
Engineering Protocells: Prospects for Self-Assembly and Nanoscale Production-Lines

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Protocells—From the Ground Up
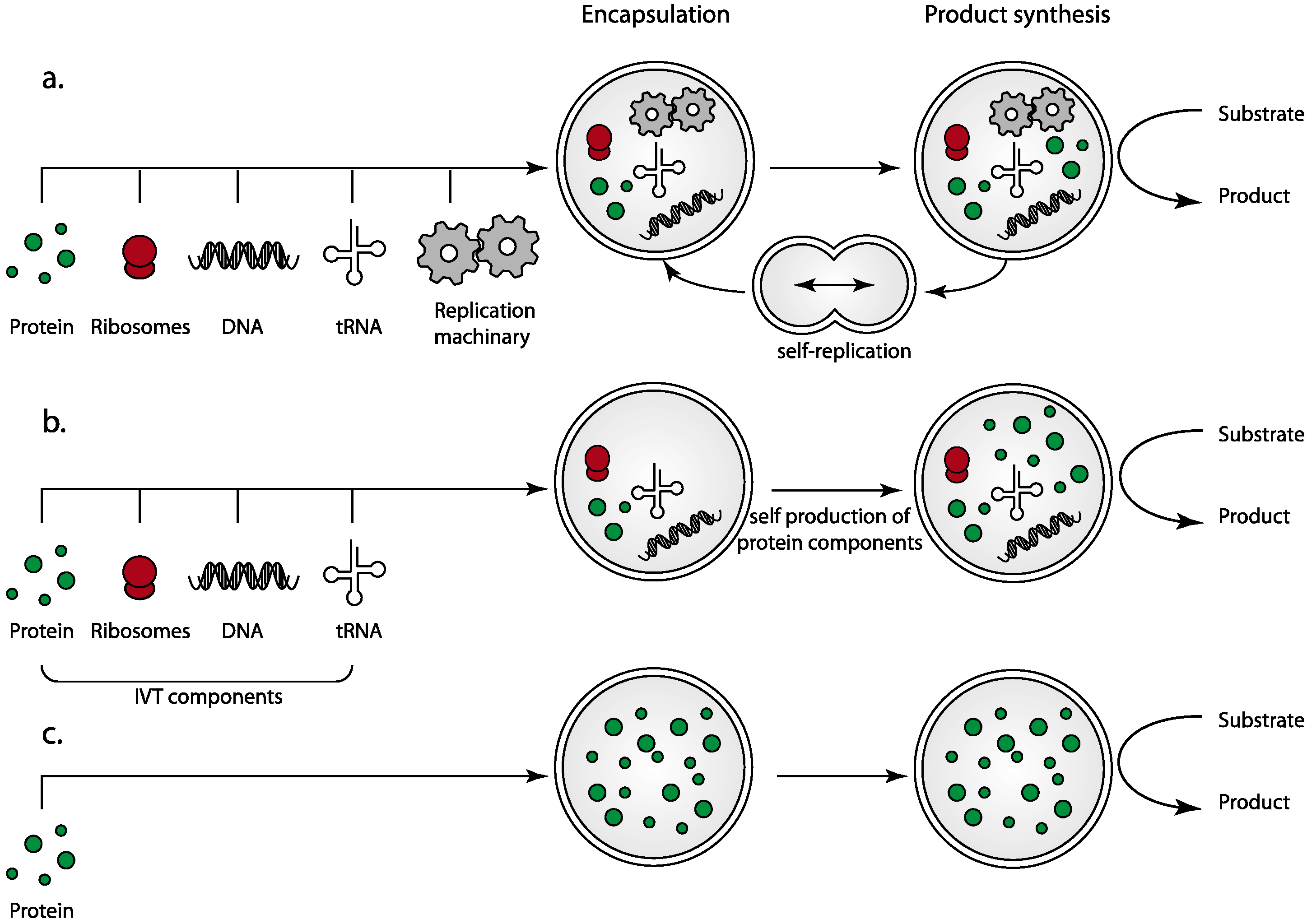
2.1. The Complexity of Replication and Cell Division: The Benefits of Simplification
2.2. Protein-Only Protocells
2.3. The Protocell Membrane and Encapsulation of Internal Components
3. Self-Assembling Protocells—The Next Step
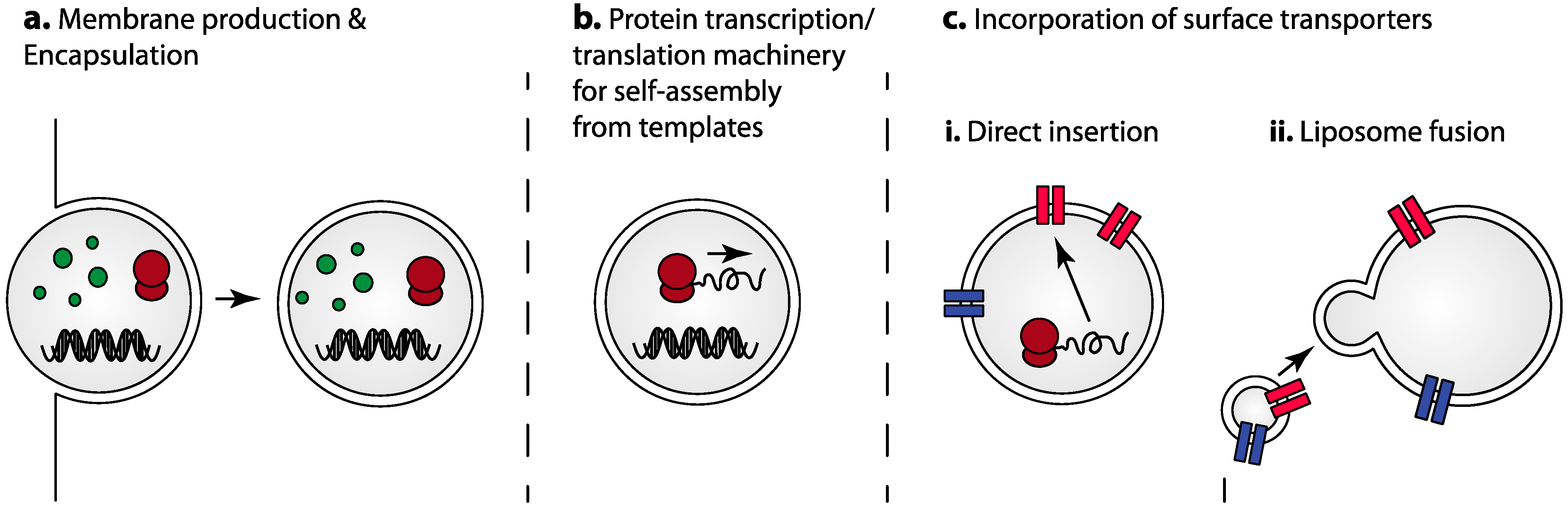
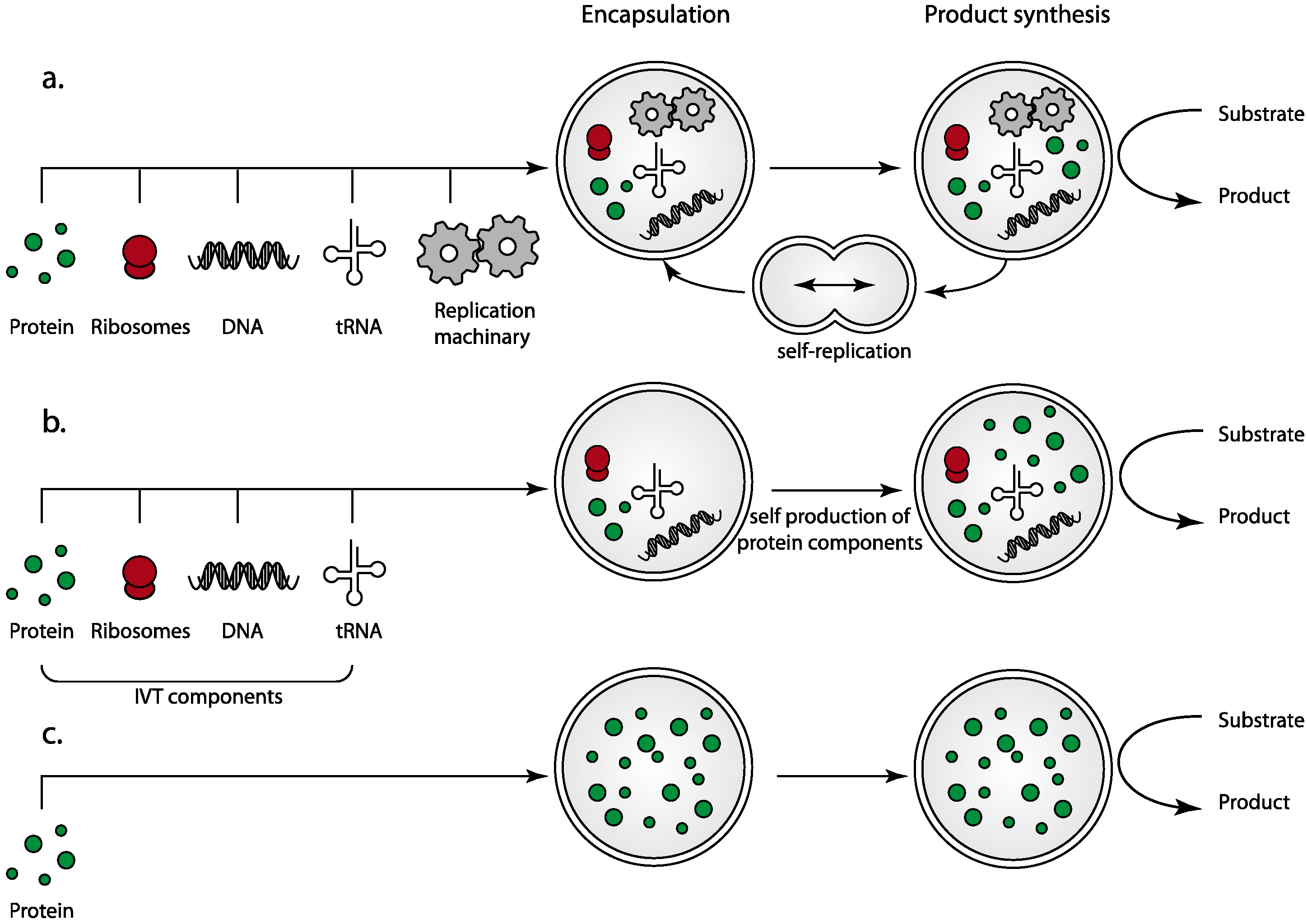
3.1. Self-Assembly Utilizing Transcription/Translation Machinery for Protein Synthesis
3.2. Incorporation of Surface Transporters and Receptors
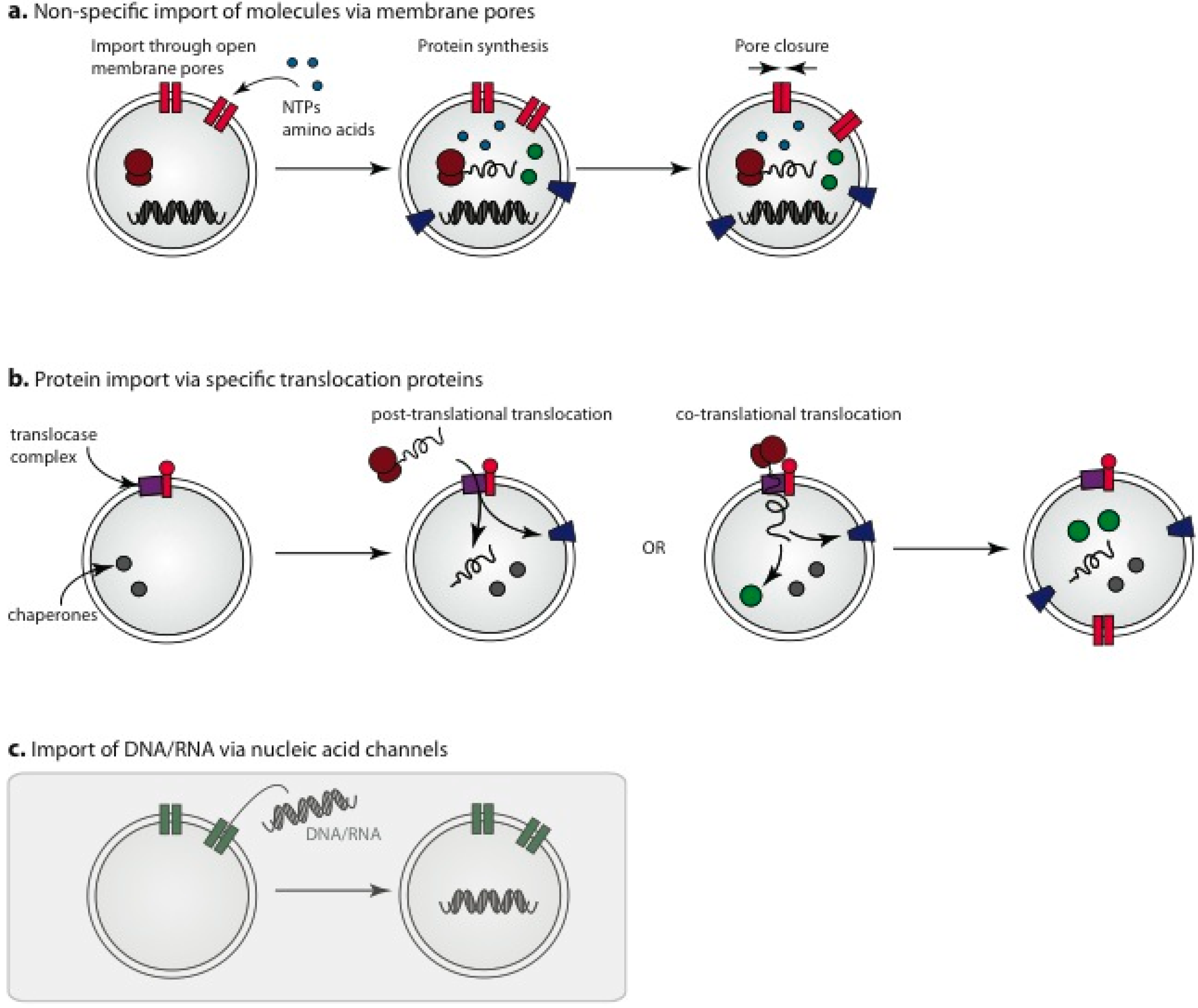
3.3. Membrane Pores for Incorporation of Internal Protocell Components
3.4. Engineered Protein Pores for Incorporation of Small Molecules into Protocells
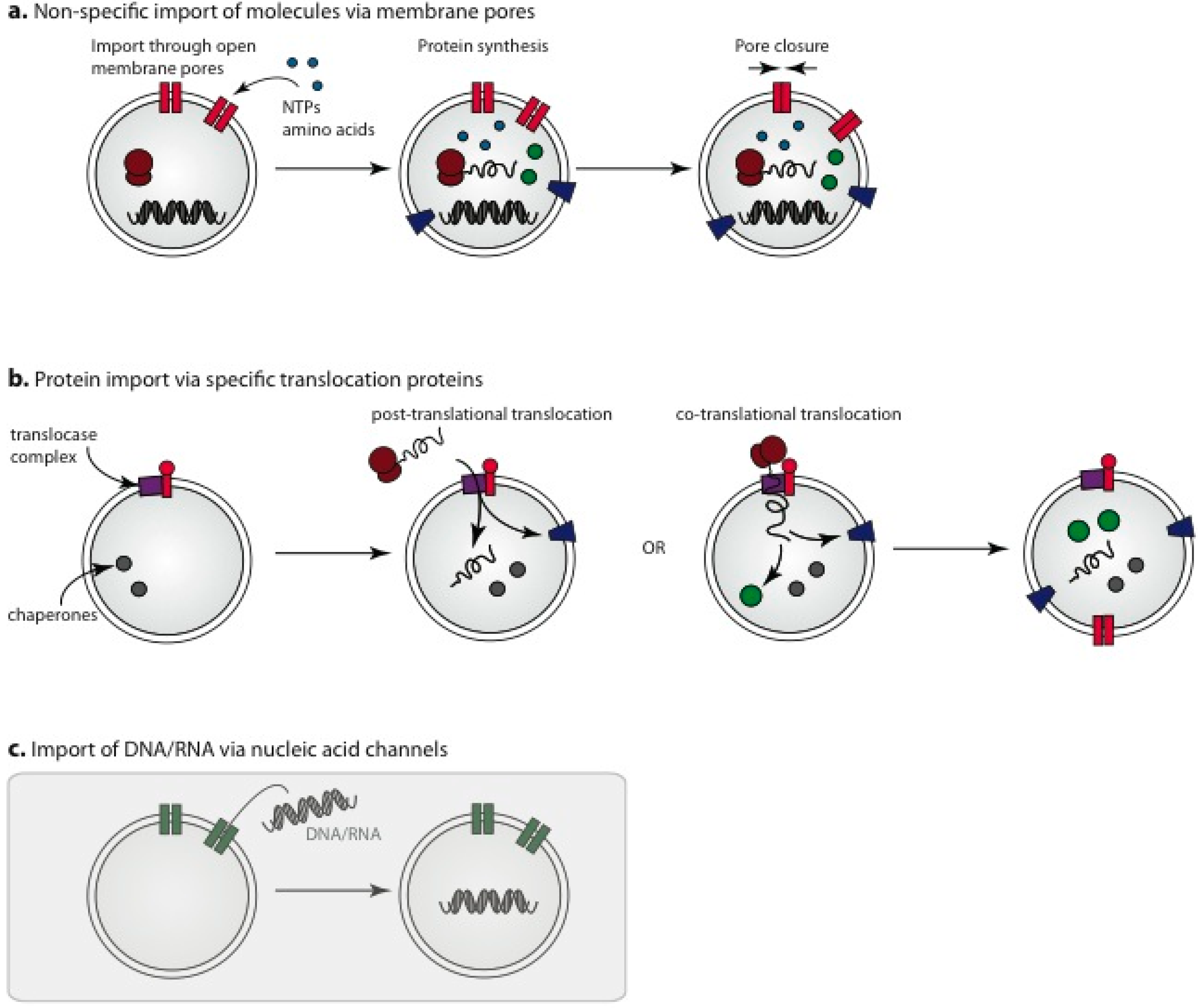
3.5. Import of Pre-Assembled Protein Components
3.6. Nucleic Acid Channels
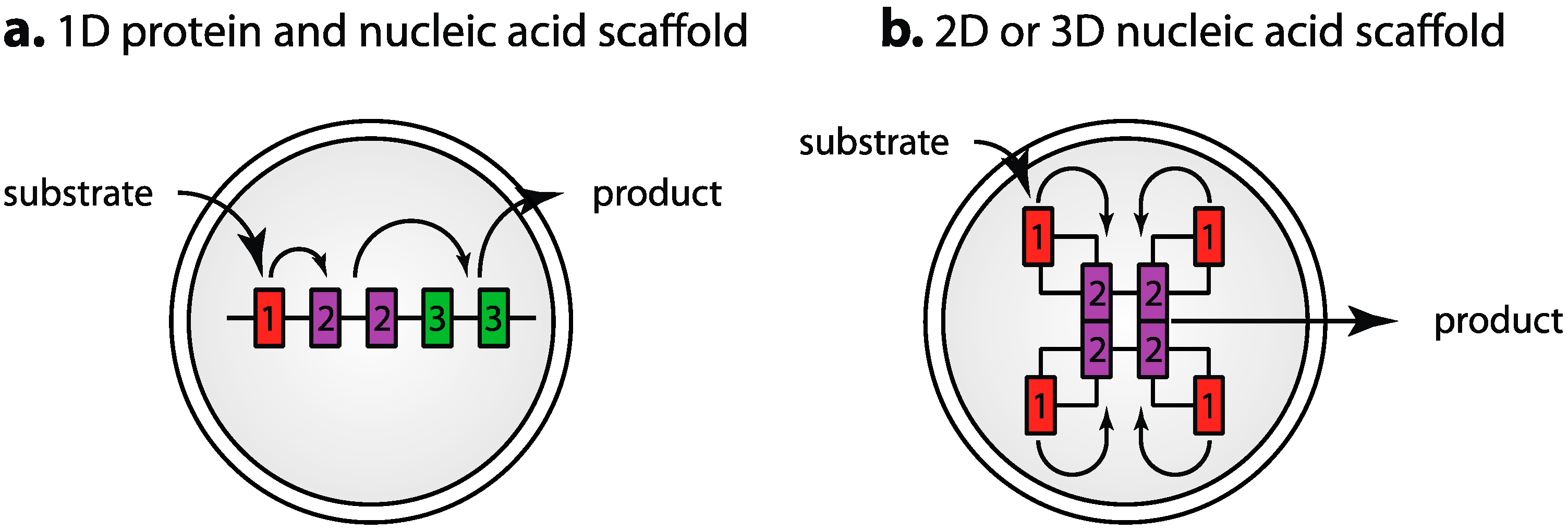
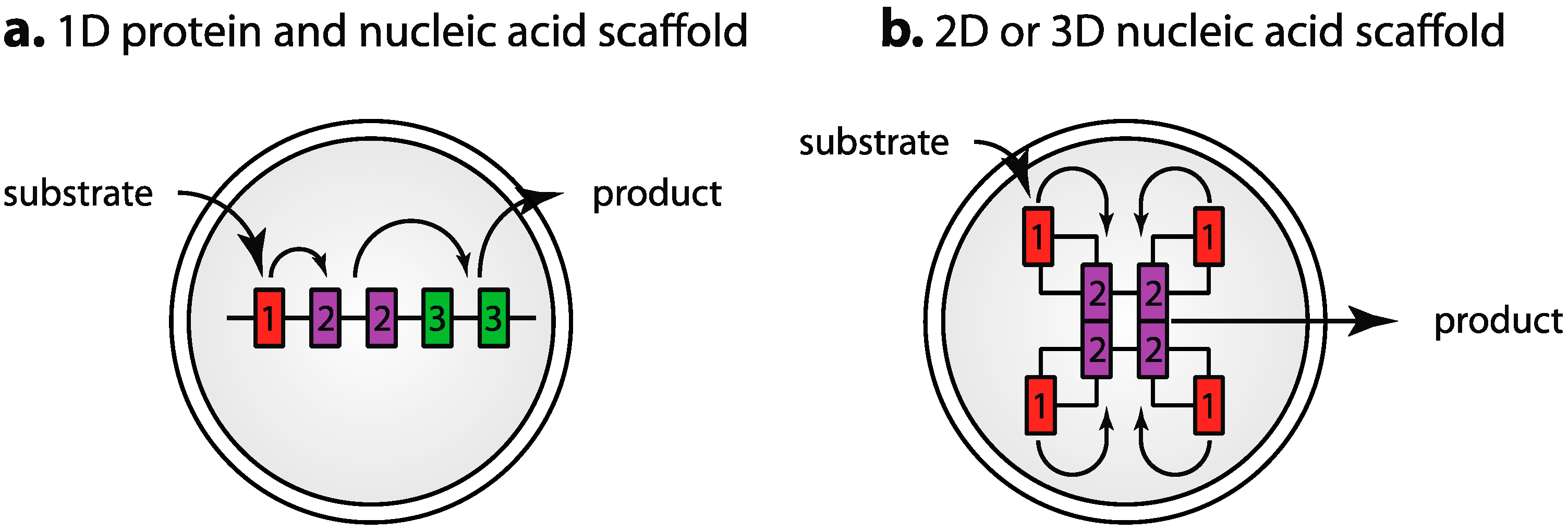
3.7. Segregation of Components: Engineered Scaffolds and Compartmentalization
4. Microfluidic Production Lines for Protocell Construction
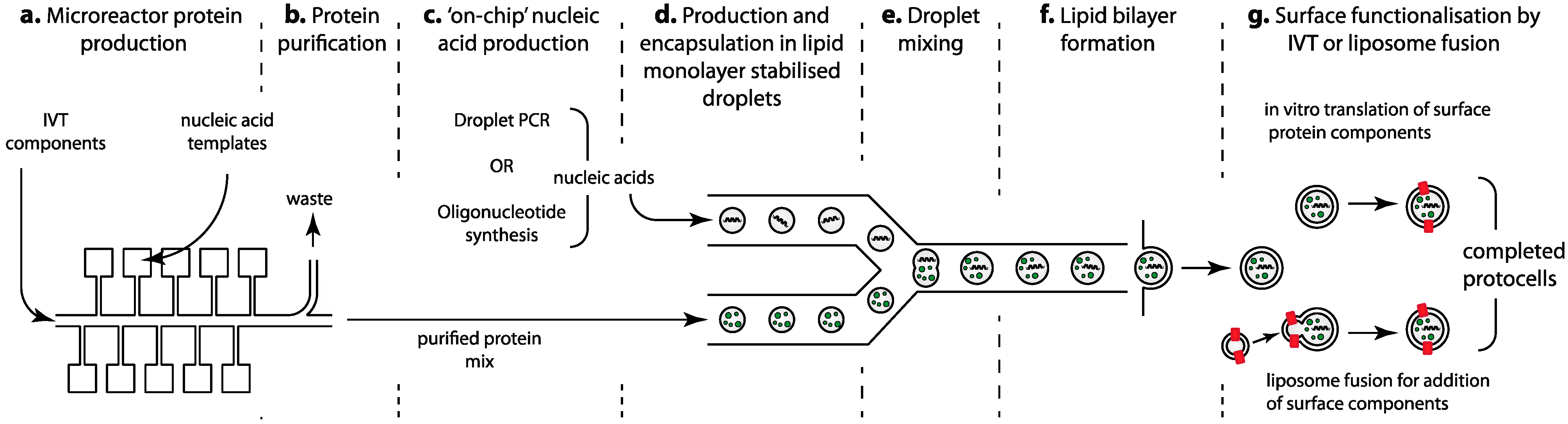
4.1. Microfluidic Protein Production and Purification

4.2. Microfluidic Production of Nucleic Acid “Genomes” and Scaffolds
4.3. Droplet Based Methods for Processing and Mixing
4.4. The Use of Microfluidics for Liposome/Protocell Formation
5. Additional Considerations in Designing Purpose-Built Protocells
5.1. Internal Crowding
5.2. Protocell Lifetime
5.3. Energy Generation
5.4. Ribosomes
5.5. Optimizing Design
5.6. Gene Regulation
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Howard, T.P.; Middelhaufe, S.; Moore, K.; Edner, C.; Kolak, D.M.; Taylor, G.N.; Parker, D.A.; Lee, R.; Smirnoff, N.; Aves, S.J.; et al. Synthesis of customized petroleum-replica fuel molecules by targeted modification of free fatty acid pools in Escherichia coli. Proc. Natl. Acad. Sci. USA 2013, 110, 7636–7641. [Google Scholar] [CrossRef]
- Atsumi, S.; Hanai, T.; Liao, J.C. Non-fermentative pathways for synthesis of branched-chain higher alcohols as biofuels. Nature 2008, 451, 86–89. [Google Scholar] [CrossRef] [PubMed]
- Avalos, J.L.; Fink, G.R.; Stephanopoulos, G. Compartmentalization of metabolic pathways in yeast mitochondria improves the production of branched-chain alcohols. Nat. Biotechnol. 2013, 31, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Tezuka, H.; Ishii, J.; Matsuda, F.; Ogino, C.; Kondo, A. Genetic engineering to enhance the Ehrlich pathway and alter carbon flux for increased isobutanol production from glucose by Saccharomyces cerevisiae. J. Biotechnol. 2012, 159, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Nielsen, K.F.; Borodina, I.; Kielland-Brandt, M.C.; Karhumaa, K. Increased isobutanol production in Saccharomyces cerevisiae by overexpression of genes in valine metabolism. Biotechnol. Biofuels 2011, 4, 2089–2090. [Google Scholar] [CrossRef]
- Lee, W.-H.; Seo, S.-O.; Bae, Y.-H.; Nan, H.; Jin, Y.-S.; Seo, J.-H. Isobutanol production in engineered Saccharomyces cerevisiae by overexpression of 2-ketoisovalerate decarboxylase and valine biosynthetic enzymes. Bioprocess Biosyst. Eng. 2012, 35, 1467–1475. [Google Scholar] [CrossRef] [PubMed]
- Atsumi, S.; Cann, A.F.; Connor, M.R.; Shen, C.R.; Smith, K.M.; Brynildsen, M.P.; Chou, K.J.Y.; Hanai, T.; Liao, J.C. Metabolic engineering of Escherichia coli for 1-butanol production. Metab. Eng. 2008, 10, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Hanai, T.; Atsumi, S.; Liao, J.C. Engineered synthetic pathway for isopropanol production in Escherichia coli. Appl. Environ. Microbiol. 2007, 73, 7814–7818. [Google Scholar] [CrossRef] [PubMed]
- Connor, M.R.; Liao, J.C. Engineering of an Escherichia coli strain for the production of 3-methyl-1-butanol. Appl. Environ. Microbiol. 2008, 74, 5769–5775. [Google Scholar] [CrossRef] [PubMed]
- Atsumi, S.; Higashide, W.; Liao, J.C. Direct photosynthetic recycling of carbon dioxide to isobutyraldehyde. Nat. Biotechnol. 2009, 27, 1177–1180. [Google Scholar] [CrossRef] [PubMed]
- Lan, E.I.; Liao, J.C. Metabolic engineering of cyanobacteria for 1-butanol production from carbon dioxide. Metab. Eng. 2011, 13, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Martin, V.J.J.; Pitera, D.J.; Withers, S.T.; Newman, J.D.; Keasling, J.D. Engineering a mevalonate pathway in Escherichia coli for production of terpenoids. Nat. Biotechnol. 2003, 21, 796–802. [Google Scholar] [CrossRef] [PubMed]
- Paddon, C.J.; Westfall, P.J.; Pitera, D.J.; Benjamin, K.; Fisher, K.; McPhee, D.; Leavell, M.D.; Tai, A.; Main, A.; Eng, D.; et al. High-level semi-synthetic production of the potent antimalarial artemisinin. Nature 2013, 496, 528–532. [Google Scholar] [CrossRef]
- Ro, D.-K.; Paradise, E.M.; Ouellet, M.; Fisher, K.J.; Newman, K.L.; Ndungu, J.M.; Ho, K.A.; Eachus, R.A.; Ham, T.S.; Kirby, J.; et al. Production of the antimalarial drug precursor artemisinic acid in engineered yeast. Nature 2006, 440, 940–943. [Google Scholar] [CrossRef]
- Ajikumar, P.K.; Xiao, W.-H.; Tyo, K.E.J.; Wang, Y.; Simeon, F.; Leonard, E.; Mucha, O.; Phon, T.H.; Pfeifer, B.; Stephanopoulos, G. Isoprenoid pathway optimization for Taxol precursor overproduction in Escherichia coli. Science 2010, 330, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Pitera, D.J.; Paddon, C.J.; Newman, J.D.; Keasling, J.D. Balancing a heterologous mevalonate pathway for improved isoprenoid production in Escherichia coli. Metab. Eng. 2007, 9, 193–207. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.M.; Lawman, P.D.; Cameron, D.C. Improving 1,3-propanediol production from glycerol in a metabolically engineered Escherichia coli by reducing accumulation of sn-glycerol-3-phosphate. Biotechnol. Prog. 2002, 18, 694–699. [Google Scholar] [CrossRef] [PubMed]
- Barbirato, F.; Grivet, J.P.; Soucaille, P.; Bories, A. 3-Hydroxypropionaldehyde, an inhibitory metabolite of glycerol fermentation to 1,3-propanediol by enterobacterial species. Appl. Environ. Microbiol. 1996, 62, 1448–1451. [Google Scholar] [PubMed]
