
Frozen Accident Pushing 50: Stereochemistry, Expansion, and Chance in the Evolution of the Genetic Code

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Why the Universal Code?
3. The Three Principal Scenarios of the Code Origin and Evolution: Achievements, Limitations, and Compatibility
4. Primordial Expansion of the Code
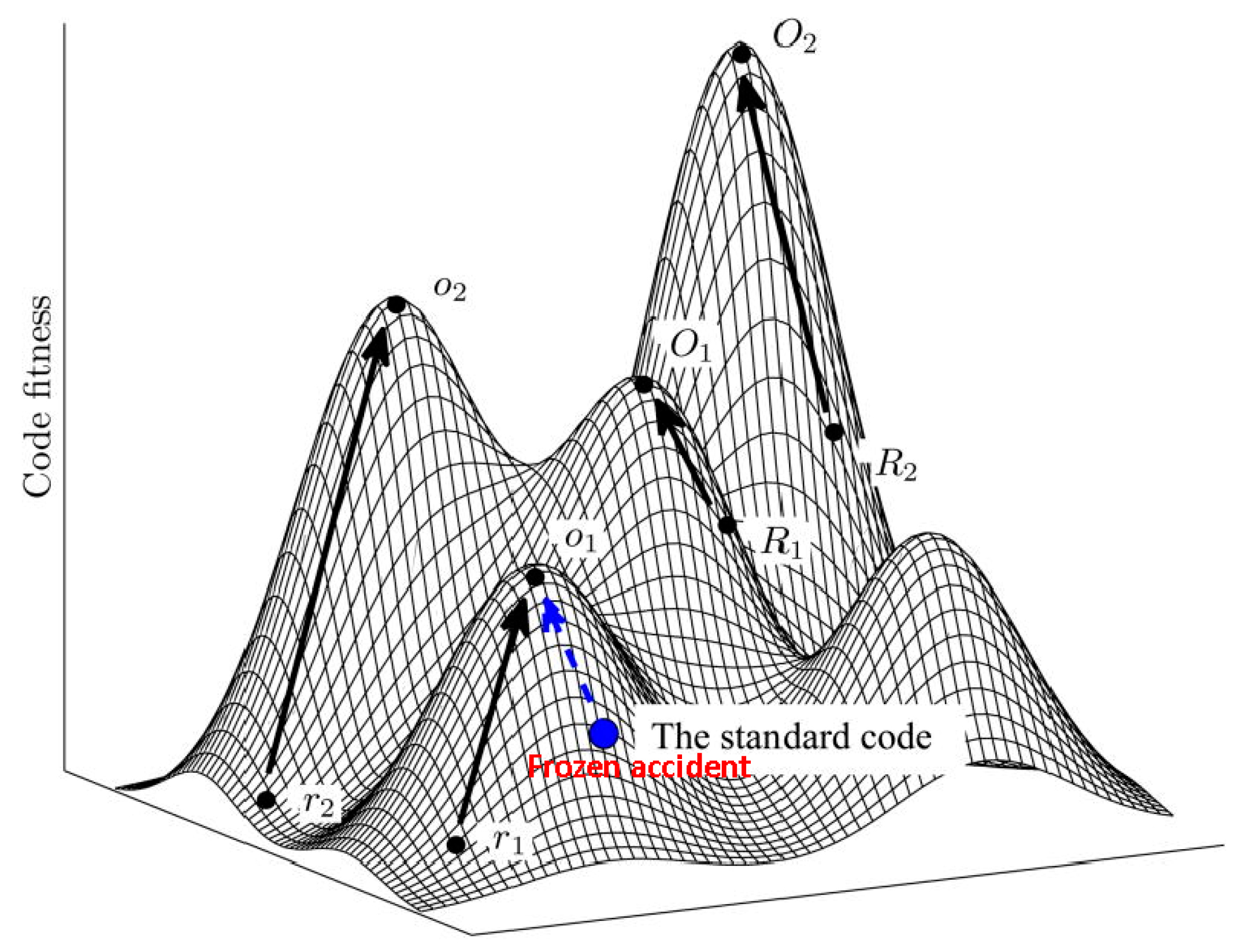
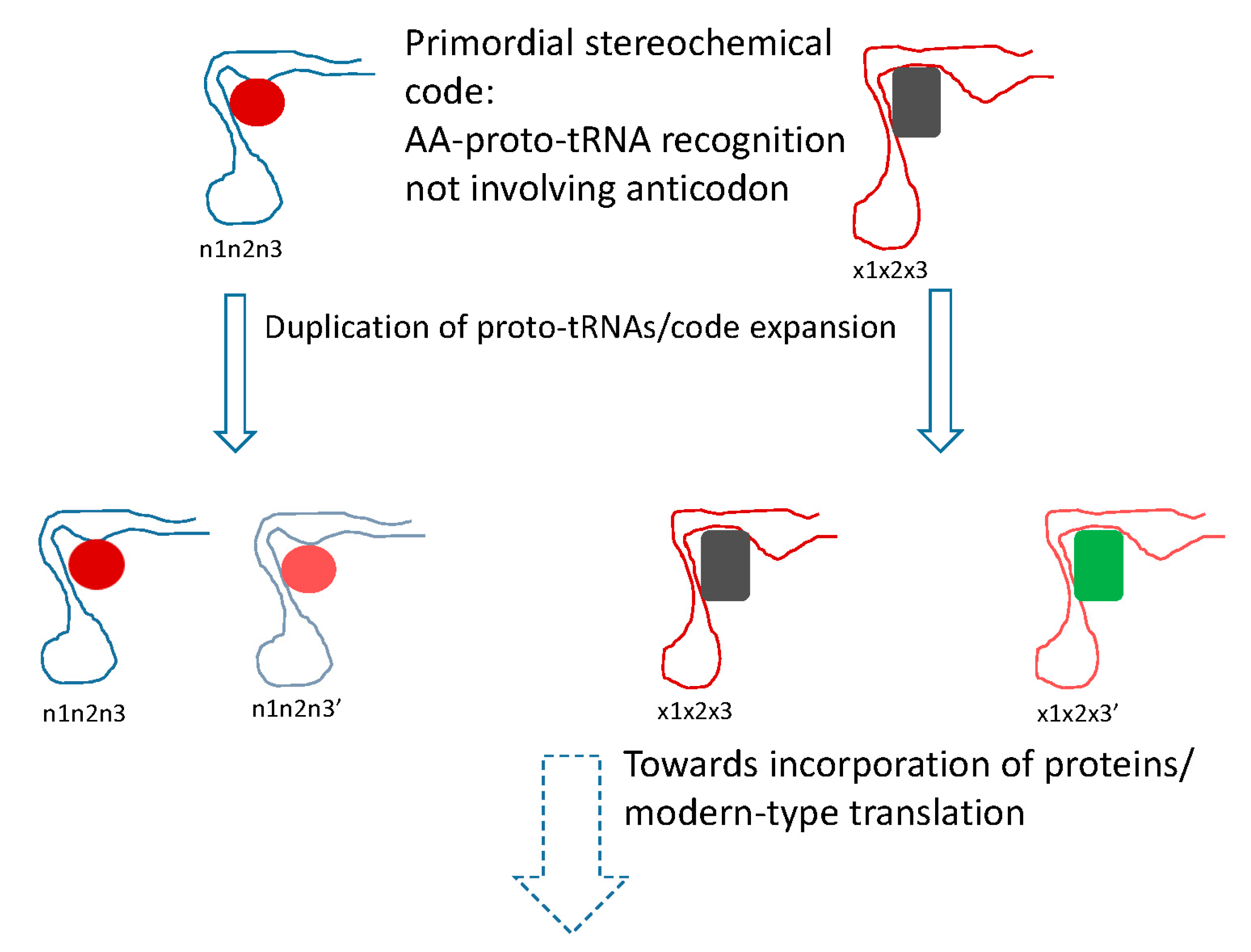
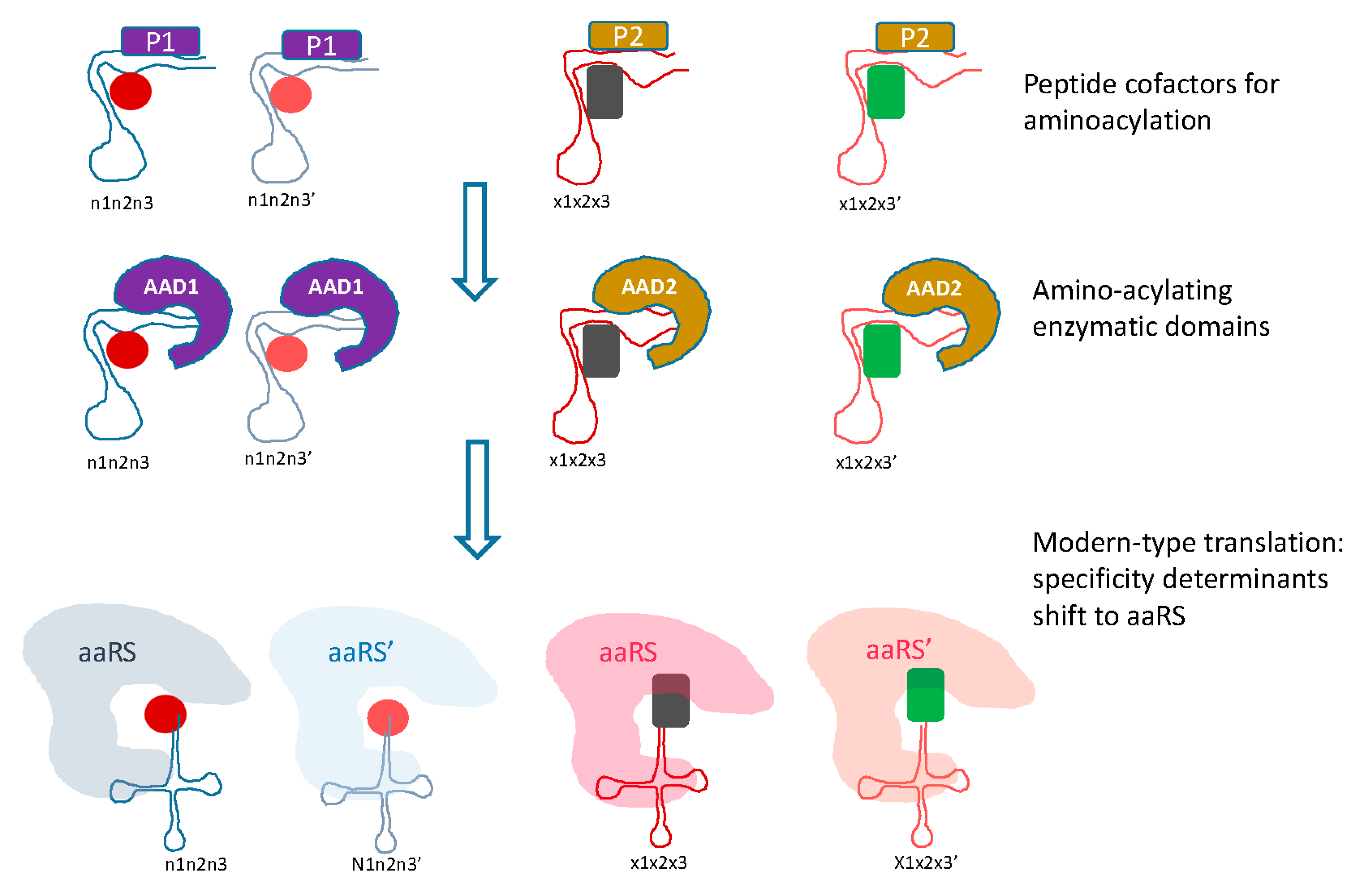
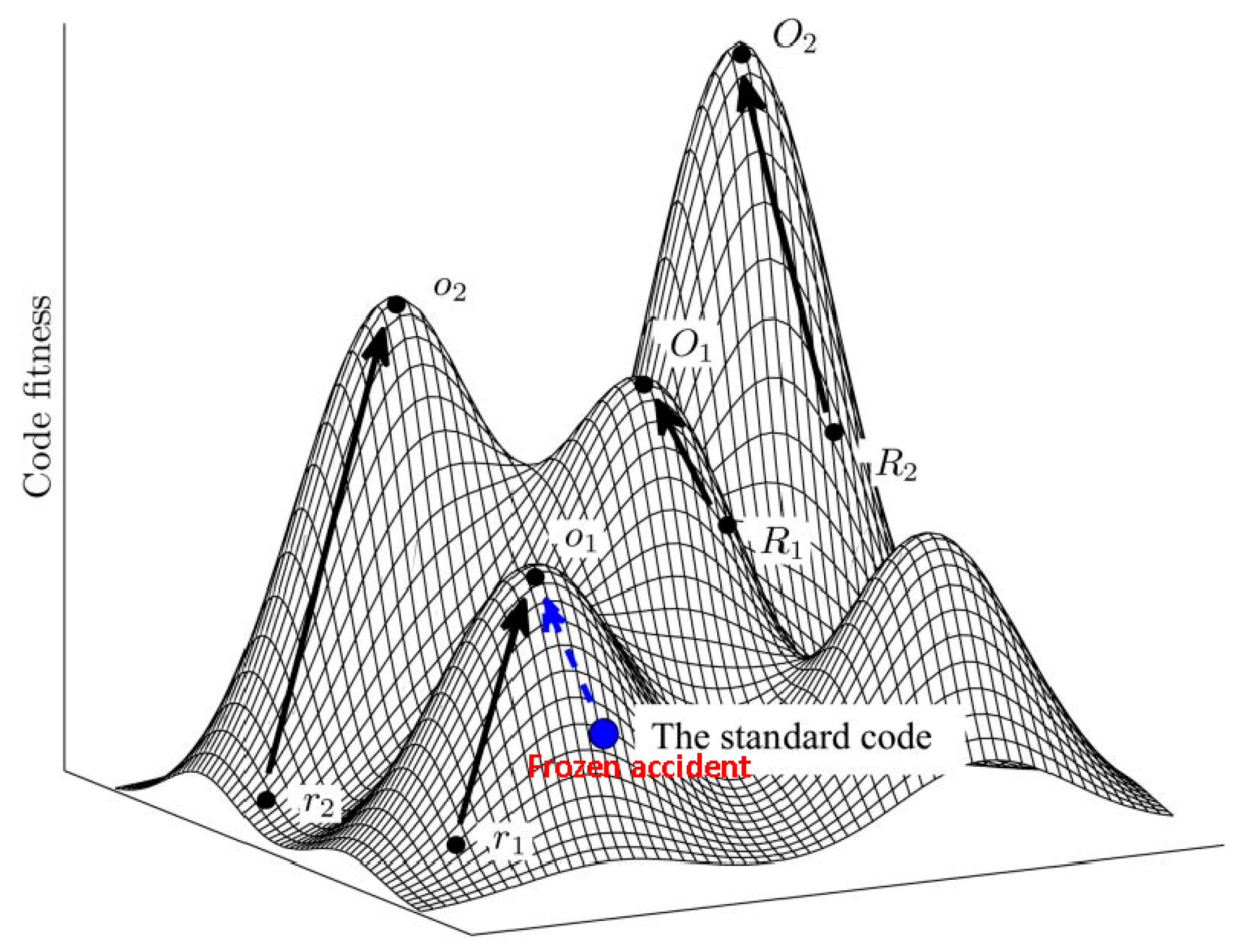
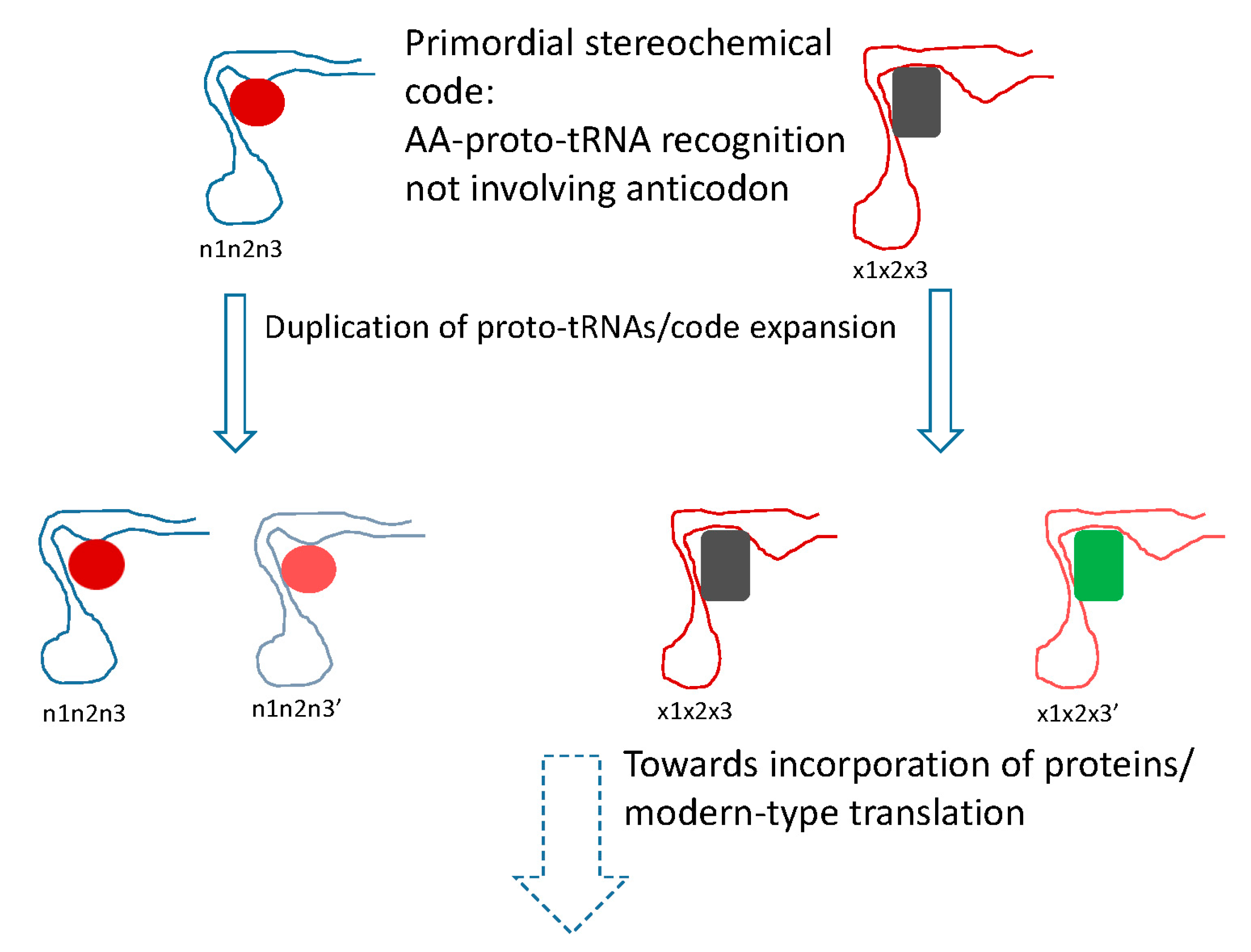
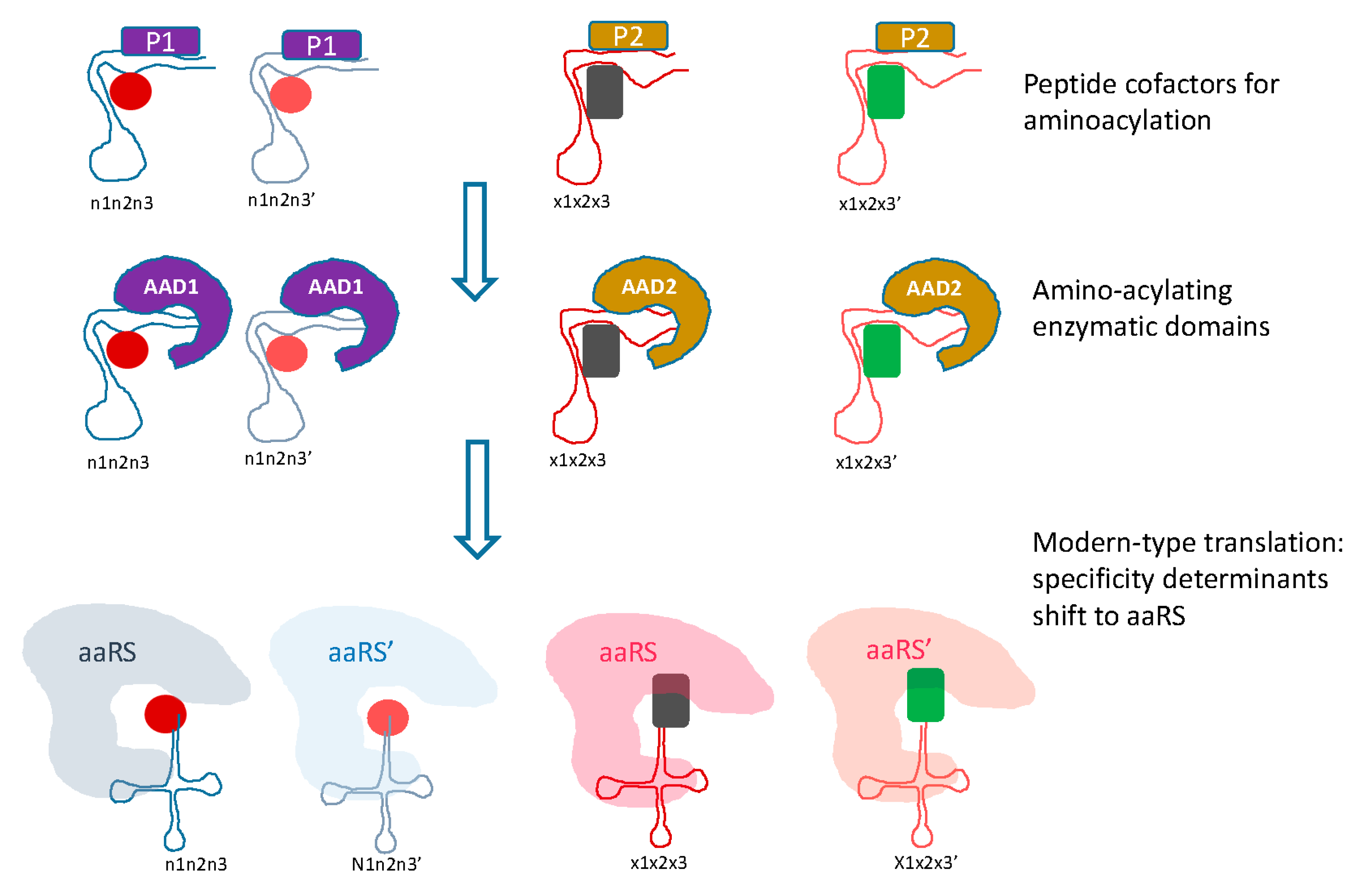
5. Protein Evolution Paradox, Extinct Primordial Stereochemical Code, Expansion of the Code, and Frozen Accident: A Coevolution Scenario for the Code and the Translation System
6. Concluding Remarks
Acknowledgments
Conflicts of Interest
References
- Crick, F.H. The origin of the genetic code. J. Mol. Biol. 1968, 38, 367–379. [Google Scholar] [CrossRef]
- Tamura, K. The Genetic Code: Francis Crick’s Legacy and Beyond. Life 2016, 6, 36. [Google Scholar] [CrossRef] [PubMed]
- Watson, J.D.; Crick, F.H. Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature 1953, 171, 737–738. [Google Scholar] [CrossRef] [PubMed]
- Watson, J.D.; Crick, F.H. Genetical implications of the structure of deoxyribonucleic acid. Nature 1953, 171, 964–967. [Google Scholar] [CrossRef] [PubMed]
- Crick, F.H. On protein synthesis. Symp. Soc. Exp. Biol. 1958, 12, 138–163. [Google Scholar] [PubMed]
- Crick, F.H.; Barnett, L.; Brenner, S.; Watts-Tobin, R.J. General nature of the genetic code for proteins. Nature 1961, 192, 1227–1232. [Google Scholar] [CrossRef] [PubMed]
- Nirenberg, M.W.; Matthaei, J.H. The dependence of cell-free protein synthesis in E. coli upon naturally occurring or synthetic polyribonucleotides. Proc. Natl. Acad. Sci. USA 1961, 47, 1588–1602. [Google Scholar] [CrossRef] [PubMed]
- Nirenberg, M. Historical review: Deciphering the genetic code—A personal account. Trends Biochem. Sci. 2004, 29, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Woese, C. The Genetic Code; Harper & Row: New York, NY, USA, 1967. [Google Scholar]
- Koonin, E.V.; Novozhilov, A.S. Origin and evolution of the genetic code: The universal enigma. IUBMB Life 2009, 61, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Higgs, P.G. Pathways of Genetic Code Evolution in Ancient and Modern Organisms. J. Mol. Evol. 2015, 80, 229–243. [Google Scholar] [CrossRef] [PubMed]
