
Pulmonary Hypertension: Let’s Take Stock!



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Epidemiology
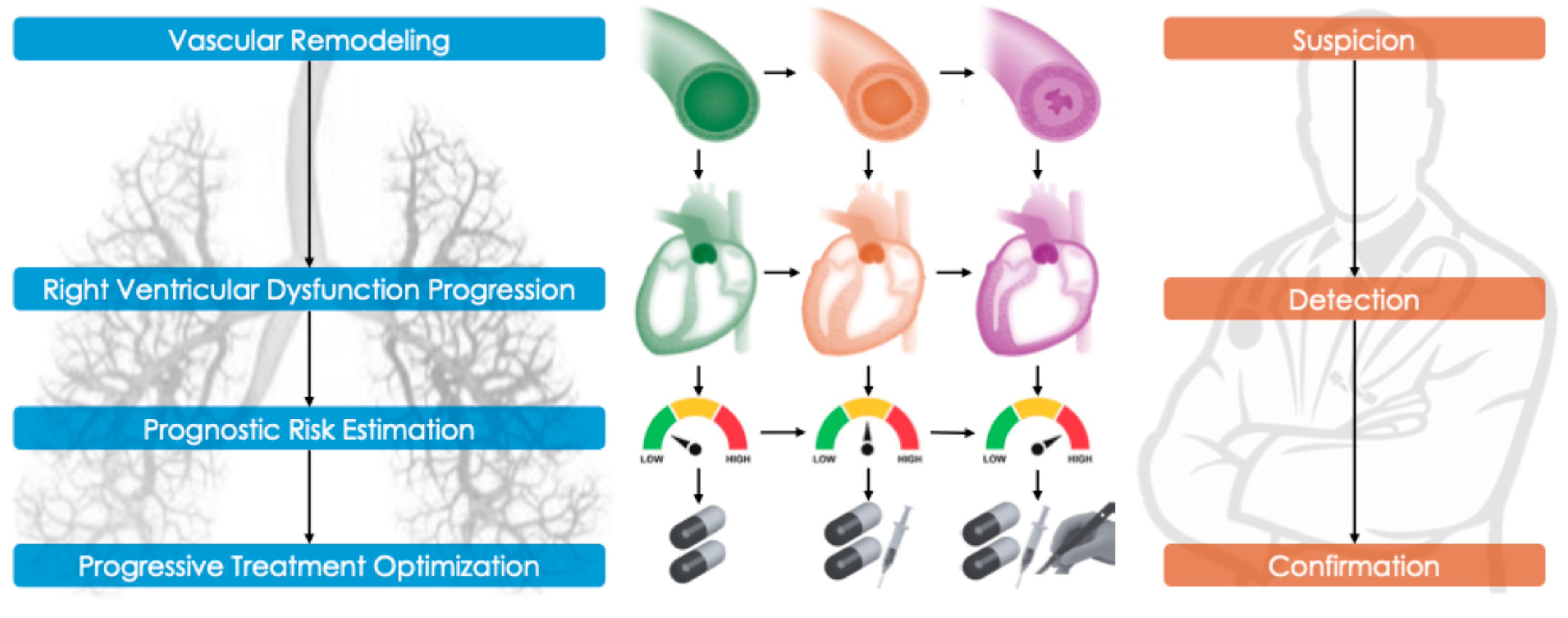
3. Pathophysiology and Disease Mechanisms
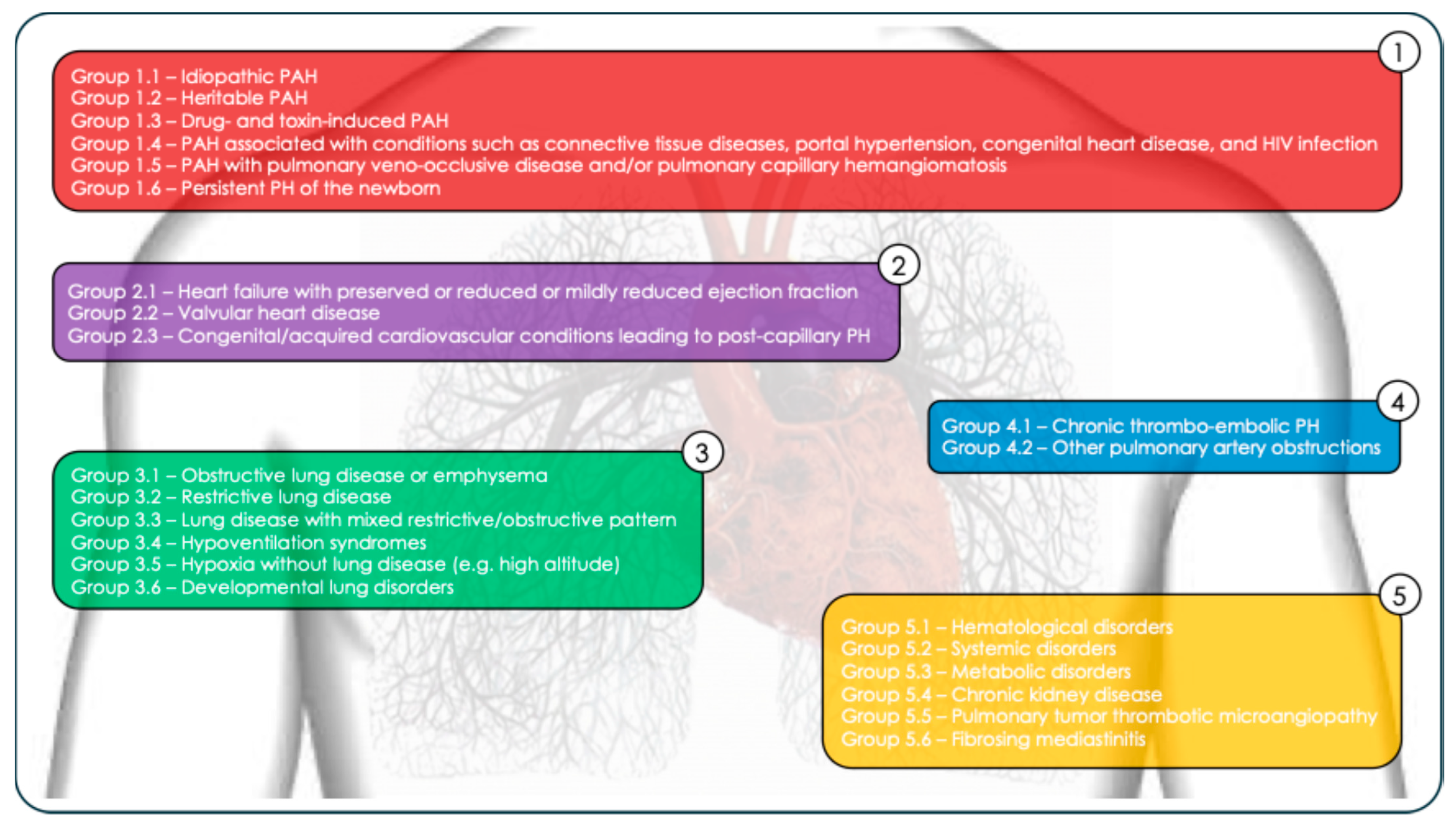
4. Classification and Hemodynamic Diagnosis of Pulmonary Hypertension
- Idiopathic PAH (Group 1.1);
- Heritable PAH (Group 1.2);
- Drug- and toxin-induced PAH (Group 1.3);
- PAH associated with conditions such as connective tissue diseases, portal hypertension, congenital heart disease, and HIV infection (Group 1.4);
- PAH with pulmonary veno-occlusive disease and/or pulmonary capillary hemangiomatosis (Group 1.5);
- Persistent PH of the newborn (Group 1.6).
