
Hybrid Genome and Clinical Impact of Emerging Extensively Drug-Resistant Priority Bacterial Pathogen Acinetobacter baumannii in Saudi Arabia


Abstract
1. Introduction
2. Materials and Methods
2.1. Ethical Approval
2.2. Bacterial Strain and DNA Isolation
2.3. Nanopore Whole-Genome Sequencing and Data Processing
2.4. Bacterial Genome Analysis
3. Results
3.1. Phenomics of A. baumannii Strain IRMCBCU95U
3.2. Genomics of IRMCBCU95U
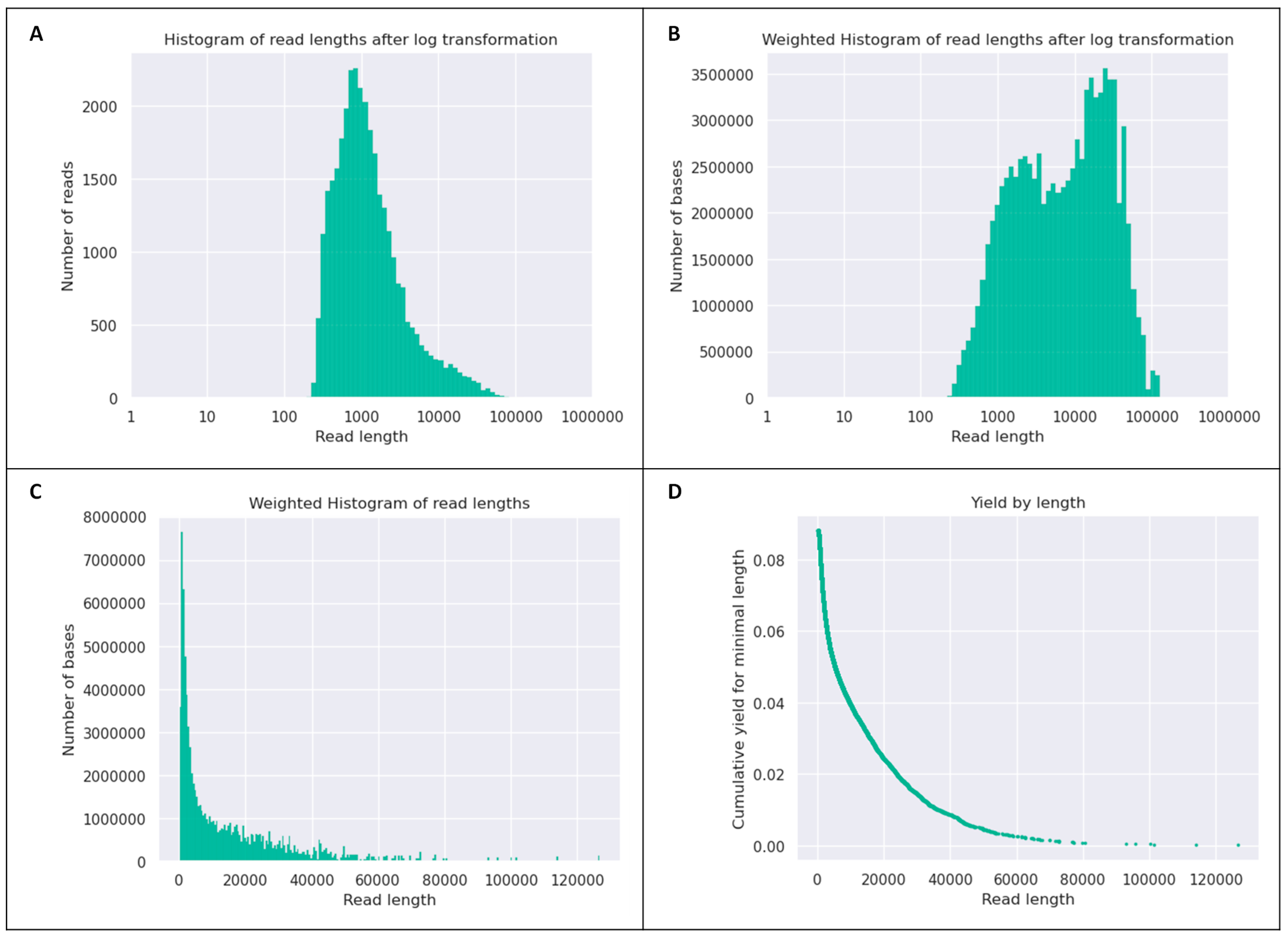
Quality of the Sequencing Data for IRMCBCU95U Genome
3.3. Assembly of the IRMCBCU9SU Dataset
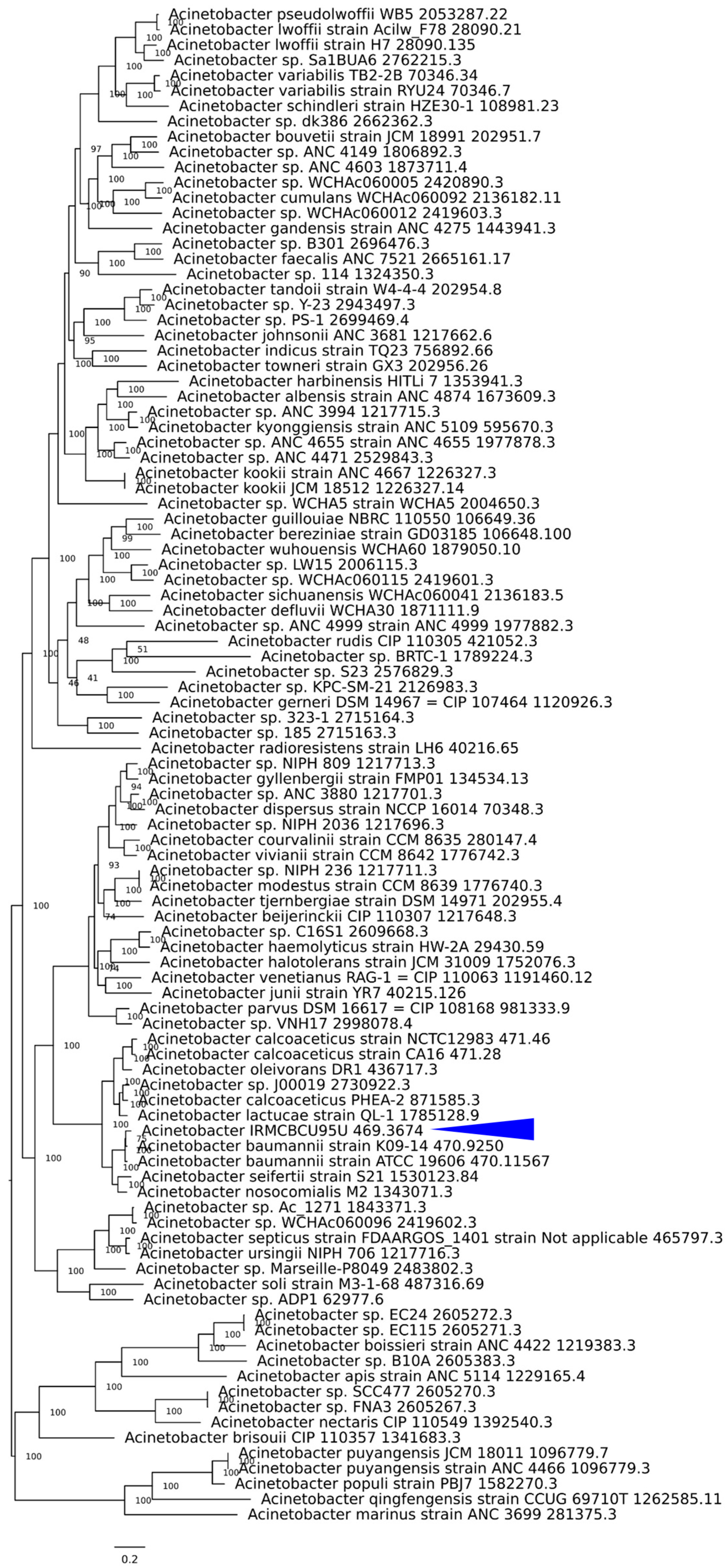
3.4. Phylogenetic Tree Based on Whole Genomes of IRMCBCU95U and Other Acinetobacter Species
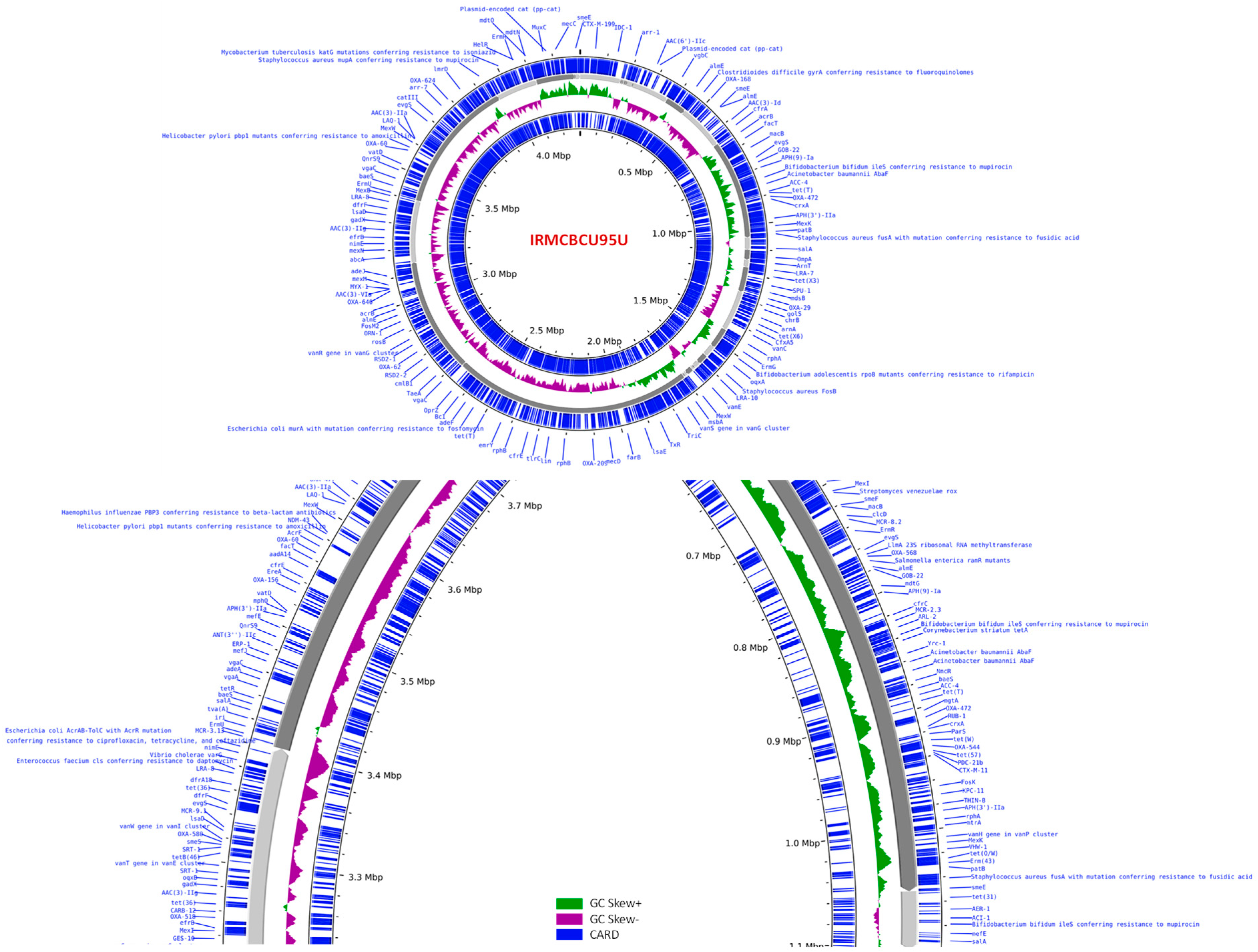
3.5. Genomic Overview of A. baumannii IRMCBCU95U
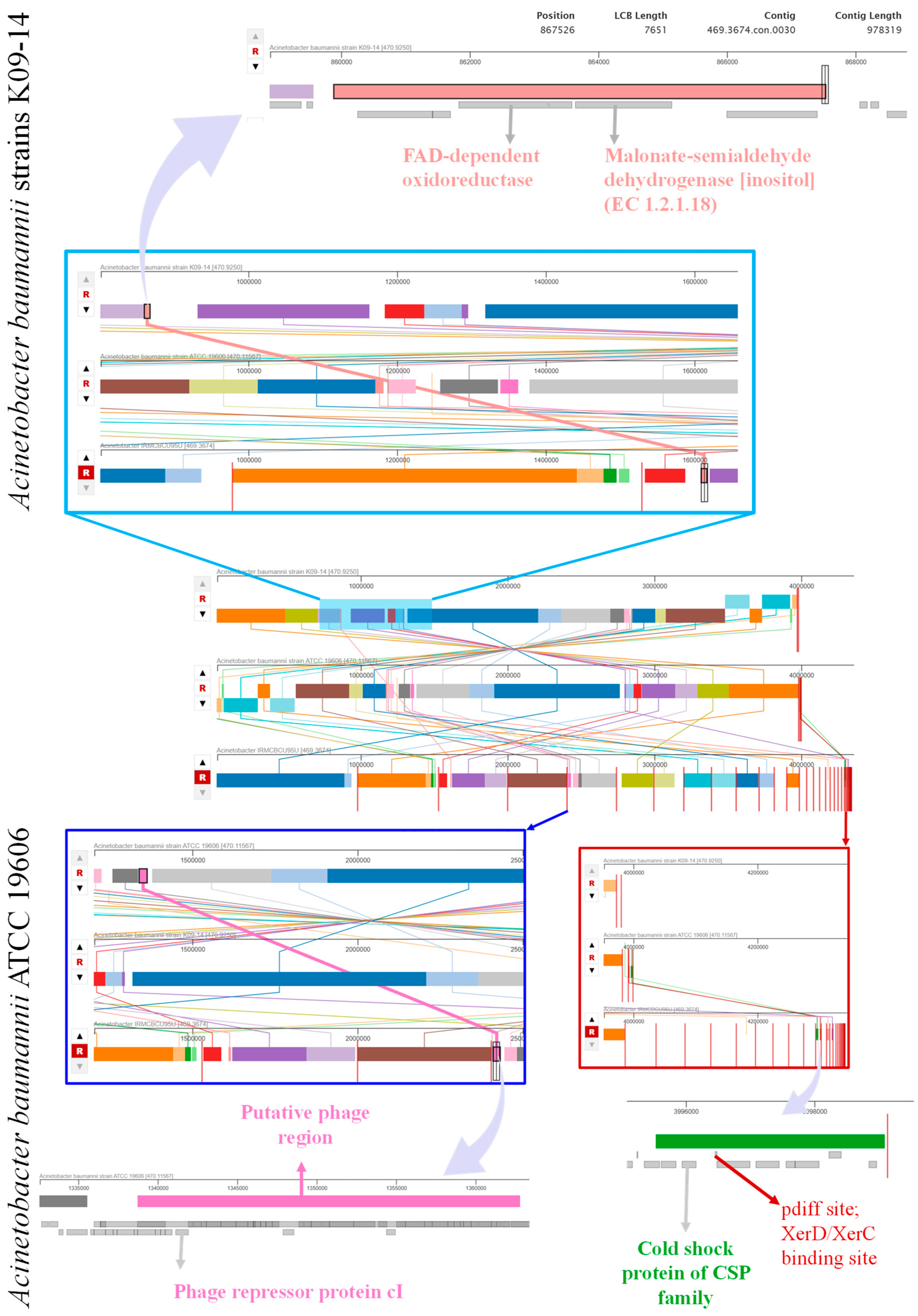
3.6. Genome Comparison
3.7. Genome-Assisted Resistant Phenotype
3.8. Virulence Factors
3.9. Mobile Elements in A. baumannii IRMCBCU95U
3.10. Pathogenic Proteins
3.11. Mycotoxin-Producing Genes
3.12. Plasmid Analysis in IRMCBCU95U
4. Discussion
5. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shi, J.; Cheng, J.; Liu, S.; Zhu, Y.; Zhu, M. Acinetobacter baumannii: An Evolving and Cunning Opponent. Front. Microbiol. 2024, 15, 1332108. [Google Scholar] [CrossRef] [PubMed]
- Howard, A.; O’Donoghue, M.; Feeney, A.; Sleator, R.D. Acinetobacter baumannii: An Emerging Opportunistic Pathogen. Virulence 2012, 3, 243–250. [Google Scholar] [CrossRef]
- Harding, C.M.; Hennon, S.W.; Feldman, M.F. Uncovering the Mechanisms of Acinetobacter baumannii Virulence. Nat. Rev. Microbiol. 2018, 16, 91–102. [Google Scholar] [CrossRef]
- Arshad, N.; Azzam, W.; Zilberberg, M.D.; Shorr, A.F. Acinetobacter baumannii Complex Infections: New Treatment Options in the Antibiotic Pipeline. Microorganisms 2025, 13, 356. [Google Scholar] [CrossRef]
- Zhang, R.; Li, D.; Fang, H.; Xie, Q.; Tang, H.; Chen, L. Iron-Dependent Mechanisms in Acinetobacter baumannii: Pathogenicity and Resistance. JAC Antimicrob. Resist. 2025, 7, dlaf039. [Google Scholar] [CrossRef]
- Sebeny, P.J.; Riddle, M.S.; Petersen, K. Acinetobacter baumannii Skin and Soft-Tissue Infection Associated with War Trauma. Clin. Infect. Dis. 2008, 47, 444–449. [Google Scholar] [CrossRef]
- Bassetti, M.; Ginocchio, F.; Mikulska, M. New Treatment Options against Gram-Negative Organisms. Crit. Care 2011, 15, 215. [Google Scholar] [CrossRef] [PubMed]
- Rice, L.B. Federal Funding for the Study of Antimicrobial Resistance in Nosocomial Pathogens: No ESKAPE. J. Infect. Dis. 2008, 197, 1079–1081. [Google Scholar] [CrossRef] [PubMed]
