Abstract
The reductive tricarboxylic acid (rTCA) cycle is among the most plausible candidates for the first autotrophic metabolism in the earliest life. Extant enzymes fixing CO2 in this cycle contain cofactors at the catalytic centers, but it is unlikely that the protein/cofactor system emerged at once in a prebiotic process. Here, we discuss the feasibility of non-enzymatic cofactor-assisted drive of the rTCA reactions in the primitive Earth environments, particularly focusing on the acetyl-CoA conversion to pyruvate. Based on the energetic and mechanistic aspects of this reaction, we propose that the deep-sea hydrothermal vent environments with active electricity generation in the presence of various sulfide catalysts are a promising setting for it to progress. Our view supports the theory of an autotrophic origin of life from primordial carbon assimilation within a sulfide-rich hydrothermal vent.
1. Introduction
The non-enzymatic processing of the reductive tricarboxylic acid (rTCA) cycle-type carbon assimilation has been among the most challenging themes in the field of the origin of life [1,2,3,4]. Various abiotic mechanisms to realize the reaction have been proposed, including the pyruvate formation from carbon monoxide (CO) and cyanide anion (CN−) in the presence of Ni2+ [5], a high pressure condensation of alkyl thiols and formic acid to pyruvate catalyzed by FeS [6], and the photo-electrochemical CO2 reduction and fixation into rTCA compounds on ZnS colloidal semiconductor under UV irradiation [7,8,9]. However, their contributions to life’s origin have been questioned [10] because large discrepancies exist between the proposed mechanisms and the corresponding metabolic processes. In the biological rTCA cycle, CO2 fixation is operated by the two enzyme cofactors (Figure 1): thiamine pyrophosphate (TPP) assists the conversion of acetyl-CoA to pyruvate and succinyl-CoA to α-ketoglutarate [11,12], whereas biotin mediates the formations of oxaloacetate and oxalosuccinate from pyruvate and α-ketoglutarate, respectively [13,14]. The two cofactors have been deduced to participate in autotrophic metabolism from the very beginning of the life’s evolution, at least from the stage of the last universal common ancestor (LUCA) that could have lived in deep-sea hydrothermal systems [15]. Remarkably, replacement of heteroatoms in their ring structures with others (e.g., O or N vs. S) does not inactivate, or in some cases even improves, their functional properties [16,17,18]. Various heterocyclic compounds with structural features resembling the two have been synthesized under simulated primitive environmental conditions [19,20,21]. Therefore, an alternative possibility is that prebiotic analogs of TPP and biotin with simpler structures that are initially formed via inorganic processes, facilitated the primordial carbon fixation that preceded the origin of life, were incorporated into proto-enzymes in the course of functional evolution, and eventually developed into the modern counterparts.
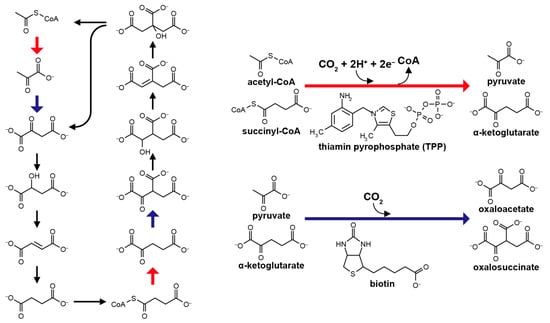
Figure 1.
Structure of the reductive tricarboxylic acid (rTCA) cycle (left), in which the CO2 fixations leading to the pyruvate and α-ketoglutarate formations are mediated by thiamin pyrophosphate (TPP), whereas to the oxaloacetate and oxalosuccinate formations are by biotin (right).
In this manuscript, we discuss the feasibility of this scenario with a special attention to the second part; the non-enzymatic cofactor-assisted CO2 fixation. Our study focused on the acetyl-CoA conversion to pyruvate on TPP because thiolated acetate derivatives (thioacids; R-COSH, thioesters; R-COS-R’), plausible ancient forms of acetyl-CoA [22], were possibly present on the primitive Earth [23,24]. Although recent geochemical surveys of the present-day submarine hydrothermal fields observed no evidence of their abiotic formations [25,26,27], the results do not necessarily deny their presence in the Hadean ocean hydrothermal ones because the geological situations are likely largely different from each other. For instance, it has been shown that a high-temperature basalt–seawater interaction in a CO2-rich condition results in the increase of solution pH to highly alkaline (pH ≥ 12; [28,29]). Owing to a denser distribution of metals in the ancient deep-ocean [30,31] derived from much greater hydrothermal activity than the present level [32], the alkaline fluid–seawater mixing in the early basalt-hosted hydrothermal systems could have precipitated metal sulfides as the main body of hydrothermal mineral deposits [33]. This environmental setting favors the abiotic production of thioester [24]. The thioester/thioacid conversion to pyruvate corresponds to the initial step of the rTCA cycle. Thus, no development of the subsequent proto-metabolism is expected unless an effective geochemical route to the pyruvate formation was established. Citrate can be a source of oxaloacetate and pyruvate [34], but a proposed abiotic synthesis of citrate requires pyruvate [35]. Note that a simple heating of thioacids and thioesters in water in a range of temperature and pH results in the hydrolysis of the thioester bond [36,37], and no experimental evidence has been reported for the mineral-promoted CO2 fixation into them in the prebiotic context [38], although approximately three decades have passed since the possibility was first proposed [1,39]. These facts motivated us to search the organic catalysts for the initiation of the primordial carbon assimilation.
2. Energetics of Pyruvate Synthesis
We initially examine the energetics of pyruvate synthesis using ethylthioacetate (ETA) as a prebiotic counterpart of acetyl-CoA to clarify the environmental condition necessary for it to be driven thermodynamically. Figure 2 shows the calculated Eh-pH relationship of the pyruvate formation (ETA + CO2 + H+ + 2e− → pyruvate + ethanethiol (EtSH)), together with those of the H2/H+, H2S/S and mackinawite/pyrite (FeS/FeS2) redox couples, at 25, 60, and 100 °C (see Appendix A for the calculation procedure). S (solid sulfur) is used as the H2S oxidation product because the H2S/S redox couple provides a major potential control in the sulfide-rich hydrothermal vent environments [40]. The concentrations of H2 and H2S were assumed to be 1 mmol·kg−1, whereas that of CO2 to be 20 mmol·kg−1. 1 mmol·kg−1 is the representative H2 and H2S concentrations in the serpentine-hosted hydrothermal systems on land [41] and on the ocean-floor [42] that have been argued to be the most plausible settings for the origin of life [33,43,44], whereas 20 mmol·kg−1 corresponds to the steady-state CO2 concentration in the early ocean [45,46]. For organic compounds, 0.1 mmol·kg−1 was arbitrarily chosen because of no definitive constraint; calculations with different initial settings (Figure B1) showed that higher organics’ concentrations result in slightly lower Eh values.
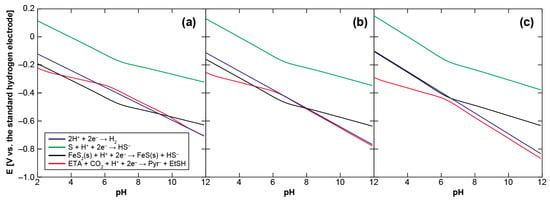
Figure 2.
Eh-pH relationships of the pyruvate formation from ethylthioacetate and CO2 (red) and of the H2/H+ (blue), H2S/S (green) and mackinawite/pyrite (black) redox couples at (a) 25, (b) 60, and (c) 100 °C. See text and Appendix A for the calculation conditions and procedures.