- Hematological disorders (Group 5.1: chronic hemolytic anemia, myeloproliferative diseases);
- Systemic disorders (Group 5.2: sarcoidosis, pulmonary Langerhans cell histiocytosis, neurofibromatosis type 1);
- Metabolic disorders (Group 5.3: glycogen storage diseases, Gaucher disease)
- Chronic kidney disease (Group 5.4);
- Pulmonary tumor thrombotic microangiopathy (Group 5.5);
- Fibrosing mediastinitis (Group 5.6).
- ○
- Pre-capillary PH (e.g., PAH, CTEPH): mPAP > 20 mmHg, PAWP ≤ 15 mmHg, PVR > 2 WU;
- ○
- Isolated post-capillary PH (due to left heart disease): mPAP > 20 mmHg, PAWP > 15 mmHg, PVR ≤ 2 WU;
- ○
- Combined pre- and post-capillary PH: mPAP > 20 mmHg, PAWP > 15 mmHg, PVR > 2 WU (as seen in heart failure patients with associated pulmonary vascular disease).
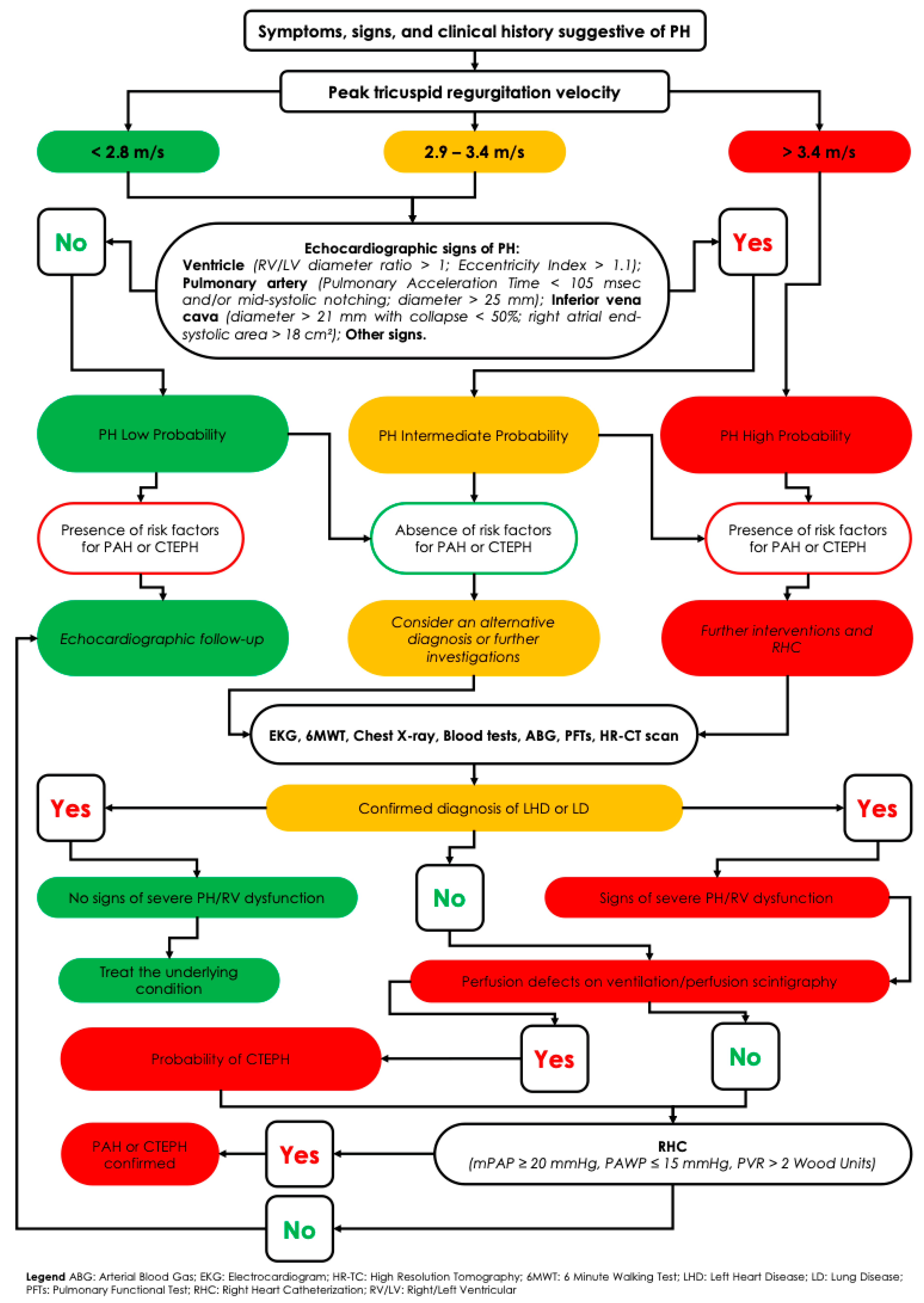
5. Diagnostic Workup of Suspected Pulmonary Hypertension
- Clinical evaluation, with a thorough history and physical examination aimed at identifying common symptoms such as exertional dyspnea, fatigue, weakness, angina, fluid retention (e.g., ankle edema), and syncope.
