

Redefining Systemic Sclerosis Classification: Anti-Topoisomerase Antibody as a Superior Predictor of Interstitial Lung Disease and Skin Progression Compared to Limited Cutaneous Systemic Sclerosis Subset
 , , ,
, , ,
Abstract
1. Introduction
2. Methods
2.1. Operational Definitions
2.2. Statistical Analysis
3. Results
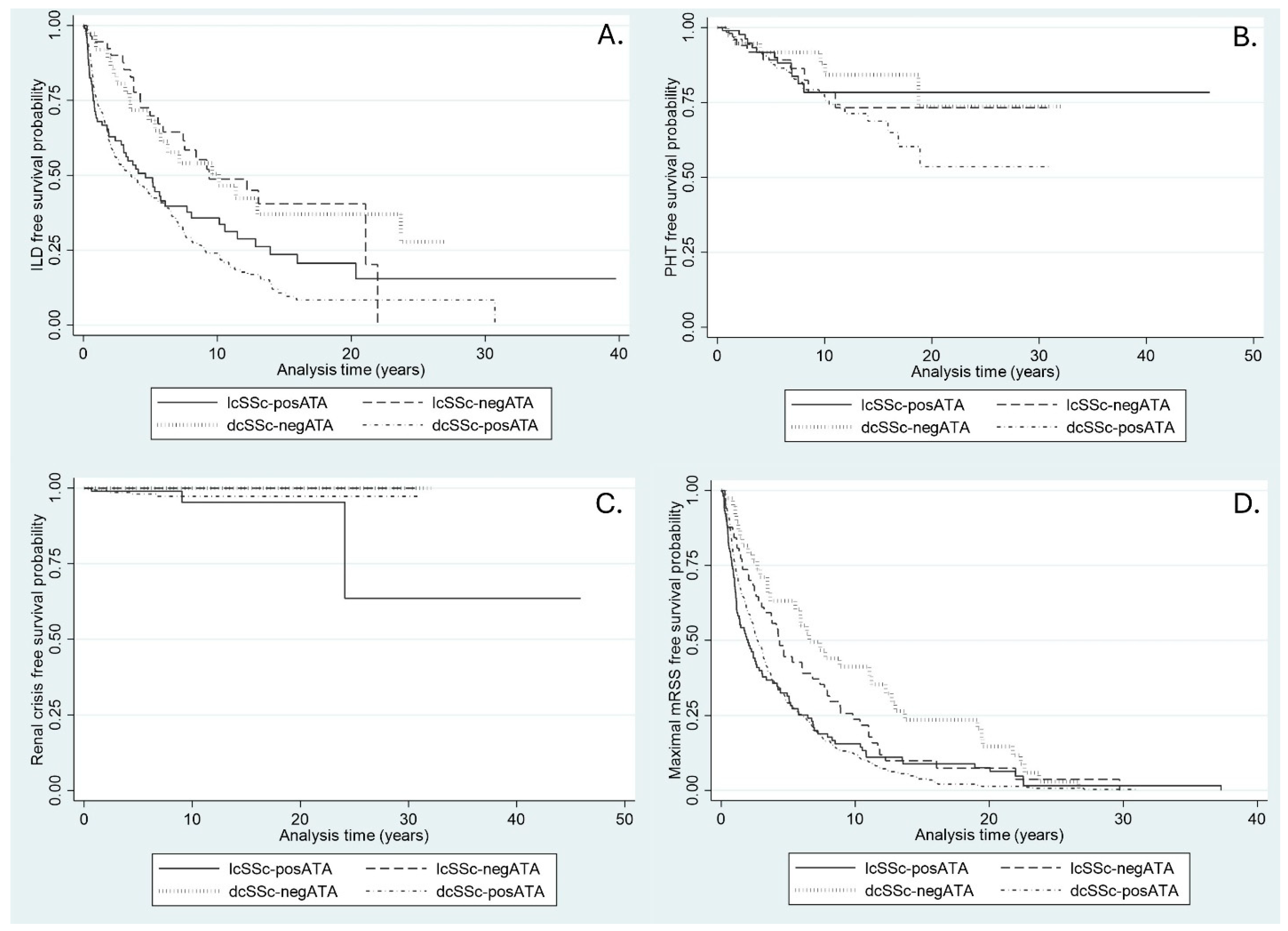
3.1. Incidence of ILD
3.2. Incidence of PHT
3.3. Incidence of Renal Crisis
3.4. Time to Maximal mRSS
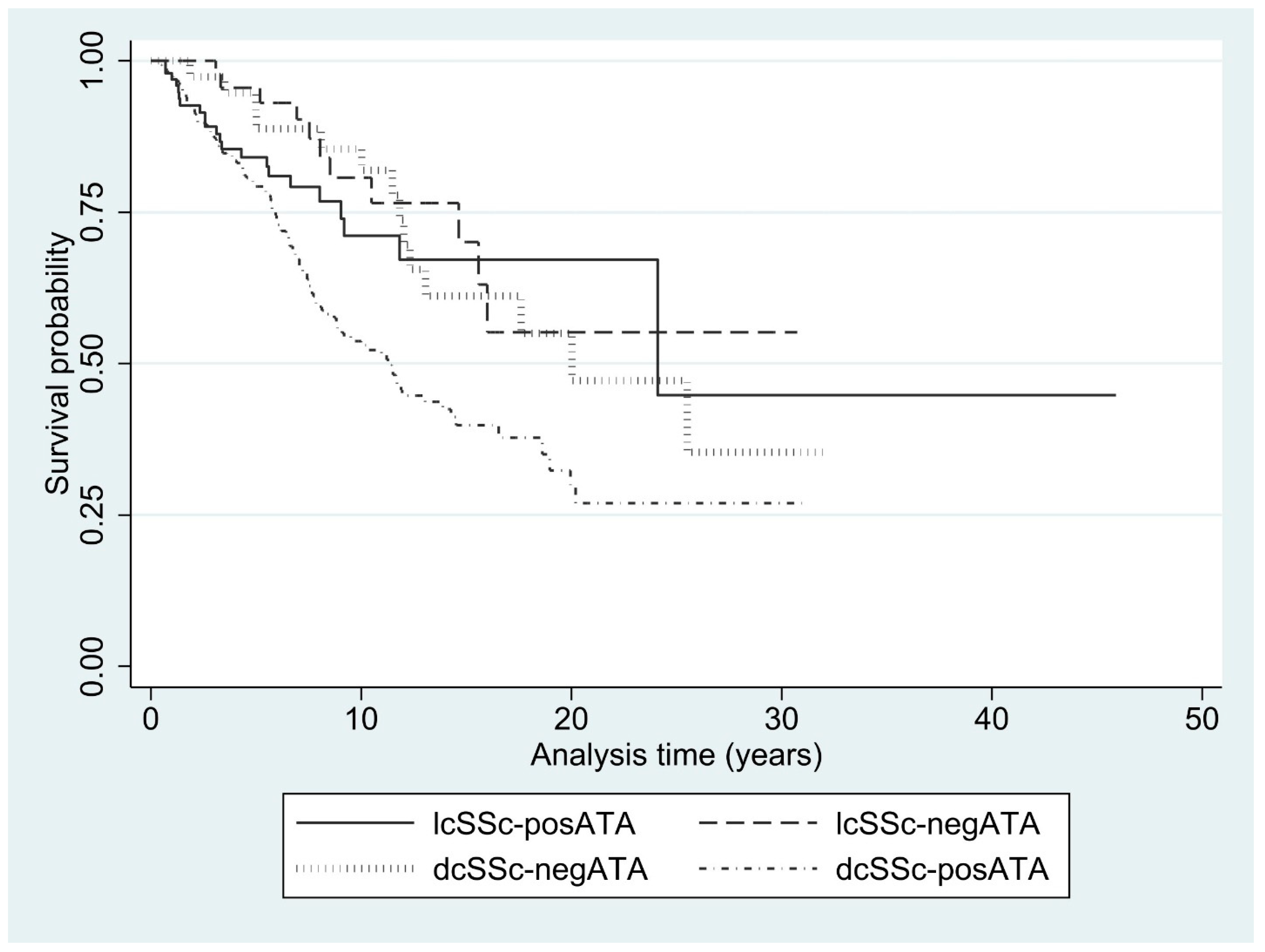
3.5. Mortality Rate
4. Discussion
5. Conclusions
6. Key Messages
- ATA is a common antibody in the dcSSc subset but can also be found in lcSSc.
- Patients with lcSSc and positive for ATA had a similar ILD risk to those with dcSSc but experienced faster pulmonary worsening.
- ATA is positive in patients with early SSc, and skin outcomes may not be favorable in this SSc subset.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Al-Dhaher, F.F.; Pope, J.E.; Ouimet, J.M. Determinants of Morbidity and Mortality of Systemic Sclerosis in Canada. Semin. Arthritis Rheum. 2010, 39, 269–277. [Google Scholar] [CrossRef] [PubMed]
- LeRoy, E.C.; Black, C.; Fleischmajer, R.; Jablonska, S.; Krieg, T.; Medsger, T.A.; Rowell, N.; Wollheim, F. Scleroderma (Systemic Sclerosis): Classification, Subsets and Pathogenesis. J. Rheumatol. 1988, 15, 202–205. [Google Scholar] [PubMed]
- Kuwana, M.; Kaburaki, J.; Okano, Y.; Tojo, T.; Homma, M. Clinical and Prognostic Associations Based on Serum Antinuclear Antibodies in Japanese Patients with Systemic Sclerosis. Arthritis Rheum. 1994, 37, 75–83. [Google Scholar] [CrossRef]
- Kuwana, M. Circulating Anti-Nuclear Antibodies in Systemic Sclerosis: Utility in Diagnosis and Disease Subsetting. J. Nippon. Med. Sch. 2017, 84, 56–63. [Google Scholar] [CrossRef] [PubMed]
- van den Hoogen, F.; Khanna, D.; Fransen, J.; Johnson, S.R.; Baron, M.; Tyndall, A.; Matucci-Cerinic, M.; Naden, R.P.; Medsger, T.A., Jr.; Carreira, P.E.; et al. 2013 Classification Criteria for Systemic Sclerosis: An American College of Rheumatology/European League against Rheumatism Collaborative Initiative. Arthritis Rheum. 2013, 65, 2737–2747. [Google Scholar] [CrossRef]
- Foocharoen, C.; Suwannachat, P.; Netwijitpan, S.; Mahakkanukrauh, A.; Suwannaroj, S.; Nanagara, R. Scleroderma Research Group Clinical Differences between Thai Systemic Sclerosis Patients with Positive versus Negative Anti-Topoisomerase I. Int. J. Rheum. Dis. 2016, 19, 312–320. [Google Scholar] [CrossRef]
- Steen, V.D. Autoantibodies in Systemic Sclerosis. Semin. Arthritis Rheum. 2005, 35, 35–42. [Google Scholar] [CrossRef]
- Graf, S.W.; Hakendorf, P.; Lester, S.; Patterson, K.; Walker, J.G.; Smith, M.D.; Ahern, M.J.; Roberts-Thomson, P.J. South Australian Scleroderma Register: Autoantibodies as Predictive Biomarkers of Phenotype and Outcome. Int. J. Rheum. Dis. 2012, 15, 102–109. [Google Scholar] [CrossRef]
- Hashimoto, A.; Endo, H.; Kondo, H.; Hirohata, S. Clinical Features of 405 Japanese Patients with Systemic Sclerosis. Mod. Rheumatol. 2012, 22, 272–279. [Google Scholar] [CrossRef]
- Wang, J.; Assassi, S.; Guo, G.; Tu, W.; Wu, W.; Yang, L.; Xiao, R.; Zhao, Y.; Chu, H.; Liu, J.; et al. Clinical and Serological Features of Systemic Sclerosis in a Chinese Cohort. Clin. Rheumatol. 2013, 32, 617–621. [Google Scholar] [CrossRef]
- Poormoghim, H.; Moghadam, A.S.; Moradi-Lakeh, M.; Jafarzadeh, M.; Asadifar, B.; Ghelman, M.; Andalib, E. Systemic Sclerosis: Demographic, Clinical and Serological Features in 100 Iranian Patients. Rheumatol. Int. 2013, 33, 1943–1950. [Google Scholar] [CrossRef] [PubMed]