- Fournier, P.-E.; Vallenet, D.; Barbe, V.; Audic, S.; Ogata, H.; Poirel, L.; Richet, H.; Robert, C.; Mangenot, S.; Abergel, C.; et al. Comparative Genomics of Multidrug Resistance in Acinetobacter baumannii. PLoS Genet. 2006, 2, e7. [Google Scholar] [CrossRef]
- Iovleva, A.; Fowler, V.G.; Doi, Y. Treatment Approaches for Carbapenem-Resistant Acinetobacter baumannii Infections. Drugs 2025, 85, 21–40. [Google Scholar] [CrossRef]
- Wright, M.S.; Iovleva, A.; Jacobs, M.R.; Bonomo, R.A.; Adams, M.D. Genome Dynamics of Multidrug-Resistant Acinetobacter baumannii during Infection and Treatment. Genome Med. 2016, 8, 26. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.P.; Sutton, G.; DePew, J.; Krishnakumar, R.; Choi, Y.; Huang, X.-Z.; Beck, E.; Harkins, D.M.; Kim, M.; Lesho, E.P.; et al. A Novel Method of Consensus Pan-Chromosome Assembly and Large-Scale Comparative Analysis Reveal the Highly Flexible Pan-Genome of Acinetobacter baumannii. Genome Biol. 2015, 16, 143. [Google Scholar] [CrossRef]
- Borgio, J.F.; Rasdan, A.S.; Sonbol, B.; Alhamid, G.; Almandil, N.B.; AbdulAzeez, S. Emerging Status of Multidrug-Resistant Bacteria and Fungi in the Arabian Peninsula. Biology 2021, 10, 1144. [Google Scholar] [CrossRef]
- Alamri, A.M.; Alsultan, A.A.; Ansari, M.A.; Alnimr, A.M. Biofilm-Formation in Clonally Unrelated Multidrug-Resistant Acinetobacter baumannii Isolates. Pathogens 2020, 9, 630. [Google Scholar] [CrossRef]
- Al-Anazi, K.A.; Abdalhamid, B.; Alshibani, Z.; Awad, K.; Alzayed, A.; Hassan, H.; Alsayiegh, M. Acinetobacter baumannii Septicemia in a Recipient of an Allogeneic Hematopoietic Stem Cell Transplantation. Case Rep. Transpl. 2012, 2012, 646195. [Google Scholar] [CrossRef]
- Aljindan, R.; Elhadi, N. Genetic Relationship of Multi-Resistant Acinetobacter baumannii Isolates in Kingdom of Saudi Arabia. J. Pure Appl. Microbiol. 2018, 12, 1951–1958. [Google Scholar] [CrossRef]
- Bhuiyan, M.S.; Ellett, F.; Murray, G.L.; Kostoulias, X.; Cerqueira, G.M.; Schulze, K.E.; Mahamad Maifiah, M.H.; Li, J.; Creek, D.J.; Lieschke, G.J.; et al. Acinetobacter baumannii Phenylacetic Acid Metabolism Influences Infection Outcome through a Direct Effect on Neutrophil Chemotaxis. Proc. Natl. Acad. Sci. USA 2016, 113, 9599–9604. [Google Scholar] [CrossRef] [PubMed]
- García-Quintanilla, M.; Pulido, M.R.; López-Rojas, R.; Pachón, J.; McConnell, M.J. Emerging Therapies for Multidrug Resistant Acinetobacter baumannii. Trends Microbiol. 2013, 21, 157–163. [Google Scholar] [CrossRef]
