
RNA-Seq of an LPS-Induced Inflammation Model Reveals Transcriptional Profile Patterns of Inflammatory Processes

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal (Mouse) Preparation
2.2. Lipopolysaccharide (LPS)-Induced Inflammation Model
2.3. Library Preparation
2.4. Public Data Preparation
2.5. RNA-seq Preprocessing
2.6. Analysis of Inflammatory Regulation
3. Results
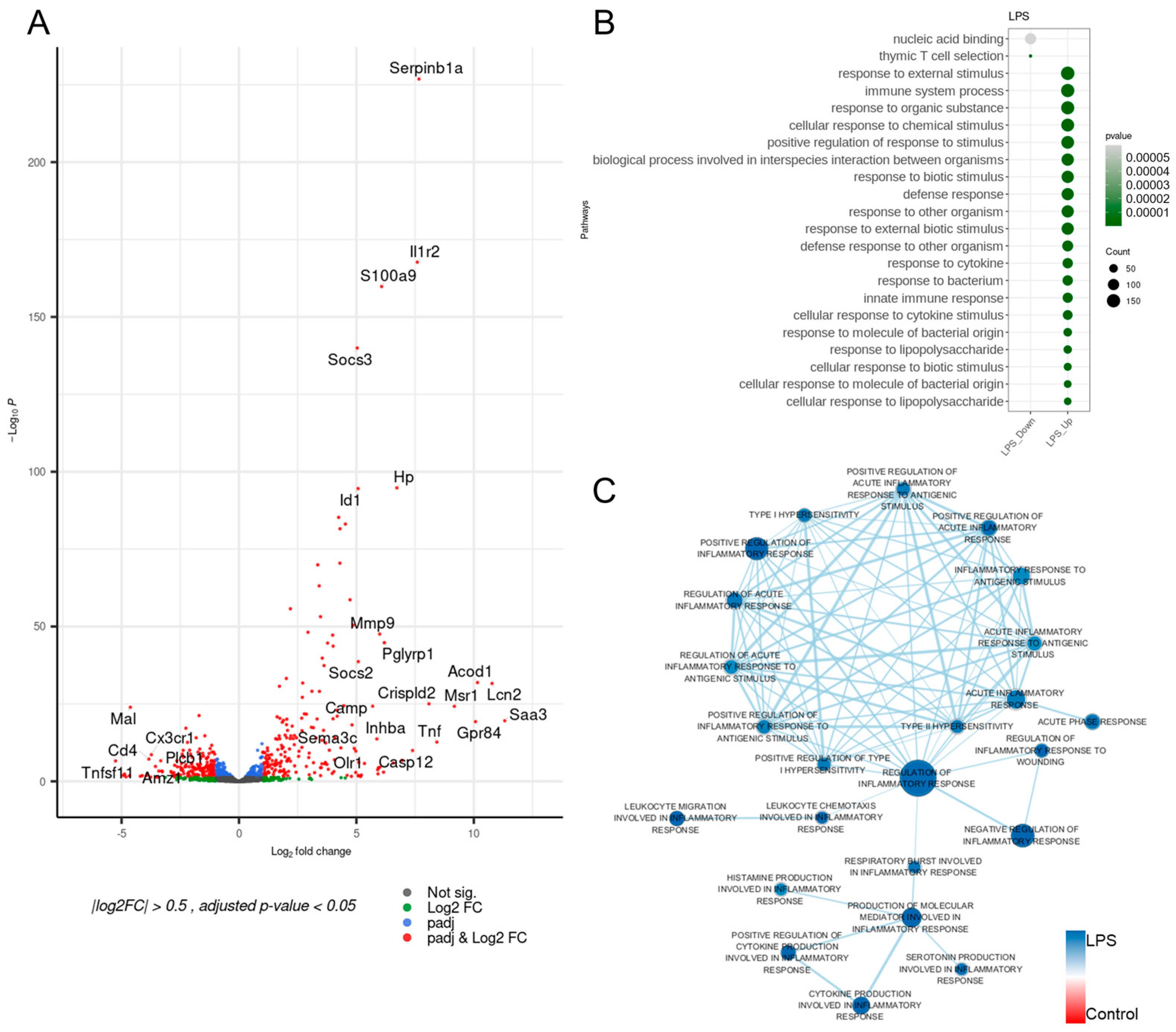
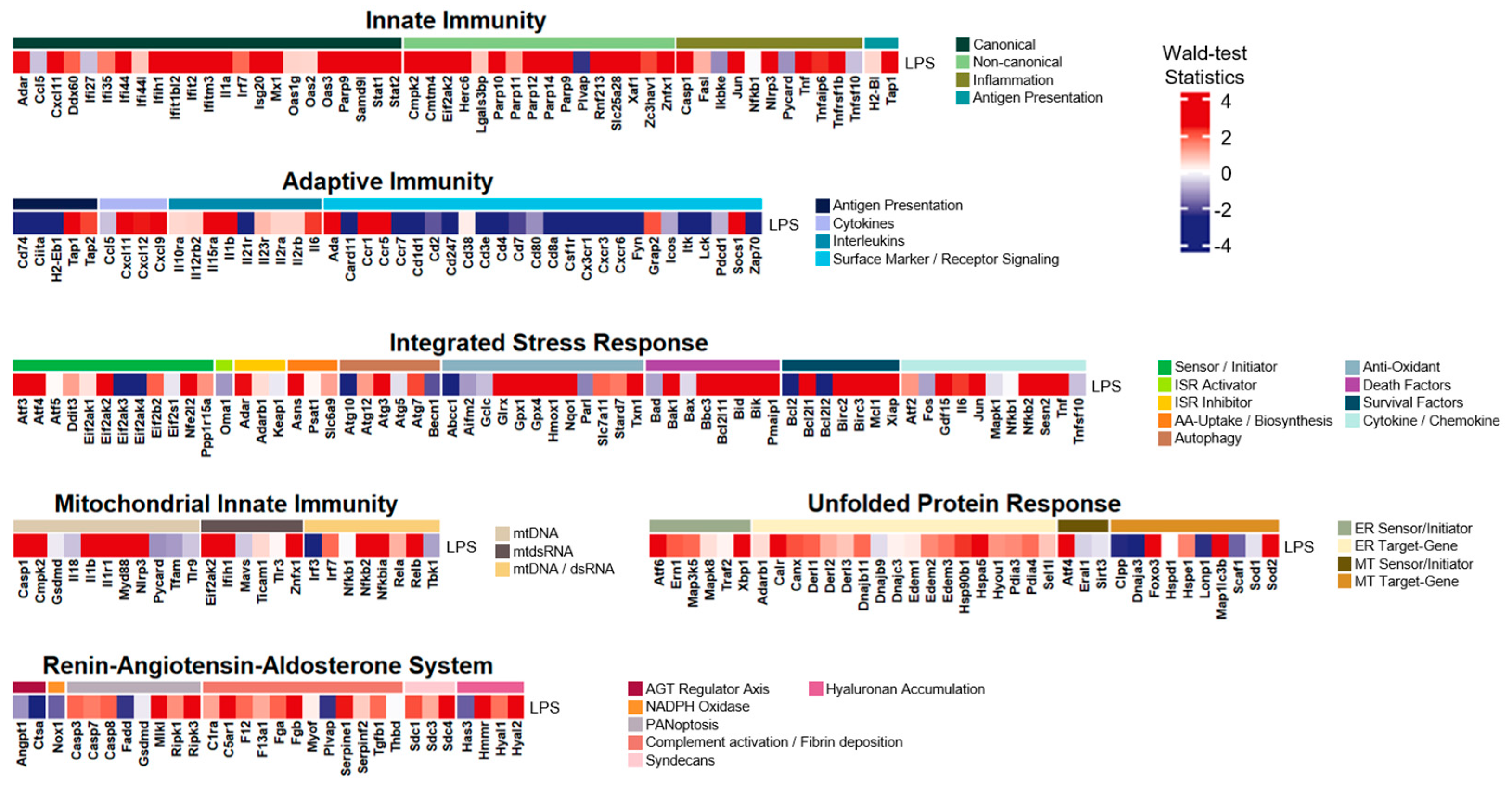
3.1. Inflammation-Associated Genes of the LPS-Induced Model Were Upregulated
3.2. The LPS-Induced Model Disclosed Considerable Upregulations in Innate Immunity
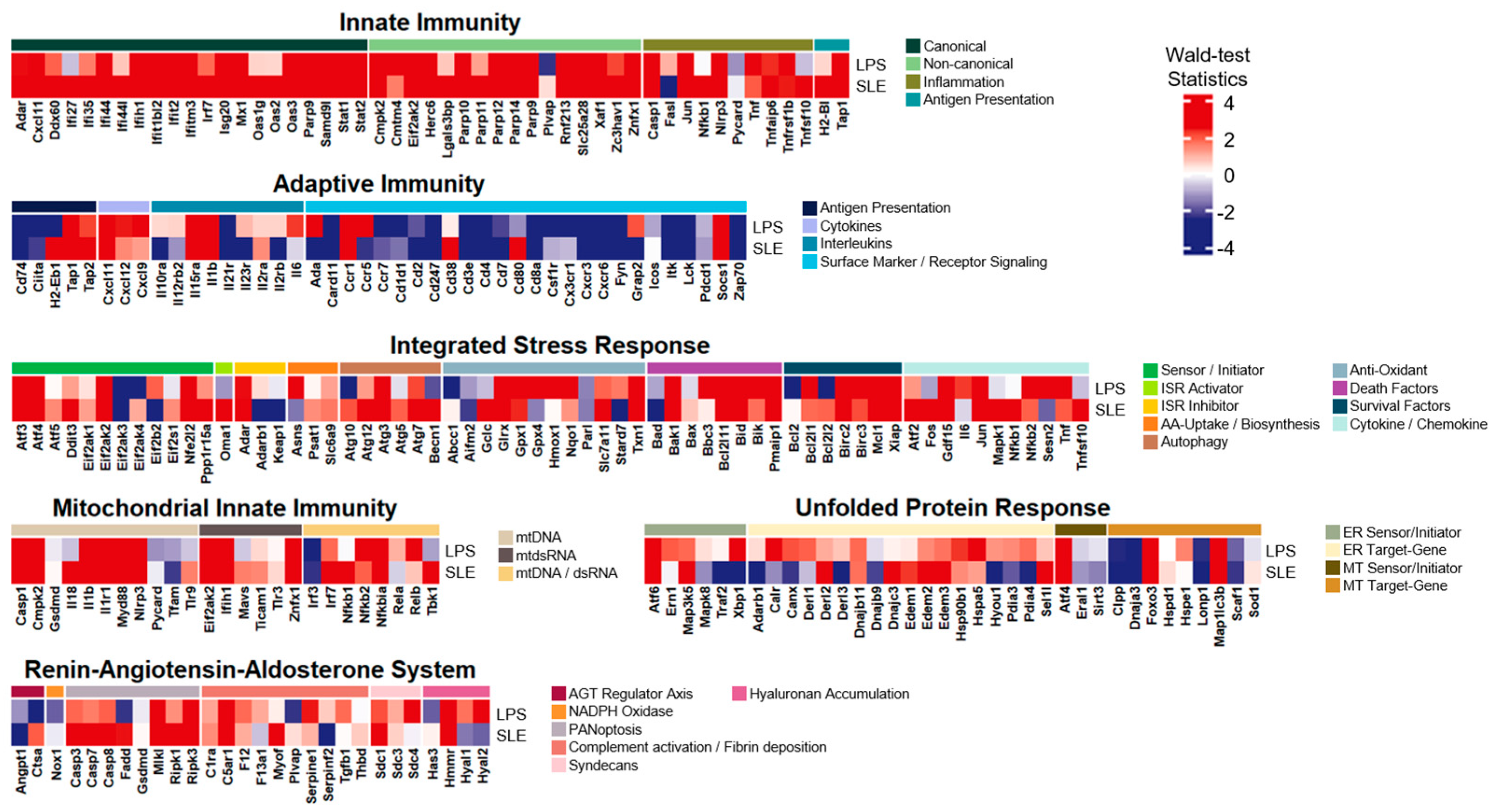
3.3. The LPS-Induced Model Exhibited Extraordinary Common Expression Patterns with SLE
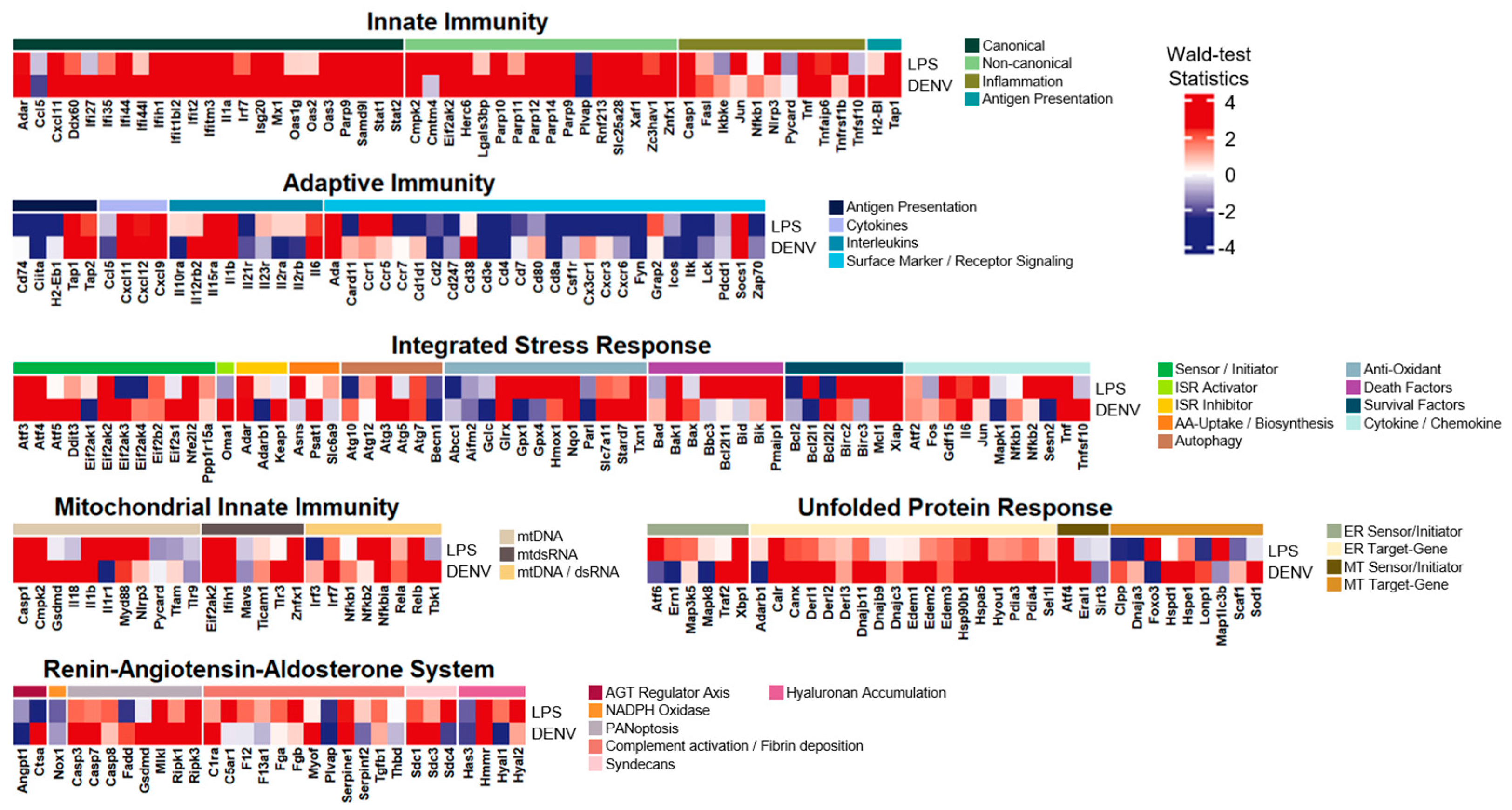
3.4. The LPS-Induced Model Revealed Exceptional Common Expression Patterns with Dengue Virus Infection
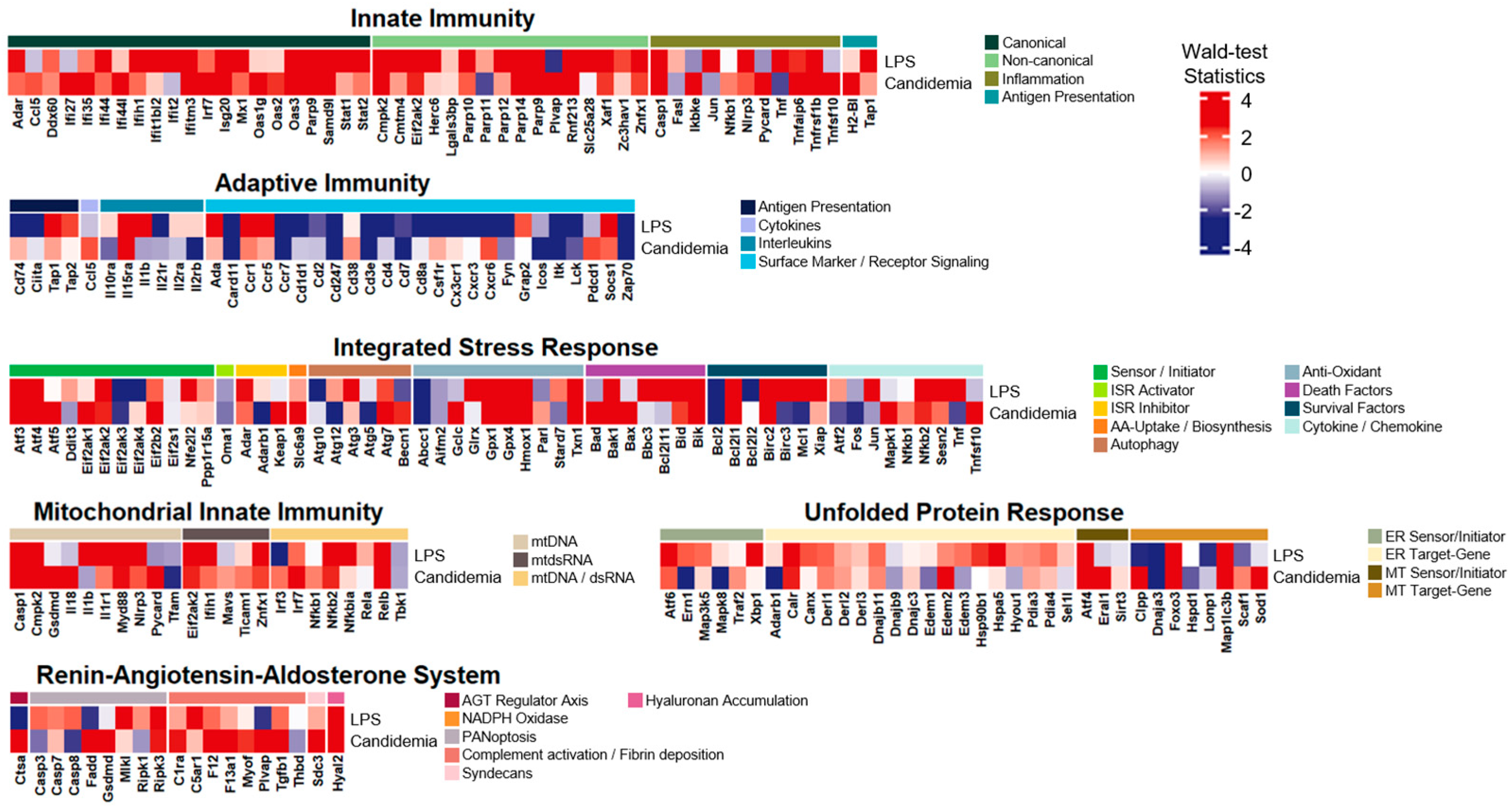
3.5. The LPS-Induced Model Disclosed Significant Common Expression Patterns with Candidemia Infection
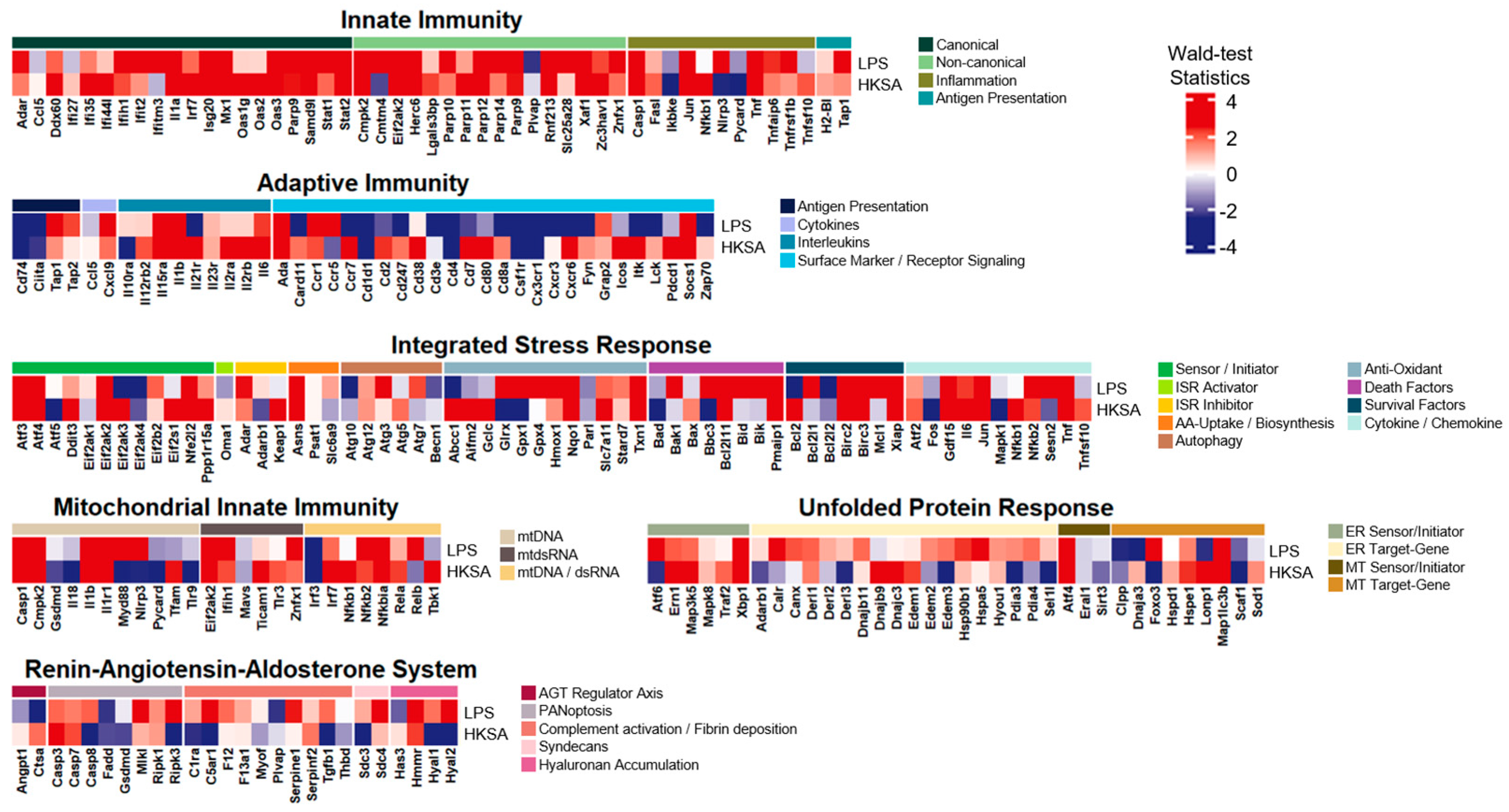
3.6. The LPS-Induced Model Divulged Considerable Common Expression Patterns with Exposure to Heat Killed S. aureus
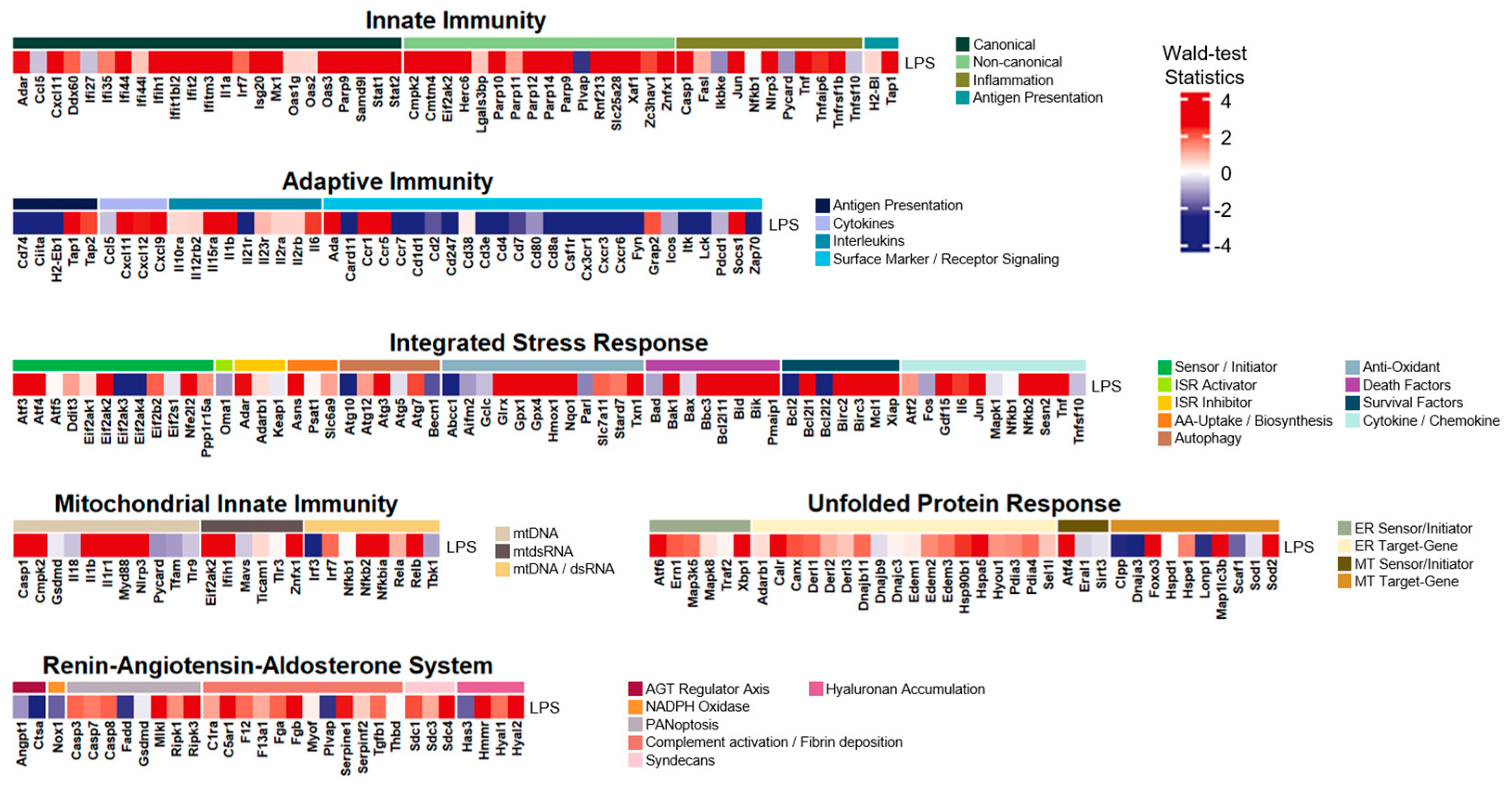
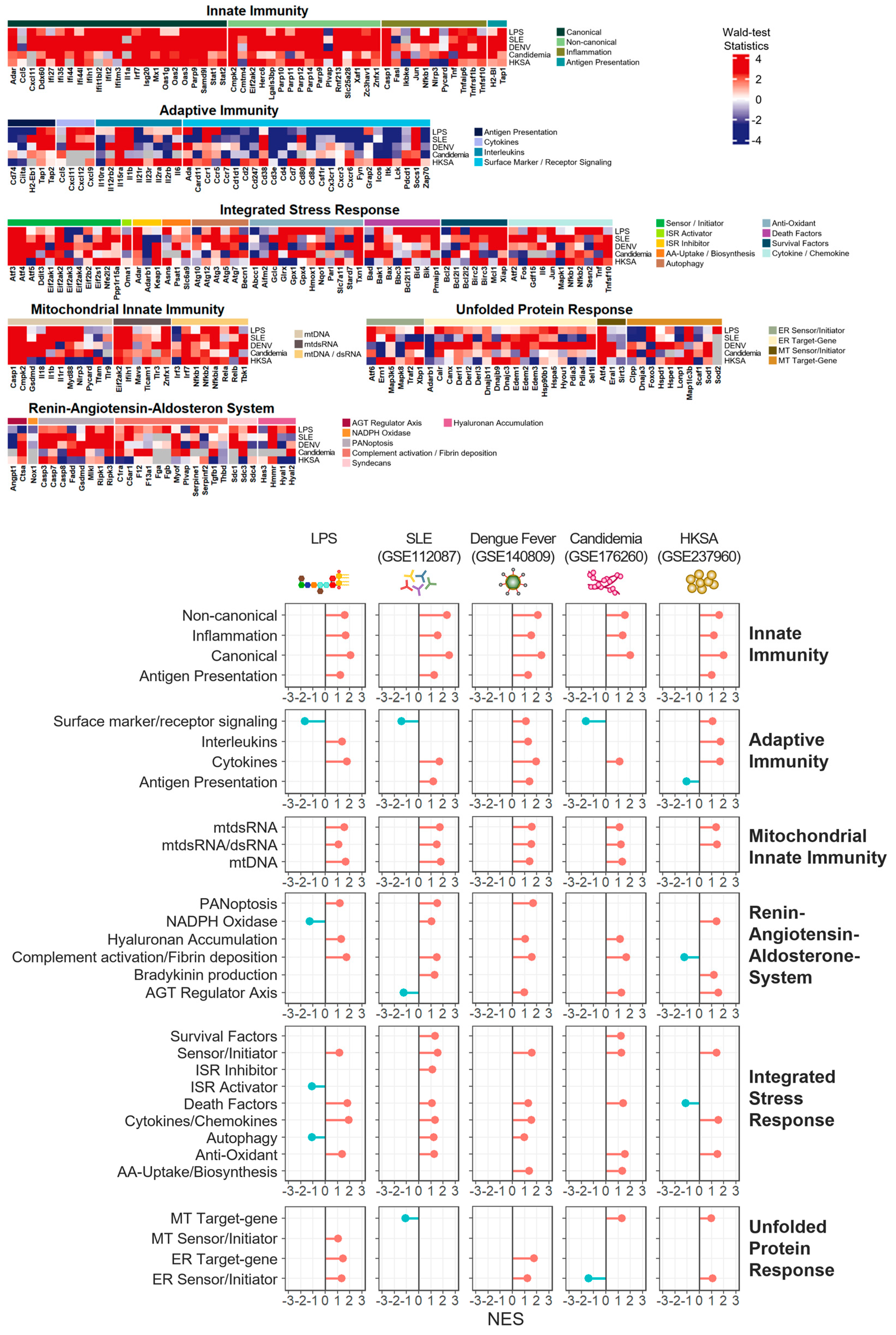
3.7. Innate Immunity and Mitochondrial Innate Immunity Demonstrated the Most Similar Activation Patterns across Four Distinct Inflammatory Conditions
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bruno, M.; Dewi, I.M.; Matzaraki, V.; Ter Horst, R.; Pekmezovic, M.; Rösler, B.; Groh, L.; Röring, R.J.; Kumar, V.; Li, Y.; et al. Comparative host transcriptome in response to pathogenic fungi identifies common and species-specific transcriptional antifungal host response pathways. Comput. Struct. Biotechnol. J. 2021, 19, 647–663. [Google Scholar] [CrossRef] [PubMed]
- Lapp, T.; Betancor, P.K.; Schlunck, G.; Auw-Hädrich, C.; Maier, P.; Lange, C.; Reinhard, T.; Wolf, J. Transcriptional profiling specifies the pathogen-specific human host response to infectious keratitis. Front. Cell. Infect. Microbiol. 2023, 13, 1285676. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, S.M.; Tarasova, O.A.; Poroikov, V.V. Transcriptome-based analysis of human peripheral blood reveals regulators of immune response in different viral infections. Front. Immunol. 2023, 14, 1199482. [Google Scholar] [CrossRef]