- Ro, D.-K.; Ouellet, M.; Paradise, E.; Burd, H.; Eng, D.; Paddon, C.; Newman, J.; Keasling, J. Induction of multiple pleiotropic drug resistance genes in yeast engineered to produce an increased level of anti-malarial drug precursor, artemisinic acid. BMC Biotechnol. 2008, 8. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.M.; Liao, J.C. An evolutionary strategy for isobutanol production strain development in Escherichia coli. Metab. Eng. 2011, 13, 674–681. [Google Scholar] [CrossRef] [PubMed]
- Fischer, C.R.; Klein-Marcuschamer, D.; Stephanopoulos, G. Selection and optimization of microbial hosts for biofuels production. Metab. Eng. 2008, 10, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Caspi, Y.; Dekker, C. Divided we stand: Splitting synthetic cells for their proliferation. Syst. Synth. Biol. 2014, 8, 249–269. [Google Scholar] [CrossRef] [PubMed]
- Nourian, Z.; Scott, A.; Danelon, C. Toward the assembly of a minimal divisome. Syst. Synth. Biol. 2014, 8, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Osawa, M.; Erickson, H.P. Liposome division by a simple bacterial division machinery. Proc. Natl. Acad. Sci. USA 2013, 110, 11000–11004. [Google Scholar] [CrossRef] [PubMed]
- Laganowsky, A.; Reading, E.; Allison, T.M.; Ulmschneider, M.B.; Degiacomi, M.T.; Baldwin, A.J.; Robinson, C.V. Membrane proteins bind lipids selectively to modulate their structure and function. Nature 2014, 510, 172–175. [Google Scholar] [CrossRef] [PubMed]
- Dowhan, W.; Bogdanov, M. Lipid-protein interactions as determinants of membrane protein structure and function. Biochem. Soc. Trans. 2011, 39, 767–774. [Google Scholar] [PubMed]
- Cantor, R.S. The influence of membrane lateral pressures on simple geometric models of protein conformational equilibria. Chem. Phys. Lipids 1999, 101, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Andersen, O.S.; Koeppe, R.E. Bilayer thickness and membrane protein function: An energetic perspective. Annu. Rev. Biophys. Biomol. Struct. 2007, 36, 107–130. [Google Scholar] [CrossRef] [PubMed]
- Curran, A.R.; Templer, R.H.; Booth, P.J. Modulation of folding and assembly of the membrane protein bacteriorhodopsin by intermolecular forces within the lipid bilayer. Biochemistry 1999, 38, 9328–9336. [Google Scholar] [CrossRef] [PubMed]
- McKibbin, C.; Farmer, N.A.; Jeans, C.; Reeves, P.J.; Khorana, H.G.; Wallace, B.A.; Edwards, P.C.; Villa, C.; Booth, P.J. Opsin stability and folding: Modulation by phospholipid bicelles. J. Mol. Biol. 2007, 374, 1319–1332. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.; Charalambous, K.; Rotem, D.; Schuldiner, S.; Curnow, P.; Booth, P.J. In vitro unfolding and refolding of the small multidrug transporter EmrE. J. Mol. Biol. 2009, 393, 815–832. [Google Scholar] [CrossRef] [PubMed]
- Cantor, R.S. Lipid composition and the lateral pressure profile in bilayers. Biophys. J. 1999, 76, 2625–2639. [Google Scholar] [CrossRef] [PubMed]
- Walde, P.; Ichikawa, S. Enzymes inside lipid vesicles: Preparation, reactivity and applications. Biomol. Eng. 2001, 18, 143–177. [Google Scholar] [CrossRef] [PubMed]
- Sessa, G.; Weissmann, G. Incorporation of lysozyme into liposomes. A model for structure-linked latency. J. Biol. Chem. 1970, 245, 3295–3301. [Google Scholar] [PubMed]
- Kaszuba, M.; Jones, M.N. Hydrogen peroxide production from reactive liposomes encapsulating enzymes. Biochim. Biophys. Acta Biomembr. 1999, 1419, 221–228. [Google Scholar] [CrossRef]
- Pereira de Souza, T.; Stano, P.; Luisi, P.L. The minimal size of liposome-based model cells brings about a remarkably enhanced entrapment and protein synthesis. Chembiochem 2009, 10, 1056–1063. [Google Scholar] [CrossRef] [PubMed]
- Luisi, P.L.; Allegretti, M.; Pereira de Souza, T.; Steiniger, F.; Fahr, A.; Stano, P. Spontaneous protein crowding in liposomes: A new vista for the origin of cellular metabolism. Chembiochem 2010, 11, 1989–1992. [Google Scholar] [CrossRef] [PubMed]
- Olson, F.; Hunt, C.A.; Szoka, F.C.; Vail, W.J.; Papahadjopoulos, D. Preparation of liposomes of defined size distribution by extrusion through polycarbonate membranes. Biochim. Biophys. Acta Biomembr. 1979, 557, 9–23. [Google Scholar] [CrossRef]
- Hub, H.H.; Zimmermann, U.; Ringsdorf, H. Preparation of large unilamellar vesicles. FEBS Lett. 1982, 140, 254–256. [Google Scholar] [CrossRef]
- Mueller, P.; Chien, T.F.; Rudy, B. Formation and properties of cell-size lipid bilayer vesicles. Biophys. J. 1983, 44, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Angelova, M.I.; Dimitrov, D.S. Liposome electroformation. Faraday Discuss. Chem. Soc. 1986, 81, 303–311. [Google Scholar] [CrossRef]
- Oberholzer, T.; Wick, R.; Luisi, P.L.; Biebricher, C.K. Enzymatic RNA replication in self-reproducing vesicles: An approach to a minimal cell. Biochem. Biophys. Res. Commun. 1995, 207, 250–257. [Google Scholar] [CrossRef] [PubMed]
- Oberholzer, T.; Albrizio, M.; Luisi, P. Polymerase chain reaction in liposomes. Chem. Biol. 1995, 2, 677–682. [Google Scholar] [CrossRef] [PubMed]
- Oberholzer, T.; Meyer, E.; Amato, I.; Lustig, A.; Monnard, P.-A. Enzymatic reactions in liposomes using the detergent-induced liposome loading method. Biochim. Biophys. Acta Biomembr. 1999, 1416, 57–68. [Google Scholar] [CrossRef]
- Tsai, F.-C.; Stuhrmann, B.; Koenderink, G.H. Encapsulation of active cytoskeletal protein networks in cell-sized liposomes. Langmuir 2011, 27, 10061–10071. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, K.; Sato, K.; Shima, Y.; Urabe, I.; Yomo, T. Expression of a cascading genetic network within liposomes. FEBS Lett. 2004, 576, 387–390. [Google Scholar] [CrossRef] [PubMed]
- Wick, R.; Luisi, P.L. Enzyme-containing liposomes can endogenously produce membrane-constituting lipids. Chem. Biol. 1996, 3, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Kuruma, Y.; Stano, P.; Ueda, T.; Luisi, P.L. A synthetic biology approach to the construction of membrane proteins in semi-synthetic minimal cells. Biochim. Biophys. Acta 2009, 1788, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Schmidli, P.K.; Schurtenberger, P.; Luisi, P.L. Liposome-mediated enzymatic synthesis of phosphatidylcholine as an approach to self-replicating liposomes. J. Am. Chem. Soc. 1991, 113, 8127–8130. [Google Scholar] [CrossRef]