- Foocharoen, C.; Watcharenwong, P.; Netwijitpan, S.; Mahakkanukrauh, A.; Suwannaroj, S.; Nanagara, R. Relevance of Clinical and Autoantibody Profiles in Systemic Sclerosis among Thais. Int. J. Rheum. Dis. 2017, 20, 1572–1581. [Google Scholar] [CrossRef] [PubMed]
- Foocharoen, C.; Peansukwech, U.; Mahakkanukrauh, A.; Suwannaroj, S.; Pongkulkiat, P.; Khamphiw, P.; Nanagara, R. Clinical Characteristics and Outcomes of 566 Thais with Systemic Sclerosis: A Cohort Study. Int. J. Rheum. Dis. 2020, 23, 945–957. [Google Scholar] [CrossRef]
- Villalta, D.; Imbastaro, T.; Di Giovanni, S.; Lauriti, C.; Gabini, M.; Turi, M.C.; Bizzaro, N. Diagnostic Accuracy and Predictive Value of Extended Autoantibody Profile in Systemic Sclerosis. Autoimmun. Rev. 2012, 12, 114–120. [Google Scholar] [CrossRef]
- Kayser, C.; Fritzler, M.J. Autoantibodies in Systemic Sclerosis: Unanswered Questions. Front. Immunol. 2015, 6, 167. [Google Scholar] [CrossRef]
- Moinzadeh, P.; Nihtyanova, S.I.; Howell, K.; Ong, V.H.; Denton, C.P. Impact of Hallmark Autoantibody Reactivity on Early Diagnosis in Scleroderma. Clin. Rev. Allergy Immunol. 2012, 43, 249–255. [Google Scholar] [CrossRef]
- Srivastava, N.; Hudson, M.; Tatibouet, S.; Wang, M.; Baron, M.; Fritzler, M.J. Canadian Scleroderma Research Group (CSRG) Thinking Outside the Box-The Associations with Cutaneous Involvement and Autoantibody Status in Systemic Sclerosis Are Not Always What We Expect. Semin. Arthritis Rheum. 2015, 45, 184–189. [Google Scholar] [CrossRef]
- Stochmal, A.; Czuwara, J.; Trojanowska, M.; Rudnicka, L. Antinuclear Antibodies in Systemic Sclerosis: An Update. Clin. Rev. Allergy Immunol. 2020, 58, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Korn, J.H.; Mayes, M.; Matucci Cerinic, M.; Rainisio, M.; Pope, J.; Hachulla, E.; Rich, E.; Carpentier, P.; Molitor, J.; Seibold, J.R.; et al. Digital Ulcers in Systemic Sclerosis: Prevention by Treatment with Bosentan, an Oral Endothelin Receptor Antagonist. Arthritis Rheum. 2004, 50, 3985–3993. [Google Scholar] [CrossRef]
- Young, A.; Namas, R.; Dodge, C.; Khanna, D. Hand Impairment in Systemic Sclerosis: Various Manifestations and Currently Available Treatment. Curr. Treatm Opt. Rheumatol. 2016, 2, 252–269. [Google Scholar] [CrossRef]
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic Definitions and Updated Clinical Classification of Pulmonary Hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef]
- Savarino, E.; Furnari, M.; de Bortoli, N.; Martinucci, I.; Bodini, G.; Ghio, M.; Savarino, V. Gastrointestinal Involvement in Systemic Sclerosis. Presse Med. 2014, 43, e279–e291. [Google Scholar] [CrossRef]
- Wong, C.J. Involuntary Weight Loss. Med. Clin. North. Am. 2014, 98, 625–643. [Google Scholar] [CrossRef]
- Sujau, I.; Ng, C.T.; Sthaneshwar, P.; Sockalingam, S.; Cheah, T.E.; Yahya, F.; Jasmin, R. Clinical and Autoantibody Profile in Systemic Sclerosis: Baseline Characteristics from a West Malaysian Cohort. Int. J. Rheum. Dis. 2015, 18, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Roberts-Thomson, P.J.; Jones, M.; Hakendorf, P.; Kencana Dharmapatni, A.A.; Walker, J.G.; MacFarlane, J.G.; Smith, M.D.; Ahern, M.J. Scleroderma in South Australia: Epidemiological Observations of Possible Pathogenic Significance. Intern. Med. J. 2001, 31, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Englert, H.; Small-McMahon, J.; Davis, K.; O’Connor, H.; Chambers, P.; Brooks, P. Systemic Sclerosis Prevalence and Mortality in Sydney 1974-88. Aust. N. Z. J. Med. 1999, 29, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Tamaki, T.; Mori, S.; Takehara, K. Epidemiological Study of Patients with Systemic Sclerosis in Tokyo. Arch. Dermatol. Res. 1991, 283, 366–371. [Google Scholar] [CrossRef]
