
Towards Personalized Therapy of Aortic Stenosis

 , ,
, ,
Abstract
:1. Introduction
2. Pathophysiology of Aortic Valve Calcification
2.1. Lipid-Lowering Therapy
2.2. Anti-Hypertensive Therapies
2.3. CAS Patients with Kidney Disorders
2.4. Anti-Diabetic Therapies
2.5. Therapies Targeting Calcification
2.6. Inflammation Related Targets
2.7. Coagulation Activation Related Targets
2.8. Inhibition of Other Calcification-Related Molecular Pathways
3. Future Perspectives
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lindman, B.R.; Clavel, M.A.; Mathieu, P.; Iung, B.; Lancellotti, P.; Otto, C.M.; Pibarot, P. Calcific aortic stenosis. Nat. Rev. Dis. Primers 2016, 2, 16006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vahanian, A.; Beyersdorf, F.; Praz, F.; Milojevic, M.; Baldus, S.; Bauersachs, J.; Capodanno, D.; Conradi, L.; De Bonis, M.; De Paulis, R.; et al. 2021 ESC/EACTS Guidelines for the management of valvular heart disease. Eur. Heart J. 2021, 60, 727–800. [Google Scholar] [CrossRef]
- Otto, C.M.; Nishimura, R.A.; Bonow, R.O.; Carabello, B.A.; Erwin, J.P., 3rd; Gentile, F.; Jneid, H.; Krieger, E.V.; Mack, M.; McLeod, C.; et al. 2020 ACC/AHA Guideline for the Management of Patients With Valvular Heart Disease: Executive Summary: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation 2021, 143, e35–e71. [Google Scholar] [CrossRef]
- Yutzey, K.E.; Demer, L.L.; Body, S.C.; Huggins, G.S.; Towler, D.A.; Giachelli, C.M.; Hofmann-Bowman, M.A.; Mortlock, D.P.; Rogers, M.B.; Sadeghi, M.M.; et al. Calcific aortic valve disease: A consensus summary from the Alliance of Investigators on Calcific Aortic Valve Disease. Arter. Thromb. Vasc. Biol. 2014, 34, 2387–2393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aronow, W.S.; Schwartz, K.S.; Koenigsberg, M. Correlation of serum lipids, calcium, and phosphorus, diabetes mellitus and history of systemic hypertension with presence or absence of calcified or thickened aortic cusps or root in elderly patients. Am. J. Cardiol. 1987, 59, 998–999. [Google Scholar] [CrossRef]
- Stewart, B.F.; Siscovick, D.; Lind, B.K.; Gardin, J.M.; Gottdiener, J.S.; Smith, V.E.; Kitzman, D.W.; Otto, C.M. Clinical factors associated with calcific aortic valve disease. Cardiovascular Health Study. J. Am. Coll Cardiol. 1997, 29, 630–634. [Google Scholar] [CrossRef] [Green Version]
- Yetkin, E.; Waltenberger, J. Molecular and cellular mechanisms of aortic stenosis. Int. J. Cardiol. 2009, 135, 4–13. [Google Scholar] [CrossRef]
- Freeman, R.V.; Otto, C.M. Spectrum of calcific aortic valve disease: Pathogenesis, disease progression, and treatment strategies. Circulation 2005, 111, 3316–3326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, P.M.; Batten, P.; Brand, N.J.; Thomas, P.S.; Yacoub, M.H. The cardiac valve interstitial cell. Int. J. Biochem. Cell Biol. 2003, 35, 113–118. [Google Scholar] [CrossRef]
- Liu, A.C.; Joag, V.R.; Gotlieb, A.I. The emerging role of valve interstitial cell phenotypes in regulating heart valve pathobiology. Am. J. Pathol. 2007, 171, 1407–1418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lis, G.J.; Dubrowski, A.; Lis, M.; Solewski, B.; Witkowska, K.; Aleksandrovych, V.; Jasek-Gajda, E.; Hołda, M.K.; Gil, K.; Litwin, J.A. Identification of CD34+/PGDFRα+ Valve Interstitial Cells (VICs) in Human Aortic Valves: Association of Their Abundance, Morphology and Spatial Organization with Early Calcific Remodeling. Int. J. Mol. Sci. 2020, 21, 6330. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, K.D. Pathogenesis of calcific aortic valve disease: A disease process comes of age (and a good deal more). Arter. Thromb. Vasc. Biol. 2006, 26, 1721–1728. [Google Scholar] [CrossRef] [Green Version]
- Nagy, E.; Eriksson, P.; Yousry, M.; Caidahl, K.; Ingelsson, E.; Hansson, G.K.; Franco-Cereceda, A.; Bäck, M. Valvular osteoclasts in calcification and aortic valve stenosis severity. Int. J. Cardiol. 2013, 168, 2264–2271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otto, C.M.; Kuusisto, J.; Reichenbach, D.D.; Gown, A.M.; O’Brien, K.D. Characterization of the early lesion of ‘degenerative’ valvular aortic stenosis. Histological and immunohistochemical studies. Circulation 1994, 90, 844–853. [Google Scholar] [CrossRef] [Green Version]
- Mohler, E.R., 3rd; Gannon, F.; Reynolds, C.; Zimmerman, R.; Keane, M.G.; Kaplan, F.S. Bone formation and inflammation in cardiac valves. Circulation 2001, 103, 1522–1528. [Google Scholar] [CrossRef]
- Wypasek, E.; Natorska, J.; Grudzien, G.; Filip, G.; Sadowski, J.; Undas, A. Mast cells in human stenotic aortic valves are associated with the severity of stenosis. Inflammation 2013, 36, 449–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopytek, M.; Kolasa-Trela, R.; Zabczyk, M.; Undas, A.; Natorska, J. NETosis is associated with the severity of aortic stenosis: Links with inflammation. Int. J. Cardiol. 2019, 286, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Cowell, S.J.; Newby, D.E.; Prescott, R.J.; Bloomfield, P.; Reid, J.; Northridge, D.B.; Boon, N.A.; Scottish Aortic, S.; Lipid Lowering Trial, I.o.R.I. A randomized trial of intensive lipid-lowering therapy in calcific aortic stenosis. J. Med. 2005, 352, 2389–2397. [Google Scholar] [CrossRef]
- Rossebo, A.B.; Pedersen, T.R.; Boman, K.; Brudi, P.; Chambers, J.B.; Egstrup, K.; Gerdts, E.; Gohlke-Barwolf, C.; Holme, I.; Kesaniemi, Y.A.; et al. Intensive lipid lowering with simvastatin and ezetimibe in aortic stenosis. N. Engl. J. Med. 2008, 359, 1343–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, K.L.; Teo, K.; Dumesnil, J.G.; Ni, A.; Tam, J.; Investigators, A. Effect of Lipid lowering with rosuvastatin on progression of aortic stenosis: Results of the aortic stenosis progression observation: Measuring effects of rosuvastatin (ASTRONOMER) trial. Circulation 2010, 121, 306–314. [Google Scholar] [CrossRef] [Green Version]
- Van der Linde, D.; Yap, S.C.; van Dijk, A.P.; Budts, W.; Pieper, P.G.; van der Burgh, P.H.; Mulder, B.J.; Witsenburg, M.; Cuypers, J.A.; Lindemans, J.; et al. Effects of rosuvastatin on progression of stenosis in adult patients with congenital aortic stenosis (PROCAS Trial). Am. J. Cardiol. 2011, 108, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Capoulade, R.; Yeang, C.; Chan, K.L.; Pibarot, P.; Tsimikas, S. Association of Mild to Moderate Aortic Valve Stenosis Progression With Higher Lipoprotein(a) and Oxidized Phospholipid Levels: Secondary Analysis of a Randomized Clinical Trial. JAMA Cardiol. 2018, 3, 1212–1217. [Google Scholar] [CrossRef] [PubMed]
- Melendez, Q.M.; Krishnaji, S.T.; Wooten, C.J.; Lopez, D. Hypercholesterolemia: The role of PCSK9. Arch. Biochem. Biophys. 2017, 625, 39–53. [Google Scholar] [CrossRef] [PubMed]
- Langsted, A.; Nordestgaard, B.G.; Benn, M.; Tybjaerg-Hansen, A.; Kamstrup, P.R. PCSK9 R46L Loss-of-Function Mutation Reduces Lipoprotein(a), LDL Cholesterol, and Risk of Aortic Valve Stenosis. J. Clin. Endocrinol. Metab. 2016, 101, 3281–3287. [Google Scholar] [CrossRef]