- Wick, R.; Angelova, M.; Walde, P.; Luisi, P. Microinjection into giant vesicles and light microscopy investigation of enzyme-mediated vesicle transformations. Chem. Biol. 1996, 3, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Chiba, M.; Miyazaki, M.; Ishiwata, S. Quantitative analysis of the lamellarity of giant liposomes prepared by the inverted emulsion method. Biophys. J. 2014, 107, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Pautot, S.; Frisken, B.J.; Weitz, D.A. Production of unilamellar vesicles using an inverted emulsion. Langmuir 2003, 19, 2870–2879. [Google Scholar] [CrossRef]
- Toyota, T.; Ohguri, N.; Maruyama, K.; Fujinami, M.; Saga, T.; Aoki, I. Giant vesicles containing superparamagnetic iron oxide as biodegradable cell-tracking MRI probes. Anal. Chem. 2012, 84, 3952–3957. [Google Scholar] [CrossRef] [PubMed]
- Noireaux, V.; Maeda, Y.T.; Libchaber, A. Development of an artificial cell, from self-organization to computation and self-reproduction. Proc. Natl. Acad. Sci. USA 2011, 108, 3473–3480. [Google Scholar] [CrossRef] [PubMed]
- Hamada, S.; Tabuchi, M.; Toyota, T.; Sakurai, T.; Hosoi, T.; Nomoto, T.; Nakatani, K.; Fujinami, M.; Kanzaki, R. Giant vesicles functionally expressing membrane receptors for an insect pheromone. Chem. Commun. 2014, 50, 2958–2961. [Google Scholar] [CrossRef]
- Elani, Y.; Law, R.V.; Ces, O. Vesicle-based artificial cells as chemical microreactors with spatially segregated reaction pathways. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Maeda, Y.T.; Nakadai, T.; Shin, J.; Uryu, K.; Noireaux, V.; Libchaber, A. Assembly of MreB filaments on liposome membranes: A synthetic biology approach. ACS Synth. Biol. 2012, 1, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Sunami, T.; Sato, K.; Matsuura, T.; Tsukada, K.; Urabe, I.; Yomo, T. Femtoliter compartment in liposomes for in vitro selection of proteins. Anal. Biochem. 2006, 357, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Sato, K.; Wakabayashi, M.; Nakaishi, T.; Ko-Mitamura, E.P.; Shima, Y.; Urabe, I.; Yomo, T. Synthesis of functional protein in liposome. J. Biosci. Bioeng. 2001, 92, 590–593. [Google Scholar] [CrossRef] [PubMed]
- Fujii, S.; Matsuura, T.; Sunami, T.; Kazuta, Y.; Yomo, T. In vitro evolution of α-hemolysin using a liposome display. Proc. Natl. Acad. Sci. USA 2013, 110, 16796–16801. [Google Scholar] [CrossRef] [PubMed]
- Noireaux, V.; Libchaber, A. A vesicle bioreactor as a step toward an artificial cell assembly. Proc. Natl. Acad. Sci. USA 2004, 101, 17669–17674. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Inoue, A.; Tomari, Y.; Suzuki, T.; Yokogawa, T.; Nishikawa, K.; Ueda, T. Cell-free translation reconstituted with purified components. Nat. Biotechnol. 2001, 19, 751–755. [Google Scholar] [CrossRef] [PubMed]
- Seddon, A.M.; Curnow, P.; Booth, P.J. Membrane proteins, lipids and detergents: Not just a soap opera. Biochim. Biophys. Acta 2004, 1666, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Aimon, S.; Manzi, J.; Schmidt, D.; Poveda Larrosa, J.A.; Bassereau, P.; Toombes, G.E.S. Functional reconstitution of a voltage-gated potassium channel in giant unilamellar vesicles. PLoS One 2011, 6, e25529. [Google Scholar] [CrossRef] [PubMed]
- Folgering, J.H.A.; Kuiper, J.M.; de Vries, A.H.; Engberts, J.B.F.N.; Poolman, B. Lipid-mediated light activation of a mechanosensitive channel of large conductance. Langmuir 2004, 20, 6985–6987. [Google Scholar] [CrossRef] [PubMed]
- Kedrov, A.; Kusters, I.; Krasnikov, V.V.; Driessen, A.J.M. A single copy of SecYEG is sufficient for preprotein translocation. EMBO J. 2011, 30, 4387–4397. [Google Scholar] [CrossRef] [PubMed]
- Girard, P.; Pécréaux, J.; Lenoir, G.; Falson, P.; Rigaud, J.-L.; Bassereau, P. A new method for the reconstitution of membrane proteins into giant unilamellar vesicles. Biophys. J. 2004, 87, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Nomura, S.-i.M.; Kondoh, S.; Asayama, W.; Asada, A.; Nishikawa, S.; Akiyoshi, K. Direct preparation of giant proteo-liposomes by in vitro membrane protein synthesis. J. Biotechnol. 2008, 133, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Matsubayashi, H.; Kuruma, Y.; Ueda, T. In vitro synthesis of the E. coli sec translocon from DNA. Angew. Chem. Int. Ed. 2014, 53, 7535–7538. [Google Scholar] [CrossRef]
- Kahya, N.; Pécheur, E.I.; de Boeij, W.P.; Wiersma, D.A.; Hoekstra, D. Reconstitution of membrane proteins into giant unilamellar vesicles via peptide-induced fusion. Biophys. J. 2001, 81, 1464–1474. [Google Scholar] [CrossRef] [PubMed]
- Robson Marsden, H.; Korobko, A.V.; Zheng, T.; Voskuhl, J.; Kros, A. Controlled liposome fusion mediated by SNARE protein mimics. Biomater. Sci. 2013, 1, 1046–1054. [Google Scholar] [CrossRef]
- Richmond, D.L.; Schmid, E.M.; Martens, S.; Stachowiak, J.C.; Liska, N.; Fletcher, D.A. Forming giant vesicles with controlled membrane composition, asymmetry, and contents. Proc. Natl. Acad. Sci. USA 2011, 108, 9431–9436. [Google Scholar] [CrossRef] [PubMed]
- Nomura, F.; Inaba, T.; Ishikawa, S.; Nagata, M.; Takahashi, S.; Hotani, H.; Takiguchi, K. Microscopic observations reveal that fusogenic peptides induce liposome shrinkage prior to membrane fusion. Proc. Natl. Acad. Sci. USA 2004, 101, 3420–3425. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, Y.; Tomohiro, T.; Kodaka, M.; Okuno, H.; Yamazaki, Y. Non-peptidic liposome-fusion compounds at acidic pH. Chem. Commun. 1999. [Google Scholar] [CrossRef]
- Dutta, D.; Pulsipher, A.; Luo, W.; Mak, H.; Yousaf, M.N. Engineering cell surfaces via liposome fusion. Bioconjug. Chem. 2011, 22, 2423–2433. [Google Scholar] [CrossRef] [PubMed]
- Woodbury, D.J.; Miller, C. Nystatin-induced liposome fusion. A versatile approach to ion channel reconstitution into planar bilayers. Biophys. J. 1990, 58, 833–839. [Google Scholar] [CrossRef] [PubMed]
- Russo, M.; Bayley, H.; Toner, M. Reversible permeabilization of plasma membranes with an engineered switchable pore. Nat. Biotechnol. 1997, 15, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Koçer, A.; Walko, M.; Feringa, B.L. Synthesis and utilization of reversible and irreversible light-activated nanovalves derived from the channel protein MscL. Nat. Protoc. 2007, 2, 1426–1437. [Google Scholar] [CrossRef] [PubMed]