- Seemann, S.; Zohles, F.; Lupp, A. Comprehensive comparison of three different animal models for systemic inflammation. J. Biomed. Sci. 2017, 24, 60. [Google Scholar] [CrossRef]
- Lethal COVID-19 Associates with RAAS-Induced Inflammation for Multiple Organ Damage Including Mediastinal Lymph Nodes. bioRxiv 2023. [CrossRef]
- Institute of Laboratory Animal Resources (US); Committee on Care, and Use of Laboratory Animals. Guide for the Care and Use of Laboratory Animals; No. 86; US Department of Health and Human Services, Public Health Service, National Institutes of Health: Washington, DC, USA, 1986. [Google Scholar]
- Ramírez, K.; Quesada-Yamasaki, D.; Fornaguera-Trías, J. A Protocol to Perform Systemic Lipopolysacharide (LPS) Challenge in Rats. Odovtos-Int. J. Dent. Sci. 2019, 21, 53–66. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python Framework to Work with High-Throughput Sequencing Data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Blighe, K.; Rana, S.; Lewis, M. EnhancedVolcano: Publication-Ready Volcano Plots with Enhanced Colouring and Labeling, R package version 1.14.0; R Core Team location: Vienna, Austria, 2022. [Google Scholar]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A Universal Enrichment Tool for Interpreting Omics Data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Subramanian, A.; Pablo, T.; Vamsi, K.M.; Sayan, M.; Benjamin, L.E.; Michael, A.G.; Amanda, P.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Merico, D.; Isserlin, R.; Stueker, O.; Emili, A.; Bader, G.D. Enrichment Map: A Network-Based Method for Gene-Set Enrichment Visualization and Interpretation. PLoS ONE 2010, 5, e13984. [Google Scholar] [CrossRef]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular Signatures Database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef]
- Korotkevich, G.; Sukhov, V.; Budin, N.; Shpak, B.; Artyomov, M.N.; Sergushichev, A. Fast Gene Set Enrichment Analysis. bioRxiv 2021. [Google Scholar] [CrossRef]
- Ganga, L.; Sharma, P.; Tiwari, S.; Satoeya, N.; Jha, R.; Srivastava, M. Immunophenotypic and Functional Characterization of Eosinophil and Migratory Dendritic Cell Subsets during Filarial Manifestation of Tropical Pulmonary Eosinophilia. ACS Infect. Dis. 2023, 9, 1105–1122. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Shi, B.; Suo, R.; Xiong, S.; Wang, X.; Liang, X.; Li, X.; Li, G. Itaconate regulates macrophage function through stressful iron-sulfur cluster disrupting and iron metabolism rebalancing. FASEB J. 2021, 35, e21936. [Google Scholar] [CrossRef]
