
The Potential of Hsp90 in Targeting Pathological Pathways in Cardiac Diseases




Abstract

1. Introduction
1.1. Hsp90 as a Chaperone
1.2. Hsp90 Structure
1.3. Hsp90 Chaperone Cycle and Function
2. Hsp90 in Cardiomyopathy
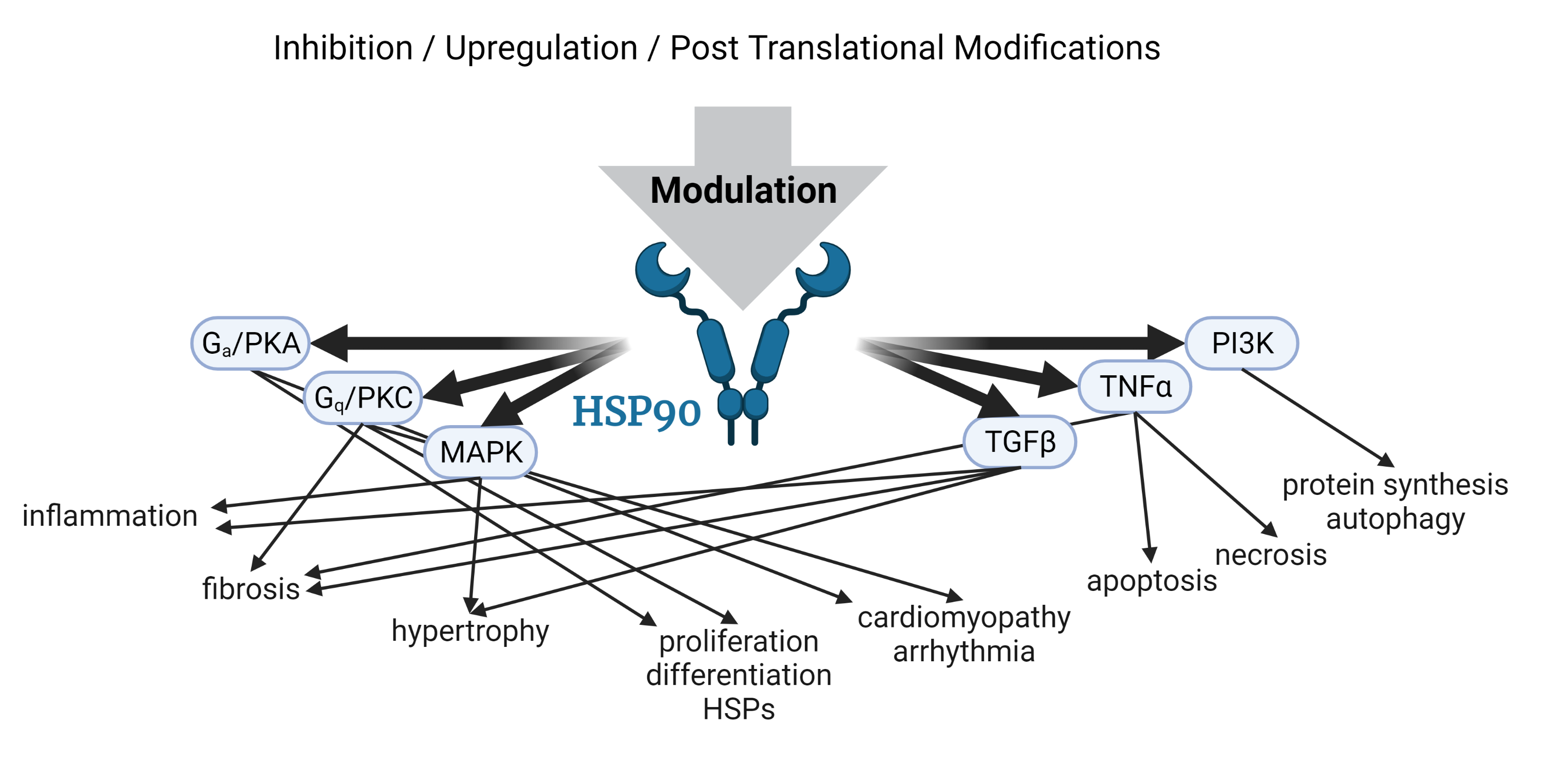
2.1. Pathways Regulated by Hsp90 in the Heart
2.1.1. TGF- Signaling
2.1.2. MAPK Signaling
2.1.3. PI3K/AKT(PKB)/mTOR Signaling
2.1.4. Gs/PKA Signaling and Calcium (Ca2+) Regulation
2.1.5. Gq/PKC Signaling
2.1.6. TNF Signaling
2.2. Pathophysiological Significance in Cardiomyopathy
3. Hsp90 Modulation
3.1. Inhibitors and Agonists
3.2. Post-Translational Modifications
4. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Richter, K.; Haslbeck, M.; Buchner, J. The Heat Shock Response: Life on the Verge of Death. Mol. Cell 2010, 40, 253–266. [Google Scholar] [CrossRef]
- Sontag, E.M.; Samant, R.S.; Frydman, J. Mechanisms and Functions of Spatial Protein Quality Control. Annu. Rev. Biochem. 2017, 86, 97–122. [Google Scholar] [CrossRef]
- Willis, M.S.; Schisler, J.C.; Portbury, A.L.; Patterson, C. Build it up–Tear it down: Protein quality control in the cardiac sarcomere. Cardiovasc. Res. 2009, 81, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.L. Evolution and function of diverse Hsp90 homologs and cochaperone proteins. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2012, 1823, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Lei, W.; Duron, D.I.; Stine, C.; Mishra, S.; Blagg, B.S.J.; Streicher, J.M. The Alpha Isoform of Heat Shock Protein 90 and the Co-chaperones p23 and Cdc37 Promote Opioid Anti-nociception in the Brain. Front. Mol. Neurosci. 2019, 12, 294. [Google Scholar] [CrossRef]
- Hoter, A.; El-Sabban, M.E.; Naim, H.Y. The HSP90 Family: Structure, Regulation, Function, and Implications in Health and Disease. Int. J. Mol. Sci. 2018, 19, 2560. [Google Scholar] [CrossRef]
- Sreedhar, A.S.; Kalmár, É.; Csermely, P.; Shen, Y.F. Hsp90 isoforms: Functions, expression and clinical importance. FEBS Lett. 2004, 562, 11–15. [Google Scholar] [CrossRef]
- Ciechanover, A.; Kwon, Y.T. Protein Quality Control by Molecular Chaperones in Neurodegeneration. Front. Neurosci. 2017, 11, 185. [Google Scholar] [CrossRef]
- Echeverría, P.C.; Bernthaler, A.; Dupuis, P.; Mayer, B.; Picard, D. An Interaction Network Predicted from Public Data as a Discovery Tool: Application to the Hsp90 Molecular Chaperone Machine. PLoS ONE 2011, 6, e26044. [Google Scholar] [CrossRef]
- Taherian, A.; Krone, P.H.; Ovsenek, N. A comparison of Hsp90α and Hsp90β interactions with cochaperones and substrates. Biochem. Cell Biol. 2008, 86, 37–45. [Google Scholar] [CrossRef]
- Wayne, N.; Bolon, D.N. Dimerization of Hsp90 Is Required for in Vivo Function. J. Biol. Chem. 2007, 282, 35386–35395. [Google Scholar] [CrossRef] [PubMed]
- Morishima, Y.; Mehta, R.K.; Yoshimura, M.; Lau, M.; Southworth, D.R.; Lawrence, T.S.; Pratt, W.B.; Nyati, M.K.; Osawa, Y. Chaperone Activity and Dimerization Properties of Hsp90α and Hsp90β in Glucocorticoid Receptor Activation by the Multiprotein Hsp90/Hsp70-Dependent Chaperone Machinery. Mol. Pharmacol. 2018, 94, 984–991. [Google Scholar] [CrossRef] [PubMed]
- Schopf, F.H.; Biebl, M.M.; Buchner, J. The HSP90 chaperone machinery. J. Biol. Chem. 2008, 18, 345–360. [Google Scholar] [CrossRef]
- Tsutsumi, S.; Mollapour, M.; Prodromou, C.; Lee, C.T.; Panaretou, B.; Yoshida, S.; Mayer, M.P.; Neckers, L.M. Charged linker sequence modulates eukaryotic heat shock protein 90 (Hsp90) chaperone activity. Proc. Natl. Acad. Sci. USA 2012, 109, 2937–2942. [Google Scholar] [CrossRef] [PubMed]
- Jahn, M.; Rehn, A.; Pelz, B.; Hellenkamp, B.; Richter, K.; Rief, M.; Buchner, J.; Hugel, T. The charged linker of the molecular chaperone Hsp90 modulates domain contacts and biological function. Proc. Natl. Acad. Sci. USA 2014, 111, 17881–17886. [Google Scholar] [CrossRef]
- López, A.; Elimelech, A.R.; Klimm, K.; Sattler, M. The Charged Linker Modulates the Conformations and Molecular Interactions of Hsp90. ChemBioChem 2021, 22, 1084–1092. [Google Scholar] [CrossRef]
- Smith, D.F. Tetratricopeptide repeat cochaperones in steroid receptor complexes. Cell Stress Chaperones 2004, 9, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Abbas-Terki, T.; Donzé, O.; Picard, D. The molecular chaperone Cdc37 is required for Ste11 function and pheromone-induced cell cycle arrest. FEBS Lett. 2000, 467, 111–116. [Google Scholar] [CrossRef]
- Lee, P.; Shabbir, A.; Cardozo, C.; Caplan, A.J. Sti1 and Cdc37 Can Stabilize Hsp90 in Chaperone Complexes with a Protein Kinase. Mol. Biol. Cell 2004, 15, 1785–1792. [Google Scholar] [CrossRef]