- Transthoracic echocardiography, the cornerstone of initial screening, provides an estimate of the probability of PH. Although it offers derived measurements (e.g., right ventricular systolic pressure), it also supplies valuable hemodynamic, morphological, and functional data—such as right atrial and ventricular dilation, pericardial effusion, deviation of the interventricular septum (indicative of right-sided pressure overload), systolic and diastolic ventricular function, and the presence of valvular disease.
- Additional investigations to determine the underlying cause may include the following:
- ○
- Chest radiography;
- ○
- Pulmonary function tests (spirometry, body plethysmography, and diffusing capacity for carbon monoxide [DLCO]);
- ○
- Arterial blood gas analysis;
- ○
- Sleep studies or overnight oximetry;
- ○
- High-resolution computed tomography (HRTC);
- ○
- Laboratory tests.
- Ventilation/perfusion (V/Q) scintigraphy, essential in all patients without confirmed left heart or primary lung disease, to rule out chronic thromboembolic pulmonary hypertension (CTEPH). Advanced imaging techniques such as single-photon emission computed tomography (SPECT V/Q) and dual-energy computed tomography (DECT) enhance the detection of perfusion defects, even in distal pulmonary vessels [20]. SPECT V/Q offers 3D perfusion imaging with improved sensitivity for detecting chronic thromboembolic obstructions while preserving high specificity. A normal V/Q scan effectively rules out CTEPH, whereas multiple segmental perfusion defects with preserved ventilation are diagnostic. If the V/Q scan is suggestive, confirmatory pulmonary angiography—either conventional (invasive) or CT-based—is required to localize and map thromboembolic lesions [21].
- DECT pulmonary angiography, for example, produces color-coded perfusion maps by tracking iodine distribution, combining high-resolution anatomical visualization with functional assessment in a single scan. Studies have shown good concordance between DECT perfusion imaging and V/Q scintigraphy, particularly in identifying small peripheral defects often missed by conventional CT [22].
- Cardiopulmonary magnetic resonance imaging (MRI) has also advanced significantly in PH diagnostics. Dynamic contrast-enhanced perfusion MRI enables the real-time visualization of lung perfusion without ionizing radiation. Recent studies report that optimized perfusion MRI achieves a sensitivity of ~95–97% for CTEPH diagnosis, comparable to V/Q scintigraphy. Non-contrast MRI techniques, such as phase-resolved functional lung imaging (PREFUL MRI), have demonstrated >90% agreement with V/Q SPECT in identifying regional perfusion abnormalities. Although MRI is not yet a first-line tool—mainly due to limited availability and cost—it offers a promising radiation-free alternative for patients ineligible for nuclear imaging and may become more widely used as technology evolves [22].
- Right heart catheterization (RHC) remains the definitive test to confirm the presence and type of PH—pre-capillary, post-capillary, or combined—particularly after non-invasive tests have excluded alternative diagnoses. RHC is indispensable for obtaining accurate pulmonary hemodynamic parameters, including mPAP, PAWP, cardiac output, and PVR. It also plays a key role in risk stratification and is mandatory before initiating targeted PAH therapy.
- Additional Investigations for Etiological and Prognostic Assessment:
- ○
- Genetic testing and counseling are recommended in cases of idiopathic, heritable, or drug/toxin-induced PAH, as outlined in the 2022 ESC/ERS guidelines.
- ○
- Mutations in the BMPR2 gene are found in approximately 75% of heritable PAH cases and over 25% of idiopathic PAH [23].
- ○
- If BMPR2 testing is negative, further evaluation for mutations in ALK1 and ENG (associated with hereditary hemorrhagic telangiectasia, or Rendu–Osler–Weber syndrome), as well as other genes such as SMAD9, CAV1, and KCNK3, is warranted.
- ○
- Genetic evaluation is also advised in patients with clinical or radiological suspicion of pulmonary veno-occlusive disease (PVOD), particularly to detect autosomal recessive EIF2AK4 mutations recently linked to heritable forms of PVOD [24].
- Biomarkers, such as brain natriuretic peptide (BNP) and N-terminal pro-brain natriuretic peptide (NT-proBNP), provide important prognostic information and help assess right ventricular strain (Figure 3).
6. Therapeutic Strategy and Follow-Up in PAH
- Endothelin receptor antagonists (ERAs): ambrisentan, bosentan, macitentan;
- Phosphodiesterase type 5 inhibitors (PDE5is): sildenafil, tadalafil;
- Soluble guanylate cyclase stimulators (sGCSs): riociguat;
- Prostacyclin analogs (PAs): beraprost, epoprostenol, treprostinil;
- Prostacyclin receptor agonists (PRAs): selexipag.