- Jacobs, A.C.; Thompson, M.G.; Black, C.C.; Kessler, J.L.; Clark, L.P.; McQueary, C.N.; Gancz, H.Y.; Corey, B.W.; Moon, J.K.; Si, Y.; et al. AB5075, a Highly Virulent Isolate of Acinetobacter baumannii, as a Model Strain for the Evaluation of Pathogenesis and Antimicrobial Treatments. mBio 2014, 5, e01076-14. [Google Scholar] [CrossRef]
- AlJindan, R.; Mahmoud, N.; AlEraky, D.M.; Almandil, N.B.; AbdulAzeez, S.; Borgio, J.F. Phenomics and Genomic Features of Enterococcus Avium IRMC1622a Isolated from a Clinical Sample of Hospitalized Patient. J. Infect. Public Health 2024, 17, 102463. [Google Scholar] [CrossRef]
- Czmil, A.; Wronski, M.; Czmil, S.; Sochacka-Pietal, M.; Cmil, M.; Gawor, J.; Wołkowicz, T.; Plewczynski, D.; Strzalka, D.; Pietal, M. NanoForms: An Integrated Server for Processing, Analysis and Assembly of Raw Sequencing Data of Microbial Genomes, from Oxford Nanopore Technology. PeerJ 2022, 10, e13056. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- De Coster, W.; D’Hert, S.; Schultz, D.T.; Cruts, M.; Van Broeckhoven, C. NanoPack: Visualizing and Processing Long-Read Sequencing Data. Bioinformatics 2018, 34, 2666–2669. [Google Scholar] [CrossRef]
- Mikheenko, A.; Prjibelski, A.; Saveliev, V.; Antipov, D.; Gurevich, A. Versatile Genome Assembly Evaluation with QUAST-LG. Bioinformatics 2018, 34, i142–i150. [Google Scholar] [CrossRef]
- Lin, Y.; Yuan, J.; Kolmogorov, M.; Shen, M.W.; Chaisson, M.; Pevzner, P.A. Assembly of Long Error-Prone Reads Using de Bruijn Graphs. Proc. Natl. Acad. Sci. USA 2016, 113, E8396–E8405. [Google Scholar] [CrossRef] [PubMed]
- Wattam, A.R.; Davis, J.J.; Assaf, R.; Boisvert, S.; Brettin, T.; Bun, C.; Conrad, N.; Dietrich, E.M.; Disz, T.; Gabbard, J.L.; et al. Improvements to PATRIC, the All-Bacterial Bioinformatics Database and Analysis Resource Center. Nucleic Acids Res. 2017, 45, D535–D542. [Google Scholar] [CrossRef]
- de Koning, W.; Miladi, M.; Hiltemann, S.; Heikema, A.; Hays, J.P.; Flemming, S.; van den Beek, M.; Mustafa, D.A.; Backofen, R.; Grüning, B.; et al. NanoGalaxy: Nanopore Long-Read Sequencing Data Analysis in Galaxy. Gigascience 2020, 9, giaa105. [Google Scholar] [CrossRef]
- AlJindan, R.; AlEraky, D.M.; Farhat, M.; Almandil, N.B.; AbdulAzeez, S.; Borgio, J.F. Genomic Insights into Virulence Factors and Multi-Drug Resistance in Clostridium Perfringens IRMC2505A. Toxins 2023, 15, 359. [Google Scholar] [CrossRef]
- Borgio, J.F.; Alhujaily, R.; Alquwaie, R.; Alabdullah, M.J.; AlHasani, E.; Alothman, W.; Alaqeel, R.K.; Alfaraj, A.S.; Kaabi, A.; Alhur, N.F.; et al. Mining the Nanotube-Forming Bacillus Amyloliquefaciens MR14M3 Genome for Determining Anti-Candida Auris and Anti-Candida Albicans Potential by Pathogenicity and Comparative Genomics Analysis. Comput. Struct. Biotechnol. J. 2023, 21, 4261–4276. [Google Scholar] [CrossRef]