- Geirsson, A.J.; Steinsson, K.; Guthmundsson, S.; Sigurthsson, V. Systemic Sclerosis in Iceland. A Nationwide Epidemiological Study. Ann. Rheum. Dis. 1994, 53, 502–505. [Google Scholar] [CrossRef]
- Ferri, C.; Valentini, G.; Cozzi, F.; Sebastiani, M.; Michelassi, C.; La Montagna, G.; Bullo, A.; Cazzato, M.; Tirri, E.; Storino, F.; et al. Systemic Sclerosis: Demographic, Clinical, and Serologic Features and Survival in 1,012 Italian Patients. Medicine 2002, 81, 139–153. [Google Scholar] [CrossRef]
- Tyndall, A.; Mueller-Ladner, U.; Matucci-Cerinic, M. Systemic Sclerosis in Europe: First Report from the EULAR Scleroderma Trials And Research (EUSTAR) Group Database. Ann. Rheum. Dis. 2005, 64, 1107. [Google Scholar] [CrossRef]
- Delisle, V.C.; Hudson, M.; Baron, M.; Thombs, B.D.; The Canadian Scleroderma Research Group A. Sex and Time to Diagnosis in Systemic Sclerosis: An Updated Analysis of 1,129 Patients from the Canadian Scleroderma Research Group Registry. Clin. Exp. Rheumatol. 2014, 32, S-10-14. [Google Scholar] [PubMed]
- Reveille, J.D.; Fischbach, M.; McNearney, T.; Friedman, A.W.; Aguilar, M.B.; Lisse, J.; Fritzler, M.J.; Ahn, C.; Arnett, F.C. GENISOS Study Group Systemic Sclerosis in 3 US Ethnic Groups: A Comparison of Clinical, Sociodemographic, Serologic, and Immunogenetic Determinants. Semin. Arthritis Rheum. 2001, 30, 332–346. [Google Scholar] [CrossRef] [PubMed]
- Perera, A.; Fertig, N.; Lucas, M.; Rodriguez-Reyna, T.S.; Hu, P.; Steen, V.D.; Medsger, T.A. Clinical Subsets, Skin Thickness Progression Rate, and Serum Antibody Levels in Systemic Sclerosis Patients with Anti-Topoisomerase I Antibody. Arthritis Rheum. 2007, 56, 2740–2746. [Google Scholar] [CrossRef]
- Martín-López, M.; Carreira, P.E. The Impact of Progressive Pulmonary Fibrosis in Systemic Sclerosis-Associated Interstitial Lung Disease. J. Clin. Med. 2023, 12, 6680. [Google Scholar] [CrossRef]
- Liakouli, V.; Ciancio, A.; Del Galdo, F.; Giacomelli, R.; Ciccia, F. Systemic Sclerosis Interstitial Lung Disease: Unmet Needs and Potential Solutions. Nat. Rev. Rheumatol. 2024, 20, 21–32. [Google Scholar] [CrossRef]
- Nihtyanova, S.I.; Denton, C.P. Pathogenesis of Systemic Sclerosis Associated Interstitial Lung Disease. J. Scleroderma Relat. Disord. 2020, 5, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Penn, H.; Howie, A.J.; Kingdon, E.J.; Bunn, C.C.; Stratton, R.J.; Black, C.M.; Burns, A.; Denton, C.P. Scleroderma Renal Crisis: Patient Characteristics and Long-Term Outcomes. QJM 2007, 100, 485–494. [Google Scholar] [CrossRef]
- Foocharoen, C.; Mahakkanukrauh, A.; Suwannaroj, S.; Nanagara, R. Prevalence and Clinical Features of Acute Kidney Injury in Thai Systemic Sclerosis Patients. KKU Med. J. 2016, 2, 29–35. [Google Scholar]
- Teixeira, L.; Mouthon, L.; Mahr, A.; Berezné, A.; Agard, C.; Mehrenberger, M.; Noël, L.-H.; Trolliet, P.; Frances, C.; Cabane, J.; et al. Mortality and Risk Factors of Scleroderma Renal Crisis: A French Retrospective Study of 50 Patients. Ann. Rheum. Dis. 2008, 67, 110–116. [Google Scholar] [CrossRef]
- Domsic, R.T.; Rodriguez-Reyna, T.; Lucas, M.; Fertig, N.; Medsger, T.A., Jr. Skin Thickness Progression Rate: A Predictor of Mortality and Early Internal Organ Involvement in Diffuse Scleroderma. Ann. Rheum. Dis. 2011, 70, 104–109. [Google Scholar] [CrossRef]