- Crick, F.H. The genetic code—Yesterday, today, and tomorrow. Cold Spring Harb. Symp. Quant. Biol. 1966, 31, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Haig, D.; Hurst, L.D. A quantitative measure of error minimization in the genetic code. J. Mol. Evol. 1991, 33, 412–417. [Google Scholar] [CrossRef] [PubMed]
- Freeland, S.J.; Hurst, L.D. The genetic code is one in a million. J. Mol. Evol. 1998, 47, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Novozhilov, A.S.; Wolf, Y.I.; Koonin, E.V. Evolution of the genetic code: Partial optimization of a random code for robustness to translation error in a rugged fitness landscape. Biol. Direct 2007, 2, 24. [Google Scholar] [CrossRef] [PubMed]
- Santos, M.A.; Moura, G.; Massey, S.E.; Tuite, M.F. Driving change: The evolution of alternative genetic codes. Trends Genet. 2004, 20, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Ling, J.; O’Donoghue, P.; Soll, D. Genetic code flexibility in microorganisms: Novel mechanisms and impact on physiology. Nat. Rev. Microbiol. 2015, 13, 707–721. [Google Scholar] [CrossRef] [PubMed]
- Ambrogelly, A.; Palioura, S.; Soll, D. Natural expansion of the genetic code. Nat. Chem. Biol. 2007, 3, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Mukai, T.; Englert, M.; Tripp, H.J.; Miller, C.; Ivanova, N.N.; Rubin, E.M.; Kyrpides, N.C.; Soll, D. Facile Recoding of Selenocysteine in Nature. Angew. Chem. Int. Ed. Engl. 2016, 55, 5337–5341. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Baranov, P.V.; Atkins, J.F.; Gladyshev, V.N. Pyrrolysine and selenocysteine use dissimilar decoding strategies. J. Biol. Chem. 2005, 280, 20740–20751. [Google Scholar] [CrossRef] [PubMed]
- Lobanov, A.V.; Turanov, A.A.; Hatfield, D.L.; Gladyshev, V.N. Dual functions of codons in the genetic code. Crit. Rev. Biochem. Mol. Biol. 2010, 45, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; O’Donoghue, P.; Ambrogelly, A.; Gundllapalli, S.; Sherrer, R.L.; Palioura, S.; Simonovic, M.; Soll, D. Distinct genetic code expansion strategies for selenocysteine and pyrrolysine are reflected in different aminoacyl-tRNA formation systems. FEBS Lett. 2010, 584, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Higgs, P.G. A unified model of codon reassignment in alternative genetic codes. Genetics 2005, 170, 831–840. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Yang, X.; Higgs, P.G. The mechanisms of codon reassignments in mitochondrial genetic codes. J. Mol. Evol. 2007, 64, 662–688. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Schultz, P.G. A chemical toolkit for proteins—An expanded genetic code. Nat. Rev. Mol. Cell Biol. 2006, 7, 775–782. [Google Scholar] [CrossRef] [PubMed]