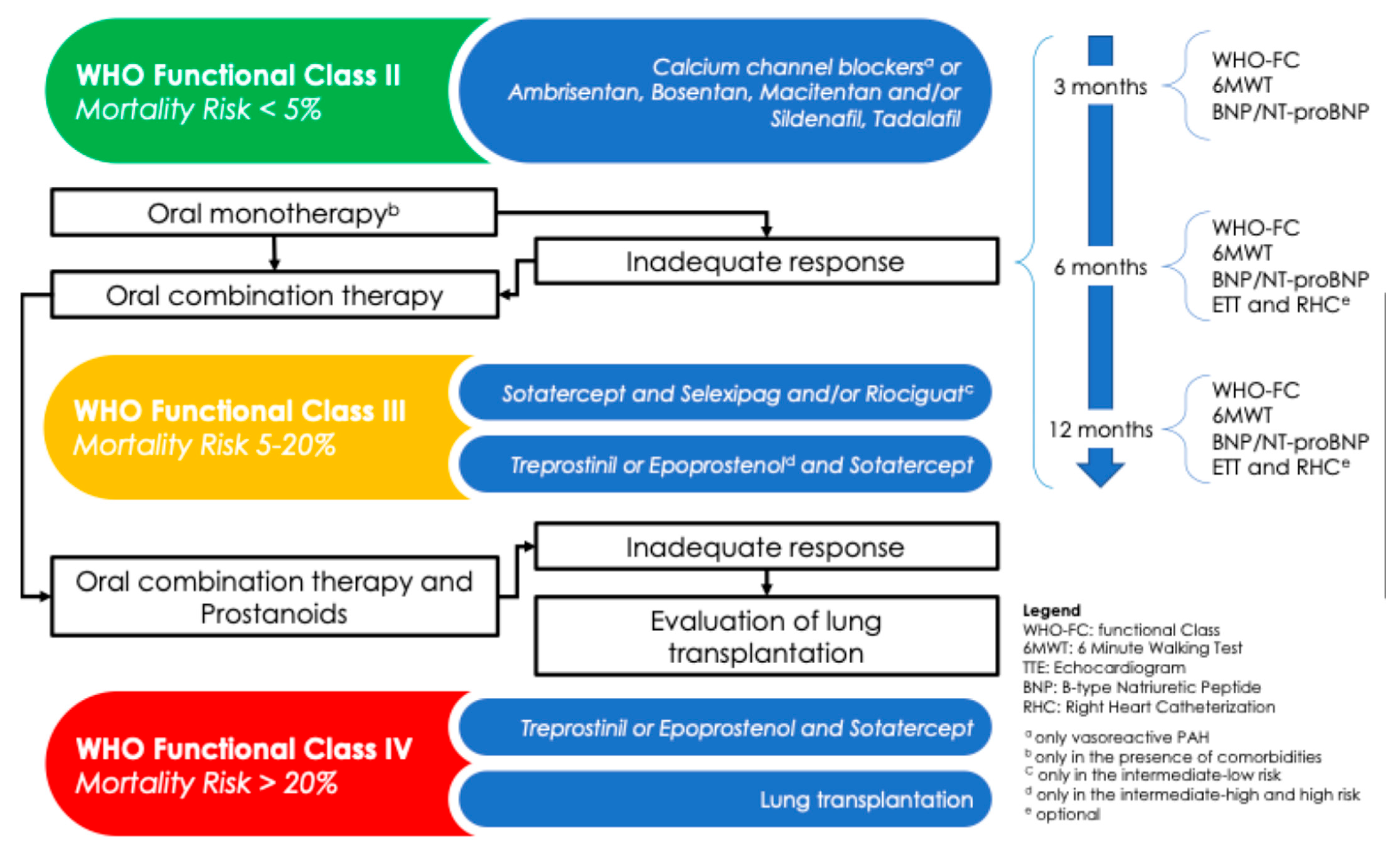
7. Risk Stratification
- At the time of diagnosis, a three-strata model is recommended, in which the variables to take in account are as follows: sign of right HF, progression of symptoms, presence of syncope, the WHO functional class, 6 min walking distance (6MWD), biomarkers (BNP or NT-proBNP), echocardiographic parameters (right atrium area, TAPSE/PAP ratio, pericardial effusion), magnetic resonance indexes (right ventricular ejection fraction, stroke volume index, right ventricular end-systolic volume index), and hemodynamic values after RHC (right atrial pressure, cardiac index, stroke volume index, mixed venous oxygen saturation). Patients can be classified at low risk (<5%), intermediate risk (5–20%), or high risk (>20%).
- At the follow-up, a simplified four-strata model including 6 min walking distance, BNP or NT-proBNP, and WHO functional class should be adopted, although additional variables like right heart imaging and hemodynamics parameters can be included. This model can divide patients into four categories: low risk, intermediate–low risk, intermediate–high risk, and high risk.
7.1. Group 1
- Pulmonary artery denervation, which uses radiofrequency ablation to disrupt stretch receptors at the pulmonary artery bifurcation, aiming to reduce vasoconstriction and vascular remodeling [26].
7.2. Group 2
7.3. Group 3
7.4. Group 4
7.5. Group 5
- In sarcoidosis-associated PH, therapy includes immunosuppressive agents such as corticosteroids and general PH management measures.
- In sickle cell disease, optimal control of the hematologic disorder with hydroxyurea and transfusion therapy is key.
8. Pediatric Pulmonary Hypertension
- WHO functional class (adapted for age);
- Growth failure;
- Episodes of syncope;
- BNP/NT-proBNP levels;
- Echocardiographic measures of right ventricular function;
- Hemodynamic indices;
- Genetic markers.
- Children without cardiopulmonary comorbidities (e.g., idiopathic/heritable PAH or PAH associated with systemic diseases);
- Children with cardiopulmonary developmental disorders (e.g., congenital heart disease, lung developmental abnormalities).
- Bosentan (ERA);
- Sildenafil (PDE5 inhibitor);
- Inhaled iloprost;
- Subcutaneous treprostinil.
- Corrective surgery for congenital shunts;
- Pulmonary therapies for lung disorders.
- Atrial septostomy;
- Potts shunt (a surgical connection between the left pulmonary artery and descending aorta), which can decompress the right heart and improve survival in selected children with suprasystemic PAH.