It can be seen in Figure 2 that H2S does not generate the potentials required to drive the pyruvate formation over the examined aqueous conditions, while the lines of the H2/H+ and FeS/FeS2 redox couples intersect with the threshold. At 25 °C, the H2 and FeS oxidations provide favorable conditions for the CO2 fixation at pH 5.5–10.5 and 3–10, respectively. The pH ranges gradually shrink at higher temperature owing to the negative shift of the necessary potential with an increasing temperature. H2 loses its thermodynamic advantage at around 60 °C (Figure 2b), whereas FeS does at around 100 °C (Figure 2c). The H2 and FeS-driven pyruvate syntheses are therefore energetically possible only in a cool to warm and near neutral aqueous solution. Plausible conditions for the accumulation of pyruvate to a proto-metabolically significant extant may be further restricted by the unstable character of pyruvate in acidic pH [47].
3. TPP-Assisted Pyruvate Synthesis: A Mechanism
Then, is pyruvate producible non-enzymatically under sufficiently reductive conditions, such as nearby a H2-rich hydrothermal vent discharging FeS precipitate continuously, aided by TPP or its prebiotic analogs? Note that the direct coupling of FeS oxidation with CO2 reduction and fixation is unlikely to occur due to the high activation energy even when the overall process is thermodynamically favorable [48].
In the biological rTCA cycle, pyruvate synthesis is catalyzed by an iron-sulfur enzyme called pyruvate:ferredoxin oxidoreductase (PFOR), or 2-oxoacid:ferrdoxin oxidoreductase (OFOR) [11,49,50,51]. The catalytic center of all the known enzymes contains TPP as an essential cofactor for the one-carbon transfer. The process starts with the deprotonation of the C2 carbon in the thiazolium ring, followed by the nucleophilic attack of the resulting carbanion on the carbonyl carbon of acetyl-CoA to form a tetrahedral intermediate (Figure 3). The intermediate then undergoes CoA release, and one electron transfer from a reduced iron-sulfur cluster yields the hydroxyethyl-TPP (HE-TPP) radical. A second electron addition reduces it to the HE-TPP C2α carbanion, and its nucleophilic attack to CO2 makes lactyl-TPP that finally produces pyruvate.
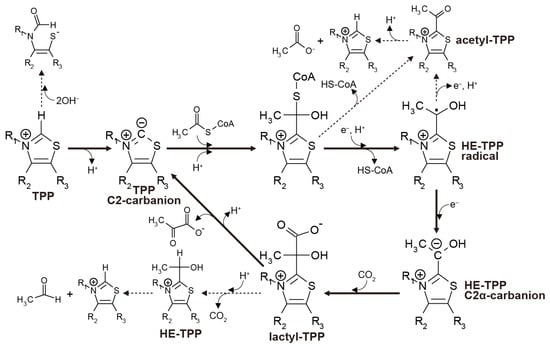
Figure 3.
Thiamin pyrophosphate (TPP)-assisted pyruvate formation operated in pyruvate:ferredoxin oxidoreductase (PFOR) (solid arrows) illustrated on the basis of the reported models [11,39,40,41] and the side reactions that disrupt the overall process (dashed arrows).
The stability and reactivity of the reaction intermediates have been examined using TPP that is unbound to enzymes. The proton dissociation from the thiazolium C2 position occurs with the pKa of 17–19 [52], while alkaline pH (>9.40) favors the opening of the thiazolium ring [53]. Acetyl-TPP, a one-electron oxidation product of the HE-TPP radical [54], hydrolyzes rapidly to acetate and TPP at neutral and alkaline pH (e.g., t1/2 = 58 s at pH 7.0 and 24 °C [55]). The pyruvate release from lactyl-TPP competes with the decarboxylation of lactyl-TPP to HE-TPP; the decarboxylation predominates at pH < 9.5 (25 °C) [56]. When the sulfur atom in the thiazolium ring is replaced with nitrogen, it increases the stability against ring-opening, while it suppresses the ylide formation [16].
In PFOR and OFOR, a conserved Glu residue stimulates the deprotonation of the thiazolium C2 at the TPP-binding site with a low dielectric constant (εr = 13–15 [57]). Electrons are provided by [4Fe-4S] ferredoxins and are transported from the external enzyme surface to the catalytic center via optimally arranged single or multiple [4Fe-4S] cluster(s) [11]. The proximal [4Fe-4S] cluster that locates within 15 Å from TPP [58,59,60] allows for rapid electron transfer to the adducts of TPP immediately after they are formed, thereby prohibiting the intermediates from decaying. Although the bacterial and archaeal enzymes differ in the subunit composition and overall structure, the proximal [4Fe-4S] cluster is conserved [60], indicating the importance of this electron transfer pathway in the enzymatic processes. The [4Fe-4S] cluster possesses the potential as low as −540 mV (vs. standard hydrogen electrode; SHE [61]) that is sufficiently low to drive the energetically up-hill acetyl-CoA carboxylation.
4. Discussion: Feasibility of Abiotic Pyruvate Synthesis in a Geological Setting
The above summary clearly indicates that the TPP-assisted pyruvate formation never takes place in single aqueous condition. In contrast, at the mineral-water interface with a low dielectric constant (εr = 26–53 [62]), the thiazolium ylide and the HE-TPP carbanion are expected to be stabilized significantly [57], while such condition accelerates the decarboxylation of lactyl-TPP [56,63]. It has been shown that imidazolium species, which contain nitrogen atom at the place of sulfur in the thiazolium structure, effectively catalyze the CO2 reduction to CO and formate on FeS2, and to ethylene glycol on gold under an externally applied negative electric potential (at −0.85 V (vs. SHE)) [64,65]. The CO2 activation was inferred to be induced by the imidazolium ylide formation on the negatively charged electrodes, followed by the CO2 binding at the C2 position [64,65]. FeS could also provide reactive surface and electric energy by coupling with its oxidation to FeS2, as was demonstrated for the H2S reduction to H2 [66,67], nitrogen oxides to ammonia [68,69], ethyne to ethane, and ethane [70], and the reductive amination of α-keto acids [71]. Interestingly, freshly precipitated FeS has the point of zero charge (pHZPC) of around 7.5 [72], whereas the pHZPC of FeS2 is around 1.5 [73,74]. FeS is thus expected to provide a wide range of surface pH in the course of its oxidation even under a constant aqueous condition, and controls the speciation of adsorbed molecules [75,76,77]. A drawback of the FeS-driven CO2 fixation is that the electron supply ceases when the FeS surface is fully oxidized. Organic molecules thus need to be transported onto fresh FeS via diffusion and/or convection to continue their reductions.
Wider and diverse electrochemical environments are available in sulfide-rich hydrothermal systems on the ocean floor [78], where the potential gradient between the hydrothermal fluids and seawater across the sulfide deposits drives the flow of electric current, and promotes redox reactions at the vent-seawater interface by the continuous electron supply in the presence of various mineral catalysts (Figure 4) [40,79,80]. If 1 mmol kg−1 H2 in hot and alkaline hydrothermal fluids serves as the electron source, it generates the potential (e.g., −0.84 V (vs. SHE) at 100 °C and pH 12) well below the desired value for the pyruvate synthesis at 25 °C and slightly acidic to neutral pH (−0.3~−0.4 V (vs. SHE); Figure 2). Water molecules in an external electric field have a low dielectric property [81,82]. The geo-electrochemical setting thus could provide reaction conditions resembling the electron transfer system in PFOR and OFOR in terms of the direct donation of low-potential electrons from metal-sulfur clusters to the catalytic center with a low dielectric constant.