- Asgari, S.; Schlapbach, L.J.; Anchisi, S.; Hammer, C.; Bartha, I.; Junier, T.; Mottet-Osman, G.; Posfay-Barbe, K.M.; Longchamp, D.; Stocker, M.; et al. Severe viral respiratory infections in children with IFIH1 loss-of-function mutations. Proc. Natl. Acad. Sci. USA 2017, 114, 8342–8347. [Google Scholar] [CrossRef] [PubMed]
- Tran, V.; Ledwith, M.P.; Thamamongood, T.; Higgins, C.A.; Tripathi, S.; Chang, M.W.; Benner, C.; García-Sastre, A.; Schwemmle, M.; Boon, A.C.M.; et al. Influenza virus repurposes the antiviral protein IFIT2 to promote translation of viral mRNAs. Nat. Microbiol. 2020, 5, 1490–1503. [Google Scholar] [CrossRef]
- Vavassori, S.; Chou, J.; Faletti, L.E.; Haunerdinger, V.; Opitz, L.; Joset, P.; Fraser, C.J.; Prader, S.; Gao, X.; Schuch, L.A.; et al. Multisystem inflammation and susceptibility to viral infections in human ZNFX1 deficiency. J. Allergy Clin. Immunol. 2021, 148, 381–393. [Google Scholar] [CrossRef]
- Schiffer, R.; Baron, J.; Dagtekin, G.; Jahnen-Dechent, W.; Zwadlo-Klarwasser, G. Differential regulation of the expression of transporters associated with antigen processing, TAP1 and TAP2, by cytokines and lipopolysaccharide in primary human macrophages. Inflamm. Res. 2002, 51, 403–408. [Google Scholar] [CrossRef]
- Vielhauer, V.; Berning, E.; Eis, V.; Kretzler, M.; Segerer, S.; Strutz, F.; Horuk, R.; Gröne, H.-J.; Schlöndorff, D.; Anders, H.-J. CCR1 blockade reduces interstitial inflammation and fibrosis in mice with glomerulosclerosis and nephrotic syndrome. Kidney Int. 2004, 66, 2264–2278. [Google Scholar] [CrossRef] [PubMed]
- Janssens, S.; Beyaert, R. A universal role for MyD88 in TLR/IL-1R-mediated signaling. Trends Biochem. Sci. 2002, 27, 474–482. [Google Scholar] [CrossRef] [PubMed]
- Zani, A.; Yount, J.S. Antiviral Protection by IFITM3 In Vivo. Curr. Clin. Microbiol. Rep. 2018, 5, 229–237. [Google Scholar] [CrossRef]
- Pei, J.; Pan, X.; Wei, G.; Hua, Y. Research progress of glutathione peroxidase family (GPX) in redoxidation. Front. Pharmacol. 2023, 14, 1147414. [Google Scholar] [CrossRef] [PubMed]