- De Luca, L.; Corsini, A.; Uguccioni, M.; Colivicchi, F. Statins plus ezetimibe in the era of proprotein convertase subtilisin/kexin type 9 inhibitors. Kardiol. Pol. 2020, 78, 850–860. [Google Scholar] [CrossRef]
- Bergmark, B.A.; O’Donoghue, M.L.; Murphy, S.A.; Kuder, J.F.; Ezhov, M.V.; Ceska, R.; Gouni-Berthold, I.; Jensen, H.K.; Tokgozoglu, S.L.; Mach, F.; et al. An Exploratory Analysis of Proprotein Convertase Subtilisin/Kexin Type 9 Inhibition and Aortic Stenosis in the FOURIER Trial. JAMA Cardiol. 2020, 5, 709–713. [Google Scholar] [CrossRef]
- Perrot, N.; Theriault, S.; Rigade, S.; Chen, H.Y.; Dina, C.; Martinsson, A.; Boekholdt, S.M.; Capoulade, R.; Le Tourneau, T.; Messika-Zeitoun, D.; et al. Lipoprotein-associated phospholipase A2 activity, genetics and calcific aortic valve stenosis in humans. Heart 2020, 106, 1407–1412. [Google Scholar] [CrossRef] [PubMed]
- Group, H.T.R.C.; Bowman, L.; Hopewell, J.C.; Chen, F.; Wallendszus, K.; Stevens, W.; Collins, R.; Wiviott, S.D.; Cannon, C.P.; Braunwald, E.; et al. Effects of Anacetrapib in Patients with Atherosclerotic Vascular Disease. N. Engl. J. Med. 2017, 377, 1217–1227. [Google Scholar] [CrossRef]
- Nicholls, S.J.; Ruotolo, G.; Brewer, H.B.; Wang, M.D.; Liu, L.; Willey, M.B.; Deeg, M.A.; Krueger, K.A.; Nissen, S.E. Evacetrapib alone or in combination with statins lowers lipoprotein(a) and total and small LDL particle concentrations in mildly hypercholesterolemic patients. J. Clin. Lipidol. 2016, 10, 519–527.e4. [Google Scholar] [CrossRef]
- Hovingh, G.K.; Kastelein, J.J.; van Deventer, S.J.; Round, P.; Ford, J.; Saleheen, D.; Rader, D.J.; Brewer, H.B.; Barter, P.J. Cholesterol ester transfer protein inhibition by TA-8995 in patients with mild dyslipidaemia (TULIP): A randomised, double-blind, placebo-controlled phase 2 trial. Lancet 2015, 386, 452–460. [Google Scholar] [CrossRef]
- Tsimikas, S.; Viney, N.J.; Hughes, S.G.; Singleton, W.; Graham, M.J.; Baker, B.F.; Burkey, J.L.; Yang, Q.; Marcovina, S.M.; Geary, R.S.; et al. Antisense therapy targeting apolipoprotein(a): A randomised, double-blind, placebo-controlled phase 1 study. Lancet 2015, 386, 1472–1483. [Google Scholar] [CrossRef]
- Viney, N.J.; van Capelleveen, J.C.; Geary, R.S.; Xia, S.; Tami, J.A.; Yu, R.Z.; Marcovina, S.M.; Hughes, S.G.; Graham, M.J.; Crooke, R.M.; et al. Antisense oligonucleotides targeting apolipoprotein(a) in people with raised lipoprotein(a): Two randomised, double-blind, placebo-controlled, dose-ranging trials. Lancet 2016, 388, 2239–2253. [Google Scholar] [CrossRef]
- Ray, K.K.; Wright, R.S.; Kallend, D.; Koenig, W.; Leiter, L.A.; Raal, F.J.; Bisch, J.A.; Richardson, T.; Jaros, M.; Wijngaard, P.L.J.; et al. Two Phase 3 Trials of Inclisiran in Patients with Elevated LDL Cholesterol. N. Engl. J. Med. 2020, 382, 1507–1519. [Google Scholar] [CrossRef]
- Greve, A.M.; Bang, C.N.; Boman, K.; Egstrup, K.; Kesaniemi, Y.A.; Ray, S.; Pedersen, T.R.; Wachtell, K. Relation of Lipid-Lowering Therapy to Need for Aortic Valve Replacement in Patients With Asymptomatic Mild to Moderate Aortic Stenosis. Am. J. Cardiol. 2019, 124, 1736–1740. [Google Scholar] [CrossRef]
- Yamamoto, K.; Yamamoto, H.; Takeuchi, M.; Kisanuki, A.; Akasaka, T.; Ohte, N.; Hirano, Y.; Yoshida, K.; Nakatani, S.; Takeda, Y.; et al. Risk Factors for Progression of Degenerative Aortic Valve Disease in the Japanese—The Japanese Aortic Stenosis Study (JASS) Prospective Analysis. Circ. J. 2015, 79, 2050–2057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancusi, C.; de Simone, G.; Brguljan Hitij, J.; Sudano, I.; Mahfoud, F.; Parati, G.; Kahan, T.; Barbato, E.; Pierard, L.A.; Garbi, M.; et al. Management of patients with combined arterial hypertension and aortic valve stenosis: A consensus document from the Council on Hypertension and Council on Valvular Heart Disease of the European Society of Cardiology, the European Association of Cardiovascular Imaging (EACVI), and the European Association of Percutaneous Cardiovascular Interventions (EAPCI). Eur. Heart J. Cardiovasc. Pharmacother. 2021, 7, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Gardikioti, V.; Terentes-Printzios, D.; Iliopoulos, D.; Aznaouridis, K.; Sigala, E.; Tsioufis, K.; Vlachopoulos, C. Arterial biomarkers in the evaluation, management and prognosis of aortic stenosis. Atherosclerosis 2021, 332, 1–15. [Google Scholar] [CrossRef]
- Pate, G.E. Association between aortic stenosis and hypertension. J. Heart Valve Dis. 2002, 11, 612–614. [Google Scholar]
- Rahimi, K.; Mohseni, H.; Kiran, A.; Tran, J.; Nazarzadeh, M.; Rahimian, F.; Woodward, M.; Dwyer, T.; MacMahon, S.; Otto, C.M. Elevated blood pressure and risk of aortic valve disease: A cohort analysis of 5.4 million UK adults. Eur. Heart J. 2018, 39, 3596–3603. [Google Scholar] [CrossRef] [PubMed]
- Iwata, S.; Russo, C.; Jin, Z.; Schwartz, J.E.; Homma, S.; Elkind, M.S.; Rundek, T.; Sacco, R.L.; Di Tullio, M.R. Higher ambulatory blood pressure is associated with aortic valve calcification in the elderly: A population-based study. Hypertension 2013, 61, 55–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tastet, L.; Capoulade, R.; Clavel, M.A.; Larose, E.; Shen, M.; Dahou, A.; Arsenault, M.; Mathieu, P.; Bedard, E.; Dumesnil, J.G.; et al. Systolic hypertension and progression of aortic valve calcification in patients with aortic stenosis: Results from the PROGRESSA study. Eur. Heart J. Cardiovasc. Imaging 2017, 18, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Helske, S.; Lindstedt, K.A.; Laine, M.; Mayranpaa, M.; Werkkala, K.; Lommi, J.; Turto, H.; Kupari, M.; Kovanen, P.T. Induction of local angiotensin II-producing systems in stenotic aortic valves. J. Am. Coll. Cardiol. 2004, 44, 1859–1866. [Google Scholar] [CrossRef] [Green Version]
- Peltonen, T.; Napankangas, J.; Ohtonen, P.; Aro, J.; Peltonen, J.; Soini, Y.; Juvonen, T.; Satta, J.; Ruskoaho, H.; Taskinen, P. (Pro)renin receptors and angiotensin converting enzyme 2/angiotensin-(1-7)/Mas receptor axis in human aortic valve stenosis. Atherosclerosis 2011, 216, 35–43. [Google Scholar] [CrossRef]
- Fujisaka, T.; Hoshiga, M.; Hotchi, J.; Takeda, Y.; Jin, D.; Takai, S.; Hanafusa, T.; Ishizaka, N. Angiotensin II promotes aortic valve thickening independent of elevated blood pressure in apolipoprotein-E deficient mice. Atherosclerosis 2013, 226, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Shen, Y.; Hu, W.; Chen, Z.; Li, Y. Angiotensin II promotes an osteoblast-like phenotype in porcine aortic valve myofibroblasts. Aging Clin. Exp. Res. 2016, 28, 181–187. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, K.D.; Shavelle, D.M.; Caulfield, M.T.; McDonald, T.O.; Olin-Lewis, K.; Otto, C.M.; Probstfield, J.L. Association of angiotensin-converting enzyme with low-density lipoprotein in aortic valvular lesions and in human plasma. Circulation 2002, 106, 2224–2230. [Google Scholar] [CrossRef] [Green Version]
- Jekell, A.; Nilsson, P.M.; Kahan, T. Treatment of Hypertensive Left Ventricular Hypertrophy. Curr. Pharm. Des. 2018, 24, 4391–4396. [Google Scholar] [CrossRef]