- Neumann, H.; Wang, K.; Davis, L.; Garcia-Alai, M.; Chin, J.W. Encoding multiple unnatural amino acids via evolution of a quadruplet-decoding ribosome. Nature 2010, 464, 441–444. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Schultz, P.G. Adding new chemistries to the genetic code. Annu. Rev. Biochem. 2010, 79, 413–444. [Google Scholar] [CrossRef] [PubMed]
- Chin, J.W. Expanding and reprogramming the genetic code of cells and animals. Annu. Rev. Biochem. 2014, 83, 379–408. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V. Comparative genomics, minimal gene-sets and the last universal common ancestor. Nat. Rev. Microbiol. 2003, 1, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Szathmary, E.; Demeter, L. Group selection of early replicators and the origin of life. J. Theor. Biol. 1987, 128, 463–486. [Google Scholar] [CrossRef]
- Szathmary, E.; Maynard Smith, J. From replicators to reproducers: The first major transitions leading to life. J. Theor. Biol. 1997, 187, 555–571. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Martin, W. On the origin of genomes and cells within inorganic compartments. Trends Genet. 2005, 21, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Vetsigian, K.; Woese, C.; Goldenfeld, N. Collective evolution and the genetic code. Proc. Natl. Acad. Sci. USA 2006, 103, 10696–10701. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Aggarwal, N.; Bandhu, A.V. Two perspectives on the origin of the standard genetic code. Orig. Life Evol. Biosph. 2014, 44, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, N.; Bandhu, A.V.; Sengupta, S. Finite population analysis of the effect of horizontal gene transfer on the origin of an universal and optimal genetic code. Phys. Biol. 2016, 13, 036007. [Google Scholar] [CrossRef] [PubMed]
- Doolittle, W.F. Phylogenetic classification and the universal tree. Science 1999, 284, 2124–2129. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Makarova, K.S.; Aravind, L. Horizontal gene transfer in prokaryotes: Quantification and classification. Annu. Rev. Microbiol. 2001, 55, 709–742. [Google Scholar] [CrossRef] [PubMed]