- Schomburg, I.; Chang, A.; Ebeling, C.; Gremse, M.; Heldt, C.; Huhn, G.; Schomburg, D. BRENDA, the Enzyme Database: Updates and Major New Developments. Nucleic Acids Res. 2004, 32, D431–D433. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a Reference Resource for Gene and Protein Annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the Unification of Biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Davis, J.J.; Gerdes, S.; Olsen, G.J.; Olson, R.; Pusch, G.D.; Shukla, M.; Vonstein, V.; Wattam, A.R.; Yoo, H. PATtyFams: Protein Families for the Microbial Genomes in the PATRIC Database. Front. Microbiol. 2016, 7, 118. [Google Scholar] [CrossRef] [PubMed]
- Overbeek, R.; Begley, T.; Butler, R.M.; Choudhuri, J.V.; Chuang, H.-Y.; Cohoon, M.; de Crécy-Lagard, V.; Diaz, N.; Disz, T.; Edwards, R.; et al. The Subsystems Approach to Genome Annotation and Its Use in the Project to Annotate 1000 Genomes. Nucleic Acids Res. 2005, 33, 5691–5702. [Google Scholar] [CrossRef]
- McArthur, A.G.; Waglechner, N.; Nizam, F.; Yan, A.; Azad, M.A.; Baylay, A.J.; Bhullar, K.; Canova, M.J.; De Pascale, G.; Ejim, L.; et al. The Comprehensive Antibiotic Resistance Database. Antimicrob. Agents Chemother. 2013, 57, 3348–3357. [Google Scholar] [CrossRef]
- Law, V.; Knox, C.; Djoumbou, Y.; Jewison, T.; Guo, A.C.; Liu, Y.; Maciejewski, A.; Arndt, D.; Wilson, M.; Neveu, V.; et al. DrugBank 4.0: Shedding New Light on Drug Metabolism. Nucleic Acids Res. 2014, 42, D1091–D1097. [Google Scholar] [CrossRef]
- Zhu, F.; Han, B.; Kumar, P.; Liu, X.; Ma, X.; Wei, X.; Huang, L.; Guo, Y.; Han, L.; Zheng, C.; et al. Update of TTD: Therapeutic Target Database. Nucleic Acids Res. 2010, 38, D787–D791. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zheng, D.; Liu, B.; Yang, J.; Jin, Q. VFDB 2016: Hierarchical and Refined Dataset for Big Data Analysis--10 Years On. Nucleic Acids Res. 2016, 44, D694–D697. [Google Scholar] [CrossRef]
- Mao, C.; Abraham, D.; Wattam, A.R.; Wilson, M.J.C.; Shukla, M.; Yoo, H.S.; Sobral, B.W. Curation, Integration and Visualization of Bacterial Virulence Factors in PATRIC. Bioinformatics 2015, 31, 252–258. [Google Scholar] [CrossRef]
- Saier, M.H.; Reddy, V.S.; Tsu, B.V.; Ahmed, M.S.; Li, C.; Moreno-Hagelsieb, G. The Transporter Classification Database (TCDB): Recent Advances. Nucleic Acids Res. 2016, 44, D372–D379. [Google Scholar] [CrossRef]
- Carattoli, A.; Zankari, E.; García-Fernández, A.; Voldby Larsen, M.; Lund, O.; Villa, L.; Møller Aarestrup, F.; Hasman, H. In Silico Detection and Typing of Plasmids Using PlasmidFinder and Plasmid Multilocus Sequence Typing. Antimicrob. Agents Chemother. 2014, 58, 3895–3903. [Google Scholar] [CrossRef] [PubMed]