- Nguyen, B.; Assassi, S.; Arnett, F.C.; Mayes, M.D. Association of RNA Polymerase III Antibodies with Scleroderma Renal Crisis. J. Rheumatol. 2010, 37, 1068. [Google Scholar] [CrossRef] [PubMed]
- Foocharoen, C.; Tonsawan, P.; Pongkulkiat, P.; Anutrakulchai, S.; Mahakkanukrauh, A.; Suwannaroj, S. Management Review of Scleroderma Renal Crisis: An Update with Practical Pointers. Mod. Rheumatol. 2023, 33, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Steen, V.D. Scleroderma Renal Crisis. Rheum. Dis. Clin. North. Am. 2003, 29, 315–333. [Google Scholar] [CrossRef] [PubMed]
- Dolnikov, K.; Milo, G.; Assady, S.; Dragu, R.; Braun-Moscovici, Y.; Balbir-Gurman, A. Scleroderma Renal Crisis as an Early Presentation of Systemic Sclerosis. Isr. Med. Assoc. J. 2020, 11, 722–723. [Google Scholar]
- Muangchan, C.; Canadian Scleroderma Research Group; Baron, M.; Pope, J. The 15% Rule in Scleroderma: The Frequency of Severe Organ Complications in Systemic Sclerosis. A Systematic Review. J. Rheumatol. 2013, 40, 1545–1556. [Google Scholar] [CrossRef]
- Elhai, M.; Meune, C.; Boubaya, M.; Avouac, J.; Hachulla, E.; Balbir-Gurman, A.; Riemekasten, G.; Airò, P.; Joven, B.; Vettori, S.; et al. Mapping and Predicting Mortality from Systemic Sclerosis. Ann. Rheum. Dis. 2017, 76, 1897–1905. [Google Scholar] [CrossRef]
- Steen, V.D.; Medsger, T.A. Changes in Causes of Death in Systemic Sclerosis, 1972-2002. Ann. Rheum. Dis. 2007, 66, 940–944. [Google Scholar] [CrossRef]
- Rubio-Rivas, M.; Royo, C.; Simeón, C.P.; Corbella, X.; Fonollosa, V. Mortality and Survival in Systemic Sclerosis: Systematic Review and Meta-Analysis. Semin. Arthritis Rheum. 2014, 44, 208–219. [Google Scholar] [CrossRef]
- Raghu, G.; Montesi, S.B.; Silver, R.M.; Hossain, T.; Macrea, M.; Herman, D.; Barnes, H.; Adegunsoye, A.; Azuma, A.; Chung, L.; et al. Treatment of Systemic Sclerosis-Associated Interstitial Lung Disease: Evidence-Based Recommendations. An Official American Thoracic Society Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2024, 209, 137–152. [Google Scholar] [CrossRef]
- Foocharoen, C.; Pussadhamma, B.; Mahakkanukrauh, A.; Suwannaroj, S.; Nanagara, R. Asymptomatic Cardiac Involvement in Thai Systemic Sclerosis: Prevalence and Clinical Correlations with Non-Cardiac Manifestations (Preliminary Report). Rheumatology 2015, 54, 1616–1621. [Google Scholar] [CrossRef]
- Pussadhamma, B.; Mahakkanukrauh, A.; Suwannaroj, S.; Nanagara, R.; Foocharoen, C. Clinical Outcomes of Asymptomatic Cardiac Involvement in Systemic Sclerosis Patients After a 2-Year Follow-Up (Extended Study). Am. J. Med. Sci. 2021, 362, 570–577. [Google Scholar] [CrossRef]
- Tipparot, T.; Foocharoen, C.; Mahakkanukrauh, A.; Suwannaroj, S.; Nanagara, R.; Pussadhamma, B.; Chaosuwannakit, N. Clinical and Laboratory Predictions of Myocardial Inflammation as Detected by Cardiac Magnetic Resonance Imaging in Patients with Systemic Sclerosis: A Pilot Study. Int. J. Rheum. Dis. 2019, 22, 2125–2133. [Google Scholar] [CrossRef] [PubMed]
- Mahakkanukrauh, A.; Foocharoen, C.; Chaosuwannakit, N.; Suwannaroj, S.; Pongkulkiat, P.; Onchan, T.; Pussadhamma, B. Outcomes of Myocarditis in Systemic Sclerosis: A 3-Year Follow-Up. Rheumatol. Immunol. Res. 2024, 5, 117–125. [Google Scholar] [CrossRef]
- Pussadhamma, B.; Tipparot, T.; Chaosuwannakit, N.; Mahakkanukrauh, A.; Suwannaroj, S.; Nanagara, R.; Foocharoen, C. Clinical Outcomes of Myocarditis after Moderate-Dose Steroid Therapy in Systemic Sclerosis: A Pilot Study. Int. J. Rheumatol. 2020, 2020, 8884442. [Google Scholar] [CrossRef] [PubMed]