- Doolittle, W.F.; Bapteste, E. Pattern pluralism and the Tree of Life hypothesis. Proc. Natl. Acad. Sci. USA 2007, 104, 2043–2049. [Google Scholar] [CrossRef] [PubMed]
- Puigbo, P.; Lobkovsky, A.E.; Kristensen, D.M.; Wolf, Y.I.; Koonin, E.V. Genomes in turmoil: Quantification of genome dynamics in prokaryote supergenomes. BMC Biol. 2014, 12, 66. [Google Scholar] [CrossRef] [PubMed]
- Treangen, T.J.; Rocha, E.P. Horizontal transfer, not duplication, drives the expansion of protein families in prokaryotes. PLoS Genet. 2011, 7, e1001284. [Google Scholar] [CrossRef] [PubMed]
- Iranzo, J.; Puigbo, P.; Lobkovsky, A.E.; Wolf, Y.I.; Koonin, E.V. Inevitability of genetic parasites. Genome Biol. Evol. 2016, 8, 2856–2869. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, N.; Kaneko, K.; Koonin, E.V. Horizontal gene transfer can rescue prokaryotes from Muller’s ratchet: Benefit of DNA from dead cells and population subdivision. G3 (Bethesda) 2014, 4, 325–339. [Google Scholar] [CrossRef] [PubMed]
- Knight, R.D.; Freeland, S.J.; Landweber, L.F. Selection, history and chemistry: The three faces of the genetic code. Trends Biochem. Sci. 1999, 24, 241–247. [Google Scholar] [CrossRef]
- Di Giulio, M. The origin of the genetic code: Theories and their relationships, a review. Biosystems 2005, 80, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Novozhilov, A.S. Origin and evolution of the universal genetic code. Annu. Rev. Genet. 2017, 51. in press. [Google Scholar]
- Woese, C.R. The fundamental nature of the genetic code: Prebiotic interactions between polynucleotides and polyamino acids or their derivatives. Proc. Natl. Acad. Sci. USA 1968, 59, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Pelc, S.R.; Welton, M.G. Stereochemical relationship between coding triplets and amino-acids. Nature 1966, 209, 868–870. [Google Scholar] [CrossRef] [PubMed]
- Root-Bernstein, R.S. On the origin of the genetic code. J. Theor. Biol. 1982, 94, 895–904. [Google Scholar] [CrossRef]
- Dunnill, P. Triplet nucleotide-amino-acid pairing; a stereochemical basis for the division between protein and non-protein amino-acids. Nature 1966, 210, 1265–1267. [Google Scholar] [CrossRef] [PubMed]
