2.66) |
| Children diagnosed with acute asthma, bronchiolitis or wheeze, n (%) | 18 (95) | 16 (100) | 9 (90) | 11 (100) |
| Exacerbation severity Z score, mean (SD) | 0.55 (0.81) | 0.37 (0.92) | 0.57 (0.73) | 0.42 (0.84) |
| Acute use of corticosteroid, n (%) | 9/11 (82) | 9/9 (100) | 8 (80) | 8/9 (89) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coleman, L.A.; Khoo, S.-K.; Franks, K.; Prastanti, F.; Le Souëf, P.; Karpievitch, Y.V.; Laing, I.A.; Bosco, A. Personal Network Inference Unveils Heterogeneous Immune Response Patterns to Viral Infection in Children with Acute Wheezing. J. Pers. Med. 2021, 11, 1293. https://doi.org/10.3390/jpm11121293
Coleman LA, Khoo S-K, Franks K, Prastanti F, Le Souëf P, Karpievitch YV, Laing IA, Bosco A. Personal Network Inference Unveils Heterogeneous Immune Response Patterns to Viral Infection in Children with Acute Wheezing. Journal of Personalized Medicine. 2021; 11(12):1293. https://doi.org/10.3390/jpm11121293
Chicago/Turabian StyleColeman, Laura A., Siew-Kim Khoo, Kimberley Franks, Franciska Prastanti, Peter Le Souëf, Yuliya V. Karpievitch, Ingrid A. Laing, and Anthony Bosco. 2021. "Personal Network Inference Unveils Heterogeneous Immune Response Patterns to Viral Infection in Children with Acute Wheezing" Journal of Personalized Medicine 11, no. 12: 1293. https://doi.org/10.3390/jpm11121293
APA StyleColeman, L. A., Khoo, S.-K., Franks, K., Prastanti, F., Le Souëf, P., Karpievitch, Y. V., Laing, I. A., & Bosco, A. (2021). Personal Network Inference Unveils Heterogeneous Immune Response Patterns to Viral Infection in Children with Acute Wheezing. Journal of Personalized Medicine, 11(12), 1293. https://doi.org/10.3390/jpm11121293