9. Emerging Therapies in PAH
- Sotatercept is a first-in-class biologic therapy as an activin receptor IIA-Fc fusion protein that modulates signaling in the BMP/TGF-β superfamily. By binding activins and growth differentiation factors, sotatercept aims to rebalance the dysregulated pro-proliferative TGF-β pathway seen in PAH and restore BMPR2 signaling. In the pivotal STELLAR trial, sotatercept—used in addition to background PAH therapy—showed significant clinical benefit, with a +34 m increase in 6 min walk distance at 24 weeks compared to placebo, along with improvements in 8 out of 9 secondary endpoints (including PVR, NT-proBNP levels, and WHO functional class). This marks a major advance, as sotatercept acts at a molecular level, targeting vascular remodeling rather than simply vasodilation. The therapy was generally well tolerated, with mostly mild adverse effects (e.g., epistaxis, telangiectasias, elevated hemoglobin, thrombocytopenia) related to its mechanism of action. Sotatercept is currently under further investigation in ongoing trials (e.g., HYPERION and ZENITH) and may soon represent a disease-modifying option in PAH, potentially transforming the treatment paradigm in combination with current vasodilators.
- Ralinepag is a next-generation oral prostacyclin receptor agonist (non-prostanoid), developed to provide potent, sustained activation of the prostacyclin pathway. With a high affinity for the IP receptor and a long plasma half-life, it is designed for once-daily dosing to improve treatment adherence and convenience. Phase II studies indicate that ralinepag improves hemodynamic parameters, and it is currently being evaluated in the ADVANCE program (Phase III), including the ADVANCE-Outcomes trial, which is assessing its ability to delay disease progression and enhance exercise capacity when added to PDE5i/ERA-based therapy. If successful, ralinepag could offer a practical oral alternative to parenteral prostacyclin analogs and support the earlier adoption of triple combination therapy. Although results are pending (as of 2024), optimism is high due to the already validated IP receptor pathway (e.g., selexipag).
- Inhaled imatinib (dry powder formulation), a tyrosine kinase inhibitor initially used in chronic myeloid leukemia, demonstrated a significant reduction in PVR in a Phase III PAH trial over a decade ago. However, its oral administration was limited by systemic side effects (e.g., peripheral edema, subdural hematomas). This led to the development of inhaled imatinib (e.g., AV-101), a dry powder formulation designed to deliver the drug directly to the pulmonary vasculature, thereby reducing systemic exposure. The IMPAHCT trial [35], an ongoing Phase IIb/III study, is evaluating its efficacy and safety. Inhaled imatinib aims to reverse vascular remodeling by inhibiting PDGF and c-Kit signaling in the lungs. Preliminary data suggest a better safety profile than the oral form, although optimal dosing is still under investigation. If successful, it could represent a repurposed cancer drug with targeted disease-modifying potential for advanced PAH.
- New Treprostinil formulations: Treprostinil, a prostacyclin analog, has long been a cornerstone of PAH therapy, typically administered via intravenous, subcutaneous, or inhaled routes. Recent innovations aim to improve patient convenience and safety, expanding access to prostacyclin-based treatment. Tyvaso DPI (dry powder inhaler): Approved by the Food and Drug Administration in 2022 for PAH (Group 1) and PH-ILD (Group 3) [36]. Tyvaso DPI delivers Treprostinil via a breath-actuated handheld device eliminating the need for nebulizers or external power sources. Clinical studies showed comparable efficacy to nebulized Treprostinil, with high patient satisfaction and no new safety concerns. It allows more convenient, outpatient-friendly administration, including in patients with comorbid lung disease. Implantable Treprostinil pump: Already in use in Europe, this device provides continuous IV Treprostinil infusion while reducing infection risk associated with external catheter lines, improving long-term safety. Treprostinil Palmitil: An inhalable prodrug formulation of Treprostinil designed for once-daily administration, currently under investigation. It may offer ultra-long-acting prostacyclin activity, further simplify dosing and improving adherence. These innovations aim to retain the robust efficacy of Treprostinil while enhancing tolerability and accessibility across different stages of disease.
- Several other investigational agents show potential as adjunctive or disease-modifying treatments: Seralutinib (inhaled): A PDGF receptor kinase inhibitor, currently under evaluation in the TORREY trial for PAH, targeting vascular remodeling pathways. Rodatristat ethyl: An oral tryptophan hydroxylase inhibitor that reduces peripheral serotonin levels—a known mediator of pulmonary vascular proliferation in PAH. Immunomodulatory agents: Including anastrozole, which reduces estrogen levels implicated in PAH pathogenesis, and tocilizumab, an IL-6 receptor antagonist under investigation for CTD-associated PAH.
10. Prognosis and Follow-Up Strategies
- WHO functional class;
- Exercise capacity (6MWD or Cardiopulmonary Exercise Testing, when feasible);
- Biomarkers (particularly NT-proBNP);
- Imaging (echocardiographic assessment of RV size and function; some centers utilize serial cardiac MRI);
- Hemodynamic measurements (via periodic RHC, particularly when considering transplant referral).
- Adverse effects of therapy;
- Routine laboratory values (e.g., liver function tests in patients on endothelin receptor antagonists);
- Comorbid conditions such as sleep apnea, anemia, or iron deficiency;
- Patient adherence and education, including recognition of worsening symptoms.
- Wearable devices and smartphone applications to track heart rate, oxygen saturation, and daily physical activity.
- Integration of smartwatch data (heart rate, step count, heart rate variability) directly into electronic health records for physician review [38].
- Trials exploring whether declines in activity levels or rising resting heart rates can predict decompensation before clinical symptoms appear.
- Supervised exercise training, which has been shown to improve functional capacity and quality of life in PAH.
- Psychological support, as many patients suffer from anxiety or depression due to chronic illness; referrals to mental health services and support groups are often beneficial.
- Palliative care, which can be introduced at any stage of advanced PH to address symptom burden and improve quality of life—not limited to end-stage disease.
11. Role of Expert Centers in Pulmonary Hypertension Management
- Specialized ETT with PH-specific protocols.
- RHC (including provocative maneuvers like exercise or fluid challenge when appropriate).
- Cardiopulmonary Exercise Testing (CPET).
- Imaging modalities such as ventilation/perfusion SPECT and cardiac MRI.
- Intravenous prostacyclin therapy.
- Titration of complex drug regimens.
- Access to clinical trials of emerging therapies.
- Multidisciplinary evaluation for lung or heart–lung transplantation.
- Patient education on lifestyle modifications and symptom recognition;
- Vaccination programs (e.g., for influenza and pneumococcal infections);
- Infection prophylaxis;
- 24/7 on-call services, which are essential for patients on continuous infusions or with implantable devices.