- Caplan, A.J.; Ma’ayan, A.; Willis, I.M. Multiple Kinases and System Robustness: A Link Between Cdc37 and Genome Integrity. Cell Cycle 2007, 6, 3145–3147. [Google Scholar] [CrossRef]
- Taipale, M.; Krykbaeva, I.; Koeva, M.; Kayatekin, C.; Westover, K.D.; Karras, G.I.; Lindquist, S. Quantitative analysis of HSP90-client interactions reveals principles of substrate recognition. Cell 2012, 150, 987–1001. [Google Scholar] [CrossRef]
- Chen, S.; Prapapanich, V.; Rimerman, R.A.; Honoré, B.; Smith, D.F. Interactions of p60, a mediator of progesterone receptor assembly, with heat shock proteins hsp90 and hsp70. Mol. Endocrinol. 1996, 10, 682–693. [Google Scholar] [CrossRef]
- Johnson, B.D.; Schumacher, R.J.; Ross, E.D.; Toft, D.O. Hop Modulates hsp70/hsp90 Interactions in Protein Folding. J. Biol. Chem. 1998, 273, 3679–3686. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, K.; Weidenauer, L.; Luengo, T.M.; Pieters, E.C.; Echeverría, P.C.; Bernasconi, L.; Wider, D.; Sadian, Y.; Koopman, M.B.; Villemin, M.; et al. The Hsp70-Hsp90 co-chaperone Hop/Stip1 shifts the proteostatic balance from folding towards degradation. Nat. Commun. 2020, 11, 5975. [Google Scholar] [CrossRef]
- Pirkl, F.; Buchner, J. Functional analysis of the hsp90-associated human peptidyl prolyl Cis/Trans isomerases FKBP51, FKBP52 and cyp4011Edited by R. Huber. J. Mol. Biol. 2001, 308, 795–806. [Google Scholar] [CrossRef]
- Oroz, J.; Blair, L.J.; Zweckstetter, M. Dynamic Aha1 co-chaperone binding to human Hsp90. Protein Sci. 2019, 28, 1545–1551. [Google Scholar] [CrossRef] [PubMed]
- Harst, A.; Lin, H.; Obermann, W.M.J. Aha1 competes with Hop, p50 and p23 for binding to the molecular chaperone Hsp90 and contributes to kinase and hormone receptor activation. Biochem. J. 2005, 387, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Mader, S.L.; Lopez, A.; Lawatscheck, J.; Luo, Q.; Rutz, D.A.; Gamiz-Hernandez, A.P.; Sattler, M.; Buchner, J.; Kaila, V.R.I. Conformational dynamics modulate the catalytic activity of the molecular chaperone Hsp90. Nat. Commun. 2020, 11, 1410. [Google Scholar] [CrossRef]
- McLaughlin, S.H.; Sobott, F.; Yao, Z.P.; Zhang, W.; Nielsen, P.R.; Grossmann, J.G.; Laue, E.D.; Robinson, C.V.; Jackson, S.E. The Co-chaperone p23 Arrests the Hsp90 ATPase Cycle to Trap Client Proteins. J. Mol. Biol. 2006, 356, 746–758. [Google Scholar] [CrossRef]
- Biebl, M.M.; Lopez, A.; Rehn, A.; Freiburger, L.; Lawatscheck, J.; Blank, B.; Sattler, M.; Buchner, J. Structural elements in the flexible tail of the co-chaperone p23 coordinate client binding and progression of the Hsp90 chaperone cycle. Nat. Commun. 2021, 12, 828. [Google Scholar] [CrossRef]
- Lee, K.; Thwin, A.C.; Nadel, C.M.; Tse, E.; Gates, S.N.; Gestwicki, J.E.; Southworth, D.R. The structure of an Hsp90-immunophilin complex reveals cochaperone recognition of the client maturation state. Mol. Cell 2021, 81, 3496–3508.e5. [Google Scholar] [CrossRef] [PubMed]
- Wexler, R.; Elton, T.; Pleister, A.; Feldman, D. Cardiomyopathy: An Overview. Am. Fam. Physician 2009, 79, 778–784. [Google Scholar]
- Maron, B.J.; Towbin, J.A.; Thiene, G.; Antzelevitch, C.; Corrado, D.; Arnett, D.; Moss, A.J.; Seidman, C.E.; Young, J.B. Contemporary Definitions and Classification of the Cardiomyopathies. Circ. Res. 2006, 113, 1807–1816. [Google Scholar] [CrossRef] [PubMed]
- Sekulic, M.; Zacharias, M.; Medalion, B. Ischemic Cardiomyopathy and Heart Failure. Circ. Res. 2019, 12, e006006. [Google Scholar] [CrossRef]
- González, A.; Schelbert, E.B.; Díez, J.; Butler, J. Myocardial Interstitial Fibrosis in Heart Failure: Biological and Translational Perspectives. J. Am. Coll. Cardiol. 2018, 71, 1696–1706. [Google Scholar] [CrossRef]
- Tham, Y.K.; Bernardo, B.C.; Ooi, J.Y.Y.; Weeks, K.L.; McMullen, J.R. Pathophysiology of cardiac hypertrophy and heart failure: Signaling pathways and novel therapeutic targets. Arch. Toxicol. 2015, 89, 1401–1438. [Google Scholar] [CrossRef]
- Heidenreich, P.A.; Albert, N.M.; Allen, L.A.; Bluemke, D.A.; Butler, J.; Fonarow, G.C.; Ikonomidis, J.S.; Khavjou, O.; Konstam, M.A.; Maddox, T.M.; et al. Forecasting the Impact of Heart Failure in the United States. Circ. Heart Fail. 2013, 6, 606–619. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.G.; Yuan, Y.P.; Wu, H.M.; Zhang, X.; Tang, Q.Z. Cardiac fibrosis: New insights into the pathogenesis. Int. J. Biol. Sci. 2018, 14, 1645–1657. [Google Scholar] [CrossRef]
- Khalil, H.; Kanisicak, O.; Prasad, V.; Correll, R.N.; Fu, X.; Schips, T.; Vagnozzi, R.J.; Liu, R.; Huynh, T.; Lee, S.J.; et al. Fibroblast-specific TGF-β-Smad2/3 signaling underlies cardiac fibrosis. J. Clin. Investig. 2017, 127, 3770–3783. [Google Scholar] [CrossRef]