- Chiu, H.-C.; Lin, Y.-W.; Huang, Y.-F.; Chuang, C.-K.; Chern, C.-S. Polymer vesicles containing small vesicles within interior aqueous compartments and pH-responsive transmembrane channels. Angew. Chem. Int. Ed. Engl. 2008, 47, 1875–1878. [Google Scholar] [CrossRef] [PubMed]
- Chaize, B.; Colletier, J.-P.; Winterhalter, M.; Fournier, D. Encapsulation of Enzymes in Liposomes: High Encapsulation Efficiency and Control of Substrate Permeability. Artif. Cells Blood Substitutes Biotechnol. 2004, 32, 67–75. [Google Scholar] [CrossRef]
- Pfanner, N.; Geissler, A. Versatility of the mitochondrial protein import machinery. Nat. Rev. Mol. Cell Biol. 2001, 2, 339–349. [Google Scholar] [CrossRef] [PubMed]
- Wiedemann, N.; Frazier, A.E.; Pfanner, N. The protein import machinery of mitochondria. J. Biol. Chem. 2004, 279, 14473–14476. [Google Scholar] [CrossRef] [PubMed]
- Dudek, J.; Rehling, P.; van der Laan, M. Mitochondrial protein import: Common principles and physiological networks. Biochim. Biophys. Acta 2013, 1833, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Neupert, W. Protein import into mitochondria. Annu. Rev. Biochem. 1997, 66, 863–917. [Google Scholar] [CrossRef] [PubMed]
- Soll, J.; Schleiff, E. Protein import into chloroplasts. Nat. Rev. Mol. Cell Biol. 2004, 5, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Agne, B.; Kessler, F. Protein transport in organelles: The Toc complex way of preprotein import. FEBS J. 2009, 276, 1156–1165. [Google Scholar] [CrossRef] [PubMed]
- Balsera, M.; Soll, J.; Bölter, B. Protein import machineries in endosymbiotic organelles. Cell. Mol. Life Sci. 2009, 66, 1903–1923. [Google Scholar] [CrossRef] [PubMed]
- Vasiljev, A.; Ahting, U.; Nargang, F.E.; Go, N.E.; Habib, S.J.; Kozany, C.; Panneels, V.; Sinning, I.; Prokisch, H.; Neupert, W.; et al. Reconstituted TOM core complex and Tim9/Tim10 complex of mitochondria are sufficient for translocation of the ADP/ATP carrier across membranes. Mol. Biol. Cell 2004, 15, 1445–1458. [Google Scholar] [CrossRef] [PubMed]
- Kang, P.J.; Ostermann, J.; Shilling, J.; Neupert, W.; Craig, E.A.; Pfanner, N. Requirement for hsp70 in the mitochondrial matrix for translocation and folding of precursor proteins. Nature 1990, 348, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Ungermann, C.; Neupert, W.; Cyr, D.M. The role of Hsp70 in conferring unidirectionality on protein translocation into mitochondria. Science 1994, 266, 1250–1253. [Google Scholar] [CrossRef] [PubMed]
- Voisine, C.; Craig, E.A.; Zufall, N.; von Ahsen, O.; Pfanner, N.; Voos, W. The Protein Import Motor of Mitochondria: Unfolding and Trapping of Preproteins are Distinct and Separable Functions of Matrix Hsp70. Cell 1999, 97, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Geissler, A.; Rassow, J.; Pfanner, N.; Voos, W. Mitochondrial import driving forces: Enhanced trapping by matrix Hsp70 stimulates translocation and reduces the membrane potential dependence of loosely folded preproteins. Mol. Cell. Biol. 2001, 21, 7097–7104. [Google Scholar] [CrossRef] [PubMed]
- Kovermann, P.; Truscott, K.N.; Guiard, B.; Rehling, P.; Sepuri, N.B.; Müller, H.; Jensen, R.E.; Wagner, R.; Pfanner, N. Tim22, the essential core of the mitochondrial protein insertion complex, forms a voltage-activated and signal-gated channel. Mol. Cell 2002, 9, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Rehling, P.; Model, K.; Brandner, K.; Kovermann, P.; Sickmann, A.; Meyer, H.E.; Kühlbrandt, W.; Wagner, R.; Truscott, K.N.; Pfanner, N. Protein insertion into the mitochondrial inner membrane by a twin-pore translocase. Science 2003, 299, 1747–1751. [Google Scholar] [CrossRef] [PubMed]
- Schleiff, E.; Jelic, M.; Soll, J. A GTP-driven motor moves proteins across the outer envelope of chloroplasts. Proc. Natl. Acad. Sci. USA 2003, 100, 4604–4609. [Google Scholar] [CrossRef] [PubMed]
- Heins, L.; Mehrle, A.; Hemmler, R.; Wagner, R.; Küchler, M.; Hörmann, F.; Sveshnikov, D.; Soll, J. The preprotein conducting channel at the inner envelope membrane of plastids. EMBO J. 2002, 21, 2616–2625. [Google Scholar] [CrossRef] [PubMed]
- Balsera, M.; Goetze, T.A.; Kovács-Bogdán, E.; Schürmann, P.; Wagner, R.; Buchanan, B.B.; Soll, J.; Bölter, B. Characterization of Tic110, a channel-forming protein at the inner envelope membrane of chloroplasts, unveils a response to Ca2+ and a stromal regulatory disulfide bridge. J. Biol. Chem. 2009, 284, 2603–2616. [Google Scholar] [CrossRef] [PubMed]
- Sanganna Gari, R.R.; Frey, N.C.; Mao, C.; Randall, L.L.; King, G.M. Dynamic structure of the translocon SecYEG in membrane: Direct single molecule observations. J. Biol. Chem. 2013, 288, 16848–16854. [Google Scholar] [CrossRef] [PubMed]
- Stiegler, N.; Dalbey, R.E.; Kuhn, A. M13 procoat protein insertion into YidC and SecYEG proteoliposomes and liposomes. J. Mol. Biol. 2011, 406, 362–370. [Google Scholar] [CrossRef] [PubMed]
- Van der Laan, M.; Houben, E.N.; Nouwen, N.; Luirink, J.; Driessen, A.J. Reconstitution of Sec-dependent membrane protein insertion: Nascent FtsQ interacts with YidC in a SecYEG-dependent manner. EMBO Rep. 2001, 2, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Price, C.E.; Kocer, A.; Kol, S.; van der Berg, J.P.; Driessen, A.J.M. In vitro synthesis and oligomerization of the mechanosensitive channel of large conductance, MscL, into a functional ion channel. FEBS Lett. 2011, 585, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Hanss, B.; Leal-Pinto, E.; Teixeira, A.; Christian, R.E.; Shabanowitz, J.; Hunt, D.F.; Klotman, P.E. Cytosolic malate dehydrogenase confers selectivity of the nucleic acid-conducting channel. Proc. Natl. Acad. Sci. USA 2002, 99, 1707–1712. [Google Scholar] [CrossRef] [PubMed]
- Hanss, B.; Leal-Pinto, E.; Bruggeman, L.A.; Copeland, T.D.; Klotman, P.E. Identification and characterization of a cell membrane nucleic acid channel. Proc. Natl. Acad. Sci. USA 1998, 95, 1921–1926. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, E.H.; Hunter, C.P. Transport of dsRNA into cells by the transmembrane protein SID-1. Science 2003, 301, 1545–1547. [Google Scholar] [CrossRef] [PubMed]