- Hendry, L.B.; Witham, F.H. Stereochemical recognition in nucleic acid-amino acid interactions and its implications in biological coding: A model approach. Perspect. Biol. Med. 1979, 22, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, M. Molecular basis for the genetic code. J. Mol. Evol. 1982, 18, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Yarus, M. Amino acids as RNA ligands: A direct-RNA-template theory for the code’s origin. J. Mol. Evol. 1998, 47, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Yarus, M. RNA-ligand chemistry: A testable source for the genetic code. RNA 2000, 6, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Yarus, M.; Caporaso, J.G.; Knight, R. Origins of the genetic code: The escaped triplet theory. Annu. Rev. Biochem. 2005, 74, 179–198. [Google Scholar] [CrossRef] [PubMed]
- Yarus, M.; Widmann, J.J.; Knight, R. RNA-amino acid binding: A stereochemical era for the genetic code. J. Mol. Evol. 2009, 69, 406–429. [Google Scholar] [CrossRef] [PubMed]
- Yarus, M. Life from RNA World: The Ancestor Within; Harvard University Press: Cambridge, MA, USA, 2010. [Google Scholar]
- Wong, J.T. A co-evolution theory of the genetic code. Proc. Natl. Acad. Sci. USA 1975, 72, 1909–1912. [Google Scholar] [CrossRef] [PubMed]
- Di Giulio, M. An extension of the coevolution theory of the origin of the genetic code. Biol. Direct 2008, 3, 37. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.T.; Ng, S.K.; Mat, W.K.; Hu, T.; Xue, H. Coevolution Theory of the Genetic Code at Age Forty: Pathway to Translation and Synthetic Life. Life 2016, 6, 12. [Google Scholar] [CrossRef] [PubMed]
- Di Giulio, M. The lack of foundation in the mechanism on which are based the physico-chemical theories for the origin of the genetic code is counterposed to the credible and natural mechanism suggested by the coevolution theory. J. Theor. Biol. 2016, 399, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Goodarzi, H.; Nejad, H.A.; Torabi, N. On the optimality of the genetic code, with the consideration of termination codons. Biosystems 2004, 77, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Freeland, S. The standard genetic code enhances adaptive evolution of proteins. J. Theor. Biol. 2006, 239, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Torabi, N.; Goodarzi, H.; Shateri Najafabadi, H. The case for an error minimizing set of coding amino acids. J. Theor. Biol. 2007, 244, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Massey, S.E. A neutral origin for error minimization in the genetic code. J. Mol. Evol. 2008, 67, 510–516. [Google Scholar] [CrossRef] [PubMed]