- Möbus, L.; Rodriguez, E.; Harder, I.; Boraczynski, N.; Szymczak, S.; Hübenthal, M.; Stölzl, D.; Gerdes, S.; Kleinheinz, A.; Abraham, S.; et al. Blood transcriptome profiling identifies 2 candidate endotypes of atopic dermatitis. J. Allergy Clin. Immunol. 2022, 150, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Nielepkowicz-Goździńska, A.; Fendler, W.; Robak, E.; Kulczycka-Siennicka, L.; Górski, P.; Pietras, T.; Brzeziańska, E.; Pietrusińska, M.; Antczak, A. The Role of CXC Chemokines in Pulmonary Fibrosis of Systemic Lupus Erythematosus Patients. Arch. Immunol. Ther. Exp. 2015, 63, 465–473. [Google Scholar] [CrossRef]
- Robinson, T.; Kariuki, S.N.; Franek, B.S.; Kumabe, M.; Kumar, A.A.; Badaracco, M.; Mikolaitis, R.A.; Guerrero, G.; Utset, T.O.; Drevlow, B.E.; et al. Autoimmune disease risk variant of IFIH1 is associated with increased sensitivity to IFN-α and serologic autoimmunity in lupus patients. J. Immunol. 2011, 187, 1298–1303. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Liu, J.; Hao, S.; Wu, P.; Zhang, X.; Xiao, Y.; Jiang, G.; Huang, X. Higher activation of the interferon-gamma signaling pathway in systemic lupus erythematosus patients with a high type I IFN score: Relation to disease activity. Clin. Rheumatol. 2018, 37, 2675–2684. [Google Scholar] [CrossRef]
- Gao, F.; Tan, Y.; Luo, H. MALAT1 is involved in type I IFNs-mediated systemic lupus erythematosus by up-regulating OAS2, OAS3, and OASL. Braz. J. Med. Biol. Res. 2020, 53, e9292. [Google Scholar] [CrossRef]
- Tian, Q.; Zhao, H.; Ling, H.; Sun, L.; Xiao, C.; Yin, G.; Wang, X.; Wu, G.; Yang, C.; Chen, M.; et al. Poly(ADP-Ribose) Polymerase Enhances Infiltration of Mononuclear Cells in Primary Sjögren’s Syndrome through Interferon-Induced Protein with Tetratricopeptide Repeats 1-Mediated Up-Regulation of CXCL10. Arthritis Rheumatol. 2020, 72, 1003–1012. [Google Scholar] [CrossRef] [PubMed]
- Iwata, H.; Goettsch, C.; Sharma, A.; Ricchiuto, P.; Goh, W.W.; Halu, A.; Yamada, I.; Yoshida, H.; Hara, T.; Wei, M.; et al. PARP9 and PARP14 cross-regulate macrophage activation via STAT1 ADP-ribosylation. Nat. Commun. 2016, 7, 12849. [Google Scholar] [CrossRef] [PubMed]
- Gallucci, S.; Meka, S.; Gamero, A.M. Abnormalities of the type I interferon signaling pathway in lupus autoimmunity. Cytokine 2021, 146, 155633. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Berthier, C.C.; Kahlenberg, J.M. Enhanced Inflammasome Activity in Systemic Lupus Erythematosus Is Mediated via Type I Interferon-Induced Up-Regulation of Interferon Regulatory Factor 1. Arthritis Rheumatol. 2017, 69, 1840–1849. [Google Scholar] [CrossRef] [PubMed]