- Watkins, S.J.; Borthwick, G.M.; Oakenfull, R.; Robson, A.; Arthur, H.M. Angiotensin II-induced cardiomyocyte hypertrophy in vitro is TAK1-dependent and Smad2/3-independent. Hypertens. Res. 2012, 35, 393–398. [Google Scholar] [CrossRef]
- Kong, P.; Christia, P.; Frangogiannis, N.G. The Pathogenesis of Cardiac Fibrosis. Cell Mol. Life Sci. 2014, 71, 549–574. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; An, Y.S.; Kim, M.R.; Kim, Y.A.; Lee, J.K.; Hwang, C.S.; Chung, E.; Park, I.C.; Yi, J.Y. Heat Shock Protein 90 Regulates Subcellular Localization of Smads in Mv1Lu Cells. J. Cell. Biochem. 2016, 117, 230–238. [Google Scholar] [CrossRef]
- Wrighton, K.H.; Lin, X.; Feng, X.H. Critical regulation of TGFβ signaling by Hsp90. Proc. Natl. Acad. Sci. USA 2008, 105, 9244–9249. [Google Scholar] [CrossRef]
- García, R.; Merino, D.; Gómez, J.M.; Nistal, J.F.; Hurlé, M.A.; Cortajarena, A.L.; Villar, A.V. Extracellular heat shock protein 90 binding to TGFβ receptor I participates in TGFβ-mediated collagen production in myocardial fibroblasts. Cell Signal. 2016, 28, 1563–1579. [Google Scholar] [CrossRef] [PubMed]
- Ota, A.; Zhang, J.; Ping, P.; Han, J.; Wang, Y. Specific Regulation of Non-canonical p38α Activation by Hsp90-Cdc37 Chaperone Complex in Cardiomyocyte. Circ. Res. 2010, 106, 1404–1412. [Google Scholar] [CrossRef]
- Liu, X.Y.; Seh, C.C.; Cheung, P.C. HSP90 is required for TAK1 stability but not for its activation in the pro-inflammatory signaling pathway. FEBS Lett. 2008, 582, 4023–4031. [Google Scholar] [CrossRef] [PubMed]
- Moustakas, A.; Heldin, C.H. Non-Smad TGF-β signals. J. Cell Sci. 2005, 118, 3573–3584. [Google Scholar] [CrossRef] [PubMed]
- Zarubin, T.; Han, J. Activation and signaling of the p38 MAP kinase pathway. Cell Res. 2005, 15, 11–18. [Google Scholar] [CrossRef]
- Wu, Z.; Woodring, P.J.; Bhakta, K.S.; Tamura, K.; Wen, F.; Feramisco, J.R.; Karin, M.; Wang, J.Y.J.; Puri, P.L. p38 and Extracellular Signal-Regulated Kinases Regulate the Myogenic Program at Multiple Steps. Mol. Cell Biol. 2000, 20, 3951–3964. [Google Scholar] [CrossRef] [PubMed]
- Parichatikanond, W.; Luangmonkong, T.; Mangmool, S.; Kurose, H. Therapeutic Targets for the Treatment of Cardiac Fibrosis and Cancer: Focusing on TGF-β Signaling. Front. Cardiovasc. Med. 2020, 7, 34. [Google Scholar] [CrossRef] [PubMed]
- ZHANG, W.; LIU, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef] [PubMed]
- ROSE, B.A.; FORCE, T.; WANG, Y. Mitogen-Activated Protein Kinase Signaling in the Heart: Angels Versus Demons in a Heart-Breaking Tale. Physiol. Rev. 2010, 90, 1507–1546. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Steelman, L.S.; Abrams, S.L.; Lee, J.T.; Chang, F.; Bertrand, F.E.; Navolanic, P.M.; Terrian, D.M.; Franklin, R.A.; D’Assoro, A.B.; et al. Roles of the RAF/MEK/ERK and PI3K/PTEN/AKT pathways in malignant transformation and drug resistance. Adv. Enzyme Regul. 2006, 46, 249–279. [Google Scholar] [CrossRef]
- Moulick, K.; Ahn, J.H.; Zong, H.; Rodina, A.; Cerchietti, L.; Gomes DaGama, E.M.; Caldas-Lopes, E.; Beebe, K.; Perna, F.; Hatzi, K.; et al. Affinity-based proteomics reveal cancer-specific networks coordinated by Hsp90. Nat. Chem. Biol. 2011, 7, 818–826. [Google Scholar] [CrossRef] [PubMed]
- Stancato, L.F.; Silverstein, A.M.; Owens-Grillo, J.K.; Chow, Y.H.; Jove, R.; Pratt, W.B. The hsp90-binding Antibiotic Geldanamycin Decreases Raf Levels and Epidermal Growth Factor Signaling without Disrupting Formation of Signaling Complexes or Reducing the Specific Enzymatic Activity of Raf Kinase. J. Biol. Chem. 1997, 272, 4013–4020. [Google Scholar] [CrossRef] [PubMed]
- Nishimoto, S.; Nishida, E. MAPK signalling: ERK5 versus ERK1/2. J. Biol. Chem. 2006, 7, 782–786. [Google Scholar] [CrossRef]
- Gallo, S.; Vitacolonna, A.; Bonzano, A.; Comoglio, P.; Crepaldi, T. ERK: A Key Player in the Pathophysiology of Cardiac Hypertrophy. J. Mol. Sci. 2019, 20, 2164. [Google Scholar] [CrossRef] [PubMed]
- Erazo, T.; Moreno, A.; Ruiz-Babot, G.; Rodríguez-Asiain, A.; Morrice, N.A.; Espadamala, J.; Bayascas, J.R.; Gómez, N.; Lizcano, J.M. Canonical and Kinase Activity-Independent Mechanisms for Extracellular Signal-Regulated Kinase 5 (ERK5) Nuclear Translocation Require Dissociation of Hsp90 from the ERK5-Cdc37 Complex. Mol. Cell. Biol. 2013, 33, 1671–1686. [Google Scholar] [CrossRef]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K pathway in human disease. J. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef]
- Nitulescu, G.M.; Van De Venter, M.; Nitulescu, G.; Ungurianu, A.; Juzenas, P.; Peng, Q.; Olaru, O.T.; Grădinaru, D.; Tsatsakis, A.; Tsoukalas, D.; et al. The Akt pathway in oncology therapy and beyond (Review). Int. J. Oncol. 2018, 53, 2319–2331. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Ohji, G.; Hidayat, S.; Nakashima, A.; Tokunaga, C.; Oshiro, N.; Yoshino, K.I.; Yokono, K.; Kikkawa, U.; Yonezawa, K. Suppression of the mTOR-raptor signaling pathway by the inhibitor of heat shock protein 90 geldanamycin. J. Biochem. 2006, 139, 129–135. [Google Scholar] [CrossRef]