- Dalsgaard, M.; Iversen, K.; Kjaergaard, J.; Grande, P.; Goetze, J.P.; Clemmensen, P.; Hassager, C. Short-term hemodynamic effect of angiotensin-converting enzyme inhibition in patients with severe aortic stenosis: A placebo-controlled, randomized study. Am. Heart J. 2014, 167, 226–234. [Google Scholar] [CrossRef]
- Bull, S.; Loudon, M.; Francis, J.M.; Joseph, J.; Gerry, S.; Karamitsos, T.D.; Prendergast, B.D.; Banning, A.P.; Neubauer, S.; Myerson, S.G. A prospective, double-blind, randomized controlled trial of the angiotensin-converting enzyme inhibitor Ramipril In Aortic Stenosis (RIAS trial). Eur. Heart J. Cardiovasc. Imaging 2015, 16, 834–841. [Google Scholar] [CrossRef]
- Helske-Suihko, S.; Laine, M.; Lommi, J.; Kaartinen, M.; Werkkala, K.; Kovanen, P.T.; Kupari, M. Is blockade of the Renin-Angiotensin system able to reverse the structural and functional remodeling of the left ventricle in severe aortic stenosis? J. Cardiovasc. Pharmacol. 2015, 65, 233–240. [Google Scholar] [CrossRef]
- Cote, N.; Couture, C.; Pibarot, P.; Despres, J.P.; Mathieu, P. Angiotensin receptor blockers are associated with a lower remodelling score of stenotic aortic valves. Eur. J. Clin. Investig. 2011, 41, 1172–1179. [Google Scholar] [CrossRef] [PubMed]
- Cote, N.; Mahmut, A.; Fournier, D.; Boulanger, M.C.; Couture, C.; Despres, J.P.; Trahan, S.; Bosse, Y.; Page, S.; Pibarot, P.; et al. Angiotensin receptor blockers are associated with reduced fibrosis and interleukin-6 expression in calcific aortic valve disease. Pathobiology 2014, 81, 15–24. [Google Scholar] [CrossRef]
- Ngo, D.T.; Stafford, I.; Sverdlov, A.L.; Qi, W.; Wuttke, R.D.; Zhang, Y.; Kelly, D.J.; Weedon, H.; Smith, M.D.; Kennedy, J.A.; et al. Ramipril retards development of aortic valve stenosis in a rabbit model: Mechanistic considerations. Br. J. Pharmacol. 2011, 162, 722–732. [Google Scholar] [CrossRef] [Green Version]
- Rosenhek, R.; Rader, F.; Loho, N.; Gabriel, H.; Heger, M.; Klaar, U.; Schemper, M.; Binder, T.; Maurer, G.; Baumgartner, H. Statins but not angiotensin-converting enzyme inhibitors delay progression of aortic stenosis. Circulation 2004, 110, 1291–1295. [Google Scholar] [CrossRef] [Green Version]
- Capoulade, R.; Clavel, M.A.; Mathieu, P.; Cote, N.; Dumesnil, J.G.; Arsenault, M.; Bedard, E.; Pibarot, P. Impact of hypertension and renin-angiotensin system inhibitors in aortic stenosis. Eur. J. Clin. Investig. 2013, 43, 1262–1272. [Google Scholar] [CrossRef]
- O’Brien, K.D.; Probstfield, J.L.; Caulfield, M.T.; Nasir, K.; Takasu, J.; Shavelle, D.M.; Wu, A.H.; Zhao, X.Q.; Budoff, M.J. Angiotensin-converting enzyme inhibitors and change in aortic valve calcium. Arch. Int. Med. 2005, 165, 858–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nadir, M.A.; Wei, L.; Elder, D.H.; Libianto, R.; Lim, T.K.; Pauriah, M.; Pringle, S.D.; Doney, A.D.; Choy, A.M.; Struthers, A.D.; et al. Impact of renin-angiotensin system blockade therapy on outcome in aortic stenosis. J. Am. Coll. Cardiol. 2011, 58, 570–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fagyas, M.; Kertesz, A.; Siket, I.M.; Banhegyi, V.; Kracsko, B.; Szegedi, A.; Szokol, M.; Vajda, G.; Racz, I.; Gulyas, H.; et al. Level of the SARS-CoV-2 receptor ACE2 activity is highly elevated in old-aged patients with aortic stenosis: Implications for ACE2 as a biomarker for the severity of COVID-19. Geroscience 2021, 43, 19–29. [Google Scholar] [CrossRef]
- Vavilis, G.; Back, M.; Occhino, G.; Trevisan, M.; Bellocco, R.; Evans, M.; Lindholm, B.; Szummer, K.; Carrero, J.J. Kidney Dysfunction and the Risk of Developing Aortic Stenosis. J. Am. Coll. Cardiol. 2019, 73, 305–314. [Google Scholar] [CrossRef]
- Maher, E.R.; Young, G.; Smyth-Walsh, B.; Pugh, S.; Curtis, J.R. Aortic and mitral valve calcification in patients with end-stage renal disease. Lancet 1987, 2, 875–877. [Google Scholar] [CrossRef]
- Guerraty, M.A.; Chai, B.; Hsu, J.Y.; Ojo, A.O.; Gao, Y.; Yang, W.; Keane, M.G.; Budoff, M.J.; Mohler, E.R., 3rd; Investigators, C.S. Relation of aortic valve calcium to chronic kidney disease (from the Chronic Renal Insufficiency Cohort Study). Am. J. Cardiol. 2015, 115, 1281–1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marwick, T.H.; Amann, K.; Bangalore, S.; Cavalcante, J.L.; Charytan, D.M.; Craig, J.C.; Gill, J.S.; Hlatky, M.A.; Jardine, A.G.; Landmesser, U.; et al. Chronic kidney disease and valvular heart disease: Conclusions from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2019, 96, 836–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Boer, I.H.; Kestenbaum, B.; Shoben, A.B.; Michos, E.D.; Sarnak, M.J.; Siscovick, D.S. 25-hydroxyvitamin D levels inversely associate with risk for developing coronary artery calcification. J. Am. Soc. Nephrol. 2009, 20, 1805–1812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hekimian, G.; Boutten, A.; Flamant, M.; Duval, X.; Dehoux, M.; Benessiano, J.; Huart, V.; Dupre, T.; Berjeb, N.; Tubach, F.; et al. Progression of aortic valve stenosis is associated with bone remodelling and secondary hyperparathyroidism in elderly patients—The COFRASA study. Eur. Heart J. 2013, 34, 1915–1922. [Google Scholar] [CrossRef] [Green Version]
- Audet, A.; Cote, N.; Couture, C.; Bosse, Y.; Despres, J.P.; Pibarot, P.; Mathieu, P. Amyloid substance within stenotic aortic valves promotes mineralization. Histopathology 2012, 61, 610–619. [Google Scholar] [CrossRef]
- Ducy, P.; Zhang, R.; Geoffroy, V.; Ridall, A.L.; Karsenty, G. Osf2/Cbfa1: A transcriptional activator of osteoblast differentiation. Cell 1997, 89, 747–754. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Tang, R.; Zhang, Y.; Chen, S.; Guo, Y.; Wang, X.; Liu, Z.; Liu, H.; Zhang, X.; Liu, B.C. PTH-induced EndMT via miR-29a-5p/GSAP/Notch1 pathway contributed to valvular calcification in rats with CKD. Cell Prolif. 2021, 54, e13018. [Google Scholar] [CrossRef]
- Ohara, T.; Hashimoto, Y.; Matsumura, A.; Suzuki, M.; Isobe, M. Accelerated progression and morbidity in patients with aortic stenosis on chronic dialysis. Circ. J. 2005, 69, 1535–1539. [Google Scholar] [CrossRef] [Green Version]
- Candellier, A.; Henaut, L.; Morelle, J.; Choukroun, G.; Jadoul, M.; Brazier, M.; Goffin, E. Aortic stenosis in patients with kidney failure: Is there an advantage for a PD-first policy? Perit. Dial. Int. 2021, 41, 158–167. [Google Scholar] [CrossRef]
- Ix, J.H.; Chertow, G.M.; Shlipak, M.G.; Brandenburg, V.M.; Ketteler, M.; Whooley, M.A. Association of fetuin-A with mitral annular calcification and aortic stenosis among persons with coronary heart disease: Data from the Heart and Soul Study. Circulation 2007, 115, 2533–2539. [Google Scholar] [CrossRef]
- Wang, A.Y.; Woo, J.; Lam, C.W.; Wang, M.; Chan, I.H.; Gao, P.; Lui, S.F.; Li, P.K.; Sanderson, J.E. Associations of serum fetuin-A with malnutrition, inflammation, atherosclerosis and valvular calcification syndrome and outcome in peritoneal dialysis patients. Nephrol. Dial. Transplant. 2005, 20, 1676–1685. [Google Scholar] [CrossRef] [Green Version]
- Kumric, M.; Borovac, J.A.; Ticinovic Kurir, T.; Martinovic, D.; Frka Separovic, I.; Baric, L.; Bozic, J. Role of Matrix Gla Protein in the Complex Network of Coronary Artery Disease: A Comprehensive Review. Life 2021, 11, 737. [Google Scholar] [CrossRef]
