Genotyping of Single Nucleotide Polymorphisms Using Allele-Specific qPCR Producing Amplicons of Small Sizes Directly from Crude Serum Isolated from Capillary Blood by a Hand-Powered Paper Centrifuge

 ,
, 
Abstract
:
1. Introduction
2. Methods
2.1. Ethics
2.2. Volunteers
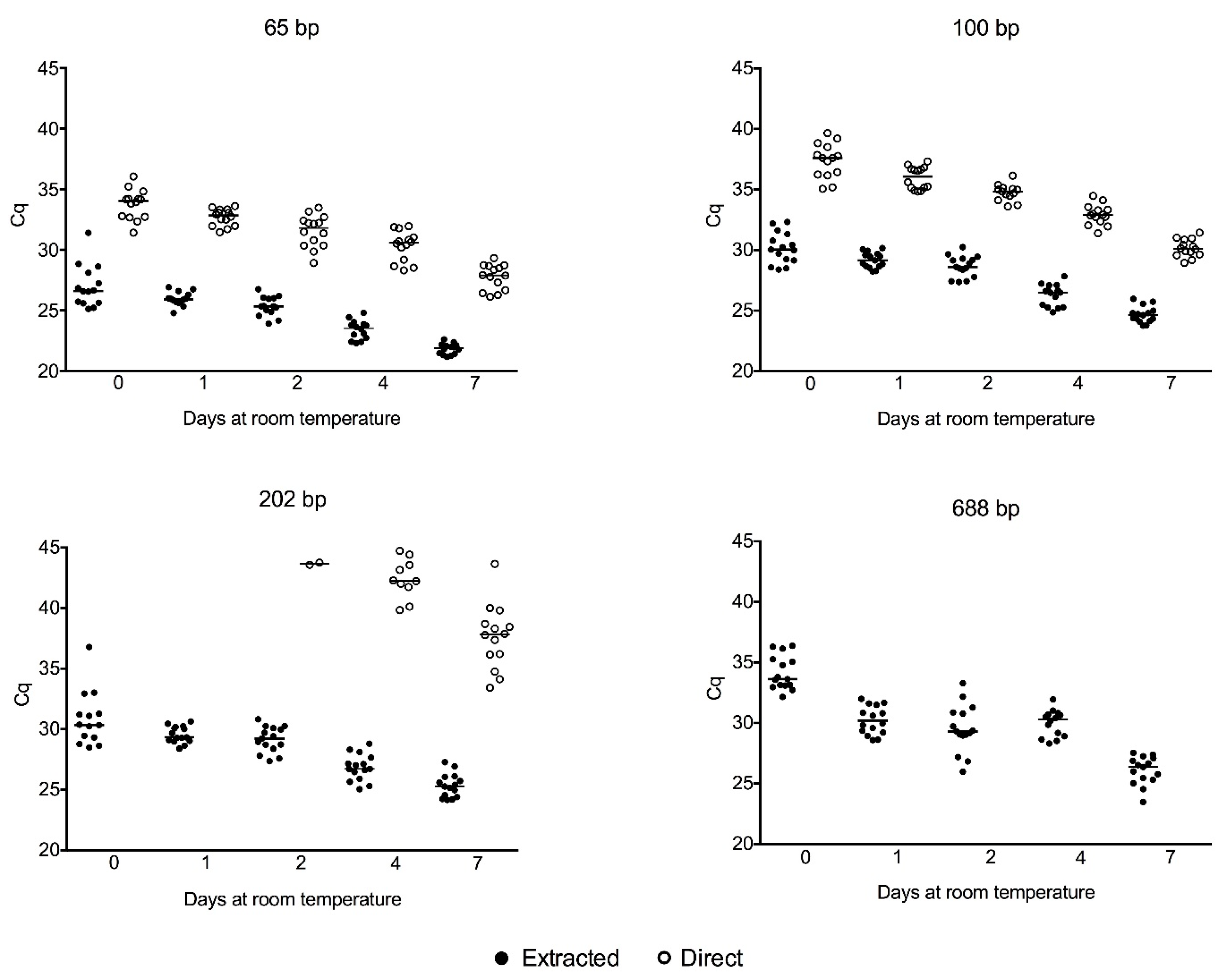
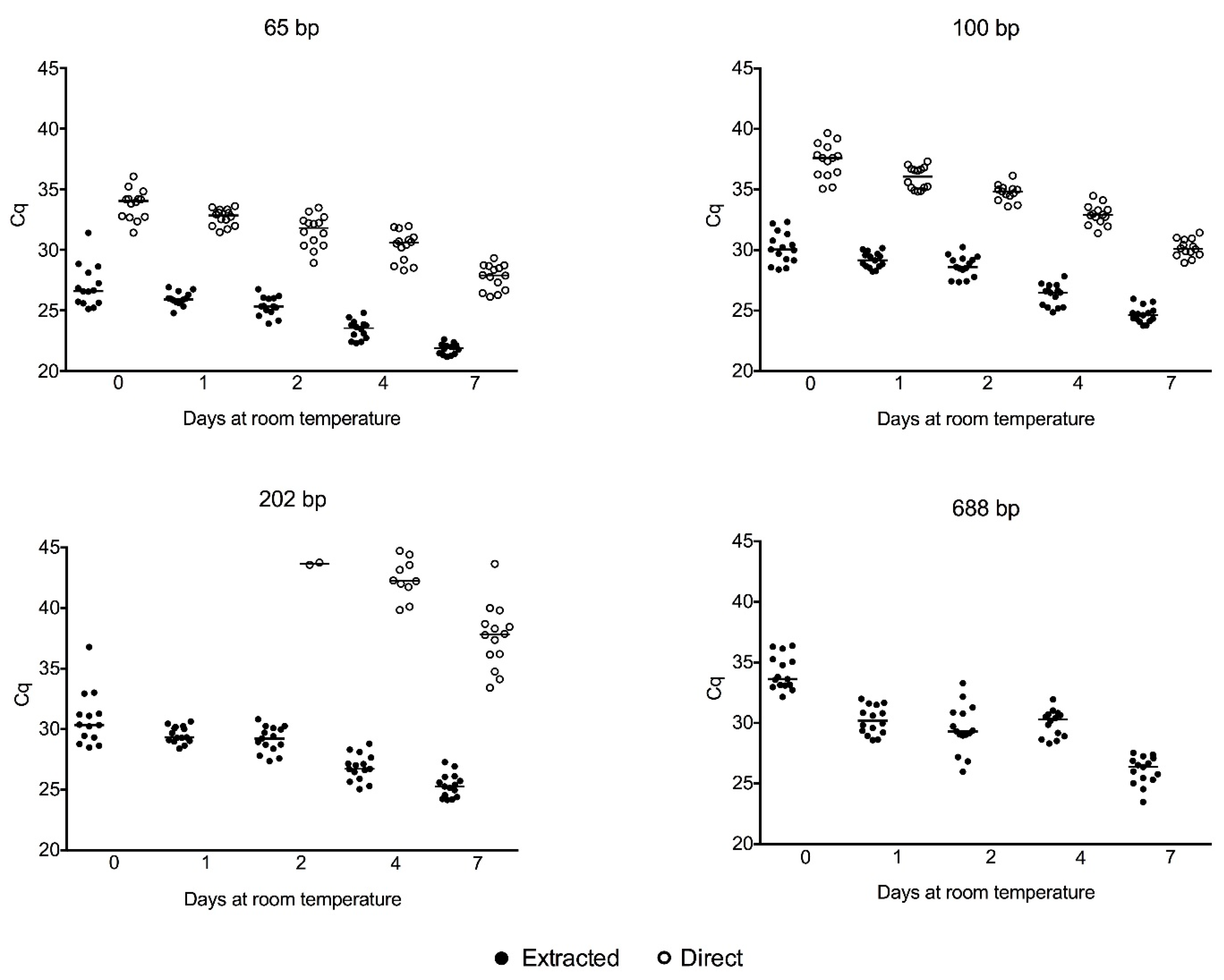
2.3. Cell-Free gDNA Size Distribution in Serum along One Week of Storage at Room Temperature and Amplification of Genomic DNA Directly from Crude Serum Exempting the DNA Extraction
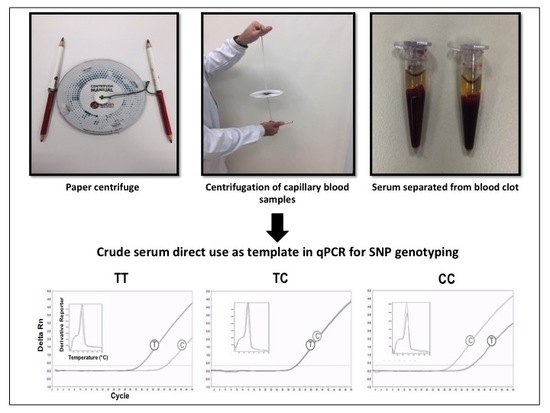
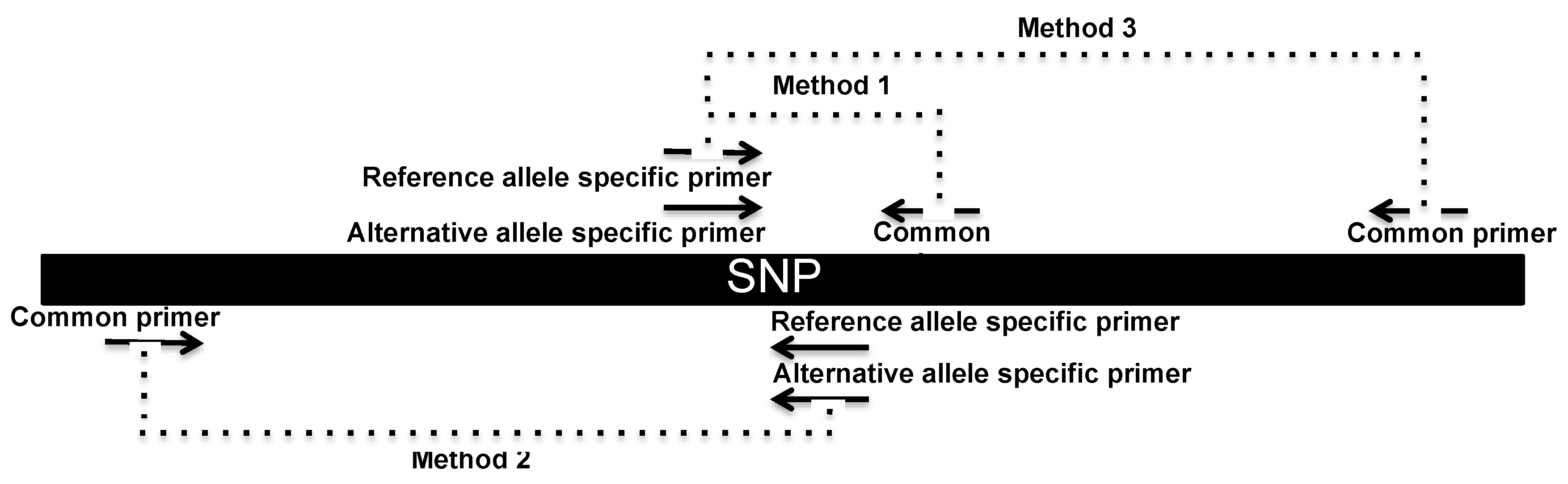
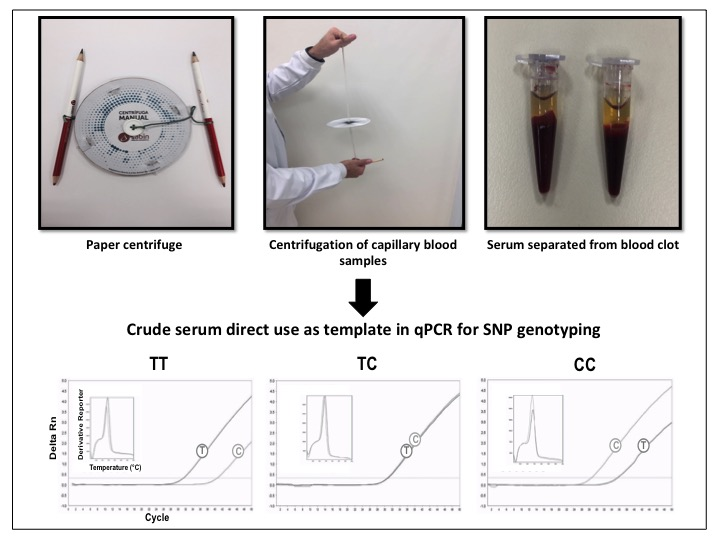
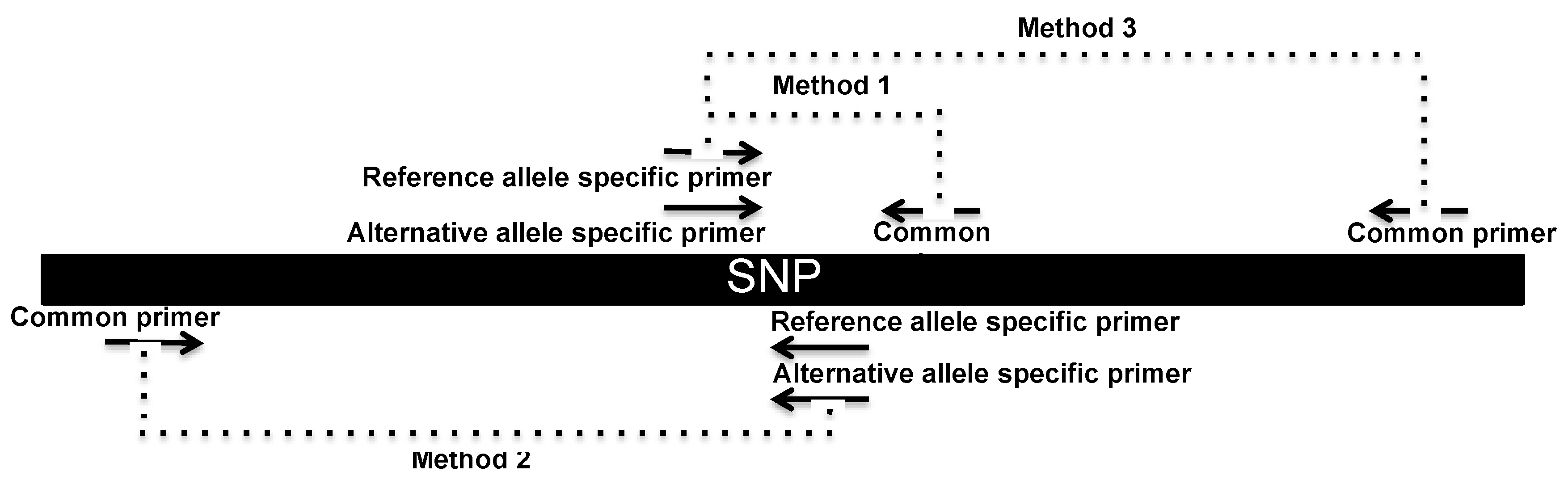
2.4. SNP Genotyping Directly from Crude Serum Isolated from Capillary Blood Using a Hand-Powered Paper Centrifuge
2.5. Statistical Analysis
3. Results