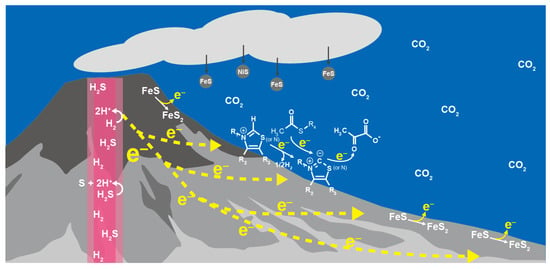
Figure 4.
Geo-electrochemical pyruvate formation in the early ocean hydrothermal vent environment as a possible initial step of the primordial carbon fixation.
The hydrothermal setting also favors the abiotic amino acid synthesis [83,84] and polymerization [19,85,86,87]. Amino acids and short peptides in some cases improve the stability and activity of electrocatalysts [88,89,90,91]. Peptides with 10–20 monomers long have the capability of recognizing TPP [92] and biotin [93]. These evidences may imply an early-stage interaction of peptides and cofactors near the vent surface that could have played a positive role in the selective and efficient progress of the primordial carbon assimilation [94,95,96]. A conclusion for the abiotic origin of the TPP-mediated pyruvate synthesis must await the time when the aforementioned possibilities are experimentally tested under simulated primordial geo-electrochemical conditions. As a future experimental study, it is of particular importance to examine whether prebiotically producible heterocyclic compounds with structural features resembling TPP [19,20,21] can assist the CO2 fixation in the proposed environment. The pyrimidine and pyrophosphate parts of TPP may be replaced with simpler structures (e.g., –CH3) without deactivation, and imidazolium and perhaps oxazolium rings could serve as electron carriers in a manner similar to the thiazolium one in TPP. If primitive analogs of TPP can catalytically provide pyruvate in a geological setting, the situation will also favor the C4 to C5 conversion (i.e., succinyl-CoA → α-ketoglutarate; Figure 1). Although extant organisms employ two distinct enzymes for the pyruvate and α-ketoglutarate syntheses, the primordial system could have used a single catalyst for the two reactions and have later developed the optimally-tuned enzymes for each, as was proposed for the evolution of many enzymes [97]. For the other CO2 fixation steps (pyruvate → oxaloacetate, α-ketoglutarate → oxalosuccinate; Figure 1), the energetically most difficult process is the tautomerization of reactants from the keto to the enol forms [98]. This problem may be overcome by borate [10,99] given the substantial amounts of boron released into ocean in the course of the early submarine hydrothermal activities (1.8–4.5 × 1010 mol·year−1 [100]) and its accumulation on seafloor clay minerals [101]. Alternatively, there is a possibility that the TPP and biotin-mediated system is a genuine biological invention with no relic of the relevant prebiotic processes [102]. Without these cofactors, no effective CO2 fixation through the rTCA cycle is expected, and thus, the early autotrophs would have had completely different metabolic strategies from those as we know [103]. In either case, the origin of life scenario must connect smoothly the current and progressively updated knowledge of the ancient geochemistry and biochemistry [104].
Finally, we discuss the suitability of other inorganic carbon compounds than CO2 as a precursor for the abiotic pyruvate production. The reaction could be facilitated in the presence of aldehydes because the usage of acetaldehyde instead of acetyl-CoA skips the route of the unstable HE-TPP radical formation [105]. Aldehydes also serve as a carbon source of thioesters through the oxidative coupling with thiols [106,107], and the reaction is catalyzed by thiazolium compounds [108]. However, the availability of aldehydes in the early-ocean hydrothermal systems remains controversial [109,110,111]. Formate may be a more realistic C1 source because formate has occasionally been observed in the present-day hydrothermal systems with high concentrations of up to ~0.7 mM [27,112,113]. Actually, the enzyme “pyruvate formate-lyase (PFL)” catalyzes the reversible conversion of pyruvate and CoA into acetyl-CoA and formate; the system plays a central role in anaerobic glucose fermentation in diverse Eukarya and Bacteria [114,115]. A drawback of the formate fixation is that it is a highly thermodynamically up-hill reaction (the standard Gibbs energy of reaction (∆rGo) = ~−21 kJ·mol−1; [116]) with the equilibrium constant of 2 × 10−4. The net PFL reaction is neither oxidation nor reduction; hence the energy barrier cannot be overcome by the geo-electrochemical mechanism discussed above. The low reactivity of formate, which is a much poorer electrophile than CO2 [117], is another problem to be solved. PFL activates the formate condensation by a radical mechanism using two cysteine and one glycine residues as radical carriers [118,119,120]. It is unclear whether such a radical process can be operated non-enzymatically in water or on minerals with the aid of prebiotically available short peptides.
5. Concluding Remarks
Abiotic CO2 fixation is among the most fundamental steps for life to originate, but no geochemically feasible process that drives the reaction has been acknowledged [121]. If the above-discussed mechanism occurs with prebiotically producible cofactor analogs, favorable conditions for it to progress could have distributed widely on the early ocean floor because the global thermal convection at that time is considered to be much greater than the present level [32]. It can also be envisioned that the geochemical CO2 fixation is a common phenomenon on terrestrial planets and satellites because hydrothermal activity is widespread in our solar system including on Europa, Enceladus, and the ancient Mars [122,123,124]. Future experimental study that mimics the conditions of the proposed model is expected to provide insights into the universality of autotrophic metabolism and its underpinning life systems in the cosmos.
Acknowledgments
This research was supported by JSPS KAKENHI (Grant Numbers; 16H04074, 16K13906, and JP26106003), and the Astrobiology Center Program of NINS (Grant Number; AB292004). As for K.F., this publication was supported by the ELSI Origins Network (EON), through a grant from the John Templeton Foundation.
Author Contributions
N.K. conceived and designed this study. N.K., M.K. and K.F. searched the chemical and biochemical aspects of the TPP-catalyzed pyruvate synthesis. N.K. conducted the thermodynamic calculation. All authors contributed to writing the paper.
Conflicts of Interest
The authors declare no conflict of interest.
Appendix A
The Eh-pH relationship of the ethylthioacetate conversion to pyruvate, and of the H2/H+, H2S/S and FeS/FeS2 redox couples (Figure 2) were calculated, respectively, using the following equations:
and
In these equations, T, R, and F stand for temperature in kelvin, the gas constant (8.31447 J·mol−1·K−1), and the Faraday constant (96,485 J mol−1 V−1), respectively. ai represents the activity of the species i. x, y, z, n, and m signify the mole fraction of H2S (), pyruvic acid (), ethanethiol (), CO2 (), and HCO3− (), respectively (Mi denotes the molality of the species i). In addition, ∆fGo(i) represents the standard molal Gibbs energy of formation of the species i at desired temperature, which were calculated according to the revised HKF equations of state [125] together with the thermodynamic data and the revised HKF parameters reported in [126] for H2 and H2S, in [127] for HS−, in [128] for ethylthioacetate, pyruvate and pyruvic acid, and in [129] for ethanethiol. The ∆fGo value for ethanethiol anion (EtS−) was estimated using the value of ∆fGo for ethanethiol in combination with its ionization constant as a function of temperature [130]. The temperature dependences of ∆fGo for S (solid sulfur) and FeS2 were calculated as follows:
where and , respectively, represent the standard molal Gibbs energy and entropy at the reference temperature (Tr = 298.15 K) and pressure (Pr = 1 bar). represents the standard molal heat capacity at Pr, and denotes the standard molal volume at the temperature of interest. In the present calculation, the values of , , and as a function of temperature for S and FeS2 were taken from [131] and [132], respectively, while the value of was assumed to be constant in the range of temperature of our interest. The ∆fGo for FeS was estimated from the equilibrium constant of FeS dissolution (FeS + 2H+ → Fe2+ + H2S [133]) together with the ∆fGo for Fe2+ (referred from [127]) and for H2S. The values of x, y, z, n, and m are expressed, respectively, as:
and
Therein, γi represents the activity coefficient of the species i (ai = Mi × γi) and Ki the dissociation constant of i (i → i− + H+), whose values were calculated as:
for H2S, pyruvic acid, and ethanethiol, and as:
and
for CO2. The ∆fGo for H2O was referred from [134]. In all calculations, the values of γi were calculated with the extended Debye–Hückel equation [135] setting the ionic strength to be 0.1 (NaCl). The pressure was set to 1 bar. S2− and the ion pair NaHS were not considered in this calculation because these are expected to be minor S and/or Na species in the examined aqueous conditions [136,137].