- Chiyoya, M.; Seya, K.; Yu, Z.; Daitoku, K.; Motomura, S.; Imaizumi, T.; Fukuda, I.; Furukawa, K.I. Matrix Gla protein negatively regulates calcification of human aortic valve interstitial cells isolated from calcified aortic valves. J. Pharmacol. Sci. 2018, 136, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Schurgers, L.J.; Barreto, D.V.; Barreto, F.C.; Liabeuf, S.; Renard, C.; Magdeleyns, E.J.; Vermeer, C.; Choukroun, G.; Massy, Z.A. The circulating inactive form of matrix gla protein is a surrogate marker for vascular calcification in chronic kidney disease: A preliminary report. Clin. J. Am. Soc. Nephrol. 2010, 5, 568–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rattazzi, M.; Bertacco, E.; Del Vecchio, A.; Puato, M.; Faggin, E.; Pauletto, P. Aortic valve calcification in chronic kidney disease. Nephrol. Dial. Transplant. 2013, 28, 2968–2976. [Google Scholar] [CrossRef] [Green Version]
- Zoppellaro, G.; Faggin, E.; Puato, M.; Pauletto, P.; Rattazzi, M. Fibroblast growth factor 23 and the bone-vascular axis: Lessons learned from animal studies. Am. J. Kidney Dis. 2012, 59, 135–144. [Google Scholar] [CrossRef]
- Jean, G.; Bresson, E.; Terrat, J.C.; Vanel, T.; Hurot, J.M.; Lorriaux, C.; Mayor, B.; Chazot, C. Peripheral vascular calcification in long-haemodialysis patients: Associated factors and survival consequences. Nephrol. Dial. Transplant. 2009, 24, 948–955. [Google Scholar] [CrossRef] [Green Version]
- Kirkpantur, A.; Balci, M.; Gurbuz, O.A.; Afsar, B.; Canbakan, B.; Akdemir, R.; Ayli, M.D. Serum fibroblast growth factor-23 (FGF-23) levels are independently associated with left ventricular mass and myocardial performance index in maintenance haemodialysis patients. Nephrol. Dial. Transplant. 2011, 26, 1346–1354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unsal, A.; Kose Budak, S.; Koc, Y.; Basturk, T.; Sakaci, T.; Ahbap, E.; Sinangil, A. Relationship of fibroblast growth factor 23 with left ventricle mass index and coronary calcificaton in chronic renal disease. Kidney Blood Press Res. 2012, 36, 55–64. [Google Scholar] [CrossRef]
- Li, F.; Yao, Q.; Ao, L.; Cleveland, J.C., Jr.; Dong, N.; Fullerton, D.A.; Meng, X. Klotho suppresses high phosphate-induced osteogenic responses in human aortic valve interstitial cells through inhibition of Sox9. J. Mol. Med. 2017, 95, 739–751. [Google Scholar] [CrossRef]
- Gu, J.; Lu, Y.; Deng, M.; Qiu, M.; Tian, Y.; Ji, Y.; Zong, P.; Shao, Y.; Zheng, R.; Zhou, B.; et al. Inhibition of acetylation of histones 3 and 4 attenuates aortic valve calcification. Exp. Mol. Med. 2019, 51, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Raggi, P.; Chertow, G.M.; Torres, P.U.; Csiky, B.; Naso, A.; Nossuli, K.; Moustafa, M.; Goodman, W.G.; Lopez, N.; Downey, G.; et al. The ADVANCE study: A randomized study to evaluate the effects of cinacalcet plus low-dose vitamin D on vascular calcification in patients on hemodialysis. Nephrol. Dial. Transplant. 2011, 26, 1327–1339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Investigators, E.T.; Chertow, G.M.; Block, G.A.; Correa-Rotter, R.; Drueke, T.B.; Floege, J.; Goodman, W.G.; Herzog, C.A.; Kubo, Y.; London, G.M.; et al. Effect of cinacalcet on cardiovascular disease in patients undergoing dialysis. N. Engl. J. Med. 2012, 367, 2482–2494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aronow, W.S.; Ahn, C.; Kronzon, I.; Goldman, M.E. Association of coronary risk factors and use of statins with progression of mild valvular aortic stenosis in older persons. Am. J. Cardiol. 2001, 88, 693–695. [Google Scholar] [CrossRef]
- Katz, R.; Wong, N.D.; Kronmal, R.; Takasu, J.; Shavelle, D.M.; Probstfield, J.L.; Bertoni, A.G.; Budoff, M.J.; O’Brien, K.D. Features of the metabolic syndrome and diabetes mellitus as predictors of aortic valve calcification in the Multi-Ethnic Study of Atherosclerosis. Circulation 2006, 113, 2113–2119. [Google Scholar] [CrossRef] [Green Version]
- Kamalesh, M.; Ng, C.; El Masry, H.; Eckert, G.; Sawada, S. Does diabetes accelerate progression of calcific aortic stenosis? Eur. J. Echocardiogr. 2009, 10, 723–725. [Google Scholar] [CrossRef] [Green Version]
- Yan, A.T.; Koh, M.; Chan, K.K.; Guo, H.; Alter, D.A.; Austin, P.C.; Tu, J.V.; Wijeysundera, H.C.; Ko, D.T. Association Between Cardiovascular Risk Factors and Aortic Stenosis: The CANHEART Aortic Stenosis Study. J. Am. Coll. Cardiol. 2017, 69, 1523–1532. [Google Scholar] [CrossRef]
- Larsson, S.C.; Wallin, A.; Hakansson, N.; Stackelberg, O.; Back, M.; Wolk, A. Type 1 and type 2 diabetes mellitus and incidence of seven cardiovascular diseases. Int. J. Cardiol. 2018, 262, 66–70. [Google Scholar] [CrossRef] [Green Version]
- Martinsson, A.; Ostling, G.; Persson, M.; Sundquist, K.; Andersson, C.; Melander, O.; Engstrom, G.; Hedblad, B.; Smith, J.G. Carotid plaque, intima-media thickness, and incident aortic stenosis: A prospective cohort study. Arter. Thromb. Vasc. Biol. 2014, 34, 2343–2348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Testuz, A.; Nguyen, V.; Mathieu, T.; Kerneis, C.; Arangalage, D.; Kubota, N.; Codogno, I.; Tubiana, S.; Estellat, C.; Cimadevilla, C.; et al. Influence of metabolic syndrome and diabetes on progression of calcific aortic valve stenosis. Int. J. Cardiol. 2017, 244, 248–253. [Google Scholar] [CrossRef] [Green Version]
- Natorska, J.; Wypasek, E.; Grudzien, G.; Sobczyk, D.; Marek, G.; Filip, G.; Sadowski, J.; Undas, A. Does diabetes accelerate the progression of aortic stenosis through enhanced inflammatory response within aortic valves? Inflammation 2012, 35, 834–840. [Google Scholar] [CrossRef] [Green Version]
- Kopytek, M.; Zabczyk, M.; Mazur, P.; Undas, A.; Natorska, J. Accumulation of advanced glycation end products (AGEs) is associated with the severity of aortic stenosis in patients with concomitant type 2 diabetes. Cardiovasc. Diabetol. 2020, 19, 92. [Google Scholar] [CrossRef]
- Li, F.; Cai, Z.; Chen, F.; Shi, X.; Zhang, Q.; Chen, S.; Shi, J.; Wang, D.W.; Dong, N. Pioglitazone attenuates progression of aortic valve calcification via down-regulating receptor for advanced glycation end products. Basic Res. Cardiol. 2012, 107, 306. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, B.; Yakobus, Y.; Indrasari, M.; Nass, N.; Santos, A.N.; Kraus, F.B.; Silber, R.E.; Simm, A. RAGE influences the development of aortic valve stenosis in mice on a high fat diet. Exp. Gerontol. 2014, 59, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Kopytek, M.; Mazur, P.; Zabczyk, M.; Undas, A.; Natorska, J. Diabetes concomitant to aortic stenosis is associated with increased expression of NF-kappaB and more pronounced valve calcification. Diabetologia 2021, 64, 2562–2574. [Google Scholar] [CrossRef]
- Yang, P.; Feng, J.; Peng, Q.; Liu, X.; Fan, Z. Advanced Glycation End Products: Potential Mechanism and Therapeutic Target in Cardiovascular Complications under Diabetes. Oxid. Med. Cell. Longev. 2019, 2019, 9570616. [Google Scholar] [CrossRef]