12. Conclusions
- Aggressive combination treatment in Group 1 PAH.
- Surgical or interventional correction for Groups 2 and 4.
- Supportive, symptom-directed care in Groups 3 and 5, with selective use of novel therapeutic options.
Funding
Conflicts of Interest
Abbreviations
| 6MWD | Six-minute walking distance |
| BPA | Balloon pulmonary angioplasty |
| BMPR2 | Bone morphogenetic protein receptor type 2 |
| BNP | Brain natriuretic peptide |
| CCBs | Calcium channel blockers |
| CPET | Cardiopulmonary Exercise Testing |
| ppm | Cases per million |
| CLD | Chronic lung disease |
| COPD | Chronic obstructive pulmonary disease |
| CTEPH | Chronic thromboembolic pulmonary hypertension |
| CPFE | Combined pulmonary fibrosis and emphysema |
| CHD | Congenital heart disease |
| CPAP | Continuous positive airway pressure |
| DECT | Dual-energy computed tomography |
| ERAs | Endothelin receptor antagonists |
| ESC | European Society of Cardiology |
| HF | Heart failure |
| HFmrEF | Heart failure with mildly reduced ejection fraction |
| HFpEF | Heart failure with preserved ejection fraction |
| HFrEF | Heart failure with reduced ejection fraction |
| HPAH | Heritable PAH |
| IPAH | Idiopathic PAH |
| IPF | Idiopathic pulmonary fibrosis |
| IL-6 | Interleukin-6 |
| ILD | Interstitial lung disease |
| LHD | Left heart disease |
| mPAP | Mean pulmonary arterial pressure |
| NO | Nitric oxide |
| PDE5i | Phosphodiesterase type 5 inhibitor |
| PDGF | Platelet-derived growth factor |
| PAs | Prostacyclin analogs |
| PRAs | Prostacyclin receptor agonists |
| PAH | Pulmonary arterial hypertension |
| PAWP | Pulmonary artery wedge pressure |
| PEA | Pulmonary endarterectomy |
| PH | Pulmonary hypertension |
| PPHN | Pulmonary hypertension of the newborn |
| PVR | Pulmonary vascular resistance |
| PVOD | Pulmonary veno-occlusive disease |
| RHC | Right heart catheterization |
| sGCS | Soluble guanylate cyclase stimulator |
| TGF-β | Transforming growth factor-beta |
| V/Q | Ventilation/perfusion |
| WU | Wood Units |
| WHO | World Health Organization |
| WSPH | World Symposium on Pulmonary Hypertension |
References
- Frost, A.E.; Badesch, D.B.; Barst, R.J.; Benza, R.L.; Elliott, C.G.; Farber, H.W.; Krichman, A.; Liou, T.G.; Raskob, G.E.; Wason, P.; et al. The changing picture of patients with pulmonary arterial hypertension in the United States: How REVEAL differs from historic and non-US Contemporary Registries. Chest 2011, 139, 128–137, Erratum in Chest 2011, 140, 1106. [Google Scholar] [CrossRef] [PubMed]
- Hoeper, M.M.; Humbert, M.; Souza, R.; Idrees, M.; Kawut, S.M.; Sliwa-Hahnle, K.; Jing, Z.-C.; Gibbs, J.S. A global view of pulmonary hypertension. Lancet Respir. Med. 2016, 4, 306–322. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Sitbon, O.; Chaouat, A.; Bertocchi, M.; Habib, G.; Gressin, V.; Yaici, A.; Weitzenblum, E.; Cordier, J.-F.; Chabot, F.; et al. Pulmonary arterial hypertension in France: Results from a national registry. Am. J. Respir. Crit. Care Med. 2006, 173, 1023–1030. [Google Scholar] [CrossRef] [PubMed]
- D’Alonzo, G.E.; Barst, R.J.; Ayres, S.M.; Bergofsky, E.H.; Brundage, B.H.; Detre, K.M.; Fishman, A.P.; Goldring, R.M.; Groves, B.M.; Kernis, J.T.; et al. Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Ann. Intern. Med. 1991, 115, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Kjellstrom, B.W.M.; Dahlerup, H.; Hesselstrand, R. Swedish Pulmonary Arterial Hypertension Registry Annual Report. Available online: http://www.ucr.uu.se/spahr/arsrapporter (accessed on 11 May 2025).