References
- Wächtershäuser, G. Evolution of the first metabolic cycles. Proc. Natl. Acad. Sci. USA 1990, 87, 200–204. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.; Morowitz, H.J. Universality in intermediary metabolism. Proc. Natl. Acad. Sci. USA 2004, 101, 13168–13173. [Google Scholar] [CrossRef] [PubMed]
- Orgel, L.E. The implausibility of metabolic cycles on the prebiotic earth. PLoS Biol. 2008, 6, e18. [Google Scholar] [CrossRef] [PubMed]
- Kitadai, N.; Aono, M.; Oono, Y. Origin of metabolism; a perspective. Chikyukagaku 2016, 50, 155–176. [Google Scholar]
- Huber, C.; Wächtershäuser, G. α-hydroxyl and α-amino acids under possible Hadean, volcanic origin-of-life conditions. Science 2006, 314, 630–632. [Google Scholar] [CrossRef] [PubMed]
- Cody, G.D.; Boctor, N.Z.; Filley, T.R.; Hazen, R.M.; Scott, J.H.; Sharma, A.; Yoder, H.S., Jr. Primordial carbonylated iron-sulfur compounds and the synthesis of pyruvate. Science 2000, 289, 1337–1340. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.V.; Martin, S.T. Driving parts of Krebs cycle in reverse through mineral photochemistry. J. Am. Chem. Soc. 2006, 128, 16032–16033. [Google Scholar] [CrossRef] [PubMed]
- Guzman, M.I.; Martin, S.T. Prebiotic metabolism: Production by mineral photoelectrochemistry of α-ketocarboxylic acids in the reductive tricarboxylic acid cycle. Astrobiology 2009, 9, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Guzman, M.I.; Martin, S.T. Photo-production of lactate from glyoxylate: How minerals can facilitate energy storage in a prebiotic world. Chem. Commun. 2010, 46, 2265–2267. [Google Scholar] [CrossRef] [PubMed]
- Aono, M.; Kitadai, N.; Oono, Y. A principled approach to the origin problem. Orig. Life Evol. Biosph. 2015, 45, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Ragsdale, S.W. Pyruvate ferredoxin oxidoreductase and its radical intermediate. Chem. Rev. 2003, 103, 2333–2346. [Google Scholar] [CrossRef] [PubMed]
- Kluger, R.; Tittmann, K. Thiamin diphosphate catalysis: Enzymic and nonenzymic covalent intermediates. Chem. Rev. 2008, 108, 1797–1833. [Google Scholar] [CrossRef] [PubMed]
- Aoshima, M.; Ishii, M.; Igarashi, Y. A novel biotin protein required for reductive carboxylation of 2-oxoglutarate by isocitrate dehydrogenase in Hydrogenobacter thermophiles TK-6. Mol. Microbiol. 2004, 51, 791–798. [Google Scholar] [CrossRef] [PubMed]
- Waldrop, G.L.; Holden, H.M.; Maurice, M.S. The enzymes of biotin dependent CO2 metabolism: What structures reveal about their reaction mechanisms. Protein Sci. 2012, 21, 1597–1619. [Google Scholar] [CrossRef] [PubMed]
- Weiss, M.C.; Sousa, F.L.; Mrnjavac, N.; Neukirchen, S.; Roettger, M.; Nelson-Sathi, S.; Martin, W.F. The physiologi and habitat of the last universal common ancestor. Nat. Microbiol. 2016, 1, 16116. [Google Scholar] [CrossRef] [PubMed]
- Duclos, J.M.; Haake, P. Ring opening of thiamine analogs. The role of ring opening in physiological function. Biochemistry 1974, 13, 5358–5362. [Google Scholar] [CrossRef] [PubMed]
- DeTitta, G.T.; Blessing, R.H.; Moss, G.R.; King, H.F.; Sukumaran, D.K.; Roskwitalski, R.L. Inherent conformation of the biotin bicyclic moiety: Searching for a role for sulfur. J. Am. Chem. Soc. 1994, 116, 6485–6493. [Google Scholar] [CrossRef]
- Holloczki, O.; Kelemen, Z.; Nyulaszi, L. On the organocatalytic activity of N-heterocyclic carbenes: Role of sulfur in thiamine. J. Org. Chem. 2012, 77, 6014–6022. [Google Scholar] [CrossRef] [PubMed]
- Huber, C.; Wächtershäuser, G. Peptide by activation of amino acids with CO on (Ni,Fe)S surfaces: Implications for the origin of life. Science 1998, 281, 670–672. [Google Scholar] [CrossRef] [PubMed]
- Orgel, L.E. Prebiotic chemistry and the origin of the RNA world. Crit. Rev. Biochem. Mol. Biol. 2004, 39, 99–123. [Google Scholar] [PubMed]
- Vazquez-Salazar, A.; Tan, G.; Stockton, A.; Fani, R.; Becerra, A.; Lazcano, A. Can an imidazole be formed from an alanyl-seryl-glycine tripeptide under possible prebiotic conditions? Orig. Life Evol. Biosph. 2017. [Google Scholar] [CrossRef] [PubMed]
- De Dube, C. Clues from present-day biology: The thioester world. In The Molecular Origins of Life; Brack, A., Ed.; Cambridge University Press: Brussels, Belgium, 1998; pp. 219–236. [Google Scholar]
- Weber, A.L. Nonenzymatic formation of “energy-rich” lactoyl and glyceroyl thioesters from glyceraldehyde and a thiol. J. Mol. Evol. 1984, 20, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Huber, C.; Wächtershäuser, G. Activated acetic acid by carbon fixation on (Fe,Ni)S under primordial conditions. Science 1997, 276, 245–247. [Google Scholar] [CrossRef] [PubMed]
- Konn, C.; Charlou, J.L.; Donval, J.P.; Holm, N.G.; Dehairs, F.; Bouillon, S. Hydrocarbons and oxidized organic compounds in hydrothermal fluids from Rainbow and Lost City ultramafic-hosted vents. Chem. Geol. 2009, 258, 299–314. [Google Scholar] [CrossRef]
- Reeves, E.P.; McDermott, J.M.; Seewald, J.S. The origin of methanethiol in midocean ridge hydrothermal fluids. Proc. Natl. Acad. Sci. USA 2014, 111, 5474–5479. [Google Scholar] [CrossRef] [PubMed]
- McDermott, J.M.; Seewald, J.S.; German, C.R.; Sylva, S.P. Pathways for abiotic organic synthesis at submarine hydrothermal fields. Proc. Natl. Acad. Sci. USA 2015, 112, 7668–7672. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, T.; Komiya, T.; Nakamura, K.; Takai, K.; Maruyama, S. Highly alkaline, high-temperature hydrothermal fluids in the early Archean ocean. Precambrian Res. 2010, 182, 230–238. [Google Scholar] [CrossRef]
- Shibuya, T.; Yoshizaki, M.; Masaki, Y.; Suzuki, K.; Takai, K.; Russell, M.J. Reactions between basalt and CO2-rich seawater at 250 and 350 °C, 500 bars: Implications for the CO2 sequestration into the modern ocean crust and the composition of hydrothermal vent fluid in the CO2-rich early ocean. Chem. Geol. 2013, 359, 1–9. [Google Scholar] [CrossRef]
- Poulton, S.W.; Canfield, D.E. Ferruginous conditions: A dominant feature of the ocean through Earth’s history. Elements 2011, 7, 107–112. [Google Scholar] [CrossRef]