- Giulino-Roth, L.; van Besien, H.J.; Dalton, T.; Totonchy, J.E.; Rodina, A.; Taldone, T.; Bolaender, A.; Erdjument-Bromage, H.; Sadek, J.; Chadburn, A.; et al. Inhibition of Hsp90 suppresses PI3K/AKT/mTOR signaling and has antitumor activity in Burkitt lymphoma. Mol. Cancer Ther. 2017, 16, 1779–1790. [Google Scholar] [CrossRef]
- Sato, S.; Fujita, N.; Tsuruo, T. Modulation of Akt kinase activity by binding to Hsp90. Proc. Natl. Acad. Sci. USA 2000, 97, 10832–10837. [Google Scholar] [CrossRef]
- Basso, A.D.; Solit, D.B.; Chiosis, G.; Giri, B.; Tsichlis, P.; Rosen, N. Akt forms an intracellular complex with heat shock protein 90 (Hsp90) and Cdc37 and is destabilized by inhibitors of Hsp90 function. J. Biol. Chem. 2002, 277, 39858–39866. [Google Scholar] [CrossRef]
- Zhang, X.H.; Wu, J.X.; Sha, J.Z.; Yang, B.; Sun, J.R.; Bao, E.D. Heat shock protein 90 relieves heat stress damage of myocardial cells by regulating Akt and PKM2 signaling in vivo. Int. J. Mol. Med. 2020, 45, 1888–1908. [Google Scholar] [CrossRef]
- Aoyagi, T.; Matsui, T. Phosphoinositide-3 kinase signaling in cardiac hypertrophy and heart failure. Curr. Pharm. Des. 2011, 17, 1818–1824. [Google Scholar] [CrossRef]
- Nagoshi, T.; Matsui, T.; Aoyama, T.; Leri, A.; Anversa, P.; Li, L.; Ogawa, W.; del Monte, F.; Gwathmey, J.K.; Grazette, L.; et al. PI3K rescues the detrimental effects of chronic Akt activation in the heart during ischemia/reperfusion injury. J. Clin. Investig. 2005, 115, 2128–2138. [Google Scholar] [CrossRef] [PubMed]
- Suwanabol, P.A.; Seedial, S.M.; Zhang, F.; Shi, X.; Si, Y.; Liu, B.; Kent, K.C. TGF-β and Smad3 modulate PI3K/Akt signaling pathway in vascular smooth muscle cells. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H2211–H2219. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Kusakari, Y.; Xiao, C.Y.; Kinsella, S.D.; Rosenberg, M.A.; Scherrer-Crosbie, M.; Hara, K.; Rosenzweig, A.; Matsui, T. mTOR attenuates the inflammatory response in cardiomyocytes and prevents cardiac dysfunction in pathological hypertrophy. Am. J. Physiol. Cell Physiol. 2010, 299, C1256–C1266. [Google Scholar] [CrossRef] [PubMed]
- Ali, D.C.; Naveed, M.; Gordon, A.; Majeed, F.; Saeed, M.; Ogbuke, M.I.; Atif, M.; Zubair, H.M.; Changxing, L. β-Adrenergic receptor, an essential target in cardiovascular diseases. Heart Fail. Rev. 2020, 25, 343–354. [Google Scholar] [CrossRef]
- Kano, H.; Toyama, Y.; Imai, S.; Iwahashi, Y.; Mase, Y.; Yokogawa, M.; Osawa, M.; Shimada, I. Structural mechanism underlying G protein family-specific regulation of G protein-gated inwardly rectifying potassium channel. Nat. Commun. 2019, 10, 2008. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, R.; Wu, Y.; McNabb, M.; Greaser, M.; Labeit, S.; Granzier, H. Protein Kinase A Phosphorylates Titin’s Cardiac-Specific N2B Domain and Reduces Passive Tension in Rat Cardiac Myocytes. Circ. Res. 2002, 90, 1181–1188. [Google Scholar] [CrossRef]
- Hanft, L.M.; Cornell, T.D.; McDonald, C.A.; Rovetto, M.J.; Emter, C.A.; McDonald, K.S. Molecule specific effects of PKA-mediated phosphorylation on rat isolated heart and cardiac myofibrillar function. Arch. Biochem. Biophys. 2016, 601, 22–31. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ponnam, S.; Sevrieva, I.; Sun, Y.B.; Irving, M.; Kampourakis, T. Site-specific phosphorylation of myosin binding protein-C coordinates thin and thick filament activation in cardiac muscle. Proc. Natl. Acad. Sci. USA. 2019, 116, 15485–15494. [Google Scholar] [CrossRef]
- Dobrev, D.; Wehrens, X.H. Role of RyR2 Phosphorylation in Heart Failure and Arrhythmias. Circ. Res. 2014, 114, 1311–1319. [Google Scholar] [CrossRef] [PubMed]
- Mattiazzi, A.; Kranias, E.G. The role of CaMKII regulation of phospholamban activity in heart disease. Front Pharmacol. 2014, 5, 5. [Google Scholar] [CrossRef]
- Lam, C.K.; Zhao, W.; Cai, W.; Vafiadaki, E.; Florea, S.M.; Ren, X.; Liu, Y.; Robbins, N.; Zhang, Z.; Zhou, X.; et al. Novel Role of HAX-1 in Ischemic Injury Protection Involvement of Heat Shock Protein 90. Circ. Res. 2013, 112, 79–89, Publisher: American Heart Association. [Google Scholar] [CrossRef]
- Bidwell, P.A.; Liu, G.S.; Nagarajan, N.; Lam, C.K.; Haghighi, K.; Gardner, G.; Cai, W.F.; Zhao, W.; Mugge, L.; Vafiadaki, E.; et al. HAX-1 regulates SERCA2a oxidation and degradation. J. Mol. Cell Cardiol. 2018, 114, 220–233. [Google Scholar] [CrossRef]