- Ondov, B.D.; Treangen, T.J.; Melsted, P.; Mallonee, A.B.; Bergman, N.H.; Koren, S.; Phillippy, A.M. Mash: Fast Genome and Metagenome Distance Estimation Using MinHash. Genome Biol. 2016, 17, 132. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple Sequence Alignment with High Accuracy and High Throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML Version 8: A Tool for Phylogenetic Analysis and Post-Analysis of Large Phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Stamatakis, A.; Hoover, P.; Rougemont, J. A Rapid Bootstrap Algorithm for the RAxML Web Servers. Syst. Biol. 2008, 57, 758–771. [Google Scholar] [CrossRef] [PubMed]
- Clausen, P.T.L.C.; Aarestrup, F.M.; Lund, O. Rapid and Precise Alignment of Raw Reads against Redundant Databases with KMA. BMC Bioinform. 2018, 19, 307. [Google Scholar] [CrossRef]
- Zhou, S.; Liu, B.; Zheng, D.; Chen, L.; Yang, J. VFDB 2025: An Integrated Resource for Exploring Anti-Virulence Compounds. Nucleic Acids Res. 2025, 53, D871–D877. [Google Scholar] [CrossRef] [PubMed]
- Hasman, H.; Clausen, P.T.L.C.; Kaya, H.; Hansen, F.; Knudsen, J.D.; Wang, M.; Holzknecht, B.J.; Samulioniené, J.; Røder, B.L.; Frimodt-Møller, N.; et al. LRE-Finder, a Web Tool for Detection of the 23S rRNA Mutations and the optrA, Cfr, Cfr(B) and poxtA Genes Encoding Linezolid Resistance in Enterococci from Whole-Genome Sequences. J. Antimicrob. Chemother. 2019, 74, 1473–1476. [Google Scholar] [CrossRef]
- Cosentino, S.; Voldby Larsen, M.; Møller Aarestrup, F.; Lund, O. PathogenFinder--Distinguishing Friend from Foe Using Bacterial Whole Genome Sequence Data. PLoS ONE 2013, 8, e77302. [Google Scholar] [CrossRef]
- Johansson, M.H.K.; Bortolaia, V.; Tansirichaiya, S.; Aarestrup, F.M.; Roberts, A.P.; Petersen, T.N. Detection of Mobile Genetic Elements Associated with Antibiotic Resistance in Salmonella Enterica Using a Newly Developed Web Tool: MobileElementFinder. J. Antimicrob. Chemother. 2021, 76, 101–109. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and Applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Yasir, M.; Shah, M.W.; Jiman-Fatani, A.A.; Almasaudi, S.B.; Ahmad, H.; Alawi, M.; Azhar, E.I. Draft Genome Sequence of a Clinical Acinetobacter baumannii Isolate of New Sequence Type ST1688 from Saudi Arabia. J. Glob. Antimicrob. Resist. 2019, 18, 151–152. [Google Scholar] [CrossRef]
- Al-Zahrani, I.A.; Brek, T.M. Comprehensive Genome Analysis of Colistin-Only-Sensitive KPC-2 and NDM1-1-Coproducing Klebsiella pneumoniae ST11 and Acinetobacter baumannii ST2 From a Critically Ill Patient With COVID-19 in Saudi Arabia: Whole Genome Sequencing (WGS) of K. pneumoniae ST11 and A. baumannii ST2. Int. J. Microbiol. 2024, 2024, 9233075. [Google Scholar] [CrossRef]