- Srere, P.A. Complexes of sequential metabolic enzymes. Annu. Rev. Biochem. 1987, 56, 89–124. [Google Scholar] [CrossRef] [PubMed]
- Campanella, M.E.; Chu, H.; Low, P.S. Assembly and regulation of a glycolytic enzyme complex on the human erythrocyte membrane. Proc. Natl. Acad. Sci. USA 2005, 102, 2402–2407. [Google Scholar] [CrossRef] [PubMed]
- Idan, O.; Hess, H. Engineering enzymatic cascades on nanoscale scaffolds. Curr. Opin. Biotechnol. 2013, 24, 606–611. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Chen, Q.; Kim, H.; Siu, K.-H.; Sun, Q.; Tsai, S.-L.; Chen, W. Biomolecular scaffolds for enhanced signaling and catalytic efficiency. Curr. Opin. Biotechnol. 2014, 28, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Wilner, O.I.; Weizmann, Y.; Gill, R.; Lioubashevski, O.; Freeman, R.; Willner, I. Enzyme cascades activated on topologically programmed DNA scaffolds. Nat. Nanotechnol. 2009, 4, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Liu, M.; Liu, Y.; Woodbury, N.W.; Yan, H. Interenzyme substrate diffusion for an enzyme cascade organized on spatially addressable DNA nanostructures. J. Am. Chem. Soc. 2012, 134, 5516–5519. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Yang, Y.R.; Johnson-Buck, A.; Liu, M.; Liu, Y.; Walter, N.G.; Woodbury, N.W.; Yan, H. Multi-enzyme complexes on DNA scaffolds capable of substrate channelling with an artificial swinging arm. Nat. Nanotechnol. 2014, 9, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Delebecque, C.J.; Lindner, A.B.; Silver, P.A.; Aldaye, F.A. Organization of intracellular reactions with rationally designed RNA assemblies. Science 2011, 333, 470–474. [Google Scholar] [CrossRef] [PubMed]
- Sachdeva, G.; Garg, A.; Godding, D.; Way, J.C.; Silver, P.A. In vivo co-localization of enzymes on RNA scaffolds increases metabolic production in a geometrically dependent manner. Nucleic Acids Res. 2014, 42, 9493–9503. [Google Scholar] [CrossRef] [PubMed]
- Bashor, C.J.; Helman, N.C.; Yan, S.; Lim, W.A. Using engineered scaffold interactions to reshape MAP kinase pathway signaling dynamics. Science 2008, 319, 1539–1543. [Google Scholar] [CrossRef] [PubMed]
- Dueber, J.E.; Wu, G.C.; Malmirchegini, G.R.; Moon, T.S.; Petzold, C.J.; Ullal, A.V.; Prather, K.L.J.; Keasling, J.D. Synthetic protein scaffolds provide modular control over metabolic flux. Nat. Biotechnol. 2009, 27, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Conrado, R.J.; Wu, G.C.; Boock, J.T.; Xu, H.; Chen, S.Y.; Lebar, T.; Turnšek, J.; Tomšič, N.; Avbelj, M.; Gaber, R.; et al. DNA-guided assembly of biosynthetic pathways promotes improved catalytic efficiency. Nucleic Acids Res. 2012, 40, 1879–1889. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.; Booth, P.J.; Seddon, J.M.; Templer, R.H.; Law, R.V.; Woscholski, R.; Ces, O.; Barter, L.M.C. Protocell design through modular compartmentalization. J. R. Soc. Interface 2013, 10. [Google Scholar] [CrossRef] [PubMed]
- Elani, Y.; Gee, A.; Law, R.V.; Ces, O. Engineering multi-compartment vesicle networks. Chem. Sci. 2013, 4, 3332–3338. [Google Scholar] [CrossRef]
- Kim, E.Y.; Tullman-Ercek, D. Engineering nanoscale protein compartments for synthetic organelles. Curr. Opin. Biotechnol. 2013, 24, 627–632. [Google Scholar] [CrossRef] [PubMed]
- Keating, C.D. Aqueous phase separation as a possible route to compartmentalization of biological molecules. Acc. Chem. Res. 2012, 45, 2114–2124. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Banjade, S.; Cheng, H.-C.; Kim, S.; Chen, B.; Guo, L.; Llaguno, M.; Hollingsworth, J.V.; King, D.S.; Banani, S.F.; et al. Phase transitions in the assembly of multivalent signalling proteins. Nature 2012, 483, 336–340. [Google Scholar] [CrossRef] [PubMed]
- Strulson, C.A.; Molden, R.C.; Keating, C.D.; Bevilacqua, P.C. RNA catalysis through compartmentalization. Nat. Chem. 2012, 4, 941–946. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Densmore, D. Integration of microfluidics into the synthetic biology design flow. Lab Chip 2014, 14, 3459–3474. [Google Scholar] [CrossRef] [PubMed]
- Gulati, S.; Rouilly, V.; Niu, X.; Chappell, J.; Kitney, R.I.; Edel, J.B.; Freemont, P.S.; deMello, A.J. Opportunities for microfluidic technologies in synthetic biology. J. R. Soc. Interface 2009, 6, S493–S506. [Google Scholar] [CrossRef] [PubMed]
- Khnouf, R.; Beebe, D.J.; Fan, Z.H. Cell-free protein expression in a microchannel array with passive pumping. Lab Chip 2009, 9, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Glick, Y.; Avrahami, D.; Michaely, E.; Gerber, D. High-throughput protein expression generator using a microfluidic platform. J. Vis. Exp. 2012, 66. [Google Scholar] [CrossRef] [PubMed]
- Mei, Q.; Fredrickson, C.K.; Simon, A.; Khnouf, R.; Fan, Z.H. Cell-free protein synthesis in microfluidic array devices. Biotechnol. Prog. 2007, 23, 1305–1311. [Google Scholar] [CrossRef] [PubMed]
- Osaki, T.; Yoshizawa, S.; Kawano, R.; Sasaki, H.; Takeuchi, S. Lipid-coated microdroplet array for in vitro protein synthesis. Anal. Chem. 2011, 83, 3186–3191. [Google Scholar] [CrossRef] [PubMed]
- Buxboim, A.; Bar-Dagan, M.; Frydman, V.; Zbaida, D.; Morpurgo, M.; Bar-Ziv, R. A single-step photolithographic interface for cell-free gene expression and active biochips. Small 2007, 3, 500–510. [Google Scholar] [CrossRef] [PubMed]
- Bracha, D.; Karzbrun, E.; Daube, S.S.; Bar-Ziv, R.H. Emergent properties of dense DNA phases toward artificial biosystems on a surface. Acc. Chem. Res. 2014, 47, 1912–1921. [Google Scholar] [CrossRef] [PubMed]
- Kinpara, T.; Mizuno, R.; Murakami, Y.; Kobayashi, M.; Yamaura, S.; Hasan, Q.; Morita, Y.; Nakano, H.; Yamane, T.; Tamiya, E. A picoliter chamber array for cell-free protein synthesis. J. Biochem. 2004, 136, 149–154. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Stoevesandt, O.; Palmer, E.A.; Khan, F.; Ericsson, O.; Taussig, M.J. Printing protein arrays from DNA arrays. Nat. Methods 2008, 5, 175–177. [Google Scholar] [CrossRef] [PubMed]
- Karzbrun, E.; Tayar, A.M.; Noireaux, V.; Bar-Ziv, R.H. Programmable on-chip DNA compartments as artificial cells. Science 2014, 345, 829–832. [Google Scholar] [CrossRef] [PubMed]