- Zhao, W.; Waggoner, J.R.; Zhang, Z.G.; Lam, C.K.; Han, P.; Qian, J.; Schroder, P.M.; Mitton, B.; Kontrogianni-Konstantopoulos, A.; Robia, S.L.; et al. The anti-apoptotic protein HAX-1 is a regulator of cardiac function. Proc. Natl. Acad. Sci. USA 2009, 106, 20776–20781. [Google Scholar] [CrossRef]
- Camors, E.; Valdivia, H.H. CaMKII regulation of cardiac ryanodine receptors and inositol triphosphate receptors. Front Pharmacol. 2014, 5, 101. [Google Scholar] [CrossRef]
- Dewenter, M.; Neef, S.; Vettel, C.; Lämmle, S.; Beushausen, C.; Zelarayan, L.C.; Katz, S.; von der Lieth, A.; Meyer-Roxlau, S.; Weber, S.; et al. Calcium/Calmodulin-Dependent Protein Kinase II Activity Persists During Chronic β-Adrenoceptor Blockade in Experimental and Human Heart Failure. Circ. Heart Fail. 2017, 10, e003840. [Google Scholar] [CrossRef] [PubMed]
- Creamer, T.P. Calcineurin. Cell Commun. Signal. 2020, 18, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Minami, Y.; Kawasaki, H.; Suzuki, K.; Yahara, I. The calmodulin-binding domain of the mouse 90-kDa heat shock protein. J. Biol. Chem. 1993, 268, 9604–9610. [Google Scholar] [CrossRef]
- Liu, Z.; Li, H.; He, L.; Xiang, Y.; Tian, C.; Li, C.; Tan, P.; Jing, J.; Tian, Y.; Du, L.; et al. Discovery of Small-Molecule Inhibitors of the HSP90-Calcineurin-NFAT Pathway against Glioblastoma. Cell Chem. Biol. 2019, 26, 352–365.e7. [Google Scholar] [CrossRef]
- Parra, V.; Rothermel, B.A. Calcineurin signaling in the heart: The importance of time and place. J. Mol. Cell Cardiol. 2017, 103, 121–136. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, B.J.; Dai, Y.S.; Bueno, O.F.; Parsons, S.A.; Xu, J.; Plank, D.M.; Jones, F.; Kimball, T.R.; Molkentin, J.D. Calcineurin/NFAT Coupling Participates in Pathological, but not Physiological, Cardiac Hypertrophy. Circ. Res. 2004, 94, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Molkentin, J.D. Calcineurin—NFAT signaling regulates the cardiac hypertrophic response in coordination with the MAPKs. Cardiovasc. Res. 2004, 63, 467–475. [Google Scholar] [CrossRef]
- Salazar, N.C.; Chen, J.; Rockman, H.A. Cardiac GPCRs: GPCR signaling in healthy and failing hearts. Biochim. Biophys. Acta 2007, 1768, 1006–1018. [Google Scholar] [CrossRef]
- Luo, J.; Benovic, J.L. G Protein-coupled Receptor Kinase Interaction with Hsp90 Mediates Kinase Maturation. J. Biol. Chem. 2003, 278, 50908–50914. [Google Scholar] [CrossRef]
- Dzimiri, N.; Al-Bahnasi, K.; Al-Halees, Z. Myocardial hypertrophy is not a prerequisite for changes in early gene expression in left ventricular volume overload. Fundam. Clin. Pharmacol. 2004, 18, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Sato, P.Y.; Chaprun, J.K.; Peroutka, R.J.; Otis, N.J.; Ibetti, J.; Pan, S.; Sheu, S.; Goa, E.; Koch, W.J. Prodeath signaling of G protein-coupled receptor kinase 2 in cardiac myocytes after ischemic stress occurs via extracellular signla-regulated kinase-dependent heat shock protein 90-mediated mitochondrial targeting. Circ. Res. 2013, 112, 1121–1134. [Google Scholar] [CrossRef]
- Salim, S.; Eikenburg, D.C. Role of 90-kDa Heat Shock Protein (Hsp 90) and Protein Degradation in Regulating Neuronal Levels of G Protein-Coupled Receptor Kinase 3. J. Pharmacol. Exp. Ther. 2006, 320, 1106–1112. [Google Scholar] [CrossRef]
- Guo, D.F.; Sun, Y.L.; Hamet, P.; Inagami, T. The angiotensin II type 1 receptor and receptor-associated proteins. Cell Res. 2001, 11, 165–180. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, S.F. Cardiac Actions of Protein Kinase C Isoforms. Physiology 2012, 27, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Braz, J.C.; Gregory, K.; Pathak, A.; Zhao, W.; Sahin, B.; Klevitsky, R.; Kimball, T.F.; Lorenz, J.N.; Nairn, A.C.; Liggett, S.B.; et al. PKC-alpha regulates cardiac contractility and propensity toward heart failure. Nat. Med. 2004, 10, 248–254. [Google Scholar] [CrossRef]
- Kranias, E.G.; Hajjar, R.J. Modulation of Cardiac Contractility by the Phopholamban/SERCA2a Regulatome. Circ. Res. 2012, 110, 1646–1660. [Google Scholar] [CrossRef]
- Braz, J.C.; Bueno, O.F.; Windt, L.J.D.; Molkentin, J.D. PKCα regulates the hypertrophic growth of cardiomyocytes through extracellular signal–regulated kinase1/2 (ERK1/2). J. Cell Biol. 2002, 156, 905–919. [Google Scholar] [CrossRef]
- Min, W.; Bin, Z.W.; Quan, Z.B.; Hui, Z.J.; Sheng, F.G. The signal transduction pathway of PKC/NF-κB/c-fos may be involved in the influence of high glucose on the cardiomyocytes of neonatal rats. Cardiovasc. Diabetol. 2009, 8, 8. [Google Scholar] [CrossRef]
- Lee, K.H.; Jang, Y.; Chung, J.H. Heat shock protein 90 regulates IκB kinase complex and NF-κB activation in angiotensin II-induced cardiac cell hypertrophy. Exp. Mol. Med. 2010, 42, 703. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.Y.; Zhang, Y.; Matkovich, S.J.; Diwan, A.; Chishti, A.H.; Dorn, G.W. Receptor-Independent Cardiac Protein Kinase Cα Activation by Calpain-Mediated Truncation of Regulatory Domains. Circ. Res. 2010, 107, 903–912. [Google Scholar] [CrossRef]