- Roth, S.H.; Danan-Gotthold, M.; Ben-Izhak, M.; Rechavi, G.; Cohen, C.J.; Louzoun, Y.; Levanon, E.Y. Increased RNA Editing May Provide a Source for Autoantigens in Systemic Lupus Erythematosus. Cell Rep. 2018, 23, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.J.; Figueras, F.; Wegener, W.A.; Goldenberg, D.M. Experience with milatuzumab, an anti-CD74 antibody against immunomodulatory macrophage migration inhibitory factor (MIF) receptor, for systemic lupus erythematosus (SLE). Ann. Rheum. Dis. 2021, 80, 954–955. [Google Scholar] [CrossRef]
- Bao, N.; Fu, B.; Zhong, X.; Jia, S.; Ren, Z.; Wang, H.; Wang, W.; Shi, H.; Li, J.; Ge, F.; et al. Role of the CXCR6/CXCL16 axis in autoimmune diseases. Int. Immunopharmacol. 2023, 121, 110530. [Google Scholar] [CrossRef] [PubMed]
- Kahlmann, D.; Davalos-Misslitz, A.C.; Ohl, L.; Stanke, F.; Witte, T.; Förster, R. Genetic variants of chemokine receptor CCR7 in patients with systemic lupus erythematosus, Sjogren’s syndrome and systemic sclerosis. BMC Genet. 2007, 8, 33. [Google Scholar] [CrossRef] [PubMed]
- Kolowos, W.; Gaipl, U.S.; Voll, R.E.; Frank, C.; Haas, J.P.; Beyer, T.D.; Kalden, J.R.; Herrmann, M. CD4 positive peripheral T cells from patients with systemic lupus erythematosus (SLE) are clonally expanded. Lupus 2001, 10, 321–331. [Google Scholar] [CrossRef]
- Kozlowska, A.; Hrycaj, P.; Lacki, J.K.; Jagodzinski, P.P. Fyn and CD70 expression in CD4+ T cells from patients with systemic lupus erythematosus. J. Rheumatol. 2010, 37, 53–59. [Google Scholar] [CrossRef]
- Jury, E.C.; Kabouridis, P.S.; Abba, A.; Mageed, R.A.; Isenberg, D.A. Increased ubiquitination and reduced expression of LCK in T lymphocytes from patients with systemic lupus erythematosus. Arthritis Rheum. 2003, 48, 1343–1354. [Google Scholar] [CrossRef] [PubMed]
- Vásquez, A.; Baena, A.; González, L.A.; Restrepo, M.; Muñoz, C.H.; Vanegas-García, A.; Ortiz-Reyes, B.; Abdoel, N.; Rojas, M.; García, L.F.; et al. Altered recruitment of Lyn, Syk and ZAP-70 into lipid rafts of activated B cells in Systemic Lupus Erythematosus. Cell Signal. 2019, 58, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Tomaselli, S.; Galeano, F.; Alon, S.; Raho, S.; Galardi, S.; Polito, V.A.; Presutti, C.; Vincenti, S.; Eisenberg, E.; Locatelli, F.; et al. Modulation of microRNA editing, expression and processing by ADAR2 deaminase in glioblastoma. Genome Biol. 2015, 16, 5. [Google Scholar] [CrossRef] [PubMed]