3.1. Cell-Free gDNA Size Distribution in Serum along One Week of Storage at Room Temperature and Amplification of Genomic DNA Directly from Crude Serum Exempting the DNA Extraction
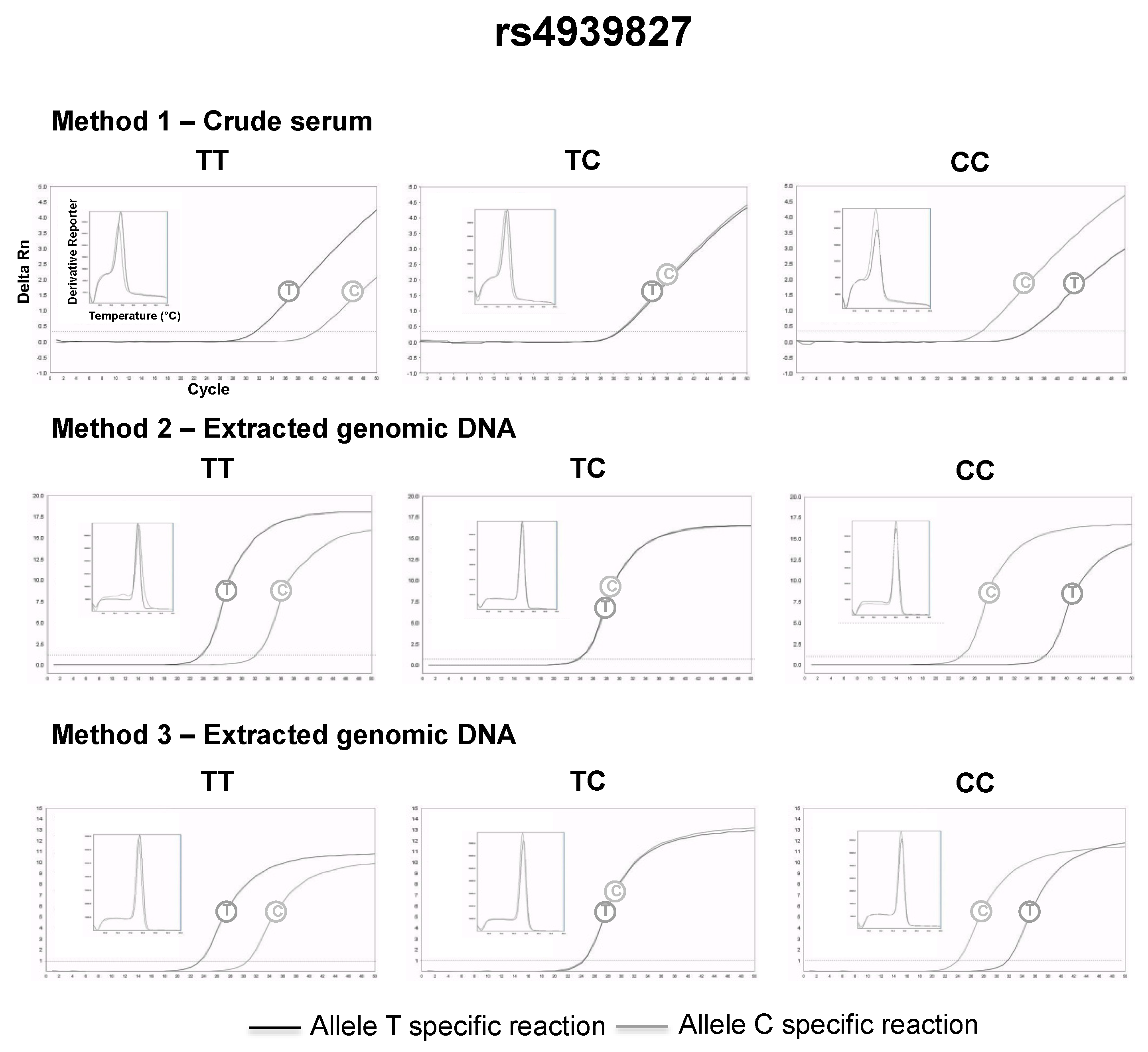
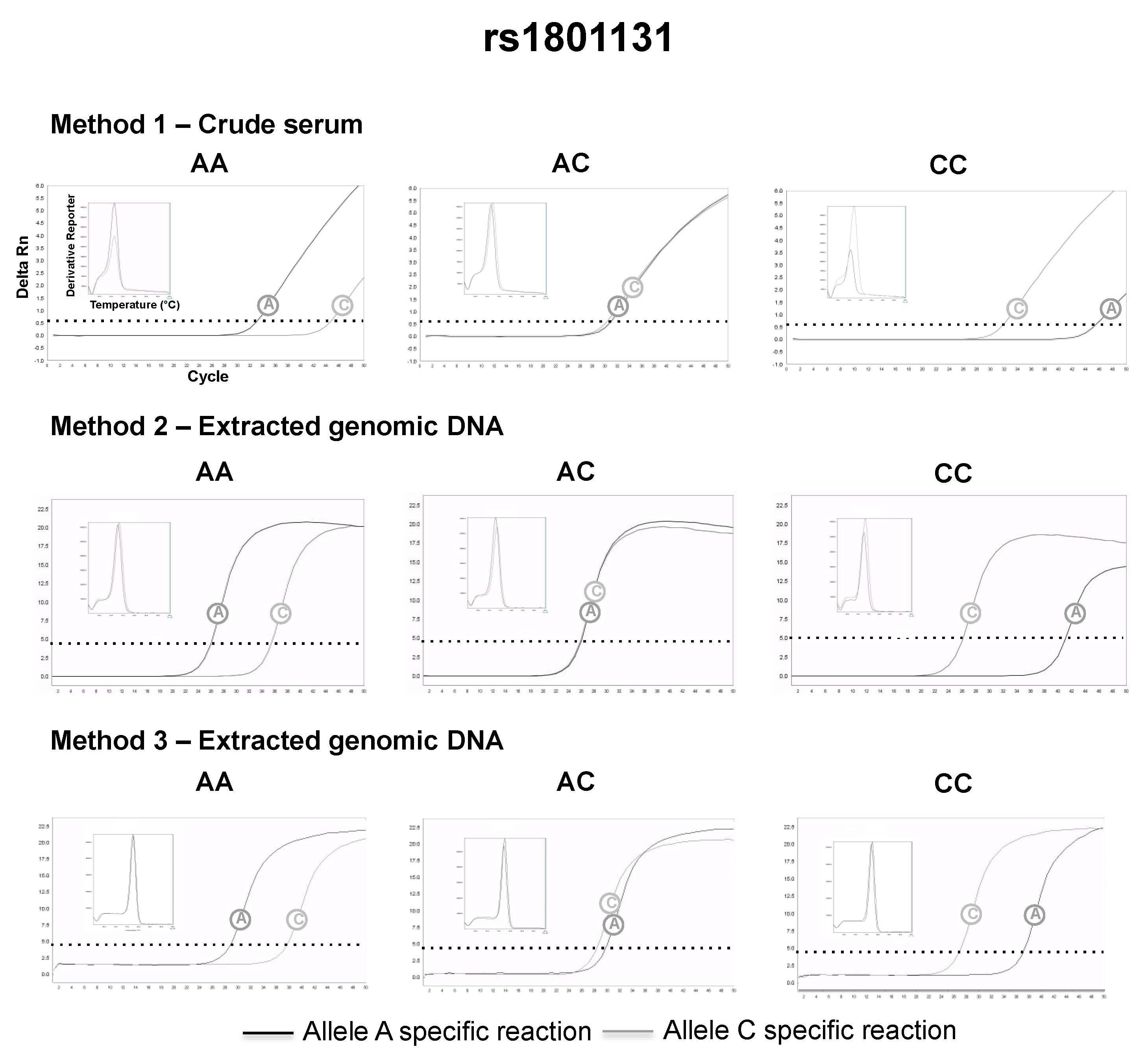
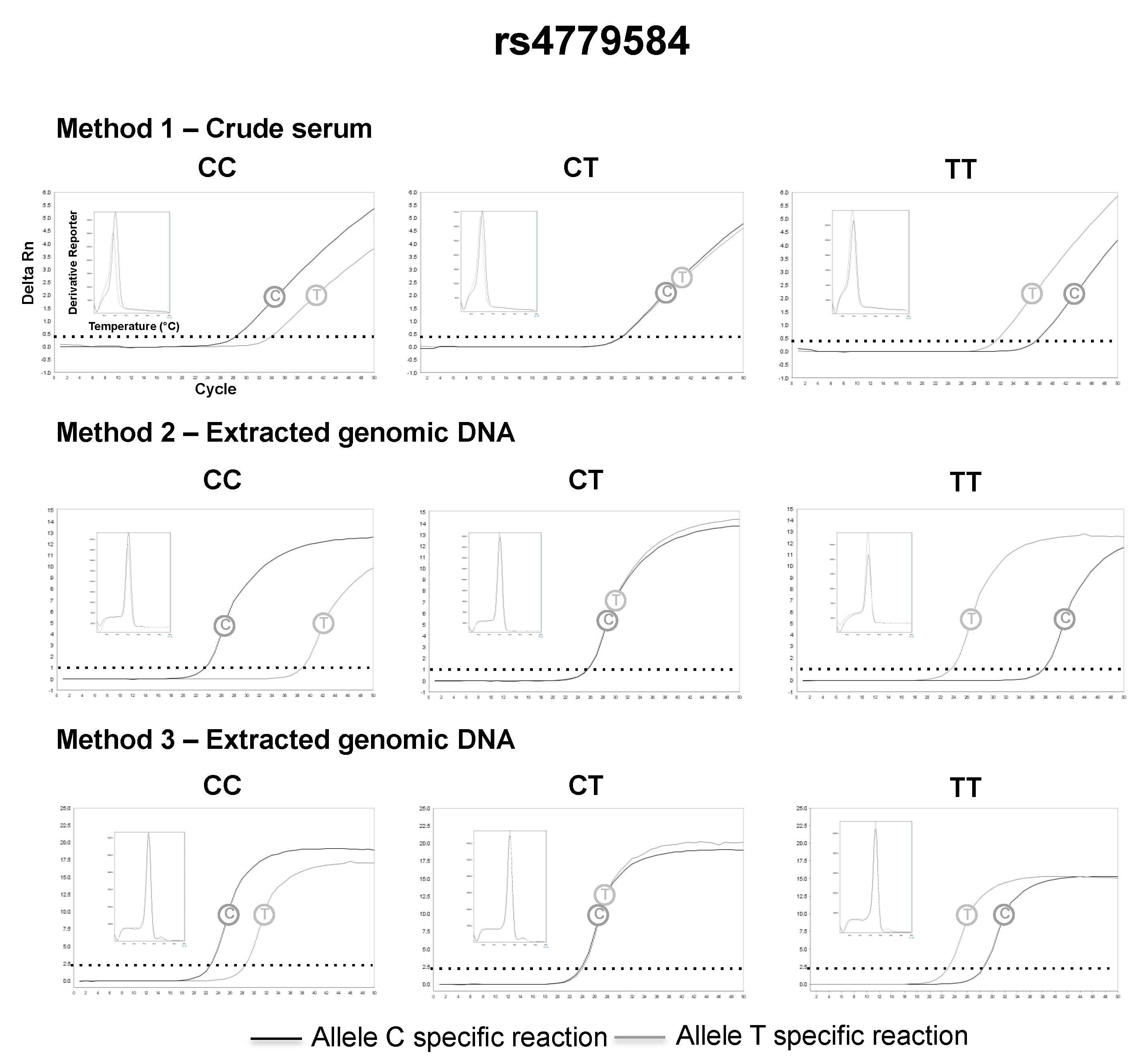
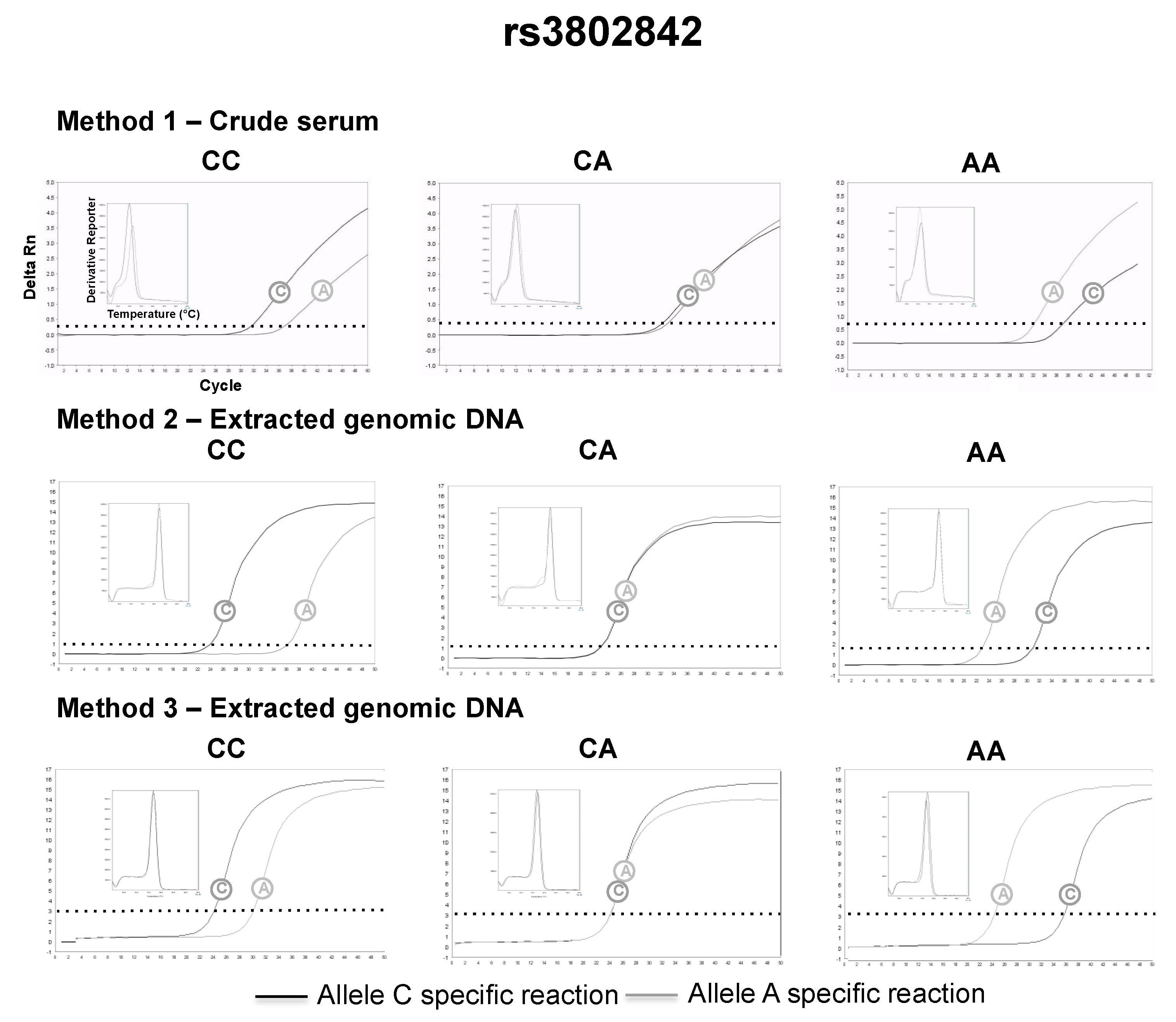
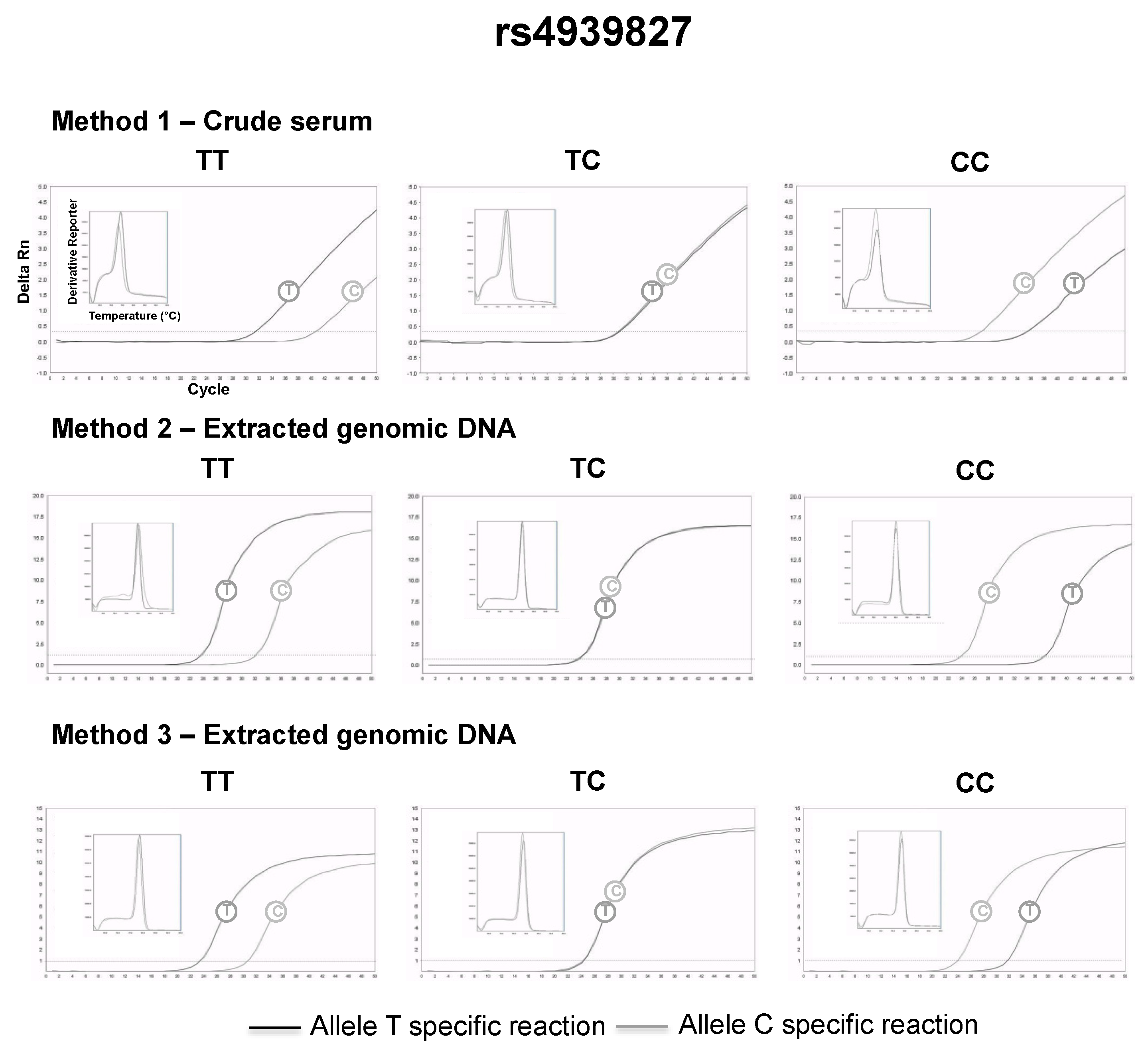
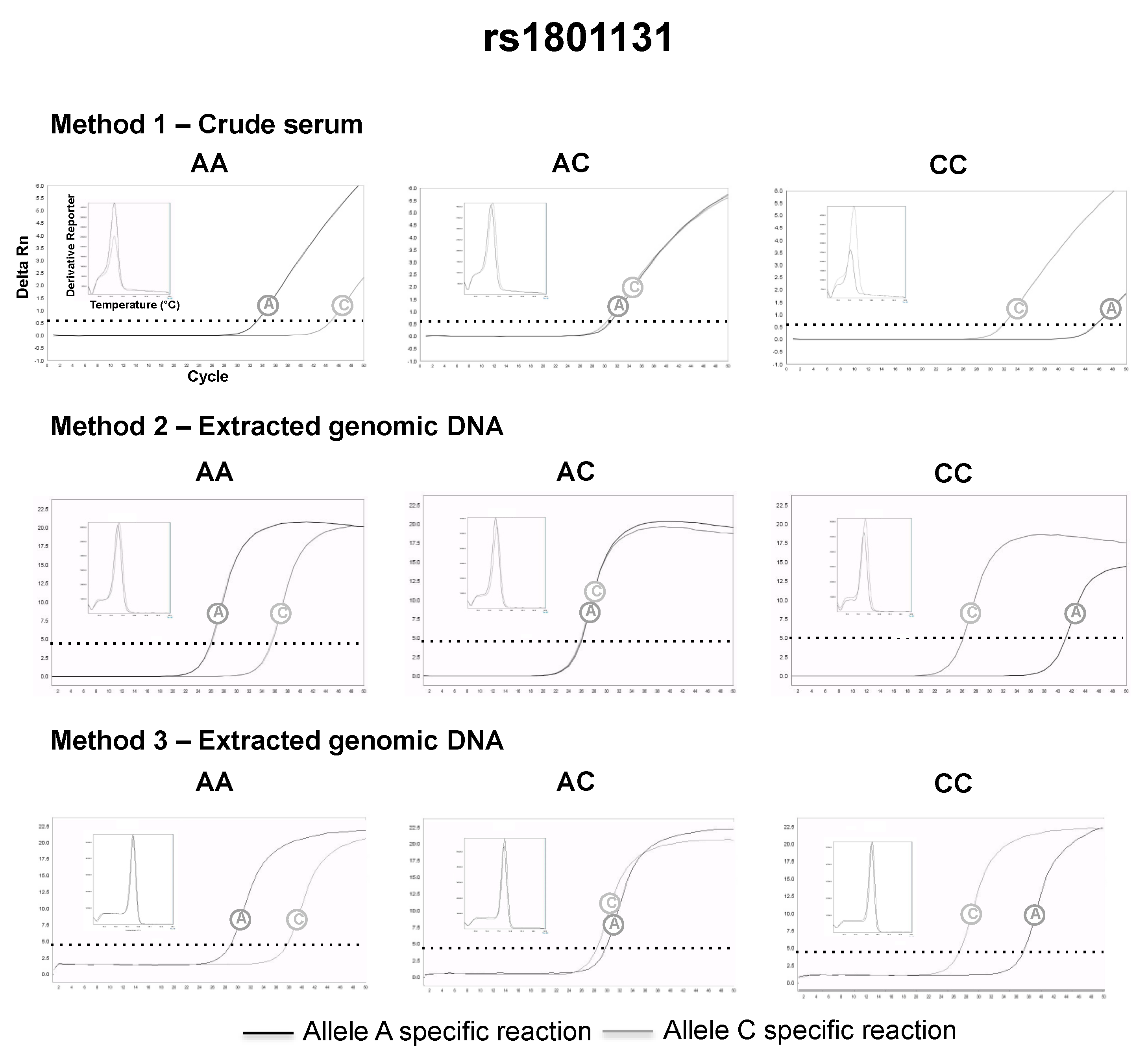
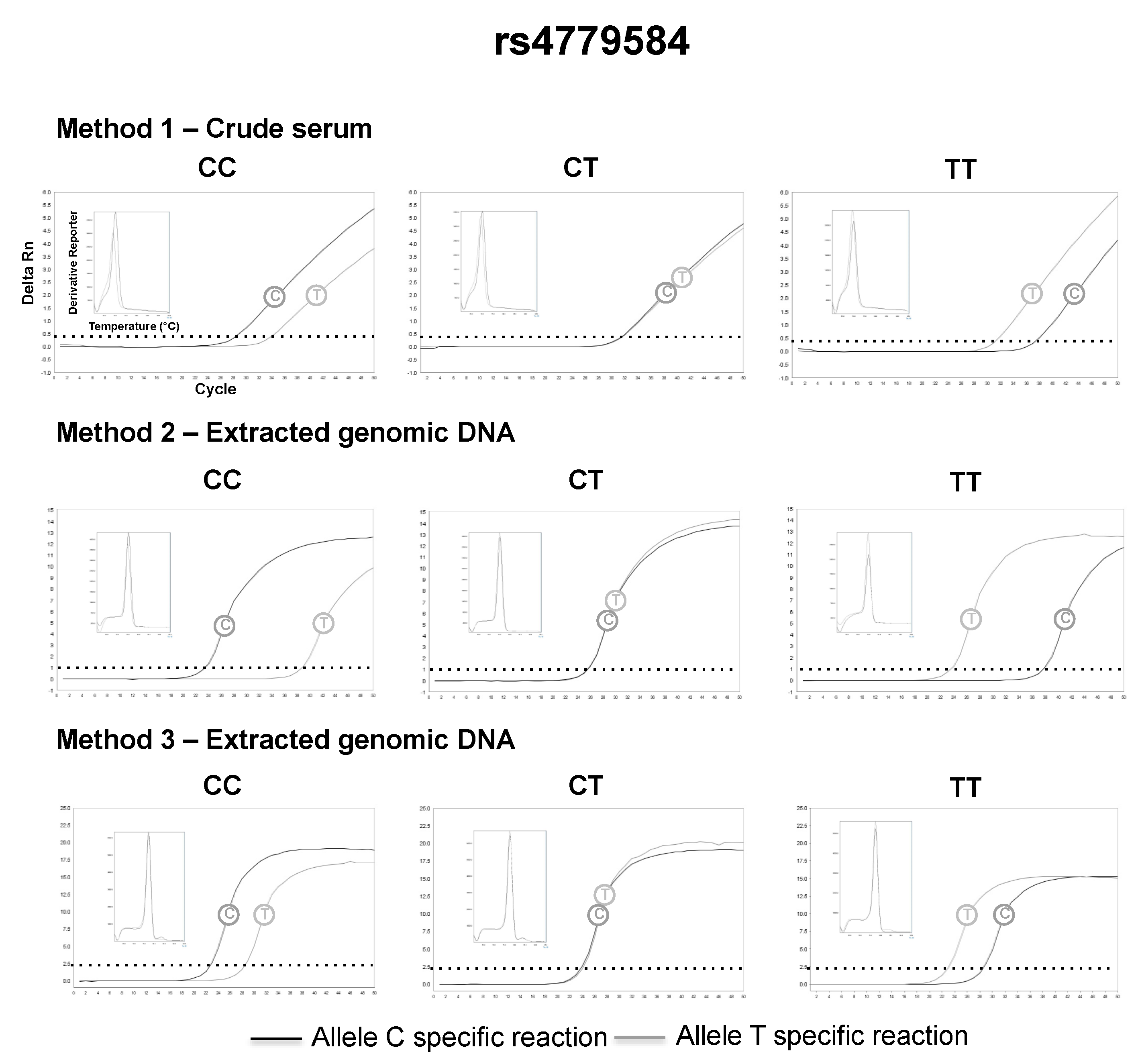
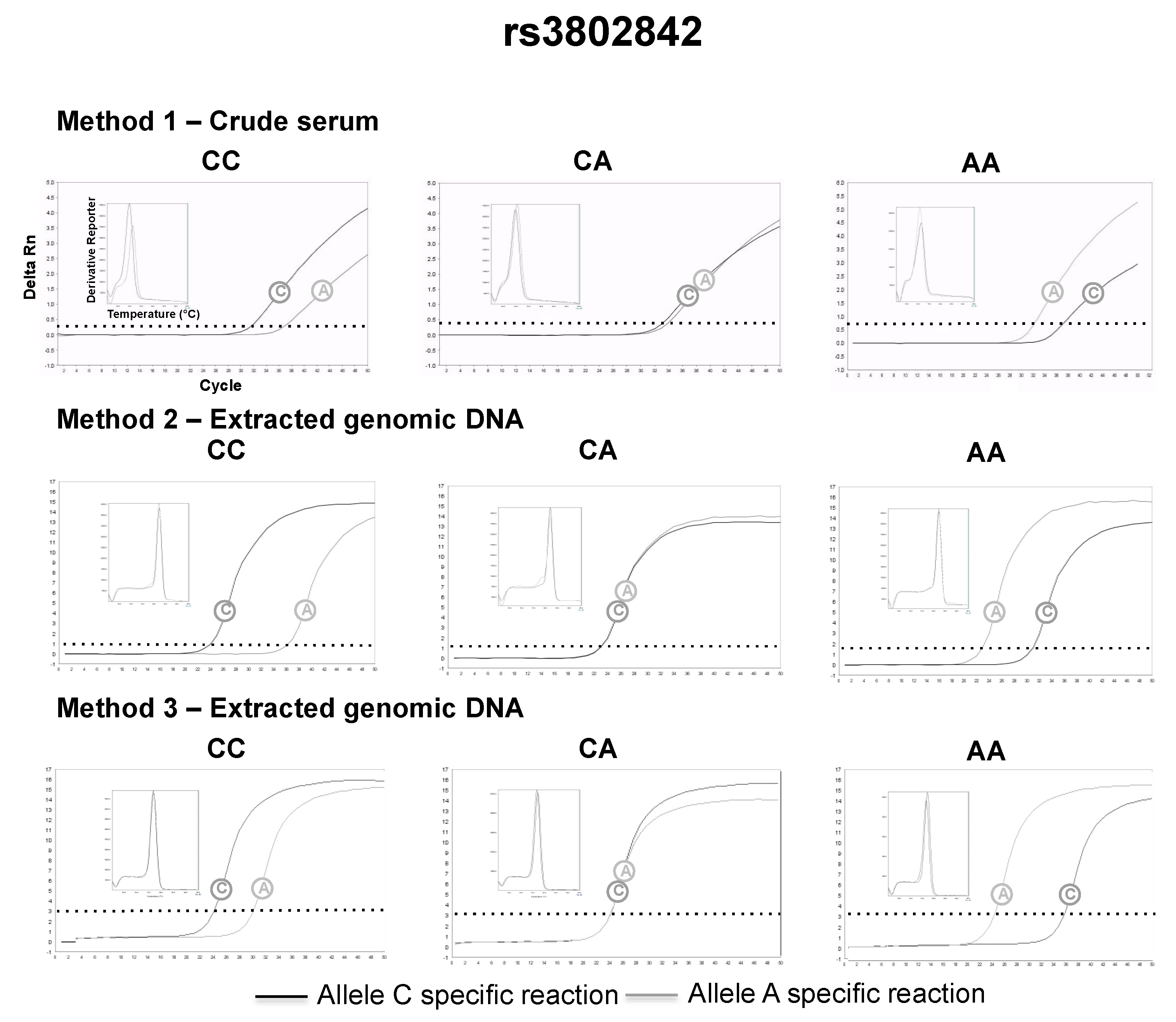
3.2. SNP Genotyping Directly from Crude Capillary Blood Serum Isolated Using a Hand-Powered Paper Centrifuge
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lee, T.H.; Montalvo, L.; Chrebtow, V.; Busch, M.P. Quantitation of genomic DNA in plasma and serum samples: Higher concentrations of genomic DNA found in serum than in plasma. Transfusion 2001, 41, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Wagner, F.F.; Flegel, W.A.; Bittner, R.; Doscher, A. Molecular