- Tian, M.; Yuan, Y.C.; Li, J.Y.; Gionfriddo, M.R.; Huang, R.C. Tumor necrosis factor-α and its role as a mediator in myocardial infarction: A brief review. Chronic Dis. Transl. Med. 2015, 1, 18–26. [Google Scholar] [CrossRef]
- Changes in Concentrations of Tumor Necrosis Factor TNF and Its Soluble Receptors Type 1 (sTNF-r1) and Type 2 (sTNF-R2) in Serum of Patients with ST-Segment Elevation Myocardial Infarction. Wiadomosci Lekarskie. 2011. Available online: https://pubmed.ncbi.nlm.nih.gov/22026268/ (accessed on 31 October 2021).
- Monden, Y.; Kubota, T.; Inoue, T.; Tsutsumi, T.; Kawano, S.; Ide, T.; Tsutsui, H.; Sunagawa, K. Tumor necrosis factor-α is toxic via receptor 1 and protective via receptor 2 in a murine model of myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H743–H753. [Google Scholar] [CrossRef]
- Bouwmeester, T.; Bauch, A.; Ruffner, H.; Angrand, P.O.; Bergamini, G.; Croughton, K.; Cruciat, C.; Eberhard, D.; Gagneur, J.; Ghidelli, S.; et al. A physical and functional map of the human TNF-α/NF-κB signal transduction pathway. Nat. Cell Biol. 2004, 6, 97–105. [Google Scholar] [CrossRef]
- Gupta, S.; Lee, C.M.; Wang, J.F.; Parodo, J.; Jia, S.H.; Hu, J.; Marshall, J.C. Heat-shock protein-90 prolongs septic neutrophil survival by protecting c-Src kinase and caspase-8 from proteasomal degradation. J. Leukoc. Biol. 2018, 103, 933–944. [Google Scholar] [CrossRef]
- Zhao, X.M.; Chen, Z.; Zhao, J.B.; Zhang, P.P.; Pu, Y.F.; Jiang, S.H.; Hou, J.J.; Cui, Y.M.; Jia, X.L.; Zhang, S.Q. Hsp90 modulates the stability of MLKL and is required for TNF-induced necroptosis. Cell Death Dis. 2016, 7, e2089. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Tong, A.; Zhang, Q.; Wei, Y.; Wei, X. The molecular mechanisms of MLKL-dependent and MLKL-independent necrosis. J. Mol. Cell Biol. 2021, 13, 3–14. [Google Scholar] [CrossRef]
- Zhu, P.; Hu, S.; Jin, Q.; Li, D.; Tian, F.; Toan, S.; Li, Y.; Zhou, H.; Chen, Y. Ripk3 promotes ER stress-induced necroptosis in cardiac IR injury: A mechanism involving calcium overload/XO/ROS/mPTP pathway. Redox Biol. 2018, 16, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Berghe, T.V.; Kalai, M.; van Loo, G.; Declercq, W.; Vandenabeele, P. Disruption of HSP90 Function Reverts Tumor Necrosis Factor-induced Necrosis to Apoptosis. J. Biol. Chem. 2003, 278, 5622–5629. [Google Scholar] [CrossRef]
- Wajant, H.; Siegmund, D. TNFR1 and TNFR2 in the Control of the Life and Death Balance of Macrophages. Front. Cell Dev. Biol. 2019, 7, 91. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Cao, P.; Goeddel, D.V. TNF-Induced Recruitment and Activation of the IKK Complex Require Cdc37 and Hsp90. Mol. Cell 2002, 9, 401–410. [Google Scholar] [CrossRef]
- Lewis, J.; Devin, A.; Miller, A.; Lin, Y.; Rodriguez, Y.; Neckers, L.; gang Liu, Z. Disruption of Hsp90 Function Results in Degradation of the Death Domain Kinase, Receptor-interacting Protein (RIP), and Blockage of Tumor Necrosis Factor-induced Nuclear Factor-κB Activation. J. Biol. Chem. 2000, 275, 10519–10526. [Google Scholar] [CrossRef]
- Cáceres, R.A.; Chavez, T.; Maestro, D.; Palanca, A.R.; Bolado, P.; Madrazo, F.; Aires, A.; Cortajarena, A.L.; Villar, A.V. Reduction of cardiac TGFβ-mediated profibrotic events by inhibition of Hsp90 with engineered protein. J. Mol. Cell Cardiol. 2018, 123, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Tomcik, M.; Zerr, P.; Pitkowski, J.; Palumbo-Zerr, K.; Avouac, J.; Distler, O.; Becvar, R.; Senolt, L.; Schett, G.; Distler, J.H. Heat shock protein 90 (Hsp90) inhibition targets canonical TGF-β signalling to prevent fibrosis. Ann. Rheum. Dis. 2014, 73, 1215–1222. [Google Scholar] [CrossRef]
- Tamura, S.; Marunouchi, T.; Tanonaka, K. Heat-shock protein 90 modulates cardiac ventricular hypertrophy via activation of MAPK pathway. J. Mol. Cell Cardiol. 2019, 127, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Marunouchi, T.; Nakashima, M.; Ebitani, S.; Umezu, S.; Karasawa, K.; Yano, E.; Tanonaka, K. Hsp90 inhibitor attenuates the development of pathophysiological cardiac fibrosis in mouse hypertrophy via suppression of the calcineurin-NFAT and c-RAF-Erk pathways. J. Cardiovasc. Pharmacol. 2021, 77, 822–829. [Google Scholar] [CrossRef]
- Wang, W.; Peng, Y.; Wang, Y.; Zhao, X.; Yuan, Z. Anti-Apoptotic Effect of Heat Shock Protein 90 on Hypoxia-Mediated Cardiomyocyte Damage Is Mediated Via the Phosphatidylinositol 3-Kinase/Akt Pathway. Clin. Exp. Pharmacol. Physiol. 2009, 36, 899–903. [Google Scholar] [CrossRef] [PubMed]