- Scott, C.; Planavsky, N.J.; Dupont, C.L.; Kendall, B.; Gill, B.C.; Robbins, L.J.; Husband, K.F.; Arnold, G.L.; Wing, B.A.; Poulton, S.W.; et al. Bioavailability of zinc in marine systems through time. Nat. Geosci. 2013, 6, 125–128. [Google Scholar] [CrossRef]
- Lowell, R.P.; Keller, S.M. High-temperature seafloor hydrothermal circulation over geologic time and Archean banded iron formations. Geophys. Res. Lett. 2003, 30, 1391. [Google Scholar] [CrossRef]
- Russell, M.J.; Hall, A.J. The emergence of life from iron monosulphide bubbles at a submarine hydrothermal redox and pH front. J. Geol. Soc. Lond. 1997, 154, 377–402. [Google Scholar] [CrossRef]
- Cody, G.D.; Boctor, N.Z.; Hazen, R.M.; Brandes, J.A.; Morowitz, H.J.; Yoder, H.S., Jr. Geochemical roots of autotrophic carbon fixation: Hydrothermal experiments in the system citric acid, H2O-(±FeS)-(±NiS). Geochim. Cosmochim. Acta 2001, 65, 3557–3576. [Google Scholar] [CrossRef]
- Cooper, G.; Reed, C.; Nguyen, D.; Carter, M.; Wang, Y. Detection and formation scenario of citric acid, pyruvic acid, and other possible metabolism precursors in carbonaceous meteorites. Proc. Natl. Acad. Sci. USA 2011, 108, 14015–14020. [Google Scholar] [CrossRef] [PubMed]
- Bracher, P.J.; Snyder, P.W.; Bohall, B.R.; Whitesides, G.M. The relative rates of thiol–thioester exchange and hydrolysis for alkyl and aryl thioalkanoates in water. Orig. Life Evol. Biosph. 2011, 41, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Chandru, K.; Gillbert, A.; Butch, C.; Aono, M.; Cleaves, H.J., II. The abiotic chemistry of thiolated acetic derivatives and the origin of life. Sci. Rep. 2016, 6, 29883. [Google Scholar] [CrossRef] [PubMed]
- Komeda, N.; Nagao, H.; Matsui, T.; Adachi, G.; Tanaka, K. Electrochemical carbon dioxide fixation to thioesters catalyzed by [Mo2Fe6S8(Set)9]3−. J. Am. Chem. Soc. 1992, 114, 3625–3630. [Google Scholar] [CrossRef]
- Camprubi, E.; Jordan, S.F.; Vasiliadou, R.; Lane, N. Iron catalysts at the origin of life. IUBMB Life 2017, 69, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Nakamura, R.; Oguri, K.; Kawagucci, S.; Suzuki, K.; Hashimoto, K.; Takai, K. Generation of electricity and illumination by an environmental fuel cell in deep-sea hydrothermal vents. Angew. Chem. Int. Ed. 2013, 52, 10758–10761. [Google Scholar] [CrossRef] [PubMed]
- Schrenk, M.O.; Brazelton, W.J.; Lang, S.Q. Serpentinization, carbon and deep life. Rev. Mineral. Geochem. 2013, 75, 575–606. [Google Scholar] [CrossRef]
- Tivey, M.K. Generation of seafloor hydrothermal vent fluids and associated mineral deposits. Oceanography 2007, 20, 50–65. [Google Scholar] [CrossRef]
- Takai, K.; Nakamura, K.; Suzuki, K.; Inagaki, F.; Nealson, K.H.; Kumagai, H. Ultramafics-Hydrothermalism-Hydrogenesis-HyperSLiME (UltraH3) linkage: A key insight into early microbial ecosystem in the Archean deep-sea hydrothermal systems. Paleontol. Res. 2006, 10, 269–282. [Google Scholar] [CrossRef]
- Mulkidjanian, A.Y.; Bychkov, A.Y.; Dibrova, D.V.; Galperin, M.Y.; Koonin, E.V. Origin of first cells at terrestrial, anoxic geothermal fields. Proc. Natl. Acad. Sci. USA 2012, 109, E821–E830. [Google Scholar] [CrossRef] [PubMed]
- Amend, J.P.; McCollom, T.M. Energetics of biomolecule synthesis on early earth. In Chemical Evolution II: From the Origin of Life to Modern Society; Zaikowski, L., Friedrich, J.M., Seidel, S.R., Eds.; American Chemical Society: Washington, DC, USA, 2009; pp. 63–94. ISBN 9780841269804. [Google Scholar]
- Shibuya, T.; Russell, M.J.; Takai, K. Free energy distribution and hydrothermal mineral precipitation in Hadean submarine alkaline vent systems: Importance of iron redox reactions under anoxic conditions. Geochim. Cosmochim. Acta 2016, 175, 1–19. [Google Scholar] [CrossRef]
- Belsky, A.J.; Maiella, P.G.; Brill, T.B. Spectroscopy of hydrothermal reactions 13. Kinetics and mechanisms of decarboxylation of acetic acid derivatives at 100–260 °C under 275 bar. J. Phys. Chem. A 1999, 103, 4253–4260. [Google Scholar] [CrossRef]
- Schoonen, M.A.A.; Xu, Y.; Bebie, J. Energetics and kinetics of the prebiotic synthesis of simple organic acids and amino acids with the FeS-H2S/FeS2 redox couple as reductant. Orig. Life Evol. Biosph. 1999, 29, 5–32. [Google Scholar] [CrossRef] [PubMed]
- Tittmann, K. Reaction mechanisms of thiamin diphosphate enzymes: Redox reactions. FEBS J. 2009, 276, 2454–2468. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, T.; Yamamoto, M.; Arai, H.; Ohmori, D.; Ishii, M.; Igarashi, Y. Enzymatic and electron paramagnetic resonance studies of anabolic pyruvate synthesis by pyruvate: Ferredoxin oxidoreductase from Hydrogenobacter thermophilus. FEBS J. 2010, 277, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Reed, G.H.; Ragsdale, S.W.; Mansoorabadi, S.O. Radical reactions of thiamin pyrophosphate in 2-oxoacid oxidoreductases. Biochim. Biophys. Acta 2012, 1824, 1291–1298. [Google Scholar] [CrossRef] [PubMed]
- Washabaugh, M.W.; Jencks, W.P. Thiazolium C(2)-proton exchange: Structure-reactivity correlations and the pKa of thiamin C(2)-H revisited. Biochemistry 1988, 27, 5044–5053. [Google Scholar] [CrossRef] [PubMed]
- Carmichael, E.C.; Geldart, V.D.; McDonald, R.S.; Moore, D.B.; Rose, S.; Colebrook, L.D.; Spiropoulos, G.D.; Tee, O.S. Kinetics and mechanism of the reversible ring-opening of thiamine and related thiazolium ions in aqueous solution. J. Chem. Soc. Perkin Trans. 1997, 2, 2609–2619. [Google Scholar] [CrossRef]
- Menon, S.; Ragsdale, S.W. Mechanism of the Clostridium thermoaceticum pyruvate:ferredoxin oxidoreductase: Evidence for the common catalytic intermediacy of the hydrosyethylthiamine phyrophosphate radical. Biochemistry 1997, 36, 8484–8494. [Google Scholar] [CrossRef] [PubMed]
- Gruys, K.J.; Halkides, C.J.; Fery, P.A. Synthesis and properties of 2-acetylthiamin pyrophosphate: An enzymatic reaction intermediate. Biochemistry 1987, 26, 7575–7585. [Google Scholar] [CrossRef] [PubMed]