typing for blood group antigens within 40 min by direct polymerase chain reaction from plasma or serum. Br. J. Haematol. 2017, 176, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Abe, K. Direct PCR from serum: Application to viral genome detection. Methods Mol. Biol. 2003, 226, 161–166. [Google Scholar] [PubMed]
- Bachofen, C.; Willoughby, K.; Zadoks, R.; Burr, P.; Mellor, D.; Russell, G.C. Direct RT-PCR from serum enables fast and cost-effective phylogenetic analysis of bovine viral diarrhoea virus. J. Virol. Methods 2013, 190, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.J.; Hu, L.H.; Fu, W.R.; Li, Y.R. Rapid quantification of hepatitis b virus DNA by direct real-time PCR from serum without DNA extraction. J. Med. Microbiol. 2007, 56, 766–771. [Google Scholar] [CrossRef] [PubMed]
- Kermekchiev, M.B.; Kirilova, L.I.; Vail, E.E.; Barnes, W.M. Mutants of taq DNA polymerase resistant to PCR inhibitors allow DNA amplification from whole blood and crude soil samples. Nucleic Acids Res. 2009, 37, e40. [Google Scholar] [CrossRef]
- Soejima, T.; Xiao, J.Z.; Abe, F. A novel mechanism for direct real-time polymerase chain reaction that does not require DNA isolation from prokaryotic cells. Sci. Rep. 2016, 6, 28000. [Google Scholar] [CrossRef] [Green Version]
- Kang, K.; Yang, K.; Zhong, J.; Tian, Y.; Zhang, L.; Zhai, J.; Zhang, L.; Song, C.; Gou, C.Y.; Luo, J.; et al. A direct real-time polymerase chain reaction assay for rapid high-throughput detection of highly pathogenic north american porcine reproductive and respiratory syndrome virus in china without rna purification. J. Anim. Sci. Biotechnol. 2014, 5, 45. [Google Scholar] [CrossRef]