- Averna, M.; De Tullio, R.; Pedrazzi, M.; Bavestrello, M.; Pellegrini, M.; Salamino, F.; Pontremoli, S.; Melloni, E. Interaction between calpain-1 and HSP90: New insights into the regulation of localization and activity of the protease. PLoS ONE 2015, 10, e0116738. [Google Scholar] [CrossRef]
- Lam, C.K.; Zhao, W.; Liu, G.S.; Cai, W.F.; Gardner, G.; Adly, G.; Kranias, E.G. HAX-1 regulates cyclophilin-D levels and mitochondria permeability transition pore in the heart. Proc. Natl. Acad. Sci. USA 2015, 112, E6466–E64675. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.X.; Xie, F.; Jia, C.C.; Yan, N.; Zeng, Y.L.; Wu, J.D.; Liu, Z.P. Synthesis and biological evaluation of selective histone deacetylase 6 inhibitors as multifunctional agents against Alzheimer’s disease. Eur. J. Med. Chem. 2021, 225, 113821. [Google Scholar] [CrossRef]
- Chatterjee, K.; Zhang, J.; Honbo, N.; Karliner, J.S. Doxorubicin Cardiomyopathy. Cardiology 2010, 115, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Lancet, J.E.; Gojo, I.; Burton, M.; Quinn, M.; Tighe, S.M.; Kersey, K.; Zhong, Z.; Albitar, M.X.; Bhalla, K.; Hannah, A.L.; et al. Phase I study of the heat shock protein 90 inhibitor alvespimycin (KOS-1022, 17-DMAG) administered intravenously twice weekly to patients with acute myeloid leukemia. Leukemia 2010, 24, 699–705. [Google Scholar] [CrossRef]
- Walker, A.R.; Klisovic, R.; Johnston, J.S.; Jiang, Y.; Geyer, S.; Kefauver, C.; Binkley, P.; Byrd, J.C.; Grever, M.R.; Garzon, R.; et al. Pharmacokinetics and dose escalation of the heat shock protein inhibitor 17-allyamino-17-demethoxygeldanamycin in combination with bortezomib in relapsed or refractory acute myeloid leukemia. Leuk. Lymphoma 2013, 54, 1996–2002. [Google Scholar] [CrossRef]
- Ficker, E.; Dennis, A.T.; Wang, L.; Brown, A.M. Role of the cytosolic chaperones Hsp70 and Hsp90 in maturation of the cardiac potassium channel HERG. Circ. Res. 2003, 92, e87–e100. [Google Scholar] [CrossRef]
- Metibemu, D.S. 3D-QSAR and Molecular Docking Approaches for the Identification of Phyto-Inhibitors of Hsp90. LIANBS 2022, 11, 3871–3886. [Google Scholar] [CrossRef]
- Xiao, Y.; Liu, Y. Recent Advances in the Discovery of Novel HSP90 Inhibitors: An Update from 2014. Curr. Drug Targets 2020, 21, 302–317. [Google Scholar] [CrossRef]
- Willis, M.S.; Patterson, C. Proteotoxicity and cardiac dysrunction–Alzheimer’s disease of the heart? N. Engl. J. Med. 2013, 368, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Chen, N.N.; You, Q.D.; Xu, X.L. An updated patent review of anticancer Hsp90 inhibitors (2013-present). Expert Opin. Ther. Patents 2021, 31, 67–80. [Google Scholar] [CrossRef]
- Kurop, M.K.; Huyen, C.M.; Kelly, J.H.; Blagg, B.S.J. The heat shock response and small molecule regulators. Eur. J. Med. Chem. 2021, 226, 113846. [Google Scholar] [CrossRef] [PubMed]
- Kliger, Y.; Levy, O.; Oren, A.; Ashkenazy, H.; Tiran, Z.; Novik, A.; Rosenberg, A.; Amir, A.; Wool, A.; Toporik, A.; et al. Peptides modulating conformational changes in secreted chaperones: From in silico design to preclinical proof of concept. Proc. Natl. Acad. Sci. USA 2009, 106, 13797–13801. [Google Scholar] [CrossRef]
- Chatterjee, B.K.; Jayaraj, A.; Kumar, V.; Blagg, B.; Davis, R.E.; Jayaram, B.; Deep, S.; Chaudhuri, T.K. Stimulation of heat shock protein 90 chaperone function through binding of a novobiocin analog KU-32. J. Biol. Chem. 2019, 294, 6450–6467. [Google Scholar] [CrossRef]
- Ma, J.; Pan, P.; Anyika, M.; Blagg, B.S.J.; Dobrowsky, R.T. Modulating Molecular Chaperones Improves Mitochondrial Bioenergetics and Decreases the Inflammatory Transcriptome in Diabetic Sensory Neurons. ACS Chem. Neurosci. 2015, 6, 1637–1648. [Google Scholar] [CrossRef]
- West, J.D.; Wang, Y.; Morano, K.A. Small Molecule Activators of the Heat Shock Response: Chemical Properties, Molecular Targets, and Therapeutic Promise. Chem. Res. Toxicol. 2012, 25, 2036–2053. [Google Scholar] [CrossRef]
- Zhang, L.; Zhao, H.; Blagg, B.S.J.; Dobrowsky, R.T. C-Terminal Heat Shock Protein 90 Inhibitor Decreases Hyperglycemia-induced Oxidative Stress and Improves Mitochondrial Bioenergetics in Sensory Neurons. J. Proteome Res. 2012, 11, 2581–2593. [Google Scholar] [CrossRef]
- Ramazi, S.; Zahiri, J. Post-translational modifications in proteins: Resources, tools and prediction methods. Database 2021, 2021, baab012. [Google Scholar] [CrossRef] [PubMed]
- Backe, S.J.; Sager, R.A.; Woodford, M.R.; Makedon, A.M.; Mollapour, M. Post-translational modifications of Hsp90 and translating the chaperone code. J. Biol. Chem. 2020, 295, 11099–11117. [Google Scholar] [CrossRef]