- Werakiat, J.; Pussadhamma, B.; Mahakkanukrauh, A.; Suwannaroj, S.; Foocharoen, C. Clinical Courses and Predictors of Left Ventricular Systolic Dysfunction in Systemic Sclerosis: A Cohort Study. Rheumatol. Immunol. Res. 2024, 5, 107–116. [Google Scholar] [CrossRef]
- Mehra, S.; Walker, J.; Patterson, K.; Fritzler, M.J. Autoantibodies in Systemic Sclerosis. Autoimmun. Rev. 2013, 12, 340–354. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, R.; Lucas, M.; Fertig, N.; Oddis, C.V.; Medsger, T.A. Anti-U3 RNP Autoantibodies in Systemic Sclerosis. Arthritis Rheum. 2009, 60, 1112–1118. [Google Scholar] [CrossRef]
- Tormey, V.J.; Bunn, C.C.; Denton, C.P.; Black, C.M. Anti-Fibrillarin Antibodies in Systemic Sclerosis. Rheumatology 2001, 40, 1157–1162. [Google Scholar] [CrossRef]
- Fertig, N.; Domsic, R.T.; Rodriguez-Reyna, T.; Kuwana, M.; Lucas, M.; Medsger, T.A.; Feghali-Bostwick, C.A. Anti-U11/U12 RNP Antibodies in Systemic Sclerosis: A New Serologic Marker Associated with Pulmonary Fibrosis. Arthritis Rheum. 2009, 61, 958–965. [Google Scholar] [CrossRef]
- Ulanet, D.B.; Wigley, F.M.; Gelber, A.C.; Rosen, A. Autoantibodies against B23, a Nucleolar Phosphoprotein, Occur in Scleroderma and Are Associated with Pulmonary Hypertension. Arthritis Rheum. 2003, 49, 85–92. [Google Scholar] [CrossRef]
- Rozman, B.; Cucnik, S.; Sodin-Semrl, S.; Czirják, L.; Varjú, C.; Distler, O.; Huscher, D.; Aringer, M.; Steiner, G.; Matucci-Cerinic, M.; et al. Prevalence and Clinical Associations of Anti-Ku Antibodies in Patients with Systemic Sclerosis: A European EUSTAR-Initiated Multi-Centre Case-Control Study. Ann. Rheum. Dis. 2008, 67, 1282–1286. [Google Scholar] [CrossRef] [PubMed]
- Mulalin, K.; Mahakkanukrauh, A.; Suwannaroj, S.; Pongkulkiat, P.; Onchan, T.; Kasa, S.; Foocharoen, C. Levels of Anti-Topoisomerase I Antibody Correlated with Short Onset of Cardiopulmonary Involvement in Thai Systemic Sclerosis Patients. Sci. Rep. 2024, 14, 10354. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Data | Overall N = 522 | lcSSc-negATA N = 58 | lcSSc-posATA N = 104 | dcSSc-negATA N = 39 | dcSSc-posATA N = 321 | p-Value Comparison Between dcSSc-negATA and lcSSc-posATA | p-Value Comparison Between dcSSc-posATA and lcSSc-posATA |
|---|---|---|---|---|---|---|---|
| Age at onset (years); mean ± SD | 52.2 ± 12.2 | 51.2 ± 11.4 | 54.2 ± 12.4 | 47.0 ± 13.6 | 52.3 ± 11.9 | 0.003 * | 0.16 |
| Age at last visit (years); mean ± SD | 59.7 ± 11.0 | 59.8 ± 10.7 | 61.5 ± 10.9 | 59.5 ± 12.0 | 59.1 ± 10.9 | 0.35 | 0.50 |
| Female sex; N (%) | 319 (61.1) | 45 (77.6) | 73 (70.2) | 27 (69.2) | 174 (54.2) | 0.91 | 0.004 * |
| Disease duration (years); mean ± SD | 7.5 ± 6.3 | 8.6 ± 6.7 | 7.2 ± 6.9 | 12.5 ± 8.2 | 6.8 ± 5.4 | <0.001 * | 0.99 |
| BMI (kg/m2); mean ± SD | 20.7 ± 3.8 | 21.3 ± 3.5 | 21.5 ± 3.7 | 19.3 ± 4.4 | 20.2 ± 3.8 | 0.95 | 0.14 |
| SSc clinical features at the last follow-up | |||||||
| Digital gangrene; N (%) | 14 (2.7) | 1 (1.72) | 0 (0) | 0 (0) | 13 (4.1) | NA | 0.04 * |
| Telangiectasia; N (%) | 187 (35.8) | 14 (24.1) | 28 (26.9) | 13 (33.3) | 132 (41.1) | 0.45 | 0.01 * |
| Calcinosis cutis; N (%) | 40 (7.67) | 4 (6.9) | 3 (2.9) | 2 (5.1) | 31 (9.7) | 0.52 | 0.03 * |
| Salt and pepper skin; N (%) | 232 (44.4) | 13 (22.4) | 30 (28.9) | 12 (30.8) | 177 (55.1) | 0.82 | <0.001 * |
| Tendon friction rub; N (%) | 71 (13.6) | 2 (3.5) | 7 (6.7) | 4 (10.3) | 58 (18.1) | 0.48 | 0.01 * |
| Hand deformities; N (%) | 199 (38.1) | 6 (10.3) | 22 (21.2) | 9 (23.1) | 162 (50.5) | 0.80 | <0.001 * |