- Choi, B.; Kim, E.Y.; Kim, J.E.; Oh, S.; Park, S.O.; Kim, S.M.; Choi, H.; Song, J.K.; Chang, E.J. Evogliptin Suppresses Calcific Aortic Valve Disease by Attenuating Inflammation, Fibrosis, and Calcification. Cells 2021, 10, 57. [Google Scholar] [CrossRef]
- Mendez-Bailon, M.; Lorenzo-Villalba, N.; Munoz-Rivas, N.; de Miguel-Yanes, J.M.; De Miguel-Diez, J.; Comin-Colet, J.; Hernandez-Barrera, V.; Jimenez-Garcia, R.; Lopez-de-Andres, A. Transcatheter aortic valve implantation and surgical aortic valve replacement among hospitalized patients with and without type 2 diabetes mellitus in Spain (2014–2015). Cardiovasc. Diabetol. 2017, 16, 144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piroli, F.; Franchin, L.; Bruno, F.; De Filippo, O.; D’Ascenzo, F.; Conrotto, F. New advances in the prevention of transcatheter aortic valve implantation failure: Current and future perspectives. Kardiol. Pol. 2020, 78, 842–849. [Google Scholar] [CrossRef]
- Sun, Y.; Liu, X.; Chen, Z.; Fan, J.; Jiang, J.; He, Y.; Zhu, Q.; Hu, P.; Wang, L.; Xu, Q.; et al. Meta-analysis of Predictors of Early Severe Bleeding in Patients Who Underwent Transcatheter Aortic Valve Implantation. Am. J. Cardiol. 2017, 120, 655–661. [Google Scholar] [CrossRef]
- Abramowitz, Y.; Jilaihawi, H.; Chakravarty, T.; Mangat, G.; Maeno, Y.; Kazuno, Y.; Takahashi, N.; Kawamori, H.; Cheng, W.; Makkar, R.R. Impact of Diabetes Mellitus on Outcomes After Transcatheter Aortic Valve Implantation. Am. J. Cardiol. 2016, 117, 1636–1642. [Google Scholar] [CrossRef]
- Lindman, B.R.; Pibarot, P.; Arnold, S.V.; Suri, R.M.; McAndrew, T.C.; Maniar, H.S.; Zajarias, A.; Kodali, S.; Kirtane, A.J.; Thourani, V.H.; et al. Transcatheter versus surgical aortic valve replacement in patients with diabetes and severe aortic stenosis at high risk for surgery: An analysis of the PARTNER Trial (Placement of Aortic Transcatheter Valve). J. Am. Coll. Cardiol. 2014, 63, 1090–1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ando, T.; Akintoye, E.; Telila, T.; Briasoulis, A.; Takagi, H.; Slovut, D.P.; Schreiber, T.; Grines, C.L.; Afonso, L. Comparison of Hospital Outcome of Transcatheter Versus Surgical Aortic Valve Replacement in Patients With Diabetes Mellitus (from the Nationwide Inpatient Sample). Am. J. Cardiol. 2017, 119, 1250–1254. [Google Scholar] [CrossRef]
- Clavel, M.A.; Pibarot, P.; Messika-Zeitoun, D.; Capoulade, R.; Malouf, J.; Aggarval, S.; Araoz, P.A.; Michelena, H.I.; Cueff, C.; Larose, E.; et al. Impact of aortic valve calcification, as measured by MDCT, on survival in patients with aortic stenosis: Results of an international registry study. J. Am. Coll. Cardiol. 2014, 64, 1202–1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clavel, M.A.; Messika-Zeitoun, D.; Pibarot, P.; Aggarwal, S.R.; Malouf, J.; Araoz, P.A.; Michelena, H.I.; Cueff, C.; Larose, E.; Capoulade, R.; et al. The complex nature of discordant severe calcified aortic valve disease grading: New insights from combined Doppler echocardiographic and computed tomographic study. J. Am. Coll. Cardiol. 2013, 62, 2329–2338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Messika-Zeitoun, D.; Aubry, M.C.; Detaint, D.; Bielak, L.F.; Peyser, P.A.; Sheedy, P.F.; Turner, S.T.; Breen, J.F.; Scott, C.; Tajik, A.J.; et al. Evaluation and clinical implications of aortic valve calcification measured by electron-beam computed tomography. Circulation 2004, 110, 356–362. [Google Scholar] [CrossRef] [Green Version]
- Hyder, J.A.; Allison, M.A.; Wong, N.; Papa, A.; Lang, T.F.; Sirlin, C.; Gapstur, S.M.; Ouyang, P.; Carr, J.J.; Criqui, M.H. Association of coronary artery and aortic calcium with lumbar bone density: The MESA Abdominal Aortic Calcium Study. Am. J. Epidemiol. 2009, 169, 186–194. [Google Scholar] [CrossRef] [Green Version]
- Sterbakova, G.; Vyskocil, V.; Linhartova, K. Bisphosphonates in calcific aortic stenosis: Association with slower progression in mild disease--a pilot retrospective study. Cardiology 2010, 117, 184–189. [Google Scholar] [CrossRef]
- Innasimuthu, A.L.; Katz, W.E. Effect of bisphosphonates on the progression of degenerative aortic stenosis. Echocardiography 2011, 28, 1–7. [Google Scholar] [CrossRef]
- Aksoy, O.; Cam, A.; Goel, S.S.; Houghtaling, P.L.; Williams, S.; Ruiz-Rodriguez, E.; Menon, V.; Kapadia, S.R.; Tuzcu, E.M.; Blackstone, E.H.; et al. Do bisphosphonates slow the progression of aortic stenosis? J. Am. Coll. Cardiol. 2012, 59, 1452–1459. [Google Scholar] [CrossRef] [Green Version]
- Lerman, D.A.; Prasad, S.; Alotti, N. Denosumab could be a Potential Inhibitor of Valvular Interstitial Cells Calcification in vitro. Int. J. Cardiovasc. Res. 2016, 5. [Google Scholar] [CrossRef] [Green Version]
- Pawade, T.A.; Doris, M.K.; Bing, R.; White, A.C.; Forsyth, L.; Evans, E.; Graham, C.; Williams, M.C.; van Beek, E.J.R.; Fletcher, A.; et al. Effect of Denosumab or Alendronic Acid on the Progression of Aortic Stenosis: A Double-Blind Randomized Controlled Trial. Circulation 2021, 143, 2418–2427. [Google Scholar] [CrossRef] [PubMed]
- Eva Sikura, K.; Combi, Z.; Potor, L.; Szerafin, T.; Hendrik, Z.; Mehes, G.; Gergely, P.; Whiteman, M.; Beke, L.; Furtos, I.; et al. Hydrogen sulfide inhibits aortic valve calcification in heart via regulating RUNX2 by NF-kappaB, a link between inflammation and mineralization. J. Adv. Res. 2021, 27, 165–176. [Google Scholar] [CrossRef]
- Borensztajn, K.; Peppelenbosch, M.P.; Spek, C.A. Factor Xa: At the crossroads between coagulation and signaling in physiology and disease. Trends Mol. Med. 2008, 14, 429–440. [Google Scholar] [CrossRef] [PubMed]
- El Husseini, D.; Boulanger, M.C.; Mahmut, A.; Bouchareb, R.; Laflamme, M.H.; Fournier, D.; Pibarot, P.; Bosse, Y.; Mathieu, P. P2Y2 receptor represses IL-6 expression by valve interstitial cells through Akt: Implication for calcific aortic valve disease. J. Mol. Cell Cardiol. 2014, 72, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Rothmeier, A.S.; Ruf, W. Protease-activated receptor 2 signaling in inflammation. Semin. Immunopathol. 2012, 34, 133–149. [Google Scholar] [CrossRef]
- Liu, H.; Wang, L.; Pan, Y.; Wang, X.; Ding, Y.; Zhou, C.; Shah, A.M.; Zhao, G.; Zhang, M. Celastrol Alleviates Aortic Valve Calcification Via Inhibition of NADPH Oxidase 2 in Valvular Interstitial Cells. JACC Basic Transl. Sci. 2020, 5, 35–49. [Google Scholar] [CrossRef]
- Kaden, J.J.; Dempfle, C.E.; Grobholz, R.; Tran, H.T.; Kilic, R.; Sarikoc, A.; Brueckmann, M.; Vahl, C.; Hagl, S.; Haase, K.K.; et al. Interleukin-1 beta promotes matrix metalloproteinase expression and cell proliferation in calcific aortic valve stenosis. Atherosclerosis 2003, 170, 205–211. [Google Scholar] [CrossRef]
- Kaden, J.J.; Vocke, D.C.; Fischer, C.S.; Grobholz, R.; Brueckmann, M.; Vahl, C.F.; Hagl, S.; Haase, K.K.; Dempfle, C.E.; Borggrefe, M. Expression and activity of matrix metalloproteinase-2 in calcific aortic stenosis. Z. Kardiol. 2004, 93, 124–130. [Google Scholar] [CrossRef]
- Kaden, J.J.; Dempfle, C.E.; Kilic, R.; Sarikoc, A.; Hagl, S.; Lang, S.; Brueckmann, M.; Borggrefe, M. Influence of receptor activator of nuclear factor kappa B on human aortic valve myofibroblasts. Exp. Mol. Pathol. 2005, 78, 36–40. [Google Scholar] [CrossRef]