- Novozhilov, A.S.; Koonin, E.V. Exceptional error minimization in putative primordial genetic codes. Biol. Direct 2009, 4, 44. [Google Scholar] [CrossRef] [PubMed]
- Salinas, D.G.; Gallardo, M.O.; Osorio, M.I. Local conditions for global stability in the space of codons of the genetic code. Biosystems 2016, 150, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Massey, S.E. Genetic code evolution reveals the neutral emergence of mutational robustness, and information as an evolutionary constraint. Life 2015, 5, 1301–1332. [Google Scholar] [CrossRef] [PubMed]
- Massey, S.E. The neutral emergence of error minimized genetic codes superior to the standard genetic code. J. Theor. Biol. 2016, 408, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Pizzarello, S. The chemistry of life’s origin: A carbonaceous meteorite perspective. Acc. Chem. Res. 2006, 39, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Zaia, D.A.; Zaia, C.T.; De Santana, H. Which amino acids should be used in prebiotic chemistry studies? Orig. Life Evol. Biosph. 2008, 38, 469–488. [Google Scholar] [CrossRef] [PubMed]
- Cleaves, H.J., 2nd. The origin of the biologically coded amino acids. J. Theor. Biol. 2010, 263, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Burton, A.S.; Stern, J.C.; Elsila, J.E.; Glavin, D.P.; Dworkin, J.P. Understanding prebiotic chemistry through the analysis of extraterrestrial amino acids and nucleobases in meteorites. Chem. Soc. Rev. 2012, 41, 5459–5472. [Google Scholar] [CrossRef] [PubMed]
- Ritson, D.J.; Sutherland, J.D. Synthesis of aldehydic ribonucleotide and amino acid precursors by photoredox chemistry. Angew. Chem. Int. Ed. Engl. 2013, 52, 5845–5847. [Google Scholar] [CrossRef] [PubMed]
- Patel, B.H.; Percivalle, C.; Ritson, D.J.; Duffy, C.D.; Sutherland, J.D. Common origins of RNA, protein and lipid precursors in a cyanosulfidic protometabolism. Nat. Chem. 2015, 7, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Higgs, P.G.; Pudritz, R.E. A thermodynamic basis for prebiotic amino acid synthesis and the nature of the first genetic code. Astrobiology 2009, 9, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Jordan, I.K.; Kondrashov, F.A.; Adzhubei, I.A.; Wolf, Y.I.; Koonin, E.V.; Kondrashov, A.S.; Sunyaev, S. A universal trend of amino acid gain and loss in protein evolution. Nature 2005, 433, 633–638. [Google Scholar] [CrossRef] [PubMed]