- Kluger, R.; Chin, J.; Smyth, T. Thiamin-catalyzed decarboxylation of pyruvate. Synthesis and reactivity analysis of the central, elusive intermediate, α-lactylthiamin. J. Am. Chem. Soc. 1981, 103, 884–888. [Google Scholar] [CrossRef]
- Jordan, F.; Li, H.; Brown, A. Remarkable stabilization of zwitterionic intermediates may account for a billon-fold rate acceleration by thiamin diphosphate-dependent decarboxylases. Biochemistry 1999, 38, 6369–6373. [Google Scholar] [CrossRef] [PubMed]
- Chabriere, E.; Charon, M.H.; Volbeda, A.; Pieulle, L.; Hatchikian, E.C.; Fontecilla-Camps, J.C. Crystal structures of the key anaerobic enzyme pyruvate:ferredoxin oxidoreductase, free and in complex with pyruvate. Nat. Struct. Biol. 1999, 6, 182–190. [Google Scholar] [PubMed]
- Gibson, M.I.; Brignole, E.J.; Pierce, E.; Can, M.; Ragsdale, S.W.; Drennan, C.L. The structure of an oxalate oxidoreductase provides insight into microbial 2-oxoacid metabolism. Biochemistry 2015, 54, 4112–4120. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Maruyama, A.; Arakawa, T.; Fushinobu, S.; Wakagi, T. Crystal structures of archaeal 2-oxoacid: Ferredoxin oxidoreductases from Sulfolobus tokodaii. Sci. Rep. 2016, 6, 33061. [Google Scholar] [CrossRef] [PubMed]
- Pieulle, L.; Guigliarelli, B.; Asso, M.; Dole, F.; Bernadac, A.; Hatchikian, E.C. Isolation and characterization of the pyruvate-ferredoxin oxidoreductase from the sulfate-reducing bacterium Desulfovibrio africanus. Biochim. Biophys. Acta 1995, 1250, 49–59. [Google Scholar] [CrossRef]
- Sverjensky, D.A. Prediction of surface charge on oxides in salt solutions: Revisions for 1:1 (M+L−) electrolytes. Geochim. Cosmochim. Acta 2005, 69, 225–257. [Google Scholar] [CrossRef]
- Crosby, J.; Stone, R.; Linenhard, G.E. Mechanisms of thiamine-catalyzed reactions. Decarboxylation of 2-(1-carboxy-1-hydroxyethyl)-3,4-dimethylthiazolium. J. Am. Chem. Soc. 1970, 92, 2891–2900. [Google Scholar] [CrossRef] [PubMed]
- Bocarsly, A.B.; Gibson, Q.D.; Morris, A.J.; L’Esperance, R.P.; Detweiler, Z.M.; Lakkaraju, P.S.; Zeitler, E.L.; Shaw, T.W. Comparative study of imidazole and pyridine catalyzed reduction of carbon dioxide at illuminated iron pyrite electrode. ACS Catal. 2012, 2, 1684–1692. [Google Scholar] [CrossRef]
- Tamura, J.; Ono, A.; Sugano, Y.; Huang, C.; Nishizawa, H.; Mikoshiba, S. Electrochemical reduction of CO2 to ethylene glycol on imidazolium ion-terminated self-assembly monolayer-modified Au electrodes in an aqueous solution. Phys. Chem. Chem. Phys. 2015, 17, 26072–26078. [Google Scholar] [CrossRef] [PubMed]
- Drobner, E.; Huber, H.; Wächtershäuser, G.; Rose, D.; Stetter, K.O. Pyrite formation linked with hydrogen evolution under anaerobic conditions. Nature 1990, 346, 742–744. [Google Scholar] [CrossRef]
- Taylor, P.; Rummery, T.E.; Owen, D.G. Reactions of iron monosulfide solids with aqueous hydrogen sulfide up to 160 °C. J. Inorg. Nucl. Chem. 1979, 41, 1683–1687. [Google Scholar] [CrossRef]
- Summers, D.P. Ammonia formation by the reduction of nitrite/nitrate by FeS: Ammonia formation under acidic conditions. Orig. Life Evol. Biosph. 2005, 35, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Summers, D.P.; Basa, R.C.B.; Khare, B.; Rodoni, D. Abiotic nitrogen fixation on terrestrial planets: Reduction of NO to ammonia by FeS. Astrobiology 2012, 12, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Blochl, E.; Keller, M.; Wächtershäuser, G.; Stetter, K.O. Reactions depending on iron sulfide and linking geochemistry with biochemistry. Proc. Natl. Acad. Sci. USA 1992, 89, 8117–8120. [Google Scholar] [CrossRef] [PubMed]
- Huber, C.; Wächtershäuser, G. Primordial reductive amination revisited. Tetrahedron Lett. 2003, 44, 1695–1697. [Google Scholar] [CrossRef]
- Wolthers, M.; Charlet, L.; van der Linde, P.R.; Rickard, D.; van der Weijden, C.H. Surface chemistry of disordered mackinawite (FeS). Geochim. Cosmochim. Acta 2005, 69, 3469–3481. [Google Scholar] [CrossRef]
- Bebie, J.; Schoonen, M.A.A.; Fuhrmann, M.; Strongin, D.R. Surface charge development on transition metal sulfides: An electrokinetic study. Geochim. Cosmochim. Acta 1998, 62, 633–642. [Google Scholar] [CrossRef]
- Weerasooria, R.; Tobschall, H.J. Pyrite-water interactions: Effects of pH and pFe on surface charge. Coll. Surf. A Physcochim. Eng. Asp. 2005, 264, 68–74. [Google Scholar] [CrossRef]
- Kitadai, N.; Yokoyama, T.; Nakashima, S. ATR-IR spectroscopic study of L-lysine adsorption on amorphous silica. J. Colloid Interface Sci. 2009, 329, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Kitadai, N.; Yokoyama, T.; Nakashima, S. In situ ATR-IR investigation of L-lysine adsorption on montmorillonite. J. Colloid Interface Sci. 2009, 338, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Kitadai, N.; Nishiuchi, K.; Nishii, A.; Fukushi, K. Amorphous silica-promoted lysine dimerization: A thermodynamic prediction. Orig. Life Evol. Biosph. 2017. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Nakamura, R.; Kasaya, T.; Kumagai, H.; Suzuki, K.; Takai, K. Spontaneous and widespread electricity generation in natural deep-sea hydrothermal fields. Angew. Chem. Int. Ed. 2017, 56, 5725–5728. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, R.; Takashima, T.; Kato, S.; Takai, K.; Yamamoto, M.; Hashimoto, K. Electrical current generation across a black smoker chimney. Angew. Chem. Int. Ed. 2010, 49, 7692–7694. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, A.; Yamamoto, M.; Takai, K.; Ishii, T.; Hashimoto, K.; Nakamura, R. Electrochemical CO2 reduction by Ni-containing iron sulfides: How is CO2 electrochemically reduced at bisulfide-bearing deep-sea hydrothermal precipitates? Electrochim. Acta 2014, 141, 311–318. [Google Scholar] [CrossRef]
- Yeh, I.C.; Berkowitz, M.L. Dielectric constant of water at high electric fields: Molecular dynamics study. J. Chem. Phys. 1999, 110, 7935–7942. [Google Scholar] [CrossRef]
- Suresh, S.J.; Satish, A.V. Influence of electric field on the hydrogen bond network of water. J. Chem. Phys. 2006, 124, 074506. [Google Scholar] [CrossRef] [PubMed]