- UK National Health Service Digital. National Audit of Pulmonary Hypertension Great Britain—Tenth Annual Report, 2018–2019; UK National Health Service Digital: London, UK, 2019. [Google Scholar]
- Kopeć, G.; Kurzyna, M.; Mroczek, E.; Chrzanowski, Ł.; Mularek-Kubzdela, T.; Skoczylas, I.; Kuśmierczyk, B.; Pruszczyk, P.; Błaszczak, P.; Lewicka, E.; et al. Characterization of Patients with Pulmonary Arterial Hypertension: Data from the Polish Registry of Pulmonary Hypertension (BNP-PL). J. Clin. Med. 2020, 9, 173. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Skride, A.; Sablinskis, K.; Lejnieks, A.; Rudzitis, A.; Lang, I. Characteristics and survival data from Latvian pulmonary hypertension registry: Comparison of prospective pulmonary hypertension registries in Europe. Pulm. Circ. 2018, 8, 2045894018780521. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hoeper, M.M.; Huscher, D.; Pittrow, D. Incidence and prevalence of pulmonary arterial hypertension in Germany. Int. J. Cardiol. 2016, 203, 612–613. [Google Scholar] [CrossRef] [PubMed]
- Kramm, T.; Wilkens, H.; Fuge, J.; Schäfers, H.-J.; Guth, S.; Wiedenroth, C.B.; Weingard, B.; Huscher, D.; Pittrow, D.; Cebotari, S.; et al. Incidence and characteristics of chronic thromboembolic pulmonary hypertension in Germany. Clin. Res. Cardiol. 2018, 107, 548–553. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Martínez-Santos, P.; Velázquez-Martín, M.T.; Barberá, J.A.; Fernández Pérez, C.; López-Meseguer, M.; López-Reyes, R.; Mar-tínez-Meñaca, A.; Lara-Padrón, A.; Domingo-Morera, J.A.; Blanco, I.; et al. Chronic throm-boembolic pulmonary hypertension in Spain: A decade of change. Rev. Esp. Cardiol. (Engl. Ed.) 2021, 74, 384–392, (In English, Spanish). [Google Scholar] [CrossRef] [PubMed]
- Leber, L.; Beaudet, A.; Muller, A. Epidemiology of pulmonary arterial hypertension and chronic thromboembolic pulmonary hypertension: Identification of the most accurate estimates from a systematic literature review. Pulm. Circ. 2021, 11, 2045894020977300. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kwiatkowska, J.; Zuk, M.; Migdal, A.; Kusa, J.; Skiba, E.; Zygielo, K.; Przetocka, K.; Werynski, P.; Banaszak, P.; Rzeznik-Bieniaszewska, A.; et al. Children and Adolescents with Pulmonary Arterial Hypertension: Baseline and Follow-Up Data from the Polish Registry of Pulmonary Hypertension (BNP-PL). J. Clin. Med. 2020, 9, 1717. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- del Cerro Marín, M.J.; Sabaté Rotés, A.; Rodriguez Ogando, A.; Mendoza Soto, A.; Quero Jiménez, M.; Gavilán Camacho, J.L.; Raposo Sonnenfeld, I.; Moya Bonora, A.; Albert Brotons, D.C.; Moreno Galdó, A.; et al. Assessing pulmonary hypertensive vascular disease in childhood. Data from the Spanish registry. Am. J. Respir. Crit. Care Med. 2014, 190, 1421–1429. [Google Scholar] [CrossRef] [PubMed]
- Bousseau, S.; Sobrano Fais, R.; Gu, S.; Frump, A.; Lahm, T. Pathophysiology and new advances in pulmonary hypertension. BMJ Med. 2023, 2, e000137. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Vachiéry, J.-L.; Tedford, R.J.; Rosenkranz, S.; Palazzini, M.; Lang, I.; Guazzi, M.; Coghlan, G.; Chazova, I.; De Marco, T. Pulmonary hypertension due to left heart disease. Eur. Respir. J. 2019, 53, 1801897. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Nathan, S.D.; Barbera, J.A.; Gaine, S.P.; Harari, S.; Martinez, F.J.; Olschewski, H.; Olsson, K.M.; Peacock, A.J.; Pepke-Zaba, J.; Provencher, S.; et al. Pulmonary hypertension in chronic lung disease and hypoxia. Eur. Respir. J. 2019, 53, 1801914. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kim, N.H.; Delcroix, M.; Jais, X.; Madani, M.M.; Matsubara, H.; Mayer, E.; Ogo, T.; Tapson, V.F.; Ghofrani, H.-A.; Jenkins, D.P. Chronic thromboembolic pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801915. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Humbert, M.; Kovacs, G.; Hoeper, M.M.; Badagliacca, R.; Berger, R.M.F.; Brida, M.; Carlsen, J.; Coats, A.J.S.; Escribano-Subias, P.; Ferrari, P.; et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur. Heart J. 2022, 43, 3618–3731, Erratum in Eur. Heart J. 2023, 44, 1312. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.H.; D’ARmini, A.M.; Delcroix, M.; Jaïs, X.; Jevnikar, M.; Madani, M.M.; Matsubara, H.; Palazzini, M.; Wiedenroth, C.B.; Simonneau, G.; et al. Chronic thromboembolic pulmonary disease. Eur. Respir. J. 2024, 64, 2401294. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ha, S.; Han, S. The Role of Lung Ventilation/Perfusion Scan in the Management of Chronic Thromboembolic Pulmonary Hypertension. Nucl. Med. Mol. Imaging 2024, 58, 449–458. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Schüssler, A.; Lug, Q.; Kremer, N.; Harth, S.; Kriechbaum, S.D.; Richter, M.J.; Guth, S.; Wiedenroth, C.B.; Tello, K.; Steiner, D.; et al. Evaluation of diagnostic accuracy of dual-energy computed tomography in patients with chronic thromboembolic pulmonary hypertension compared to V/Q-SPECT and pulmonary angiogram. Front. Med. 2023, 10, 1194272. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Evans, J.D.; Girerd, B.; Montani, D.; Wang, X.-J.; Galiè, N.; Austin, E.D.; Elliott, G.; Asano, K.; Grünig, E.; Yan, Y.; et al. BMPR2 mutations and survival in pulmonary arterial hypertension: An individual participant data meta-analysis. Lancet Respir. Med. 2016, 4, 129–137. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Eyries, M.; Montani, D.; Girerd, B.; Perret, C.; Leroy, A.; Lonjou, C.; Chelghoum, N.; Coulet, F.; Bonnet, D.; Dorfmüller, P.; et al. EIF2AK4 mutations cause pulmonary veno-occlusive disease, a recessive form of pulmonary hypertension. Nat. Genet. 2014, 46, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Badesch, D.B.; Raskob, G.E.; Elliott, C.G.; Krichman, A.M.; Farber, H.W.; Frost, A.E.; Barst, R.J.; Benza, R.L.; Liou, T.G.; Turner, M.; et al. Pulmonary arterial hypertension: Baseline characteristics from the REVEAL Registry. Chest 2010, 137, 376–387. [Google Scholar] [CrossRef] [PubMed]