- Power, D.; Santoso, N.; Dieringer, M.; Yu, J.; Huang, H.; Simpson, S.; Seth, I.; Miao, H.; Zhu, J. IFI44 suppresses HIV-1 LTR promoter activity and facilitates its latency. Virology 2015, 481, 142–150. [Google Scholar] [CrossRef]
- Fensterl, V.; Wetzel, J.L.; Ramachandran, S.; Ogino, T.; Stohlman, S.A.; Bergmann, C.C.; Diamond, M.S.; Virgin, H.W.; Sen, G.C. Interferon-induced Ifit2/ISG54 protects mice from lethal VSV neuropathogenesis. PLoS Pathog. 2012, 8, e1002712. [Google Scholar] [CrossRef] [PubMed]
- Deymier, S.; Louvat, C.; Fiorini, F.; Cimarelli, A. ISG20: An enigmatic antiviral RNase targeting multiple viruses. FEBS Open Bio 2022, 12, 1096–1111. [Google Scholar] [CrossRef] [PubMed]
- Deguine, J.; Barton, G.M. MyD88: A central player in innate immune signaling. F1000prime Rep. 2014, 6, 97. [Google Scholar] [CrossRef] [PubMed]
- Colina, R.; Costa-Mattioli, M.; Dowling, R.J.; Jaramillo, M.; Tai, L.H.; Breitbach, C.J.; Martineau, Y.; Larsson, O.; Rong, L.; Svitkin, Y.V.; et al. Translational control of the innate immune response through IRF-7. Nature 2008, 452, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Place, D.E.; Kanneganti, T.D. Cell death-mediated cytokine release and its therapeutic implications. J. Exp. Med. 2019, 216, 1474–1486. [Google Scholar] [CrossRef]
- Meraz, M.A.; White, J.M.; Sheehan, K.C.; Bach, E.A.; Rodig, S.J.; Dighe, A.S.; Kaplan, D.H.; Riley, J.K.; Greenlund, A.C.; Campbell, D.; et al. Targeted disruption of the Stat1 gene in mice reveals unexpected physiologic specificity in the JAK-STAT signaling pathway. Cell 1996, 84, 431–442. [Google Scholar] [CrossRef]
- Bai, P. Biology of Poly(ADP-Ribose) Polymerases: The Factotums of Cell Maintenance. Mol. Cell 2015, 58, 947–958. [Google Scholar] [CrossRef]
- Guo, X.; Aviles, G.; Liu, Y.; Tian, R.; Unger, B.A.; Lin, Y.T.; Wiita, A.P.; Xu, K.; Correia, M.A.; Kampmann, M. Mitochondrial stress is relayed to the cytosol by an OMA1-DELE1-HRI pathway. Nature 2020, 579, 427–432. [Google Scholar] [CrossRef]
- Costa-Mattioli, M.; Walter, P. The integrated stress response: From mechanism to disease. Science 2020, 368, eaat5314. [Google Scholar] [CrossRef]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Domachowske, J.B.; Bonville, C.A.; Rosenberg, H.F. Cytokeratin 17 is expressed in cells infected with respiratory syncytial virus via NF-kappaB activation and is associated with the formation of cytopathic syncytia. J. Infect. Dis. 2000, 182, 1022–1028. [Google Scholar] [CrossRef]