- Ulvik, A.; Ueland, P.M. Single nucleotide polymorphism (SNP) genotyping in unprocessed whole blood and serum by real-time PCR: Application to snps affecting homocysteine and folate metabolism. Clin. Chem. 2001, 47, 2050–2053. [Google Scholar]
- Abu Al-Soud, W.; Radstrom, P. Effects of amplification facilitators on diagnostic PCR in the presence of blood, feces, and meat. J. Clin. Microbiol. 2000, 38, 4463–4470. [Google Scholar]
- Kreader, C.A. Relief of amplification inhibition in PCR with bovine serum albumin or t4 gene 32 protein. Appl. Environ. Microbiol. 1996, 62, 1102–1106. [Google Scholar] [PubMed]
- Wirth, F.; Zahra, G.; Xuereb, R.G.; Barbara, C.; Fenech, A.; Azzopardi, L.M. Comparison of a rapid point-of-care and two laboratory-based cyp2c19*2 genotyping assays for personalisation of antiplatelet therapy. Int. J. Clin. Pharm. 2016, 38, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Marziliano, N.; Notarangelo, M.F.; Cereda, M.; Caporale, V.; Coppini, L.; Demola, M.A.; Guidorossi, A.; Crocamo, A.; Pigazzani, F.; Boffetti, F.; et