| Muscle weakness; N (%) | 26 (5.0) | 2 (3.5) | 6 (5.8) | 1 (2.6) | 17 (5.3) | 0.43 | 0.85 |
| Esophageal involvement; N (%) | 220 (42.2) | 19 (32.8) | 40 (38.5) | 16 (41.0) | 145 (45.2) | 0.78 | 0.23 |
| Stomach involvement; N (%) | 87 (16.7) | 10 (17.2) | 14 (13.5) | 3 (7.7) | 60 (18.7) | 0.34 | 0.22 |
| Intestine involvement; N (%) | 89 (17.1) | 10 (17.2) | 15 (14.4) | 9 (23.1) | 55 (17.3) | 0.22 | 0.52 |
| mRSS (points); median (IQR) | 3 (0–10) | 0 (0–2) | 2 (0–6) | 2 (0–4) | 6 (2–16) | 0.83 | <0.001 * |
| Immunosuppressants | |||||||
| Cyclophosphamide; N (%) | 61 (11.7) | 2 (3.5) | 7 (6.7) | 0 (0) | 52 (16.2) | 0.01 * | 0.02 * |
| Mycophenolate; N (%) | 77 (14.8) | 4 (6.9) | 24 (23.1) | 4 (10.3) | 45 (14.0) | 0.09 | 0.03 * |
| Methotrexate; N (%) | 27 (5.2) | 2 (3.5) | 7 (6.7) | 1 (2.6) | 17 (5.3) | 0.33 | 0.58 |
| Interested events | |||||||
| Maximum mRSS (points); mean ± SD | 15.6 ± 11.2 | 6.0 ± 5.9 | 9.5 ± 7.4 | 12.2 ± 9.8 | 19.7 ± 11.1 | 0.95 | <0.001 * |
| Duration of disease at maximum mRSS (years); median (IQR) | 2.7 (1.0–6.7) | 4.2 (1.5–8.9) | 1.8 (0.8–5.5) | 6.4 (2.4–13.0) | 2.5 (0.9–5.7) | <0.001 * | 0.17 |
| ILD; N (%) | 315 (60.3) | 25 (43.1) | 61 (58.6) | 20 (51.3) | 209 (65.1) | 0.43 | 0.24 |
| Duration of disease at onset of ILD (years); median (IQR) | 1.9 (0.7–5.6) | 4.2 (1.1–7.6) | 1.0 (0.4–5.2) | 4.8 (2.1–11.3) | 1.8 (0.7–5.1) | 0.001 * | 0.21 |
| PHT; N (%) | 78 of 521 (15.0) | 9 (15.5) | 12 (11.5) | 6 (15.4) | 51 of 320 (15.9) | 0.54 | 0.27 |
| Duration of disease at onset of PHT (years); median (IQR) | 4.08 (1.7–8.0) | 4.3 (2.8–8.5) | 3.0 (1.7–6.9) | 4.9 (1.6–10.0) | 4.2 (1.7–8.0) | 0.08 | 0.30 |
| Renal crisis; N (%) | 9 of 522 (1.7) | 0 (0) | 3 (2.9) | 0 (0) | 8 (1.9) | 0.28 | 0.62 |
| Duration of disease at onset of renal crisis (years); median (IQR) | 2.2 (1.0–9.0) | - | 16.6 (9.0–24.1) | - | 2.1 (1.0–2.2) | NA | 0.05 |
| Data | Overall N = 522 | lcSSc-negATA N = 58 | lcSSc-posATA N = 104 | dcSSc-negATA N = 39 | dcSSc-posATA N = 321 | p-Value Comparison Between dcSSc-negATA and lcSSc-posATA | p-Value Comparison Between dcSSc-posATA and lcSSc-posATA |
|---|---|---|---|---|---|---|---|
| ILD development | |||||||
| Incidence rate of ILD (per 100 persons-year) | 12.8 (11.5–14.4) | 6.4 (4.3–9.6) | 12.4 (9.6–16.0) | 6.3 (4.1–9.7) | 16.7 (14.5–19.1) | ||
| Median survival time to ILD development (year) | 4.9 (1.3–12.3) | 9.3 (5.9–NA) | 4.6 (1.0–2.8) | 10.1 (4.9–23.7) | 3.6 (2.4–4.9) | ||
| ILD-free survival (%) | 0.03 * | 0.22 | |||||
| At 2 years | 66.5 | 92.3 | 62.9 | 89.2 | 60.2 | ||
| At 5 years | 49.6 | 69.8 | 49.2 | 68.3 | 43.9 | ||
| At 10 years | 31.3 | 48.7 | 35.7 | 50.5 | 24.1 | ||
| At 15 years | 19.3 | 40.5 | 23.6 | 37.0 | 10.8 | ||
| At 20 years | 17.3 | 40.5 | 20.6 | 37.0 | 8.4 | ||
| PHT development | |||||||
| Incidence rate of PHT (per 100 persons-year) | 2.1 (1.7–2.7) | 2.0 (1.0–3.8) | 1.7 (0.9–3.0) | 1.3 (0.5–3.0) | 2.5 (1.9–3.2) | ||
| Median survival time to PHT development (year) | NA | NA | NA | NA | NA | ||
| PHT-free survival (%) | 0.68 | 0.29 | |||||
| At 2 years | 96.3 | 94.3 | 99.0 | 94.7 | 96.1 | ||
| At 5 years | 89.8 | 89.2 | 91.3 | 91.7 | 88.9 | ||
| At 10 years | 78.6 | 78.5 | 78.4 | 88.0 | 76.9 | ||
| At 15 years | 73.2 | 73.3 | 78.4 | 84.2 | 68.8 | ||
| At 20 years | 64.4 | 73.3 | 78.4 | 73.7 | 53.6 | ||
| Scleroderma renal crisis (SRC) development | |||||||
| Incidence rate of SRC (per 100 persons-year) | 0.2 (0.1–0.4) | NA | 0.4 (0.01–0.1) | NA | 0.02 (0.01–0.1) | ||
| Median survival time to SRC development (year) | NA | NA | NA | NA | NA | ||