- Meagher, R.J.; Light, Y.K.; Singh, A.K. Rapid, continuous purification of proteins in a microfluidic device using genetically-engineered partition tags. Lab Chip 2008, 8, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Lu, Y.; Zhou, Y.-G.; Wang, F.-B.; He, F.-Y.; Xia, X.-H. A simple, disposable microfluidic device for rapid protein concentration and purification via direct-printing. Lab Chip 2008, 8, 1496–1501. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ozdemir, P. Microfluidic DNA amplification—A review. Anal. Chim. Acta 2009, 638, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Schaerli, Y.; Wootton, R.C.; Robinson, T.; Stein, V.; Dunsby, C.; Neil, M.A.A.; French, P.M.W.; deMello, A.J.; Abell, C.; Hollfelder, F. Continuous-flow polymerase chain reaction of single-copy DNA in microfluidic microdroplets. Anal. Chem. 2009, 81, 302–306. [Google Scholar] [CrossRef] [PubMed]
- Beer, N.R.; Hindson, B.J.; Wheeler, E.K.; Hall, S.B.; Rose, K.A.; Kennedy, I.M.; Colston, B.W. On-chip, real-time, single-copy polymerase chain reaction in picoliter droplets. Anal. Chem. 2007, 79, 8471–8475. [Google Scholar] [CrossRef] [PubMed]
- Kiss, M.M.; Ortoleva-Donnelly, L.; Beer, N.R.; Warner, J.; Bailey, C.G.; Colston, B.W.; Rothberg, J.M.; Link, D.R.; Leamon, J.H. High-Throughput Quantitative Polymerase Chain Reaction in Picoliter Droplets. Anal. Chem. 2008, 80, 8975–8981. [Google Scholar] [CrossRef] [PubMed]
- Beer, N.R.; Wheeler, E.K.; Lee-Houghton, L.; Watkins, N.; Nasarabadi, S.; Hebert, N.; Leung, P.; Arnold, D.W.; Bailey, C.G.; Colston, B.W. On-chip single-copy real-time reverse-transcription PCR in isolated picoliter droplets. Anal. Chem. 2008, 80, 1854–1858. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Cai, S.; Hong, A.; You, Q.; Yu, P.; Sheng, N.; Srivannavit, O.; Muranjan, S.; Rouillard, J.M.; Xia, Y.; et al. Microfluidic PicoArray synthesis of oligodeoxynucleotides and simultaneous assembling of multiple DNA sequences. Nucleic Acids Res. 2004, 32, 5409–5417. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-C.; Snyder, T.M.; Quake, S.R. A microfluidic oligonucleotide synthesizer. Nucleic Acids Res. 2010, 38, 2514–2521. [Google Scholar] [CrossRef] [PubMed]
- Srivannavit, O.; Gulari, M.; Hua, Z.; Gao, X.; Zhou, X.; Hong, A.; Zhou, T.; Gulari, E. Microfluidic Reactor Array Device for Massively Parallel in-situ Synthesis of Oligonucleotides. Sens. Actuators. B Chem. 2009, 140, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Tawfik, D.S.; Griffiths, A.D. Man-made cell-like compartments for molecular evolution. Nat. Biotechnol. 1998, 16, 652–656. [Google Scholar] [CrossRef] [PubMed]
- Agresti, J.J.; Antipov, E.; Abate, A.R.; Ahn, K.; Rowat, A.C.; Baret, J.-C.; Marquez, M.; Klibanov, A.M.; Griffiths, A.D.; Weitz, D.A. Ultrahigh-throughput screening in drop-based microfluidics for directed evolution. Proc. Natl. Acad. Sci. USA 2010, 107, 4004–4009. [Google Scholar] [CrossRef] [PubMed]
- Kintses, B.; Hein, C.; Mohamed, M.F.; Fischlechner, M.; Courtois, F.; Lainé, C.; Hollfelder, F. Picoliter cell lysate assays in microfluidic droplet compartments for directed enzyme evolution. Chem. Biol. 2012, 19, 1001–1009. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Courtois, F.; Surjadi, R.; Oakeshott, J.; Peat, T.S.; Easton, C.J.; Abell, C.; Zhu, Y. Enzyme synthesis and activity assay in microfluidic droplets on a chip. Eng. Life Sci. 2011, 11, 157–164. [Google Scholar] [CrossRef]
- Seemann, R.; Brinkmann, M.; Pfohl, T.; Herminghaus, S. Droplet based microfluidics. Rep. Prog. Phys. 2012, 75, 016601. [Google Scholar] [CrossRef] [PubMed]
- Stanley, C.E.; Elvira, K.S.; Niu, X.Z.; Gee, A.D.; Ces, O.; Edel, J.B.; deMello, A.J. A microfluidic approach for high-throughput droplet interface bilayer (DIB) formation. Chem. Commun. 2010, 46, 1620–1622. [Google Scholar] [CrossRef]
- Elani, Y.; deMello, A.J.; Niu, X.; Ces, O. Novel technologies for the formation of 2-D and 3-D droplet interface bilayer networks. Lab Chip 2012, 12, 3514–3520. [Google Scholar] [CrossRef] [PubMed]
- Van Swaay, D.; deMello, A. Microfluidic methods for forming liposomes. Lab Chip 2013, 13, 752–767. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Lee, R.J.; Lee, L.J. Microfluidic methods for production of liposomes. Methods Enzymol. 2009, 465, 129–141. [Google Scholar] [PubMed]
- Teh, S.; Khnouf, R.; Fan, H.; Lee, A. Stable, biocompatible lipid vesicle generation by solvent extraction-based droplet microfluidics. Biomicrofluidics 2011, 5. [Google Scholar] [CrossRef]
- Shum, H.C.; Lee, D.; Yoon, I.; Kodger, T.; Weitz, D.A. Double emulsion templated monodisperse phospholipid vesicles. Langmuir 2008, 24, 7651–7653. [Google Scholar] [CrossRef] [PubMed]
- Matosevic, S.; Paegel, B.M. Stepwise synthesis of giant unilamellar vesicles on a microfluidic assembly line. J. Am. Chem. Soc. 2011, 133, 2798–2800. [Google Scholar] [CrossRef] [PubMed]
- Karamdad, K.; Law, R.V.; Seddon, J.M.; Brooks, N.J.; Ces, O. Preparation and mechanical characterisation of giant unilamellar vesicles by a microfluidic method. Lab Chip 2015, 15, 557–562. [Google Scholar] [CrossRef] [PubMed]
- Stachowiak, J.C.; Richmond, D.L.; Li, T.H.; Liu, A.P.; Parekh, S.H.; Fletcher, D.A. Unilamellar vesicle formation and encapsulation by microfluidic jetting. Proc. Natl. Acad. Sci. USA 2008, 105, 4697–4702. [Google Scholar] [CrossRef] [PubMed]
- Li, T.H.; Stachowiak, J.C.; Fletcher, D.A. Mixing solutions in inkjet formed vesicles. Methods Enzymol. 2009, 465, 75–94. [Google Scholar] [PubMed]
- Ota, S.; Yoshizawa, S.; Takeuchi, S. Microfluidic formation of monodisperse, cell-sized, and unilamellar vesicles. Angew. Chem. Int. Ed. Engl. 2009, 48, 6533–6537. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.; Saurabh, S.; Bruchez, M.P.; Schwartz, R.; Leduc, P. Molecular crowding shapes gene expression in synthetic cellular nanosystems. Nat. Nanotechnol. 2013, 8, 602–608. [Google Scholar] [CrossRef] [PubMed]
- Asial, I.; Cheng, Y.X.; Engman, H.; Dollhopf, M.; Wu, B.; Nordlund, P.; Cornvik, T. Engineering protein thermostability using a generic activity-independent biophysical screen inside the cell. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef]
- Ahmad, S.; Kamal, M.Z.; Sankaranarayanan, R.; Rao, N.M. Thermostable Bacillus subtilis lipases: In vitro evolution and structural insight. J. Mol. Biol. 2008, 381, 324–340. [Google Scholar] [CrossRef] [PubMed]