- Brek, T.M.; Muhajir, A.A.; Alkuwaity, K.K.; Haddad, M.A.; Alattas, E.M.; Eisa, Z.M.; Al-Thaqafy, M.S.; Albarraq, A.M.; Al-Zahrani, I.A. Genomic Insights of Predominant International High-Risk Clone ST2 Acinetobacter baumannii Isolates in Saudi Arabia. J. Glob. Antimicrob. Resist. 2025, 42, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Okdah, L.; AlDosary, M.S.; AlMazyed, A.; Alkhurayb, H.M.; Almossallam, M.; Al Obaisi, Y.S.; Marie, M.A.; Abdelrahman, T.; Diene, S.M. Genomic Characterization of Colistin-Resistant Isolates from the King Fahad Medical City, Kingdom of Saudi Arabia. Antibiotics 2022, 11, 1597. [Google Scholar] [CrossRef]
- Yasir, M.; Subahi, A.M.; Shukri, H.A.; Bibi, F.; Sohrab, S.S.; Alawi, M.; Sindi, A.A.; Jiman-Fatani, A.A.; Azhar, E.I. Bacterial Community and Genomic Analysis of Carbapenem-Resistant Acinetobacter baumannii Isolates from the Environment of a Health Care Facility in the Western Region of Saudi Arabia. Pharmaceuticals 2022, 15, 611. [Google Scholar] [CrossRef] [PubMed]
- Bakour, S.; Alsharapy, S.A.; Touati, A.; Rolain, J.-M. Characterization of Acinetobacter baumannii Clinical Isolates Carrying Bla(OXA-23) Carbapenemase and 16S rRNA Methylase armA Genes in Yemen. Microb. Drug Resist. 2014, 20, 604–609. [Google Scholar] [CrossRef]
- Clausen, P.T.L.C.; Zankari, E.; Aarestrup, F.M.; Lund, O. Benchmarking of Methods for Identification of Antimicrobial Resistance Genes in Bacterial Whole Genome Data. J. Antimicrob. Chemother. 2016, 71, 2484–2488. [Google Scholar] [CrossRef]
- Shankar, C.; Vasudevan, K.; Jacob, J.J.; Baker, S.; Isaac, B.J.; Neeravi, A.R.; Sethuvel, D.P.M.; George, B.; Veeraraghavan, B. Hybrid Plasmids Encoding Antimicrobial Resistance and Virulence Traits Among Hypervirulent Klebsiella pneumoniae ST2096 in India. Front. Cell. Infect. Microbiol. 2022, 12, 875116. [Google Scholar] [CrossRef]
- Zeng, Z.; Lei, L.; Li, L.; Hua, S.; Li, W.; Zhang, L.; Lin, Q.; Zheng, Z.; Yang, J.; Dou, X.; et al. In Silico Characterization of blaNDM-Harboring Plasmids in Klebsiella pneumoniae. Front. Microbiol. 2022, 13, 1008905. [Google Scholar] [CrossRef]
- Fomda, B.A.; Khan, A.; Zahoor, D. NDM-1 (New Delhi Metallo Beta Lactamase-1) Producing Gram-Negative Bacilli: Emergence & Clinical Implications. Indian J. Med. Res. 2014, 140, 672–678. [Google Scholar] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibiotic | Interpretation | MIC (µg/mL) |
|---|---|---|
| Cefepime | Resistant | NaN |