- Trifonov, E.N. The triplet code from first principles. J. Biomol. Struct. Dyn. 2004, 22, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Massey, S.E. A sequential “2-1-3” model of genetic code evolution that explains codon constraints. J. Mol. Evol. 2006, 62, 809–810. [Google Scholar] [CrossRef] [PubMed]
- Higgs, P.G. A four-column theory for the origin of the genetic code: Tracing the evolutionary pathways that gave rise to an optimized code. Biol. Direct 2009, 4, 16. [Google Scholar] [CrossRef] [PubMed]
- Francis, B.R. Evolution of the genetic code by incorporation of amino acids that improved or changed protein function. J. Mol. Evol. 2013, 77, 134–158. [Google Scholar] [CrossRef] [PubMed]
- Fitch, W.M.; Upper, K. The phylogeny of tRNA sequences provides evidence for ambiguity reduction in the origin of the genetic code. Cold Spring Harb. Symp. Quant. Biol. 1987, 52, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Wolf, Y.I.; Aravind, L.; Grishin, N.V.; Koonin, E.V. Evolution of aminoacyl-tRNA synthetases—Analysis of unique domain architectures and phylogenetic trees reveals a complex history of horizontal gene transfer events. Genome Res. 1999, 9, 689–710. [Google Scholar] [PubMed]
- Woese, C.R.; Olsen, G.J.; Ibba, M.; Soll, D. Aminoacyl-tRNA synthetases, the genetic code, and the evolutionary process. Microbiol. Mol. Biol. Rev. 2000, 64, 202–236. [Google Scholar] [CrossRef] [PubMed]
- Aravind, L.; Anantharaman, V.; Koonin, E.V. Monophyly of class I aminoacyl tRNA synthetase, USPA, ETFP, photolyase, and PP-ATPase nucleotide-binding domains: Implications for protein evolution in the RNA. Proteins 2002, 48, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Aravind, L.; Mazumder, R.; Vasudevan, S.; Koonin, E.V. Trends in protein evolution inferred from sequence and structure analysis. Curr. Opin. Struct. Biol. 2002, 12, 392–399. [Google Scholar] [CrossRef]
- Artymiuk, P.J.; Rice, D.W.; Poirrette, A.R.; Willet, P. A tale of two synthetases. Nat. Struct. Biol. 1994, 1, 758–760. [Google Scholar] [CrossRef] [PubMed]
- Anantharaman, V.; Koonin, E.V.; Aravind, L. Comparative genomics and evolution of proteins involved in RNA metabolism. Nucleic Acids Res. 2002, 30, 1427–1464. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V. The Logic of Chance: The Nature and Origin of Biological Evolution; FT Press: Upper Saddle River, NJ, USA, 2011. [Google Scholar]
- Tamura, K. Origins and Early Evolution of the tRNA Molecule. Life 2015, 5, 1687–1699. [Google Scholar] [CrossRef] [PubMed]