- Kaden, J.J.; Dempfle, C.E.; Grobholz, R.; Fischer, C.S.; Vocke, D.C.; Kilic, R.; Sarikoc, A.; Pinol, R.; Hagl, S.; Lang, S.; et al. Inflammatory regulation of extracellular matrix remodeling in calcific aortic valve stenosis. Cardiovasc. Pathol. 2005, 14, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Bond, M.; Chase, A.J.; Baker, A.H.; Newby, A.C. Inhibition of transcription factor NF-kappaB reduces matrix metalloproteinase-1, -3 and -9 production by vascular smooth muscle cells. Cardiovasc. Res. 2001, 50, 556–565. [Google Scholar] [CrossRef]
- Edep, M.E.; Shirani, J.; Wolf, P.; Brown, D.L. Matrix metalloproteinase expression in nonrheumatic aortic stenosis. Cardiovasc. Pathol. 2000, 9, 281–286. [Google Scholar] [CrossRef]
- Perrotta, I.; Russo, E.; Camastra, C.; Filice, G.; Di Mizio, G.; Colosimo, F.; Ricci, P.; Tripepi, S.; Amorosi, A.; Triumbari, F.; et al. New evidence for a critical role of elastin in calcification of native heart valves: Immunohistochemical and ultrastructural study with literature review. Histopathology 2011, 59, 504–513. [Google Scholar] [CrossRef]
- Matilla, L.; Roncal, C.; Ibarrola, J.; Arrieta, V.; Garcia-Pena, A.; Fernandez-Celis, A.; Navarro, A.; Alvarez, V.; Gainza, A.; Orbe, J.; et al. A Role for MMP-10 (Matrix Metalloproteinase-10) in Calcific Aortic Valve Stenosis. Arter. Thromb. Vasc. Biol. 2020, 40, 1370–1382. [Google Scholar] [CrossRef]
- Zhou, K.; Guo, T.; Xu, Y.; Guo, R. Correlation Between Plasma Matrix Metalloproteinase-28 Levels and Severity of Calcific Aortic Valve Stenosis. Med. Sci. Monit. 2020, 26, e925260. [Google Scholar] [CrossRef]
- Natorska, J.; Marek, G.; Hlawaty, M.; Sobczyk, D.; Sadowski, J.; Tracz, W.; Undas, A. Evidence for tissue factor expression in aortic valves in patients with aortic stenosis. Pol. Arch. Med. Wewnętrznej 2009, 119, 636–643. [Google Scholar] [CrossRef]
- Breyne, J.; Juthier, F.; Corseaux, D.; Marechaux, S.; Zawadzki, C.; Jeanpierre, E.; Ung, A.; Ennezat, P.V.; Susen, S.; Van Belle, E.; et al. Atherosclerotic-like process in aortic stenosis: Activation of the tissue factor-thrombin pathway and potential role through osteopontin alteration. Atherosclerosis 2010, 213, 369–376. [Google Scholar] [CrossRef]
- Natorska, J.; Marek, G.; Hlawaty, M.; Sadowski, J.; Tracz, W.; Undas, A. Fibrin presence within aortic valves in patients with aortic stenosis: Association with in vivo thrombin generation and fibrin clot properties. Thromb. Haemost. 2011, 105, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Kapusta, P.; Wypasek, E.; Natorska, J.; Grudzien, G.; Sobczyk, D.; Sadowski, J.; Undas, A. Factor XIII expression within aortic valves and its plasma activity in patients with aortic stenosis: Association with severity of disease. Thromb. Haemost. 2012, 108, 1172–1179. [Google Scholar] [CrossRef]
- Wypasek, E.; Natorska, J.; Mazur, P.; Kopytek, M.; Gaweda, B.; Kapusta, P.; Madeja, J.; Iwaniec, T.; Kapelak, B.; Undas, A. Effects of rivaroxaban and dabigatran on local expression of coagulation and inflammatory factors within human aortic stenotic valves. Vasc. Pharmacol. 2020, 130, 106679. [Google Scholar] [CrossRef]
- Borissoff, J.I.; Spronk, H.M.; ten Cate, H. The hemostatic system as a modulator of atherosclerosis. N. Engl. J. Med. 2011, 364, 1746–1760. [Google Scholar] [CrossRef] [PubMed]
- Spronk, H.M.; de Jong, A.M.; Crijns, H.J.; Schotten, U.; Van Gelder, I.C.; Ten Cate, H. Pleiotropic effects of factor Xa and thrombin: What to expect from novel anticoagulants. Cardiovasc. Res. 2014, 101, 344–351. [Google Scholar] [CrossRef] [Green Version]
- Schurgers, L.J.; Uitto, J.; Reutelingsperger, C.P. Vitamin K-dependent carboxylation of matrix Gla-protein: A crucial switch to control ectopic mineralization. Trends Mol. Med. 2013, 19, 217–226. [Google Scholar] [CrossRef]
- Koos, R.; Mahnken, A.H.; Muhlenbruch, G.; Brandenburg, V.; Pflueger, B.; Wildberger, J.E.; Kuhl, H.P. Relation of oral anticoagulation to cardiac valvular and coronary calcium assessed by multislice spiral computed tomography. Am. J. Cardiol. 2005, 96, 747–749. [Google Scholar] [CrossRef]
- Koos, R.; Krueger, T.; Westenfeld, R.; Kuhl, H.P.; Brandenburg, V.; Mahnken, A.H.; Stanzel, S.; Vermeer, C.; Cranenburg, E.C.; Floege, J.; et al. Relation of circulating Matrix Gla-Protein and anticoagulation status in patients with aortic valve calcification. Thromb. Haemost. 2009, 101, 706–713. [Google Scholar] [PubMed] [Green Version]
- Tastet, L.; Pibarot, P.; Shen, M.; Clisson, M.; Cote, N.; Salaun, E.; Arsenault, M.; Bedard, E.; Capoulade, R.; Puri, R.; et al. Oral Anticoagulation Therapy and Progression of Calcific Aortic Valve Stenosis. J. Am. Coll. Cardiol. 2019, 73, 1869–1871. [Google Scholar] [CrossRef]
- Ueland, T.; Gullestad, L.; Dahl, C.P.; Aukrust, P.; Aakhus, S.; Solberg, O.G.; Vermeer, C.; Schurgers, L.J. Undercarboxylated matrix Gla protein is associated with indices of heart failure and mortality in symptomatic aortic stenosis. J. Int. Med. 2010, 268, 483–492. [Google Scholar] [CrossRef]
- Pan, M.H.; Maresz, K.; Lee, P.S.; Wu, J.C.; Ho, C.T.; Popko, J.; Mehta, D.S.; Stohs, S.J.; Badmaev, V. Inhibition of TNF-alpha, IL-1alpha, and IL-1beta by Pretreatment of Human Monocyte-Derived Macrophages with Menaquinone-7 and Cell Activation with TLR Agonists In Vitro. J. Med. Food 2016, 19, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Ohsaki, Y.; Shirakawa, H.; Hiwatashi, K.; Furukawa, Y.; Mizutani, T.; Komai, M. Vitamin K suppresses lipopolysaccharide-induced inflammation in the rat. Biosci. Biotechnol. Biochem. 2006, 70, 926–932. [Google Scholar] [CrossRef] [Green Version]
- Lupo, M.G.; Biancorosso, N.; Brilli, E.; Tarantino, G.; Adorni, M.P.; Vivian, G.; Salvalaio, M.; Dall’Acqua, S.; Sut, S.; Neutel, C.; et al. Cholesterol-Lowering Action of a Novel Nutraceutical Combination in Uremic Rats: Insights into the Molecular Mechanism in a Hepatoma Cell Line. Nutrients 2020, 12, 436. [Google Scholar] [CrossRef] [Green Version]
- Brandenburg, V.M.; Reinartz, S.; Kaesler, N.; Kruger, T.; Dirrichs, T.; Kramann, R.; Peeters, F.; Floege, J.; Keszei, A.; Marx, N.; et al. Slower Progress of Aortic Valve Calcification With Vitamin K Supplementation: Results From a Prospective Interventional Proof-of-Concept Study. Circulation 2017, 135, 2081–2083. [Google Scholar] [CrossRef] [PubMed]