| Ceftazidime | Resistant | NaN |
| Ciprofloxacin | Resistant | NaN |
| Imipenem | Resistant | NaN |
| Meropenem | Resistant | NaN |
| Piperacillin/Tazobactam | Resistant | NaN |
| Tigecycline | Sensitive | 2.0 |
| Trimethoprim/Sulfamethoxazole | Sensitive | NaN |
| Resistance Gene | Identity (%) | Alignment Length | Position in Reference | Contig or Depth | Position in Contig | Resistant Phenotype | Notes |
|---|---|---|---|---|---|---|---|
| aph(3′)-Ia | 99.51 | 818 | 1...816 | contig_25_segment0 contig_25:1.0-336501.0 | 85910...86726 | (‘kanamycin’, ‘neomycin’, ‘lividomycin’, ‘paromomycin’, ‘ribostamycin’) | |
| armA | 99.35 | 774 | 1...772 | contig_5_segment0 contig_5:1.0-977170.0 | 774608...775381 | (‘gentamicin’, ‘tobramycin’, ‘amikacin’, ‘isepamicin’, ‘netilmicin’) | |
| blaOXA-66 | 99.76 | 825 | 1...824 | contig_2_segment0 contig_2:1.0-102924.0 | 85778...86602 | (‘unknown beta-lactam’) | Class D; OXA-51-like; naturally occurring in A. baumannii |
| blaTEM-1D | 99.77 | 863 | 1...861 | contig_25_segment0 contig_25:1.0-336501.0 | 79447...80309 | (‘amoxicillin’, ‘ampicillin’, ‘piperacillin’, ‘ticarcillin’, ‘cephalothin’) | Class A |
| blaADC-25 | 99.57 | 1153 | 1...1152 | contig_5_segment2 contig_5:979486.0-1449651.0 | 20168...21319 | (‘unknown beta-lactam’) | Chromosomal |
| blaOXA-23 | 99.88 | 822 | 1...822 | contig_6_segment1 contig_6:253978.0-804087.0 | 537440...538260 | (‘imipenem’, ‘meropenem’) | Class D; OXA-23-like; naturally occurring in Acinetobacter radioresistens; alternative name: blaARI-1 |
| blaOXA-422 | 99.88 | 822 | 1...822 | contig_6_segment1 contig_6:253978.0-804087.0 | 537440...538260 | (‘unknown beta-lactam’) | Class D; OXA-23-like; natural occurring in A. baumannii |
| msr(E) | 99.80 | 1477 | 1...1476 | contig_5_segment0 contig_5:1.0-977170.0 | 770829...772303 | (‘erythromycin’, ‘azithromycin’, ‘quinupristin’, ‘pristinamycin ia’, ‘virginiamycin s’) | ABC transporter |
| mph(E) | 99.77 | 886 | 1...885 | contig_5_segment0 contig_5:1.0-977170.0 | 769889...770773 | (‘erythromycin’) | Macrolide phosphotransferase |
| Template | Score | Template Length | Q Value | p Value | Template Coverage | Query Coverage | Depth | Depth Corr |
|---|---|---|---|---|---|---|---|---|
| NZ_CP091346.1 A. baumannii strain AB217-VUB chromosome, complete genome | 137,198 | 152,863 | 137,185.02 | 1 × 1026 | 89.66 | 86.57 | 0.90 | 0.6321 |
| aph(3′)-Ia_7_X62115 | 806 | 816 | 800.34 | 1 × 1026 | 100.12 | 99.88 | 1.00 | 0.6708 |
| armA_1_AY220558 | 759 | 774 | 753.59 | 1 × 1026 | 100.26 | 99.74 | 1.00 | 0.6708 |
| blaOXA-23_1_AY795964 | 821 | 822 | 815.31 | 1 × 1026 | 99.88 | 100.12 | 1.00 | 0.6708 |