- Leisching, G.; Wiid, I.; Baker, B. OAS1, 2, and 3: Significance during Active Tuberculosis? J. Infect. Dis. 2018, 217, 1517–1521. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Fodor, E.; Keown, J.R. A structural understanding of influenza virus genome replication. Trends Microbiol. 2023, 31, 308–319. [Google Scholar] [CrossRef]
- Zhang, X.J.; Xu, H.S.; Li, C.H.; Fu, Y.R.; Yi, Z.J. Up-regulated SAMD9L modulated by TLR2 and HIF-1α as a promising biomarker in tuberculosis. J. Cell Mol. Med. 2022, 26, 2935–2946. [Google Scholar] [CrossRef]
- Hato, T.; Maier, B.; Syed, F.; Myslinski, J.; Zollman, A.; Plotkin, Z.; Eadon, M.T.; Dagher, P.C. Bacterial sepsis triggers an antiviral response that causes translation shutdown. J. Clin. Investig. 2019, 129, 296–309. [Google Scholar] [CrossRef]
- Otten, E.G.; Werner, E.; Crespillo-Casado, A.; Boyle, K.B.; Dharamdasani, V.; Pathe, C.; Santhanam, B.; Randow, F. Ubiquitylation of lipopolysaccharide by RNF213 during bacterial infection. Nature 2021, 594, 111–116. [Google Scholar] [CrossRef]
- Uchiyama, R.; Tsutsui, H. Caspases as the key effectors of inflammatory responses against bacterial infection. Arch. Immunol. Ther. Exp. 2015, 63, 1–13. [Google Scholar] [CrossRef]
- Zaidi, T.; Reidy, T.; D‘Ortona, S.; Fichorova, R.; Pier, G.; Gadjeva, M. CD74 deficiency ameliorates Pseudomonas aeruginosa-induced ocular infection. Sci. Rep. 2011, 1, 58. [Google Scholar] [CrossRef] [PubMed]
- Nishi, K.; Oda, T.; Takabuchi, S.; Oda, S.; Fukuda, K.; Adachi, T.; Semenza, G.L.; Shingu, K.; Hirota, K. LPS induces hypoxia-inducible factor 1 activation in macrophage-differentiated cells in a reactive oxygen species-dependent manner. Antioxid. Redox. Signal. 2008, 10, 983–995. [Google Scholar] [CrossRef]
- Monaci, S.; Coppola, F.; Rossi, D.; Giuntini, G.; Filippi, I.; Marotta, G.; Sozzani, S.; Carraro, F.; Naldini, A. Hypoxia Induces Autophagy in Human Dendritic Cells: Involvement of Class III PI3K/Vps34. Cells 2022, 11, 1695. [Google Scholar] [CrossRef]
- Marinkovic, D.; Zhang, X.; Yalcin, S.; Luciano, J.P.; Brugnara, C.; Huber, T.; Ghaffari, S. Foxo3 is required for the regulation of oxidative stress in erythropoiesis. J. Clin. Investig. 2007, 117, 2133–2144. [Google Scholar] [CrossRef]