- Wolf, Y.I.; Koonin, E.V. On the origin of the translation system and the genetic code in the RNA world by means of natural selection, exaptation, and subfunctionalization. Biol. Direct 2007, 2, 14. [Google Scholar] [CrossRef] [PubMed]
- Woese, C.R. On the evolution of the genetic code. Proc. Natl. Acad. Sci. USA 1965, 54, 1546–1552. [Google Scholar] [CrossRef] [PubMed]
- Szathmary, E. Coding coenzyme handles: A hypothesis for the origin of the genetic code. Proc. Natl. Acad. Sci. USA 1993, 90, 9916–9920. [Google Scholar] [CrossRef] [PubMed]
- Yarus, M. The meaning of a minuscule ribozyme. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2011, 366, 2902–2909. [Google Scholar] [CrossRef] [PubMed]
- Chumachenko, N.V.; Novikov, Y.; Yarus, M. Rapid and simple ribozymic aminoacylation using three conserved nucleotides. J. Am. Chem. Soc. 2009, 131, 5257–5263. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.K.; Yarus, M. RNA-catalyzed amino acid activation. Biochemistry 2001, 40, 6998–7004. [Google Scholar] [CrossRef] [PubMed]
- Serganov, A.; Patel, D.J. Amino acid recognition and gene regulation by riboswitches. Biochim. Biophys. Acta 2009, 1789, 592–611. [Google Scholar] [CrossRef] [PubMed]
- Serganov, A.; Patel, D.J. Metabolite recognition principles and molecular mechanisms underlying riboswitch function. Annu. Rev. Biophys. 2012, 41, 343–370. [Google Scholar] [CrossRef] [PubMed]
- Monod, J. Chance and Necessity: An Essay on the Natural Philosophy of Modern Biology; Vintage: New York, NY, USA, 1972. [Google Scholar]
- Lynch, M. The origins of Genome Archiecture; Sinauer Associates: Sunderland, MA, USA, 2007. [Google Scholar]
© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koonin, E.V. Frozen Accident Pushing 50: Stereochemistry, Expansion, and Chance in the Evolution of the Genetic Code. Life 2017, 7, 22. https://doi.org/10.3390/life7020022
Koonin EV. Frozen Accident Pushing 50: Stereochemistry, Expansion, and Chance in the Evolution of the Genetic Code. Life. 2017; 7(2):22. https://doi.org/10.3390/life7020022
Chicago/Turabian StyleKoonin, Eugene V. 2017. "Frozen Accident Pushing 50: Stereochemistry, Expansion, and Chance in the Evolution of the Genetic Code" Life 7, no. 2: 22. https://doi.org/10.3390/life7020022
APA StyleKoonin, E. V. (2017). Frozen Accident Pushing 50: Stereochemistry, Expansion, and Chance in the Evolution of the Genetic Code. Life, 7(2), 22. https://doi.org/10.3390/life7020022