- Kitadai, N. Energetics of amino acid synthesis in alkaline hydrothermal environments. Orig. Life Evol. Biosph. 2015, 45, 377–409. [Google Scholar] [CrossRef] [PubMed]
- Kitadai, N. Predicting thermodynamic behaviors of non-protein amino acids as a function of temperature and pH. Orig. Life Evol. Biosph. 2016, 46, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Sakata, K.; Kitadai, N.; Yokoyama, T. Effects of pH and temperature on dimerization rate of glycine: Evaluation of favorable environmental conditions for chemical evolution of life. Geochim. Cosmochim. Acta 2010, 74, 6841–6851. [Google Scholar] [CrossRef]
- Kitadai, N. Thermodynamic prediction of glycine polymerization as a function of temperature and pH consistent with experimentally obtained results. J. Mol. Evol. 2014, 78, 171–187. [Google Scholar] [CrossRef] [PubMed]
- Kitadai, N. Dissolved divalent metal and pH effects on amino acid polymerization: A thermodynamic evaluation. Orig. Life Evol. Biosph. 2017, 47, 13–37. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.T.; Chem, Z.; Kang, P.; Meyer, T.J. Electrocatalytic water oxidation with a copper(II) polypeptide complex. J. Am. Chem. Soc. 2013, 135, 2048–2051. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Ghirlanda, G.; Allen, J.P. Water oxidation by a nickel-glycine catalyst. J. Am. Chem. Soc. 2014, 136, 10198–10201. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Wang, J.; Chen, Z. Water oxidation by copper-amino acid catalysts at low overpotential. ChemCatChem 2016, 8, 2165–2170. [Google Scholar] [CrossRef]
- Yoshida, M.; Onishi, S.; Mitsutomi, Y.; Yamamoto, F.; Nagasaka, M.; Yuzawa, H.; Kosugi, N.; Kondoh, H. Integration of active nickel oxide clusters by amino acids for water oxidation. J. Phys. Chem. C 2017, 121, 255–260. [Google Scholar] [CrossRef]
- Robinson, B.H.; Chun, K. The relationships between transketolase, yeast pyruvate decarboxylase and pyruvate dehydrogenase of the pyruvate dehydrogenase complex. FEBS Lett. 1993, 328, 99–102. [Google Scholar] [CrossRef]
- Beckett, D.; Kovaleva, E.; Schatz, P.J. A minimal peptide substrate in biotin holoenzyme synthetase-catalyzed biotinylation. Protein Sci. 1999, 8, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Milner-White, E.J.; Russell, M.J. Sites for phosphates and iron-sulfur thiolates in the first membranes: 3 to 6 residue anion-binding motifs (nests). Orig. Life Evol. Biosph. 2005, 35, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Miner-White, E.J.; Russell, M.J. Functional capabilities of the earliest peptides and the emergence of life. Genes 2011, 2, 671–688. [Google Scholar] [CrossRef] [PubMed]
- Nitschke, W.; McGlynn, S.E.; Milner-White, E.J.; Russell, M.J. On the antiquity of metalloenzymes and their substrates in bioenergetics. Biochim. Biophys. Acta 2013, 1827, 871–881. [Google Scholar] [CrossRef] [PubMed]
- Jensen, R.A. Enzyme recruitment in evolution of new function. Annu. Rev. Microbiol. 1976, 30, 409–425. [Google Scholar] [CrossRef] [PubMed]
- Voet, D.; Voet, J.G. Biochemistry, 4th ed.; Wiley: Somerset, NJ, USA, 2011; ISBN 9780470570951. [Google Scholar]
- Knox, W.E.; Pitt, B.M. Enzymatic catalysis of the keto-enol tautomerization of phenylpyruvic acids. J. Biol. Chem. 1957, 225, 675–688. [Google Scholar] [PubMed]
- Kakegawa, T.; Noda, M.; Nannri, H. Geochemical cycles of bio-essential elements on the early Earth and their relationships to origin of life. Resour. Geol. 2002, 52, 83–89. [Google Scholar] [CrossRef]
- Mishima, S.; Ohtomo, Y.; Kakegawa, T. Occurrence of tourmaline in metasedimentary rocks of the Isua supracrustal belt, Greenland: Implications for ribose stabilization in Hadean marine sediments. Orig. Life Evol. Biosph. 2016, 46, 247–271. [Google Scholar] [CrossRef] [PubMed]
- Lazcano, A.; Miller, S.L. On the origin of metabolic pathways. J. Mol. Evol. 1999, 49, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Wächtershäuser, G. Life as we don’t know it. Science 2000, 289, 1307–1308. [Google Scholar] [CrossRef] [PubMed]
- Schonheit, P.; Buckel, W.; Martin, W.F. On the origin of heterotorophy. Trends Microbiol. 2016, 24, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Foppen, M.A.E.; de Lange, Y.M.; van Rantwijk, F.; Maat, L.; Kieboom, A.P.G. Reversal of an enzymatic decarboxylation: Thiamin mediated carboxylation of acetaldehyde into pyruvic acid. Recl. Trav. Chim. Pays-Bas 1990, 109, 359–360. [Google Scholar] [CrossRef]
- Yi, C.L.; Huang, Y.T.; Lee, C.F. Synthesis of thioesters through copper-catalyzed coupling of aldehydes with thiols in water. Green Chem. 2013, 15, 2476–2484. [Google Scholar] [CrossRef]
- Huang, Y.T.; Lu, S.Y.; Yi, C.L.; Lee, C.F. Iron-catalyzed synthesis of thioesters from thiols and aldehydes in water. J. Org. Chem. 2014, 79, 4561–4568. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, K.A.; Boydston, A.J. Organocatalyzed anodic oxidation of aldehydes to thioesters. Org. Lett. 2014, 16, 1928–1931. [Google Scholar] [CrossRef] [PubMed]
- LaRowe, D.E.; Regnier, P. Thermodynamic potential for the abiotic synthesis of adenine, cytosine, guanine, thyamine, uracil, ribose, and deoxyribose in hydrothermal systems. Orig. Life Evol. Biosph. 2008, 38, 383–397. [Google Scholar] [CrossRef] [PubMed]
- Herschy, B.; Whicher, A.; Camprubi, E.; Watson, C.; Dartnell, L.; Ward, J.; Evans, J.R.G.; Lane, N. An origin-of-life reactor to simulate alkaline hydrothermal vents. J. Mol. Evol. 2014, 79, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Jackson, J.B. The “origin-of-life reactor” and reduction of CO2 by H2 in inorganic precipitates. J. Mol. Evol. 2017, 85, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Lang, S.Q.; Butterfield, D.A.; Schulte, M.; Kelley, D.S.; Lilley, M.D. Elevated concentration of formate, acetate and dissolved organic carbon found at the Lost City hydrothermal field. Geochim. Cosmochim. Acta 2010, 74, 941–952. [Google Scholar] [CrossRef]