| blaOXA-66_1_AY750909 | 819 | 825 | 813.28 | 1 × 1026 | 100.12 | 99.88 | 1.00 | 0.6708 |
| blaTEM-1D_1_AF188200 | 858 | 861 | 852.07 | 1 × 1026 | 100.23 | 99.77 | 1.00 | 0.6708 |
| blaADC-25_1_EF016355 | 1139 | 1152 | 1131.40 | 1 × 1026 | 100.00 | 100.00 | 1.00 | 0.6708 |
| msr(E)_1_FR751518 | 1468 | 1476 | 1458.76 | 1 × 1026 | 99.93 | 100.07 | 1.00 | 0.6708 |
| mph(E)_1_DQ839391 | 881 | 885 | 874.93 | 1 × 1026 | 100.00 | 100.00 | 1.00 | 0.6708 |
| VFDB ID | Gene Name/Description | Identity (%) | E-Value | Score (bits) | Length | Identities | Gaps | Strand |
|---|---|---|---|---|---|---|---|---|
| VFG037497 | (ACICU_RS04580) TonB-dependent receptor | 94 | 0 | 4811 | 3105 | 2943/3108 | 7/3108 | +/+ |
| VFG037513 | (ACICU_RS04590) porin family protein | 94 | 0 | 2268 | 1467 | 1386/1468 | 4/1468 | +/+ |
| VFG050400 | (pilU) PilT/Pilu family type 4a pilus ATPase | 99 | 0 | 2141 | 1119 | 1108/1119 | 1/1119 | +/− |
| VFG050386 | (pilT) type IV pilus twitching motility protein PilT | 99 | 0 | 2030 | 1038 | 1038/1040 | 2/1040 | +/− |
| VFG037490 | (ACICU_RS04575) FecR domain-containing protein | 94 | 0 | 1584 | 1017 | 966/1020 | 3/1020 | +/+ |
| VFG037470 | (ACICU_RS04565) LysR family transcriptional regulator | 94 | 0 | 1392 | 891 | 839/887 | N/A | +/+ |
| VFG037505 | (ACICU_RS04585) transferrin-binding protein-like | 96 | 0 | 1332 | 792 | 763/792 | 1/792 | +/+ |
| VFG037521 | (ACICU_RS04595) TonB family protein | 92 | 0 | 1279 | 921 | 853/918 | 5/918 | +/+ |
| VFG037529 | (hemo) biliverdin-producing heme oxygenase | 97 | 0 | 1035 | 600 | 585/602 | 3/602 | +/+ |
| VFG037482 | (ACICU_RS04570) sigma-70 family RNA polymerase sigma factor | 97 | 0 | 924 | 522 | 508/522 | N/A | +/+ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borgio, J.F. Hybrid Genome and Clinical Impact of Emerging Extensively Drug-Resistant Priority Bacterial Pathogen Acinetobacter baumannii in Saudi Arabia. Life 2025, 15, 1094. https://doi.org/10.3390/life15071094
Borgio JF. Hybrid Genome and Clinical Impact of Emerging Extensively Drug-Resistant Priority Bacterial Pathogen Acinetobacter baumannii in Saudi Arabia. Life. 2025; 15(7):1094. https://doi.org/10.3390/life15071094
Chicago/Turabian StyleBorgio, J. Francis. 2025. "Hybrid Genome and Clinical Impact of Emerging Extensively Drug-Resistant Priority Bacterial Pathogen Acinetobacter baumannii in Saudi Arabia" Life 15, no. 7: 1094. https://doi.org/10.3390/life15071094
APA StyleBorgio, J. F. (2025). Hybrid Genome and Clinical Impact of Emerging Extensively Drug-Resistant Priority Bacterial Pathogen Acinetobacter baumannii in Saudi Arabia. Life, 15(7), 1094. https://doi.org/10.3390/life15071094