- Rothman, A.M.K.; Vachiery, J.-L.; Howard, L.S.; Mikhail, G.W.; Lang, I.M.; Jonas, M.; Kiely, D.G.; Shav, D.; Shabtay, O.; Avriel, A.; et al. Intravascular Ultrasound Pulmonary Artery Denervation to Treat Pulmonary Arterial Hypertension (TROPHY1): Multicenter, Early Feasibility Study. JACC Cardiovasc. Interv. 2020, 13, 989–999. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.S.; Memon, M.M.; Amin, E.; Yamani, N.; Khan, S.U.; Figueredo, V.M.; Deo, S.; Rich, J.D.; Benza, R.L.; Krasuski, R.A. Use of Balloon Atrial Septostomy in Patients with Advanced Pulmonary Arterial Hypertension: A Systematic Review and Meta-Analysis. Chest 2019, 156, 53–63. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Grady, R.M.; Canter, M.W.; Wan, F.; Shmalts, A.A.; Coleman, R.D.; Beghetti, M.; Berger, R.M.; Del Cerro Marin, M.J.; Fletcher, S.E.; Hirsch, R.; et al. Pulmonary-to-Systemic Arterial Shunt to Treat Children with Severe Pulmonary Hypertension. J. Am. Coll. Cardiol. 2021, 78, 468–477. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Waxman, A.; Restrepo-Jaramillo, R.; Thenappan, T.; Ravichandran, A.; Engel, P.; Bajwa, A.; Allen, R.; Feldman, J.; Argula, R.; Smith, P.; et al. Inhaled Treprostinil in Pulmonary Hypertension Due to Interstitial Lung Disease. N. Engl. J. Med. 2021, 384, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Delcroix, M.; Pepke-Zaba, J.; D’Armini, A.M.; Fadel, E.; Guth, S.; Hoole, S.P.; Jenkins, D.P.; Kiely, D.G.; Kim, N.H.; Madani, M.M.; et al. Worldwide CTEPH Registry: Long-Term Outcomes With Pulmonary Endarterectomy, Balloon Pulmonary Angioplasty, and Medical Therapy. Circulation 2024, 150, 1354–1365. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lang, I.M.; Andreassen, A.K.; Andersen, A.; Bouvaist, H.; Coghlan, G.; Escribano-Subias, P.; Jansa, P.; Kopec, G.; Kurzyna, M.; Matsubara, H.; et al. Balloon pulmonary angioplasty for chronic thromboembolic pulmonary hypertension: A clinical consensus statement of the ESC working group on pulmonary circulation and right ventricular function. Eur. Heart J. 2023, 44, 2659–2671. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yang, J.Z.; Poch, D.S.; Ang, L.; Mahmud, E.; Kim, N.H. Balloon pulmonary angioplasty in the current era of CTEPH treatment: How did we get here? Pulm. Circ. 2023, 13, e12312. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wacker, J.; Beghetti, M. Pulmonary hypertension in pediatrics: From clinical suspicion to management. Eur. J. Pediatr. 2025, 184, 288. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hoeper, M.M.; Badesch, D.B.; Ghofrani, H.A.; Gibbs, J.S.R.; Gomberg-Maitland, M.; McLaughlin, V.V.; Preston, I.R.; Souza, R.; Waxman, A.B.; Grünig, E.; et al. Phase 3 Trial of Sotatercept for Treatment of Pulmonary Arterial Hypertension. N. Engl. J. Med. 2023, 388, 1478–1490. [Google Scholar] [CrossRef] [PubMed]
- Gillies, H.; Chakinala, M.M.; Dake, B.T.; Feldman, J.P.; Hoeper, M.M.; Humbert, M.; Jing, Z.; Langley, J.; McLaughlin, V.V.; Niven, R.W.; et al. IMPAHCT: A randomized phase 2b/3 study of inhaled imatinib for pulmonary arterial hypertension. Pulm. Circ. 2024, 14, e12352. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- McEvoy, C.; Argula, R.; Sahay, S.; Shapiro, S.; Eagan, C.; Hickey, A.J.; Smutney, C.; Dillon, C.; Winkler, T.; Davis, B.N.; et al. Tyvaso DPI: Drug-device characteristics and patient clinical considerations. Pulm. Pharmacol. Ther. 2023, 83, 102266. [Google Scholar] [CrossRef] [PubMed]
- Reinders, S.; Didden, E.-M.; Ong, R. Survival, morbidity, and quality of life in pulmonary arterial hypertension patients: A systematic review of outcomes reported by population-based observational studies. Respir. Res. 2024, 25, 373. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ørbæk Andersen, M.; Carlsen, J. Continuous heart monitoring in patients with pulmonary hypertension smartwatches and direct transmission to their electronic health records: A trial design. Contemp. Clin. Trials 2024, 142, 107548. [Google Scholar] [CrossRef] [PubMed]
- Labrandero, C.; Deiros, L.; Abelleira, C.; Arreo, V.; Balbacid, E.J.; Gutiérrez-Larraya, F. Hemodynamic Monitoring of Pediatric Patients with Heart Failure and Pulmonary Hypertension Using CardioMEMS. J. Soc. Cardiovasc. Angiogr. Interv. 2024, 3, 101933. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Virsinskaite, R.; Karia, N.; Kotecha, T.; Schreiber, B.E.; Coghlan, J.G.; Knight, D.S. Pulmonary hypertension—The latest updates for physicians. Clin. Med. 2023, 23, 449–454. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Vatrano, M.; Manzi, G.; Picariello, C.; D’Alto, M.; Enea, I.; Ghio, S.; Caravita, S.; Argiento, P.; Garascia, A.; Vitulo, P.; et al. Documento di consenso ANMCO/SIC sull’ipertensione arteriosa polmonare [ANMCO/SIC Consensus statement on pulmonary arterial hypertension]. G. Ital. Cardiol. 2024, 25, 192–201. (In Italian) [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cacia, M.; Imbalzano, E.; Ciconte, V.A.; Vatrano, M. Pulmonary Hypertension: Let’s Take Stock! Life 2025, 15, 1137. https://doi.org/10.3390/life15071137
Cacia M, Imbalzano E, Ciconte VA, Vatrano M. Pulmonary Hypertension: Let’s Take Stock! Life. 2025; 15(7):1137. https://doi.org/10.3390/life15071137
Chicago/Turabian StyleCacia, Michele, Egidio Imbalzano, Vincenzo Antonio Ciconte, and Marco Vatrano. 2025. "Pulmonary Hypertension: Let’s Take Stock!" Life 15, no. 7: 1137. https://doi.org/10.3390/life15071137
APA StyleCacia, M., Imbalzano, E., Ciconte, V. A., & Vatrano, M. (2025). Pulmonary Hypertension: Let’s Take Stock! Life, 15(7), 1137. https://doi.org/10.3390/life15071137