- Mollapour, M.; Neckers, L. Post-translational modifications of Hsp90 and their contributions to chaperone regulation. Biochim. Biophys. Acta 2012, 1823, 648–655. [Google Scholar] [CrossRef] [PubMed]
- Voelkel, T.; Andresen, C.; Unger, A.; Just, S.; Rottbauer, W.; Linke, W.A. Lysine methyltransferase Smyd2 regulates Hsp90-mediated protection of the sarcomeric titin springs and cardiac function. Biochim. Biophys. Acta 2013, 1833, 812–822. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, Y.; Miao, Q.; Shi, Z.; Hu, L.; Liu, S.; Gao, J.; Zhao, S.; Chen, H.; Huang, Z.; et al. Inhibition of HSP90 S-nitrosylation alleviates cardiac fibrosis via TGFβ/SMAD3 signalling pathway. Br. J. Pharmacol. 2021, 178, 4608–4625. [Google Scholar] [CrossRef] [PubMed]
- Dagar, M.; Singh, J.P.; Dagar, G.; Tyagi, R.K.; Bagchi, G. Phosphorylation of HSP90 by protein kinase A is essential for the nuclear translocation of androgen receptor. J. Mol. Biol. 2019, 294, 8699–8710. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.T.; Knieß, R.A.; Daturpalli, S.; Breton, L.L.; Ke, X.; Chen, X.; Mayer, M.P. Isoform-Specific Phosphorylation in Human Hsp90β Affects Interaction with Clients and the Cochaperone Cdc37. J. Mol. Biol. 2017, 429, 732–752. [Google Scholar] [CrossRef] [PubMed]
- Scroggins, B.T.; Robzyk, K.; Wang, D.; Marcu, M.G.; Tsutsumi, S.; Beebe, K.; Cotter, R.J.; Felts, S.; Toft, D.; Karnitz, L.; et al. An Acetylation Site in the Middle Domain of Hsp90 Regulates Chaperone Function. Mol. Cell 2007, 25, 151–159. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Pathway | Hsp90 Effect on Pathway | Cardiac Phenotype | Reference |
|---|---|---|---|
| TGF- | |||
| Protect TGF- receptor from degradation allowing fibrosis signaling. | myocardial fibrosis | [44,45] | |
| Inhibition prevents TGF- fibrotic signaling. | myocardial fibrosis | [115,116] | |
| MAPK | |||
| Stablize TAK1 and p38 to allow hypertrophic signaling. | hypertrophic responses and apoptosis | [46,47] | |
| Inhibition protects rats from cardiac hypertrophy and failure by inhibiting MAPK signaling. | hypertrophic response | [117] | |
| Inhibition prevents c-Raf-Erk induced fibrosis. | hypertrophic response and myocardial fibrosis | [118] | |
| PI3K/AKT/mTOR | |||
| Hsp90 overexpression in cardiomyocytes preserves mitochondrial function via AKT and PKM2 signaling in vitro. | apoptosis | [67] | |
| Inhibition increases apoptosis in cardiomyocytes under hypoxia conditions. | apoptosis | [119] | |
| Gs/PKA | |||
| Mediates PLN, SERCA2, and HAX1 interactions to affect Ca2+ signaling and contractility. | cardiac contractility | [80] | |
| Inhibition prevents calcineurin-NFAT-induced fibrosis. | hypertrophic response and myocardial fibrosis | [119] | |
| Gq/PKC | |||
| Inhibition causes reduced angiotensin II-induced hypertrophy and NF-kB signaling. | hypertrophic response | [101] | |
| Stabilizes caplain which can cleave PKC to PKM. PKM can cause dilated cardiomyopathy. | dilated cardiomyopathy | [102,120] | |
| TNF | |||
| Stabilizes cyclophilin D to regulate cell death via mPTP. | necrosis | [80,121] | |
| Required for TNF induced NF-kB signaling. | hypertrophic response | [101,113] | |
| HAX-1 binds to Hsp90 to mediate cardioprotection. | necrosis and apoptosis | [79,121] | |
| Source | Enzyme | PTM | Hsp90 site | Effects | Pathology |
|---|---|---|---|---|---|
| [140] | Smyd2 | Methylation | K616 | methyl-hsp90 needed in proper sarcomere function | arrhythmia and myocardial stiffening |
| [141] | Nitrosylation | C589 | sno-hsp90 stablizes TGF- receptor and promotes fibrosis | DCM, HCM | |
| [142] | PKA | Phosphorylation | T89 | reduces Hsp90-androgen receptor interaction | N/A |
| [143] | CK2 | Phosphorylation | T36 | inhibition of cdc37-hsp90 complex | N/A |
| [139] | SUMOylation | K191 | recruits Aha1 | N/A | |
| [144] | Acetylation | K294 | reduced client and cochaperone interactions | N/A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roberts, R.J.; Hallee, L.; Lam, C.K. The Potential of Hsp90 in Targeting Pathological Pathways in Cardiac Diseases. J. Pers. Med. 2021, 11, 1373. https://doi.org/10.3390/jpm11121373
Roberts RJ, Hallee L, Lam CK. The Potential of Hsp90 in Targeting Pathological Pathways in Cardiac Diseases. Journal of Personalized Medicine. 2021; 11(12):1373. https://doi.org/10.3390/jpm11121373
Chicago/Turabian StyleRoberts, Richard J., Logan Hallee, and Chi Keung Lam. 2021. "The Potential of Hsp90 in Targeting Pathological Pathways in Cardiac Diseases" Journal of Personalized Medicine 11, no. 12: 1373. https://doi.org/10.3390/jpm11121373
APA StyleRoberts, R. J., Hallee, L., & Lam, C. K. (2021). The Potential of Hsp90 in Targeting Pathological Pathways in Cardiac Diseases. Journal of Personalized Medicine, 11(12), 1373. https://doi.org/10.3390/jpm11121373