- Peeters, F.; van Mourik, M.J.W.; Meex, S.J.R.; Bucerius, J.; Schalla, S.M.; Gerretsen, S.C.; Mihl, C.; Dweck, M.R.; Schurgers, L.J.; Wildberger, J.E.; et al. Bicuspid Aortic Valve Stenosis and the Effect of Vitamin K2 on Calcification Using (18)F-Sodium Fluoride Positron Emission Tomography/Magnetic Resonance: The BASIK2 Rationale and Trial Design. Nutrients 2018, 10, 386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindholt, J.S.; Frandsen, N.E.; Fredgart, M.H.; Ovrehus, K.A.; Dahl, J.S.; Moller, J.E.; Folkestad, L.; Urbonaviciene, G.; Becker, S.W.; Lambrechtsen, J.; et al. Effects of menaquinone-7 supplementation in patients with aortic valve calcification: Study protocol for a randomised controlled trial. BMJ Open 2018, 8, e022019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Lullo, L.; Tripepi, G.; Ronco, C.; D’Arrigo, G.; Barbera, V.; Russo, D.; Di Iorio, B.R.; Uguccioni, M.; Paoletti, E.; Ravera, M.; et al. Cardiac valve calcification and use of anticoagulants: Preliminary observation of a potentially modifiable risk factor. Int. J. Cardiol. 2019, 278, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Bea, F.; Preusch, M.; Wang, H.; Isermann, B.; Shahzad, K.; Katus, H.A.; Blessing, E. Evaluation of plaque stability of advanced atherosclerotic lesions in apo E-deficient mice after treatment with the oral factor Xa inhibitor rivaroxaban. Mediat. Inflamm. 2011, 2011, 432080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, I.O.; Kratz, M.T.; Schirmer, S.H.; Baumhakel, M.; Bohm, M. The effects of direct thrombin inhibition with dabigatran on plaque formation and endothelial function in apolipoprotein E-deficient mice. J. Pharmacol. Exp. Ther 2012, 343, 253–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadoglou, N.P.; Moustardas, P.; Katsimpoulas, M.; Kapelouzou, A.; Kostomitsopoulos, N.; Schafer, K.; Kostakis, A.; Liapis, C.D. The beneficial effects of a direct thrombin inhibitor, dabigatran etexilate, on the development and stability of atherosclerotic lesions in apolipoprotein E-deficient mice: Dabigatran etexilate and atherosclerosis. Cardiovasc. Drugs Ther. 2012, 26, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Fukuda, D.; Tanaka, K.; Higashikuni, Y.; Hirata, Y.; Nishimoto, S.; Yagi, S.; Yamada, H.; Soeki, T.; Wakatsuki, T.; et al. Rivaroxaban, a novel oral anticoagulant, attenuates atherosclerotic plaque progression and destabilization in ApoE-deficient mice. Atherosclerosis 2015, 242, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Van Gorp, R.H.; Dijkgraaf, I.; Broker, V.; Bauwens, M.; Leenders, P.; Jennen, D.; Dweck, M.R.; Bucerius, J.; Briede, J.J.; van Ryn, J.; et al. Off-target effects of oral anticoagulants-vascular effects of vitamin K antagonist and non-vitamin K antagonist oral anticoagulant dabigatran etexilate. J. Thromb. Haemost. 2021, 19, 1348–1363. [Google Scholar] [CrossRef]
- Bukowska, A.; Zacharias, I.; Weinert, S.; Skopp, K.; Hartmann, C.; Huth, C.; Goette, A. Coagulation factor Xa induces an inflammatory signalling by activation of protease-activated receptors in human atrial tissue. Eur. J. Pharmacol. 2013, 718, 114–123. [Google Scholar] [CrossRef] [Green Version]
- Connolly, S.J.; Eikelboom, J.W.; Bosch, J.; Dagenais, G.; Dyal, L.; Lanas, F.; Metsarinne, K.; O’Donnell, M.; Dans, A.L.; Ha, J.W.; et al. Rivaroxaban with or without aspirin in patients with stable coronary artery disease: An international, randomised, double-blind, placebo-controlled trial. Lancet 2018, 391, 205–218. [Google Scholar] [CrossRef]
- Rattazzi, M.; Bertacco, E.; Iop, L.; D’Andrea, S.; Puato, M.; Buso, G.; Causin, V.; Gerosa, G.; Faggin, E.; Pauletto, P. Extracellular pyrophosphate is reduced in aortic interstitial valve cells acquiring a calcifying profile: Implications for aortic valve calcification. Atherosclerosis 2014, 237, 568–576. [Google Scholar] [CrossRef]
- Cote, N.; El Husseini, D.; Pepin, A.; Guauque-Olarte, S.; Ducharme, V.; Bouchard-Cannon, P.; Audet, A.; Fournier, D.; Gaudreault, N.; Derbali, H.; et al. ATP acts as a survival signal and prevents the mineralization of aortic valve. J. Mol. Cell Cardiol. 2012, 52, 1191–1202. [Google Scholar] [CrossRef] [PubMed]
- Cote, N.; El Husseini, D.; Pepin, A.; Bouvet, C.; Gilbert, L.A.; Audet, A.; Fournier, D.; Pibarot, P.; Moreau, P.; Mathieu, P. Inhibition of ectonucleotidase with ARL67156 prevents the development of calcific aortic valve disease in warfarin-treated rats. Eur. J. Pharmacol. 2012, 689, 139–146. [Google Scholar] [CrossRef]
- Bowen, C.J.; Zhou, J.; Sung, D.C.; Butcher, J.T. Cadherin-11 coordinates cellular migration and extracellular matrix remodeling during aortic valve maturation. Dev. Biol. 2015, 407, 145–157. [Google Scholar] [CrossRef] [Green Version]
- Bosse, K.; Hans, C.P.; Zhao, N.; Koenig, S.N.; Huang, N.; Guggilam, A.; LaHaye, S.; Tao, G.; Lucchesi, P.A.; Lincoln, J.; et al. Endothelial nitric oxide signaling regulates Notch1 in aortic valve disease. J. Mol. Cell Cardiol. 2013, 60, 27–35. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.D.; Chu, Y.; Brooks, R.M.; Richenbacher, W.E.; Pena-Silva, R.; Heistad, D.D. Dysregulation of antioxidant mechanisms contributes to increased oxidative stress in calcific aortic valvular stenosis in humans. J. Am. Coll. Cardiol. 2008, 52, 843–850. [Google Scholar] [CrossRef] [Green Version]
- Rattazzi, M.; Donato, M.; Bertacco, E.; Millioni, R.; Franchin, C.; Mortarino, C.; Faggin, E.; Nardin, C.; Scarpa, R.; Cinetto, F.; et al. l-Arginine prevents inflammatory and pro-calcific differentiation of interstitial aortic valve cells. Atherosclerosis 2020, 298, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Majumdar, U.; Manivannan, S.; Basu, M.; Ueyama, Y.; Blaser, M.C.; Cameron, E.; McDermott, M.R.; Lincoln, J.; Cole, S.E.; Garg, V.; et al. Nitric oxide prevents aortic valve calcification by S-nitrosylation of USP9X to activate NOTCH signaling. Sci. Adv. 2021, 7, eabe3706. [Google Scholar] [CrossRef]
- Van Driel, B.O.; Schuldt, M.; Algül, S.; Levin, E.; Güclü, A.; Germans, T.; van Rossum, A.C.; Pei, J.; Harakalova, M.; Baas, A.; et al. Metabolomics in Severe Aortic Stenosis Reveals Intermediates of Nitric Oxide Synthesis as Most Distinctive Markers. Int. J. Mol. Sci. 2021, 22, 3569. [Google Scholar] [CrossRef] [PubMed]
- Moşteoru, S.; Gaiţă, D.; Banach, M. An update on PCSK9 inhibitors-pharmacokinetics, drug interactions, and toxicity. Expert Opin Drug Metab. Toxicol. 2020, 16, 1199–1205. [Google Scholar] [CrossRef] [PubMed]
- Tall, A.R.; Rader, D.J. Trials and Tribulations of CETP Inhibitors. Circ. Res. 2018, 122, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Subrahmanyan, N.A.; Koshy, R.M.; Jacob, K.; Pappachan, J.M. Efficacy and Cardiovascular Safety of DPP-4 Inhibitors. Curr. Drug Saf. 2021, 16, 154–164. [Google Scholar] [CrossRef]
- Eby, G.; Halcomb, W.W. Elimination of cardiac arrhythmias using oral taurine with l-arginine with case histories: Hypothesis for nitric oxide stabilization of the sinus node. Med. Hypotheses. 2006, 67, 1200–1204. [Google Scholar] [CrossRef] [PubMed]
- Diebold, B.A.; Smith, S.M.; Li, Y.; Lambeth, J.D. NOX2 as a Target for Drug Development: Indications, Possible Complications, and Progress. Antioxid. Redox Signal. 2015, 23, 375–405. [Google Scholar] [CrossRef] [Green Version]
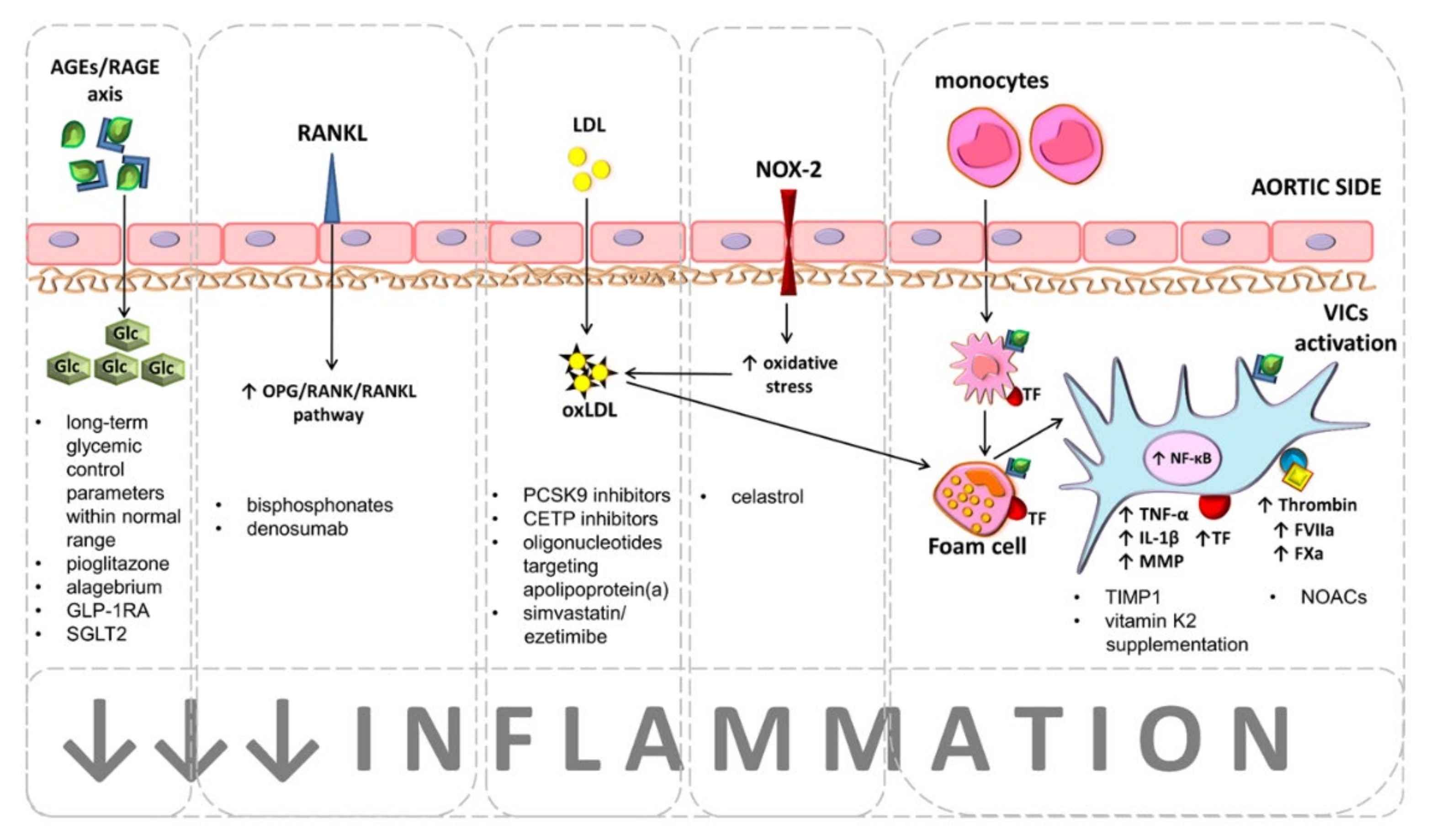

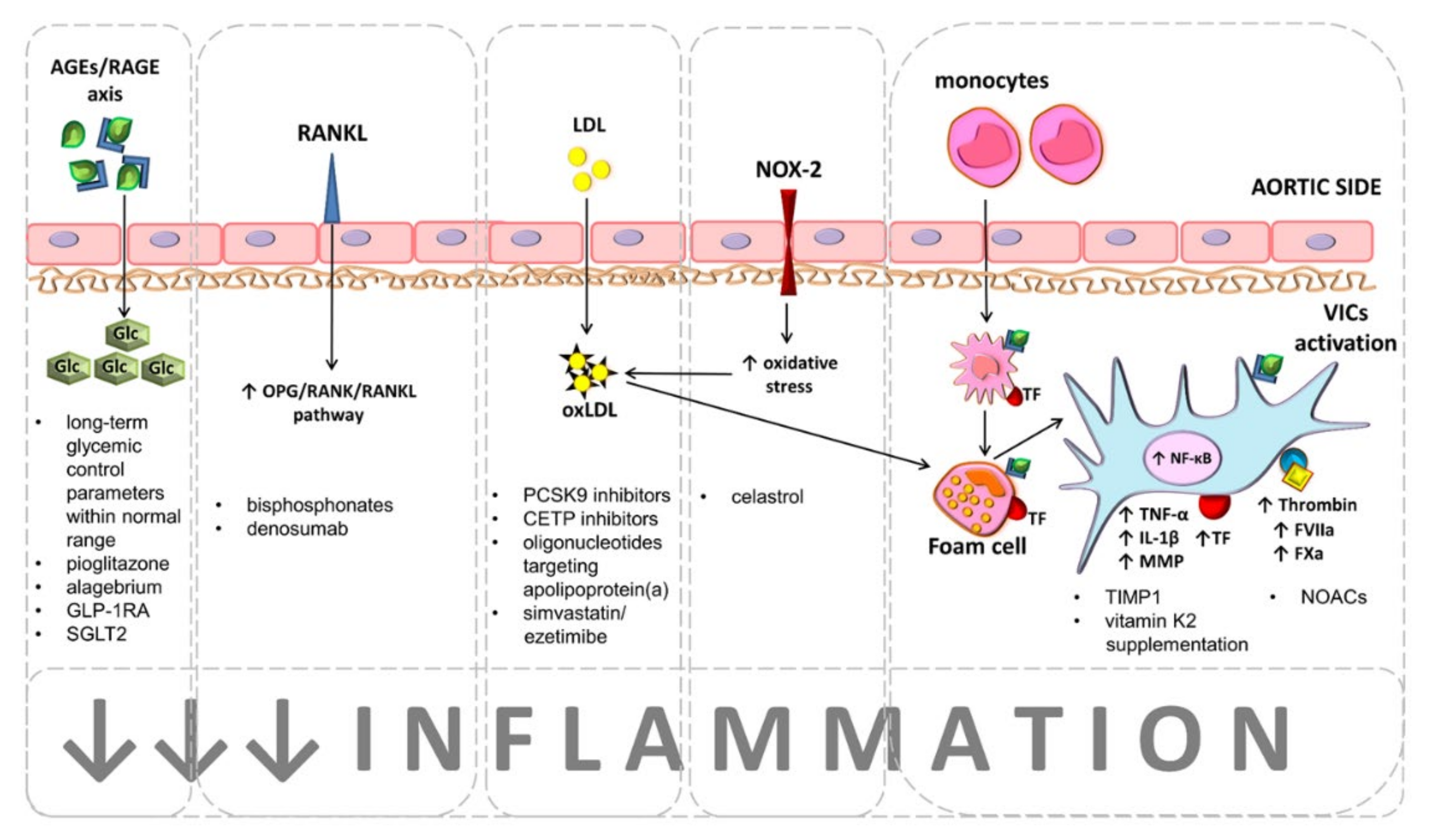{kind=link}
| Author, Year of Publication, and Trial Acronym | Study Type | Study Design | Main Findings |
|---|---|---|---|
| Bergmark et al., 2020, the FOURIER trial [26] | randomized clinical trial | 13,784 atherosclerotic vascular disease patients taking PCSK9 inhibitor evolocumab (mean age, 63 ± 9 years) and 13,784 taking placebo (mean age, 62 ± 9 years), including 63 patients who developed CAS in the full cohort | Long-term therapy with evolocumab (>1 year) may reduce AS events (HR 0.48; 95% CI, 0.25–0.93) |