- Velarde, M.C.; Flynn, J.M.; Day, N.U.; Melov, S.; Campisi, J. Mitochondrial oxidative stress caused by Sod2 deficiency promotes cellular senescence and aging phenotypes in the skin. Aging 2012, 4, 3–12. [Google Scholar] [CrossRef]
- Seifert, U.; Bialy, L.P.; Ebstein, F.; Bech-Otschir, D.; Voigt, A.; Schröter, F.; Prozorovski, T.; Lange, N.; Steffen, J.; Rieger, M.; et al. Immunoproteasomes preserve protein homeostasis upon interferon-induced oxidative stress. Cell 2010, 142, 613–624. [Google Scholar] [CrossRef]
- Chen, Y.; Zhou, Z.; Min, W. Mitochondria, Oxidative Stress and Innate Immunity. Front. Physiol. 2018, 9, 1487. [Google Scholar] [CrossRef]
- Nowowiejska, J.; Baran, A.; Hermanowicz, J.M.; Pryczynicz, A.; Sieklucka, B.; Pawlak, D.; Flisiak, I. Gasdermin C (GSDMC) is Overexpressed in Psoriatic Tissue and Elevated in Psoriatic Serum: A Potential Marker of Cell Proliferation and Local Hypoxia in Psoriasis? Dermatol. Ther. 2023, 2023, 7813287. [Google Scholar] [CrossRef]
- Grootjans, J.; Kaser, A.; Kaufman, R.J.; Blumberg, R.S. The unfolded protein response in immunity and inflammation. Nat. Rev. Immunol. 2016, 16, 469–484. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sheen, K.; Myung, S.; Lee, D.-M.; Yu, S.; Choi, Y.; Kim, T.; Kim, J.; Ji, S.-G.; Kim, M.-S.; Kim, W.; et al. RNA-Seq of an LPS-Induced Inflammation Model Reveals Transcriptional Profile Patterns of Inflammatory Processes. Life 2024, 14, 558. https://doi.org/10.3390/life14050558
Sheen K, Myung S, Lee D-M, Yu S, Choi Y, Kim T, Kim J, Ji S-G, Kim M-S, Kim W, et al. RNA-Seq of an LPS-Induced Inflammation Model Reveals Transcriptional Profile Patterns of Inflammatory Processes. Life. 2024; 14(5):558. https://doi.org/10.3390/life14050558
Chicago/Turabian StyleSheen, Kisung, Seokho Myung, Dong-Min Lee, Sanghyeon Yu, Yueun Choi, Taeyoon Kim, Jihan Kim, Sang-Gu Ji, Myung-Seo Kim, Wonnam Kim, and et al. 2024. "RNA-Seq of an LPS-Induced Inflammation Model Reveals Transcriptional Profile Patterns of Inflammatory Processes" Life 14, no. 5: 558. https://doi.org/10.3390/life14050558
APA StyleSheen, K., Myung, S., Lee, D.-M., Yu, S., Choi, Y., Kim, T., Kim, J., Ji, S.-G., Kim, M.-S., Kim, W., Lee, Y., Kim, M. S., & Park, Y.-C. (2024). RNA-Seq of an LPS-Induced Inflammation Model Reveals Transcriptional Profile Patterns of Inflammatory Processes. Life, 14(5), 558. https://doi.org/10.3390/life14050558