| SRC-free survival (%) | 0.11 | 0.73 | |||||
| At 2 years | 99.4 | 100 | 98.9 | 100 | 99.3 | ||
| At 5 years | 98.6 | 100 | 98.9 | 100 | 98.0 | ||
| At 10 years | 97.6 | 100 | 95.3 | 100 | 97.3 | ||
| At 15 years | 97.6 | 100 | 95.3 | 100 | 97.3 | ||
| At 20 years | 97.6 | 100 | 95.3 | 100 | 97.3 | ||
| Maximum mRSS | |||||||
| Median survival time to maximum mRSS development (year) | 2.9 (1.1–6.8) | 4.3 (2.8–6.8) | 2.0 (1.2–2.8) | 6.6 (3.5–12.3) | 2.7 (2.4–3.1) | ||
| Maximum mRSS-free survival (%) | 0.002 * | 0.77 | |||||
| At 2 years | 60.4 | 71.8 | 49.2 | 81.6 | 59.4 | ||
| At 5 years | 33.9 | 44.5 | 31.5 | 63.2 | 29.2 | ||
| At 10 years | 16.3 | 23.7 | 15.6 | 41.2 | 12.2 | ||
| At 15 years | 6.9 | 9.9 | 8.9 | 23.5 | 3.8 | ||
| At 20 years | 4.1 | 7.4 | 7.6 | 14.7 | 1.4 |
| Landmark (Years) | RMST (95%CI) | RMST Difference Between Groups 1 and 2 | RMST Difference Between Groups 3 and 2 | RMST Difference Between Groups 4 and 2 | |||
|---|---|---|---|---|---|---|---|
| Group 1 lcSSc-negATA | Group 2 lcSSc-posATA | Group 3 dcSSc-negATA | Group 4 dcSSc-posATA | ||||
| ILD | |||||||
| 5 years | 4.1 (3.7 to 4.5) | 2.9 (2.5 to 3.3) | 3.8 (3.3 to 4.4) | 3.2 (3.0 to 3.4) | 1.2 (0.6 to 1.8) * | 0.9 (0.3 to 1.6) * | 0.3 (−1.1 to 0.7) |
| 10 years | 6.7 (5.7 to 7.7) | 4.5 (3.8 to 5.3) | 6.3 (5.1 to 7.5) | 4.8 (4.4 to 5.2) | 2.1 (0.9 to 3.4) * | 1.8 (0.4 to 3.2) * | 0.3 (−0.6 to 1.2) |
| 20 years | 9.2 (7.1 to 11.2) | 6.3 (4.9 to 7.7) | 8.9 (6.6 to 11.3) | 6.3 (5.5 to 7.0) | 2.8 (0.4 to 5.2) * | 2.6 (−0.1 to 5.3) | −0.1 (−1.7 to 1.6) |
| 40 years | 10.4 (7.3 to 13.5) | 7.7 (5.4 to 10.1) | 10.8 (7.0 to 14.0) | 7.0 (5.8 to 8.2) | 2.6 (−1.3 to 6.5) | 3.0 (−1.5 to 7.6) | −0.8 (−3.4 to 1.9) |
| PHT | |||||||
| 5 years | 4.6 (4.3 to 5.0) | 4.8 (4.6 to 4.9) | 4.7 (4.4 to 5.1) | 4.7 (4.6 to 4.8) | −0.1 (−0.5 to 0.2) | −0.1 (−0.5 to 0.3) | −0.5 (−0.2 to 0.1) |
| 10 years | 8.6 (7.7 to 9.5) | 9.1 (8.6 to 9.6) | 8.8 (7.8 to 9.8) | 8.7 (8.4 to 9.1) | −0.4 (−1.5 to 0.6) | −0.2 (−1.3 to 0.8) | −0.3 (−1.0 to 0.3) |
| 20 years | 15.4 (12.9 to 17.9) | 16.6 (14.8 to 18.4) | 15.9 (13.2 to 18.6) | 14.9 (13.7 to 16.1) | −1.1 (−4.1 to 1.9) | −0.7 (−3.8 to 2.5) | −1.7 (−3.8 to 0.5) |
| 40 years | 26.0 (18.2 to 33.7) | 28.3 (21.5 to 35.2) | 26.8 (18.5 to 35.2) | 22.5 (18.0 to 21.0) | −2.4 (−11.7 to 7.0) | −1.5 (−11.6 to 8.6) | −5.8 (−13.1 to 1.5) |
| Renal crisis | |||||||
| 5 years | 5.0 (5.0 to 5.0) | 4.9 (4.8 to 5.0) | 5.0 (5.0 to 5.0) | 4.9 (4.9 to 5.0) | 0.1 (−0.04 to −0.2) | 0.1 (−0.04 to −0.2) | 0.02 (−0.1 to 0.2) |
| 10 years | 10.0 (10.0 to 10.0) | 9.7 (9.3 to 10.1) | 10.0 (10.0 to 10.0) | 9.8 (9.7 to 10.0) | 0.3 (−0.1 to 0.7) | 0.3 (−0.1 to 0.7) | 0.2 (−0.3 to 0.6) |
| 20 years | 20.0 (20.0 to 20.0) | 18.2 (16.3 to 20.0) | 20.0 (20.0 to 20.0) | 19.1 (18.3 to 20.0) | 1.8 (0.1 to 3.6) * | 1.8 (0.1 to 3.6) * | 1.0 (−1.0 to 2.9) |
| 40 years | 40.0 (40.0 to 40.0) | 30.7 (21.7 to 39.7) | 40.0 (40.0 to 40.0) | 35.6 (28.9 to 42.3) | 9.3 (0.4 to 18.2) * | 9.3 (0.4 to 18.2) * | 4.9 (−4.3 to 14.1) |
| Maximum mRSS | |||||||
| 5 years | 3.3 (2.8 to 3.7) | 2.7 (2.3 to 3.0) | 3.8 (3.3 to 4.4) | 2.8 (2.7 to 3.0) | 0.6 (0.1 to 1.2) * | 1.2 (0.6 to 1.8) * | 0.2 (−0.2 to 0.6) |
| 10 years | 4.8 (3.9 to 5.6) | 3.8 (3.2 to 4.4) | 6.0 (4.8 to 7.1) | 3.8 (3.4 to 4.1) | 1.0 (−0.1 to 2.0) | 2.2 (0.9 to 3.4) * | −0.03 (−0.7 to 0.7) |
| 20 years | 5.8 (4.5 to 7.1) | 4.8 (3.8 to 5.8) | 7.7 (5.9 to 9.5) | 4.2 (3.8 to 4.7) | 1.1 (−0.6 to 2.7) | 2.9 (0.8 to 5.0) * | −0.5 (−1.6 to 0.6) |