- Windman, T.; Zolotova, N.; Schwandner, F.; Shock, E.L. Formate as an energy source for microbial metabolism in chemosynthetic zones of hydrothermal ecosystems. Astrobiology 2007, 7, 873–890. [Google Scholar] [CrossRef] [PubMed]
- Stairs, C.W.; Roger, A.J.; Hampl, V. Eukaryotic pyruvate formate lyase and its activating enzyme were acquired laterally from a firmicute. Mol. Biol. Evol. 2011, 28, 2087–2099. [Google Scholar] [CrossRef] [PubMed]
- Zelcbuch, L.; Lindner, S.N.; Zegman, Y.; Slutskin, I.V.; Antonovsky, N.; Gleizer, S.; Milo, R.; Bar-Even, A. Pyruvate formate-lyase enables efficient growth of Escherichia coil on acetate and formate. Biochemistry 2016, 55, 2423–2426. [Google Scholar] [CrossRef] [PubMed]
- Flamholz, A.; Noor, E.; Bar-Even, A.; Milo, R. eQuilibrator—the biochemical thermodynamics calculator. Nucleic Acids Res. 2012, 40, D770–D775. [Google Scholar] [CrossRef] [PubMed]
- Bar-Even, A. Formate assimilation: The metabolic architecture of natural and synthetic pathways. Biochemistry 2016, 55, 3851–3863. [Google Scholar] [CrossRef] [PubMed]
- Himo, F.; Eriksson, L.A. Catalytic mechanism of pyruvate formate-lyase (PFL). A theoretical study. J. Am. Chem. Soc. 1998, 120, 11449–11455. [Google Scholar] [CrossRef]
- Becker, A.; Fritz-Wolf, K.; Kabsch, W.; Knappe, J.; Schultz, S.; Wagner, A.F.V. Structure and mechanism of the glycyl radical enzyme pyruvate formate-lyase. Nat. Struc. Biol. 1999, 6, 969–975. [Google Scholar]
- Buis, J.M.; Broderick, J.B. Pyruvate formate-lyase activating enzyme: Elucidation of a novel mechanism for glycyl radical formation. Arch. Biochem. Biophys. 2005, 433, 288–296. [Google Scholar] [CrossRef] [PubMed]
- McCollom, T.M. Abiotic methane formation during experimental serpentinization of olivine. Proc. Natl. Acad. Sci. USA 2016, 113, 13965–13970. [Google Scholar] [CrossRef] [PubMed]
- Ruff, S.W.; Farmer, J.D.; Clavin, W.M.; Herkenhoff, K.E.; Johnson, J.R.; Morris, R.V.; Rice, M.S.; Arvidson, R.E.; Bell, J.F., III; Christensen, P.R.; et al. Characteristics, distribution, origin, and significance of opaline silica observed by the Spirit rover in Gusev crater, Mars. J. Geophys. Res. 2011, 116, E00F23. [Google Scholar] [CrossRef]
- Hsu, H.W.; Postberg, F.; Sekine, Y.; Shibuya, T.; Kempf, S.; Horanyi, M.; Juhasz, A.; Altobelli, N.; Suzuki, K.; Masaki, Y.; et al. Ongoing hydrothermal activities within Enceladus. Nature 2015, 519, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Sekine, Y.; Shibuya, T.; Postberg, F.; Hsu, H.W.; Suzuki, K.; Masaki, Y.; Kuwatani, T.; Mori, M.; Hong, P.K.; Yoshizaki, M.; et al. High-temperature water-rock interactions and hydrothermal environments in the chondrite-like core of Enceladus. Nat. Commun. 2015, 6, 8604. [Google Scholar] [CrossRef] [PubMed]
- Shock, E.L.; Oelkers, E.H.; Johnson, J.W.; Sverjensky, D.A.; Helgeson, H.C. Calculation of the thermodynamic properties of aqueous species at high pressures and temperatures. J. Chem. Soc. Faraday Trans. 1992, 88, 803–826. [Google Scholar] [CrossRef]
- Shock, E.L.; Helgeson, H.C.; Sverjensky, D.A. Calculation of the thermodynamic and transport properties of aqueous species at high pressures and temperatures: Standard partial molal properties of inorganic neutral species. Geochim. Cosmochim. Acta 1989, 53, 2157–2183. [Google Scholar] [CrossRef]
- Shock, E.L.; Sassani, D.C.; Willis, M.; Sverjensky, D.A. Inorganic species in geologic fluids: Correlations among standard molal thermodynamic properties of aqueous ions and hydroxide complexes. Geochim. Cosmochim. Acta 1997, 61, 907–950. [Google Scholar] [CrossRef]
- Dalla-Betta, P.; Schulte, M. Calculation of the aqueous thermodynamic properties of citric acid cycle intermediates and precursors and the estimation of high temperature and pressure equation of state parameters. Int. J. Mol. Sci. 2009, 10, 2809–2837. [Google Scholar] [CrossRef] [PubMed]
- Schulte, M.D.; Rogers, K.L. Thiols in hydrothermal solution: Standard partial molal properties and their role in the organic geochemistry of hydrothermal environments. Geochim. Cosmochim. Acta 2004, 68, 1087–1097. [Google Scholar] [CrossRef]
- Tsonopoulos, C.; Coulson, D.M.; Inman, L.B. Ionization constants of water pollutants. J. Chem. Eng. Data 1976, 21, 190–193. [Google Scholar] [CrossRef]
- Robie, R.A.; Hemingway, B.S.; Fisher, J.R. Thermodynamic Properties of Minerals and Related Substances at 298.15 K and 1 bar (105 Pascals) Pressure and at Higher Temperatures; U.S. Govt. Print. Office: Washington, DC, USA, 1979.
- Helgeson, H.C.; Delany, J.M.; Nesbitt, H.W.; Bird, D.K. Summary and critique of the thermodynamic properties of rock-forming minerals. Am. J. Sci. 1978, 278, 1–229. [Google Scholar]
- Benning, L.G.; Wilkin, R.T.; Barnes, H.L. Reaction pathways in the Fe–S system below 100 °C. Chem. Geol. 2000, 167, 25–51. [Google Scholar] [CrossRef]
- Helgeson, H.C.; Kirkham, D.H. Theoretical prediction of the thermodynamic behavior of aqueous electrolytes at high pressures and temperatures: I. summary of the thermodynamic/electrostatic properties of the solvent. Am. J. Sci. 1974, 274, 1089–1198. [Google Scholar] [CrossRef]
- Helgeson, H.C.; Kirkham, D.H.; Flowers, G.C. Theoretical prediction of the thermodynamic behavior of aqueous electrolytes at high pressures and temperatures: IV. Calculation of activity coefficients, osmotic coefficients, and appalent molal and standard and relative partial molal properties to 600 °C and 5 KB. Am. J. Sci. 1981, 281, 1249–1516. [Google Scholar]
- Migdisov, A.A.; Williams-Jones, A.E.; Lakshtanov, L.Z.; Alekhin, Y.V. Estimates of the second dissociation constant of H2S from the surface sulfidation of crystalline sulfur. Geochim. Cosmochim. Acta 2002, 66, 1713–1725. [Google Scholar] [CrossRef]
- Stefansson, A.; Seward, T.M. Experimental determination of the stability and stoichiometry of sulphide complexes of silver(I) in hydrothermal solutions to 400 °C. Geochim. Cosmochim. Acta 2003, 67, 1395–1413. [Google Scholar] [CrossRef]




© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).