al. Rapid and portable, lab-on-chip, point-of-care genotyping for evaluating clopidogrel metabolism. Clin. Chim. Acta Int. J. Clin. Chem. 2015, 451, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Bhamla, M.S.; Benson, B.; Chai, C.; Katsikis, G.; Johri, A.; Prakash, M. Hand-powered ultralow-cost paper centrifuge. Nat. Biomed. Eng. 2017, 1, 0009. [Google Scholar] [CrossRef] [Green Version]
- Barra, G.B.; Santa Rita, T.H.; de Almeida Vasques, J.; Chianca, C.F.; Nery, L.F.; Santana Soares Costa, S. Edta-mediated inhibition of dnases protects circulating cell-free DNA from ex vivo degradation in blood samples. Clin. Biochem. 2015, 48, 976–981. [Google Scholar] [CrossRef]
- Walsh, P.S.; Metzger, D.A.; Higuchi, R. Chelex 100 as a medium for simple extraction of DNA for PCR-based typing from forensic material. BioTechniques 1991, 10, 506–513. [Google Scholar] [CrossRef]
- Sabin Medicina Diagnostica: Melhor Trabalho da Categoria Estudante de Patologia Molecular do AACC. Available online: https://www.youtube.com/watch?v=sYnqwb12R3k (accessed on 8 August 2018).
- Newton, C.R.; Graham, A.; Heptinstall, L.E.; Powell, S.J.; Summers, C.; Kalsheker, N.; Smith, J.C.; Markham, A.F. Analysis of any point mutation in DNA. The amplification refractory mutation system (ARMS). Nucleic Acids Res. 1989, 17, 2503–2516. [Google Scholar] [CrossRef] [Green Version]
- Stadhouders, R.; Pas, S.D.; Anber, J.; Voermans, J.; Mes, T.H.; Schutten, M. The effect of primer-template mismatches on the detection and quantification of nucleic acids using the 5′ nuclease assay. J. Mol. Diagn. JMD 2010, 12, 109–117. [Google Scholar] [CrossRef]
- GraphPad QuickCalcs Online. Available online: https://www.graphpad.com/quickcalcs/kappa1/?K=3 (accessed on 8 August 2018).