- Dumon, C.; Varvak, A.; Wall, M.A.; Flint, J.E.; Lewis, R.J.; Lakey, J.H.; Morland, C.; Luginbühl, P.; Healey, S.; Todaro, T.; et al. Engineering hyperthermostability into a GH11 xylanase is mediated by subtle changes to protein structure. J. Biol. Chem. 2008, 283, 22557–22564. [Google Scholar] [CrossRef] [PubMed]
- Giver, L.; Gershenson, A.; Freskgard, P.-O.; Arnold, F.H. Directed evolution of a thermostable esterase. Proc. Natl. Acad. Sci. USA 1998, 95, 12809–12813. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Berry, A. A thermostable variant of fructose bisphosphate aldolase constructed by directed evolution also shows increased stability in organic solvents. Protein Eng. Des. Sel. 2004, 17, 689–697. [Google Scholar] [CrossRef] [PubMed]
- Warne, T.; Serrano-Vega, M.J.; Baker, J.G.; Moukhametzianov, R.; Edwards, P.C.; Henderson, R.; Leslie, A.G.W.; Tate, C.G.; Schertler, G.F.X. Structure of a beta1-adrenergic G-protein-coupled receptor. Nature 2008, 454, 486–491. [Google Scholar] [CrossRef] [PubMed]
- Warne, T.; Serrano-Vega, M.J.; Tate, C.G.; Schertler, G.F.X. Development and crystallization of a minimal thermostabilised G protein-coupled receptor. Protein Expr. Purif. 2009, 65, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Malawski, G.A.; Hillig, R.C.; Monteclaro, F.; Eberspaecher, U.; Schmitz, A.A.P.; Crusius, K.; Huber, M.; Egner, U.; Donner, P.; Müller-Tiemann, B. Identifying protein construct variants with increased crystallization propensity—A case study. Protein Sci. 2006, 15, 2718–2728. [Google Scholar] [CrossRef] [PubMed]
- Shibata, Y.; White, J.F.; Serrano-Vega, M.J.; Magnani, F.; Aloia, A.L.; Grisshammer, R.; Tate, C.G. Thermostabilization of the neurotensin receptor NTS1. J. Mol. Biol. 2009, 390, 262–277. [Google Scholar] [CrossRef] [PubMed]
- Suplatov, D.; Panin, N.; Kirilin, E.; Shcherbakova, T.; Kudryavtsev, P.; Švedas, V. Computational design of a pH stable enzyme: Understanding molecular mechanism of penicillin acylase’s adaptation to alkaline conditions. PLoS One 2014, 9, e100643. [Google Scholar] [CrossRef] [PubMed]
- Van Beek, H.L.; Wijma, H.J.; Fromont, L.; Janssen, D.B.; Fraaije, M.W. Stabilization of cyclohexanone monooxygenase by a computationally designed disulfide bond spanning only one residue. FEBS Open Bio 2014, 4, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Beliën, T.; Joye, I.J.; Delcour, J.A.; Courtin, C.M. Computational design-based molecular engineering of the glycosyl hydrolase family 11 B. subtilis XynA endoxylanase improves its acid stability. Protein Eng. Des. Sel. 2009, 22, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Balazs, D.A.; Godbey, W. Liposomes for use in gene delivery. J. Drug Deliv. 2011, 2011, 326497. [Google Scholar] [CrossRef] [PubMed]
- Felnerova, D.; Viret, J.-F.; Glück, R.; Moser, C. Liposomes and virosomes as delivery systems for antigens, nucleic acids and drugs. Curr. Opin. Biotechnol. 2004, 15, 518–529. [Google Scholar] [CrossRef] [PubMed]
- Allen, T.M.; Cullis, P.R. Liposomal drug delivery systems: From concept to clinical applications. Adv. Drug Deliv. Rev. 2013, 65, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Discher, B.M.; Won, Y.-Y.; Ege, D.S.; Lee, J.C.-M.; Bates, F.S.; Discher, D.E.; Hammer, D.A. Polymersomes: Tough vesicles made from diblock copolymers. Science 1999, 284, 1143–1146. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-M.; Chen, H.; Dettmer, C.M.; O’Halloran, T.V.; Nguyen, S.T. Polymer-caged lipsomes: A pH-responsive delivery system with high stability. J. Am. Chem. Soc. 2007, 129, 15096–15097. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Yan, F.; Guo, Z.; Marchant, R.E. Surface modification of liposomes by saccharides: Vesicle size and stability of lactosyl liposomes studied by photon correlation spectroscopy. J. Colloid Interface Sci. 2005, 289, 542–550. [Google Scholar] [CrossRef] [PubMed]
- Milla, P.; Dosio, F.; Cattel, L. PEGylation of Proteins and Liposomes: A Powerful and Flexible Strategy to Improve the Drug Delivery. Curr. Drug Metab. 2012, 13, 105–119. [Google Scholar] [CrossRef] [PubMed]
- Torchilin, V.P.; Levchenko, T.S.; Whiteman, K.R.; Yaroslavov, A.A.; Tsatsakis, A.M.; Rizos, A.K.; Michailova, E.V.; Shtilman, M.I. Amphiphilic poly-N-vinylpyrrolidones: Synthesis, properties and liposome surface modification. Biomaterials 2001, 22, 3035–3044. [Google Scholar] [CrossRef] [PubMed]
- Riché, E.L.; Erickson, B.W.; Cho, M.J. Novel long-circulating liposomes containing peptide library-lipid conjugates: Synthesis and in vivo behavior. J. Drug Target. 2004, 12, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Klibanov, A.L.; Maruyama, K.; Torchilin, V.P.; Huang, L. Amphipathic polyethyleneglycols effectively prolong the circulation time of liposomes. FEBS Lett. 1990, 268, 235–237. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-K.; Choi, S.-H.; Kim, C.-O.; Park, J.-S.; Ahn, W.-S.; Kim, C.-K. Enhancement of polyethylene glycol (PEG)-modified cationic liposome-mediated gene deliveries: Effects on serum stability and transfection efficiency. J. Pharm. Pharmacol. 2003, 55, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Montemagno, C.D. Artificial organelle: ATP synthesis from cellular mimetic polymersomes. Nano Lett. 2005, 5, 2538–2542. [Google Scholar] [CrossRef] [PubMed]
- Meier, W.; Nardin, C.; Winterhalter, M. Reconstitution of Channel Proteins in (Polymerized) ABA Triblock Copolymer Membranes. Angew. Chem. Int. Ed. Engl. 2000, 39, 4599–4602. [Google Scholar] [CrossRef] [PubMed]
- Nardin, C.; Thoeni, S.; Widmer, J.; Winterhalter, M.; Meier, W. Nanoreactors based on (polymerized) ABA-triblock copolymer vesicles. Chem. Commun. 2000. [Google Scholar] [CrossRef]
- Hvasanov, D.; Peterson, J.R.; Thordarson, P. Self-assembled light-driven photosynthetic-respiratory electron transport chain hybrid proton pump. Chem. Sci. 2013, 4, 3833–3838. [Google Scholar] [CrossRef]
- Zhu, Z.; Tam, T.K.; Sun, F.; You, C.; Zhang, Y.-H.P. A high-energy-density sugar biobattery based on a synthetic enzymatic pathway. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Jewett, M.C.; Fritz, B.R.; Timmerman, L.E.; Church, G.M. In vitro integration of ribosomal RNA synthesis, ribosome assembly, and translation. Mol. Syst. Biol. 2013, 9. [Google Scholar] [CrossRef]