| HPS3/TIMI55-REVEAL Collaborative Group; Bowman et al., 2017, the REVEAL trial [28] | randomized clinical trial | 15,225 atherosclerotic vascular disease patients assigned to receive CEPT inhibitor anacetrapib (mean age, 67.8 ± 8 years) and 15,224 patients taking placebo (mean age, 67.8 ± 8 years) | Long-term therapy with anacetrapib was associated with reduced Lp(a) (mean level, 43 vs. 58 nmol/L) and LDL cholesterol levels (mean level, 38 vs. 64 mg/dL), and increased HDL cholesterol levels (mean level, 85 vs. 42 mg/dL) |
| Nicholls et al., 2016 [29] | randomized clinical trial | 398 patients with elevated LDL cholesterol or low HDL cholesterol, including 118 patients taking evacetrapib (mean age, 58.6 ± 10.8 years) for 12 weeks as monotherapy or in combination with statins compared to placebo (mean age, 58.9 ± 11.4 years) | Short-term therapy with evacetrapib reduced Lp(a) levels up to 40% and LDL cholesterol up to 54% |
| Hovingh et al., 2015, the TULIP trial [30] | randomized clinical trial | 149 patients with mild dyslipidemia taking CETP inhibitor TA-8995 as monotherapy, 151 patients taking TA-8995 in combination with statins for 12 weeks, and 37 patients receiving placebo | Short term therapy with TA-8995 reduced the concentrations of LDL cholesterol up to 68.2% and increased the levels of HDL cholesterol up to 179% |
| Tsimikas et al., 2015 [31] | randomized clinical trial | 47 healthy volunteers (mean age 35 ± 16.9) with BMI < 32 kg/m2 and Lp(a) ≥ 25 nmol/l taking antisense oligonucleotide ISIS-APO(a)Rx for 4 weeks | ISIS-APO(a)Rx reduced Lp(a) concentrations in a dose-dependent manner up to 77.8% |
| Viney et al., 2016 [32] | randomized clinical trial | 64 healthy volunteers (mean age 58 ± 8 years) with elevated Lp(a) taking antisense oligonucleotide IONIS-APO(a)Rx and 58 individuals (mean age 56 ± 5 years) taking IONIS-APO(a)-LRx or placebo for 12 weeks | IONIS-APO(a)Rx reduced Lp(a) concentrations up to 71.6% and IONIS-APO(a)-LRx up to 92% |
| Ray et al., 2020, the ORION trial [33] | randomized clinical trial | 1591 patients at high risk for cardiovascular disease and increased LDL cholesterol levels taking PCSK9 inhibitor inclisiran and 1587 patients taking placebo for 18 months | Inclisiran reduced LDL cholesterol levels approximately by 50% |
| Greve et al., 2019, secondary analysis of the SEAS trail [34] | randomized clinical trial | 1687 asymptomatic patients with mild to moderate CAS taking simvastatin/ezetimibe combination vs. placebo for median time of 4.3 years | Simvastatin in combination with ezetimibe reduced the rate of aortic valve replacement in patients with mild AS (HR 0.4; 95% CI, 0.2–0.9) |
| Therapy | Target of Therapy | Risk of Therapy | |
|---|---|---|---|
| Lipid-lowering therapies | PCSK9 inhibitors (including siRNA) | Reduction in Lp(a) and LDL cholesterol, increased HDL levels associated with reduced valvular inflammation leading to reduced calcium accumulation. | PCSK9 inhibitors may increase the risk of neurocognitive effects, new onset DM or statin-associated muscle symptoms or other adverse events [162]. |
| CETP inhibitors | Reduction in the concentrations of Lp(a), LDL cholesterol, and other lipoproteins, increased HDL levels resulting in decreased valvular inflammation. | CETP inhibitors activate the renin–angiotensin system increasing blood pressure with its attendant cardiovascular risks, thus in patients with hypertension and CAS further investigations of a possible interaction between the use of antihypertensive drugs and CETP inhibitors are needed [163]. | |
| Antisense therapy: IONIS-APO(a)Rx, IONIS-APO(a)-LRx | Reduction in Lp(a) concentrations leading to decreased valvular inflammation. | No serious side effects [32], however, long-term follow-up is needed to confirm this observation. | |
| Therapies used in patients with CKD | HAT inhibitor: C646 | C646 attenuated aortic valve calcification both in vitro and in vivo. | HAT inhibition is not selective and it suppresses osteoblast-related gene expression leading to decreased osteogenic differentiation. |
| Calcimimetic: cinacalcet + low dose vitamin D | Cinacalcet + low-dose vitamin D sterols attenuated vascular and cardiac valve calcification. | Adverse gastrointestinal effects associated with cinacalcet treatment occurring in about 10% of patients [82,83]. | |
| Inflammation related targets | NF-κB inhibition | NF-κB inhibition prevented VICs calcification in cultures treated with high concentrations of glucose. | A better understanding of the molecular regulation that determines the point of conversion of NF-κB responses from protective to damaging effects is needed for therapeutic intervention in humans. |
| Glucagon-like peptide-1 receptor agonists | Decreased valvular inflammation, cytokine expression, fibrosis, and calcification in a CAS animal model. | Allergic reactions, upper respiratory tract infections, and urinary tract infection have been reported [164]. To date, not investigated in CAS patients. | |
| Precursor of NO: L-Arginine | L-Arginine inhibited induced VICs calcification. | Known to worse asthma symptoms [165]. To date, not investigates in CAS patients. | |
| NOX2 inhibitors | Celastrol prevented VICs calcification, ROS generation, valve fibrosis and left ventricular remodeling in a rabbit model of CAS. | Non-selective substances, which theoretically may exert pro-inflammatory and autoimmune effects [166]. A proof of concept for efficacy with minimal side effects is needed for the acceptance of NOX2 inhibitors as therapeutic agents in CAS patients. | |
| Tissue inhibitor of metalloproteinases:TIMPs | TIMP-1 prevented VICs inflammation and calcification. | Non-selective substances, the next generation of MMPs inhibitors must be selective against MMPs, such as MMP-3, -9, -10 or -12. | |
| Therapies targeting coagulation | NOACs | Rivaroxaban and dabigatran inhibited VICs calcification and inflammation. | Bleeding, anemia. To date, not investigated in CAS patients with regard to CAS progression. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mazur, P.; Kopytek, M.; Ząbczyk, M.; Undas, A.; Natorska, J. Towards Personalized Therapy of Aortic Stenosis. J. Pers. Med. 2021, 11, 1292. https://doi.org/10.3390/jpm11121292
Mazur P, Kopytek M, Ząbczyk M, Undas A, Natorska J. Towards Personalized Therapy of Aortic Stenosis. Journal of Personalized Medicine. 2021; 11(12):1292. https://doi.org/10.3390/jpm11121292
Chicago/Turabian StyleMazur, Piotr, Magdalena Kopytek, Michał Ząbczyk, Anetta Undas, and Joanna Natorska. 2021. "Towards Personalized Therapy of Aortic Stenosis" Journal of Personalized Medicine 11, no. 12: 1292. https://doi.org/10.3390/jpm11121292
APA StyleMazur, P., Kopytek, M., Ząbczyk, M., Undas, A., & Natorska, J. (2021). Towards Personalized Therapy of Aortic Stenosis. Journal of Personalized Medicine, 11(12), 1292. https://doi.org/10.3390/jpm11121292