| 40 years | 6.2 (4.6 to 7.7) | 5.2 (3.9 to 6.6) | 8.3 (6.1 to 10.5) | 4.3 (3.8 to 4.8) | 0.9 (−1.1 to 3.0) | 3.1 (0.5 to 5.7) * | −0.9 (−2.3 to 0.5) |
| Data | Overall N = 171 | lcSSc-negATA N = 11 | lcSSc-posATA N = 22 | dcSSc-negATA N = 14 | dcSSc-posATA N = 124 |
|---|---|---|---|---|---|
| Mortality rate per 100-person-years (95%CI) | 4.3 (3.7-5.0) | 2.2 (1.2-3.9) | 2.9 (1.9-4.4) | 2.8 (1.7-4.8) | 5.7 (4.7-6.7) |
| 2-year survival (%) | 93.5 | 100 | 92.7 | 100 | 91.9 |
| 5-year survival (%) | 83.0 | 95.6 | 84.2 | 91.6 | 79.0 |
| 10-year survival (%) | 63.0 | 80.7 | 71.3 | 85.4 | 53.4 |
| 15-year survival (%) | 49.6 | 70.1 | 67.3 | 61.1 | 39.6 |
| 20-year survival (%) | 41.3 | 55.2 | 67.3 | 55.0 | 29.5 |
| Causes of death | |||||
| SSc related death; N (%) | 79 (46.2) | 1 (9.1) | 9 (40.1) | 9 (64.3) | 60 (48.4) |
| Nonspecific organ; N (%) | 28 (16.4) | 0 (0.0) | 1 (4.6) | 3 (21.3) | 24 (19.4) |
| ILD; N (%) | 11 (6.4) | 1 (9.1) | 1 (4.6) | 1 (7.2) | 8 (6.5) |
| Cardiac involvement; N (%) | 24 (14.0) | 0 (0.0) | 2 (9.1) | 5 (35.7) | 17 (13.7) |
| PHT; N (%) | 7 (4.1) | 0 (0.0) | 1 (4.6) | 0 (0.0) | 6 (4.8) |
| Renal crisis; N (%) | 8 (4.7) | 0 (0.0) | 3 (16.4) | 0 (0.0) | 5 (4.0) |
| Gastrointestinal; N (%) | 1 (0.6) | 0 (0.0) | 1 (4.6) | 0 (0.0) | 0 (0.0) |
| SSc non related death | 92 (53.8) | 10 (90.9) | 13 (59.1) | 5 (35.7) | 64 (51.6) |
| Cancer; N (%) | 2 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 2 (1.6) |
| Pneumonia; N (%) | 19 (11.1) | 0 (0.0) | 2 (8.7) | 1 (7.1) | 16 (12.9) |
| Sepsis; N (%) | 11 (6.4) | 2 (18.2) | 1 (4.4) | 1 (7.1) | 6 (5.7) |
| CAD; N (%) | 6 (3.5) | 1 (9.1) | 0 (0.0) | 0 (0.0) | 5 (4.0) |
| Natural death; N (%) | 1 (0.6) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (0.8) |
| CKD; N (%) | 2 (1.2) | 0 (0.0) | 1 (4.4) | 0 (0.0) | 1 (0.8) |
| Liver disease; N (%) | 3 (1.7) | 1 (9.1) | 0 (0.0) | 0 (0.0) | 2 (1.6) |
| ICH; N (%) | 2 (1.2) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 2 (1.6) |
| other cause; N (%) | 46 (26.9) | 6 (54.5) | 9 (39.1) | 3 (21.4) | 28 (22.6) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chaovanitkul, C.; Onchan, T.; Pongkulkiat, P.; Mahakkanukrauh, A.; Suwannaroj, S.; Foocharoen, C. Redefining Systemic Sclerosis Classification: Anti-Topoisomerase Antibody as a Superior Predictor of Interstitial Lung Disease and Skin Progression Compared to Limited Cutaneous Systemic Sclerosis Subset. Life 2025, 15, 1067. https://doi.org/10.3390/life15071067
Chaovanitkul C, Onchan T, Pongkulkiat P, Mahakkanukrauh A, Suwannaroj S, Foocharoen C. Redefining Systemic Sclerosis Classification: Anti-Topoisomerase Antibody as a Superior Predictor of Interstitial Lung Disease and Skin Progression Compared to Limited Cutaneous Systemic Sclerosis Subset. Life. 2025; 15(7):1067. https://doi.org/10.3390/life15071067
Chicago/Turabian StyleChaovanitkul, Chana, Tippawan Onchan, Patnarin Pongkulkiat, Ajanee Mahakkanukrauh, Siraphop Suwannaroj, and Chingching Foocharoen. 2025. "Redefining Systemic Sclerosis Classification: Anti-Topoisomerase Antibody as a Superior Predictor of Interstitial Lung Disease and Skin Progression Compared to Limited Cutaneous Systemic Sclerosis Subset" Life 15, no. 7: 1067. https://doi.org/10.3390/life15071067
APA StyleChaovanitkul, C., Onchan, T., Pongkulkiat, P., Mahakkanukrauh, A., Suwannaroj, S., & Foocharoen, C. (2025). Redefining Systemic Sclerosis Classification: Anti-Topoisomerase Antibody as a Superior Predictor of Interstitial Lung Disease and Skin Progression Compared to Limited Cutaneous Systemic Sclerosis Subset. Life, 15(7), 1067. https://doi.org/10.3390/life15071067