- Rodriguez, S.; Gaunt, T.R.; Day, I.N. Hardy-weinberg equilibrium testing of biological ascertainment for mendelian randomization studies. Am. J. Epidemiol. 2009, 169, 505–514. [Google Scholar] [CrossRef]
- Schrader, C.; Schielke, A.; Ellerbroek, L.; Johne, R. Pcr inhibitors—Occurrence, properties and removal. J. Appl. Microbiol. 2012, 113, 1014–1026. [Google Scholar] [CrossRef]
- Staynov, D.Z. Dnase i digestion reveals alternating asymmetrical protection of the nucleosome by the higher order chromatin structure. Nucleic Acids Res. 2000, 28, 3092–3099. [Google Scholar] [CrossRef] [PubMed]
- Nagata, S.; Nagase, H.; Kawane, K.; Mukae, N.; Fukuyama, H. Degradation of chromosomal DNA during apoptosis. Cell Death Differ. 2003, 10, 108–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Amplicon | Primer/Probe Sequence 5′-3′ | Target |
|---|---|---|
| 65 bp | GAGCGGCTGTCTCCACAAGT | |
| HEX/TTCTGACCT/ZEN/GAAGGCTCTGCGCG/IABKFQ | RNase P gene | |
| AGATTTGGACCTGCGAGCG | ||
| 100 bp | GTAGTTTTACTTACTCTCGTCTCCACATAA | |
| HEX/TGAGCAAGC/ZEN/TTTCTCACAAGCATTTGGTTT/3IABKFQ | JAK2 gene | |
| CTTTGAAGCAGCAAGTATGA | ||
| 202 bp | CGCTTTGCTGTGACGCACTT | |
| HEX/CTTGCCGGA/ZEN/CAGACAAAGCGTTTC/IABKFQ | SBP2 gene | |
| GCAGGCGGACGGACTGAG | ||
| 688 bp | ATGGACCCTTGGCTGTCAGAATTA | |
| HEX/AGCAGAAGG/ZEN/TAGAAGGCAAAGCCA/IABKFQ | ALMS gene | |
| AATGGTGTTTCCTCACATGGTCATC |
| Variant | Primer Sequence 5′-3′ | Commentary |
|---|---|---|
| rs1801131 | Method 1 (serum DNA) | |
| GGG AGG AGC TGA CCA GTG AAa A | Reference AS primer | |
| GGG AGG AGC TGA CCA GTG AAa C | Alternative AS primer | |
| GGT AAA GAA CGA AGA CTT CAA AGA CA | Common primer | |
| Method 2 (gDNA) | ||
| GAA CGA AGA CTT CAA AGA CAC TcT | Reference AS primer | |
| GAA CGA AGA CTT CAA AGA CAC TcG | Alternative AS primer | |
| CTC TTC TAC CTG AAG AGC AAG TCC | Common primer | |
| Method 3 (gDNA) | ||
| CAG CAT CAC TCA CTT TGT GAC CAT T | Common primer | |
| Used together with method 1 specific primers | ||
| rs4939827 | Method 1 (serum DNA) | |
| TCA CAG CCT CAT CCA AAA GAG GAA tT | Reference AS primer | |
| TCA CAG CCT CAT CCA AAA GAG GAA tC | Alternative AS primer | |
| AGTCTGAGGGAGCTCTGGGGT | Common primer | |
| Method 2 (gDNA) | ||
| TGA GGG AGC TCT GGG GTC CaA | Reference AS primer | |
| TGA GGG AGC TCT GGG GTC CaG | Alternative AS primer | |
| CCA GTG CCA ATC CAT CCC ATC TAT TC | Common primer | |
| Method 3 (gDNA) | ||
| GTT TCC TCC ATG AGG AAC TCA CTC TAA AC | Common primer | |
| Used together with method 1 specific primers | ||
| rs4779584 | Method 1 (serum DNA) | |
| TCC TGT GTG TAT AGT TAT GGT TTC TGT TgG | Reference AS primer | |
| TCC TGT GTG TAT AGT TAT GGT TTC TGT TgA | Alternative AS primer | |
| CAG TAG AAC TTG TTG ATA AGC CAT TCT TC | Common primer | |
| Method 2 (gDNA) | ||
| CAG TAG AAC TTG TTG ATA AGC CAT TCT TtC | Reference AS primer | |
| CAG TAG AAC TTG TTG ATA AGC CAT TCT TtT | Alternative AS primer | |
| GAT GAG TCC TAA CAA GGA AGG TGAC | Common primer | |
| Method 3 (gDNA) | Common primer | |
| GAG CTG CTA TAA GAT GGG CTG AGT T | ||
| Used together with method 1 specific primers | ||
| rs3802842 | Method 1 (serum DNA) | |
| CCC TAA AAT GAG GTG AAT TTC TGG GtG | Reference AS primer | |
| CCC TAA AAT GAG GTG AAT TTC TGG GtT | Alternative AS primer | |
| CCC TTG CAG ACC CAT AGA AAA TCT | Common primer | |
| Method 2 (gDNA) | ||
| CCC TTG CAG ACC CAT AGA AAA TCc C | Reference AS primer | |
| CCC TTG CAG ACC CAT AGA AAA TCc A | Alternative AS primer | |
| AGG ATG TTC CAC ACA GAT GCT ATC C | Common primer | |
| Method 3 (gDNA) | ||
| CTT CCT CTG CTG TTC CTA TGA CTT C | Common primer | |
| Used together with method 1 specific primers |
| Variant | Gene | Global MAF | Clinical Association | Method 1 (Serum DNA) | Method 2 (gDNA) | Method 3 (gDNA) |
|---|---|---|---|---|---|---|
| rs1801131 | MTHFR | 0.25 | Homocystinuria | 51 bp | 97 bp | 96 bp |
| rs4939827 | SMAD7 | 0.35 | Colorectal cancer | 50 bp | 213 bp | 105 bp |
| rs4779584 | SMAD7 | 0.49 | Colorectal cancer | 59 bp | 191 bp | 147 bp |
| rs3802842 | COLCA1 | 0.28 | Colorectal cancer | 51 bp | 86 bp | 274 bp |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barra, G.B.; Santa Rita, T.H.; Jardim, D.P.; Mesquita, P.G.; Nobre, C.S.; Jácomo, R.H.; Abdalla Nery, L.F. Genotyping of Single Nucleotide Polymorphisms Using Allele-Specific qPCR Producing Amplicons of Small Sizes Directly from Crude Serum Isolated from Capillary Blood by a Hand-Powered Paper Centrifuge. Diagnostics 2019, 9, 9. https://doi.org/10.3390/diagnostics9010009
Barra GB, Santa Rita TH, Jardim DP, Mesquita PG, Nobre CS, Jácomo RH, Abdalla Nery LF. Genotyping of Single Nucleotide Polymorphisms Using Allele-Specific qPCR Producing Amplicons of Small Sizes Directly from Crude Serum Isolated from Capillary Blood by a Hand-Powered Paper Centrifuge. Diagnostics. 2019; 9(1):9. https://doi.org/10.3390/diagnostics9010009
Chicago/Turabian StyleBarra, Gustavo Barcelos, Ticiane Henriques Santa Rita, Daniella Paniago Jardim, Pedro Góes Mesquita, Camila Santos Nobre, Rafael Henriques Jácomo, and Lídia Freire Abdalla Nery. 2019. "Genotyping of Single Nucleotide Polymorphisms Using Allele-Specific qPCR Producing Amplicons of Small Sizes Directly from Crude Serum Isolated from Capillary Blood by a Hand-Powered Paper Centrifuge" Diagnostics 9, no. 1: 9. https://doi.org/10.3390/diagnostics9010009