- Ederth, J.; Mandava, C.S.; Dasgupta, S.; Sanyal, S. A single-step method for purification of active His-tagged ribosomes from a genetically engineered Escherichia coli. Nucleic Acids Res. 2009, 37, e15. [Google Scholar] [CrossRef] [PubMed]
- Boehm, C.R.; Freemont, P.S.; Ces, O. Design of a prototype flow microreactor for synthetic biology in vitro. Lab Chip 2013, 13, 3426–3432. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.S.; Palsson, B.O. The Escherichia coli MG1655 in silico metabolic genotype: Its definition, characteristics, and capabilities. Proc. Natl. Acad. Sci. USA 2000, 97, 5528–5533. [Google Scholar] [CrossRef] [PubMed]
- Reed, J.L.; Vo, T.D.; Schilling, C.H.; Palsson, B.O. An expanded genome-scale model of Escherichia coli K-12 (iJR904 GSM/GPR). Genome Biol. 2003, 4, R54. [Google Scholar] [CrossRef] [PubMed]
- Feist, A.M.; Henry, C.S.; Reed, J.L.; Krummenacker, M.; Joyce, A.R.; Karp, P.D.; Broadbelt, L.J.; Hatzimanikatis, V.; Palsson, B.Ø. A genome-scale metabolic reconstruction for Escherichia coli K-12 MG1655 that accounts for 1260 ORFs and thermodynamic information. Mol. Syst. Biol. 2007, 3. [Google Scholar] [CrossRef]
- Orth, J.D.; Conrad, T.M.; Na, J.; Lerman, J.A.; Nam, H.; Feist, A.M.; Palsson, B.Ø. A comprehensive genome-scale reconstruction of Escherichia coli metabolism—2011. Mol. Syst. Biol. 2011, 7. [Google Scholar] [CrossRef]
- Duarte, N.C.; Herrgård, M.J.; Palsson, B.Ø. Reconstruction and validation of Saccharomyces cerevisiae iND750, a fully compartmentalized genome-scale metabolic model. Genome Res. 2004, 14, 1298–1309. [Google Scholar] [CrossRef] [PubMed]
- Nogales, J.; Palsson, B.Ø.; Thiele, I. A genome-scale metabolic reconstruction of Pseudomonas putida KT2440: iJN746 as a cell factory. BMC Syst. Biol. 2008, 2. [Google Scholar] [CrossRef] [PubMed]
- Duarte, N.C.; Jamshidi, N.; Thiele, I.; Mo, M.L.; Vo, T.D.; Srivas, R.; Palsson, B.Ø. Global reconstruction of the human metabolic network based on genomic and bibliomic data. Proc. Natl. Acad. Sci. USA 2007, 104, 1777–1782. [Google Scholar] [CrossRef] [PubMed]
- Lelli, K.M.; Slattery, M.; Mann, R.S. Disentangling the many layers of eukaryotic transcriptional regulation. Annu. Rev. Genet. 2012, 46, 43–68. [Google Scholar] [CrossRef] [PubMed]
- Eberhardy, S.R.; Goncalves, J.; Coelho, S.; Segal, D.J.; Berkhout, B.; Barbas, C.F. Inhibition of human immunodeficiency virus type 1 replication with artificial transcription factors targeting the highly conserved primer-binding site. J. Virol. 2006, 80, 2873–2883. [Google Scholar] [CrossRef] [PubMed]
- Sohn, J.H.; Yeh, B.-I.; Choi, J.-W.; Yoon, J.; Namkung, J.; Park, K.-K.; Kim, H.-W. Repression of human telomerase reverse transcriptase using artificial zinc finger transcription factors. Mol. Cancer Res. 2010, 8, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Beerli, R.R.; Dreier, B.; Barbas, C.F. Positive and negative regulation of endogenous genes by designed transcription factors. Proc. Natl. Acad. Sci. USA 2000, 97, 1495–1500. [Google Scholar] [CrossRef]
- Morbitzer, R.; Römer, P.; Boch, J.; Lahaye, T. Regulation of selected genome loci using de novo-engineered transcription activator-like effector (TALE)-type transcription factors. Proc. Natl. Acad. Sci. USA 2010, 107, 21617–21622. [Google Scholar] [CrossRef] [PubMed]
- Moscou, M.J.; Bogdanove, A.J. A simple cipher governs DNA recognition by TAL effectors. Science 2009, 326, 1501. [Google Scholar] [CrossRef] [PubMed]
- Purcell, O.; Peccoud, J.; Lu, T.K. Rule-based design of synthetic transcription factors in eukaryotes. ACS Synth. Biol. 2014, 3, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Beerli, R.R.; Barbas, C.F. Engineering polydactyl zinc-finger transcription factors. Nat. Biotechnol. 2002, 20, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Magnenat, L.; Blancafort, P.; Barbas, C.F. In vivo selection of combinatorial libraries and designed affinity maturation of polydactyl zinc finger transcription factors for ICAM-1 provides new insights into gene regulation. J. Mol. Biol. 2004, 341, 635–649. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Segal, D.J.; Ghiara, J.B.; Barbas, C.F. Design of polydactyl zinc-finger proteins for unique addressing within complex genomes. Proc. Natl. Acad. Sci. USA 1997, 94, 5525–5230. [Google Scholar] [CrossRef] [PubMed]
- Semenov, S.N.; Wong, A.S.Y.; van der Made, R.M.; Postma, S.G.J.; Groen, J.; van Roekel, H.W.H.; de Greef, T.F.A.; Huck, W.T.S. Rational design of functional and tunable oscillating enzymatic networks. Nat. Chem. 2015, 7, 160–165. [Google Scholar] [CrossRef]
- Weitz, M.; Kim, J.; Kapsner, K.; Winfree, E.; Franco, E.; Simmel, F.C. Diversity in the Dynamical Behaviour of a Compartmentalized Programmable Biochemical Oscillator. Nat. Chem. 2014, 6, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Franco, E.; Friedrichs, E.; Kim, J.; Jungmann, R.; Murray, R.; Winfree, E.; Simmel, F.C. PNAS Plus: Timing molecular motion and production with a synthetic transcriptional clock. Proc. Natl. Acad. Sci. USA 2011, 108, E784–E793. [Google Scholar] [CrossRef] [PubMed]
- Elowitz, M.B.; Levine, A.J.; Siggia, E.D.; Swain, P.S. Stochastic gene expression in a single cell. Science 2002, 297, 1183–1186. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miller, D.M.; Gulbis, J.M. Engineering Protocells: Prospects for Self-Assembly and Nanoscale Production-Lines. Life 2015, 5, 1019-1053. https://doi.org/10.3390/life5021019
Miller DM, Gulbis JM. Engineering Protocells: Prospects for Self-Assembly and Nanoscale Production-Lines. Life. 2015; 5(2):1019-1053. https://doi.org/10.3390/life5021019
Chicago/Turabian StyleMiller, David M., and Jacqueline M. Gulbis. 2015. "Engineering Protocells: Prospects for Self-Assembly and Nanoscale Production-Lines" Life 5, no. 2: 1019-1053. https://doi.org/10.3390/life5021019
APA StyleMiller, D. M., & Gulbis, J. M. (2015). Engineering Protocells: Prospects for Self-Assembly and Nanoscale Production-Lines. Life, 5(2), 1019-1053. https://doi.org/10.3390/life5021019