


Novel Activity of Oral Hypoglycemic Agents Linked with Decreased Formation of Tryptophan Metabolite, Kynurenic Acid



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Materials
2.3. Production of Kynurenic Acid In Vitro
2.4. Analyses of the Activity of Kynurenine Aminotransferases I and II
2.5. Quantification of Kynurenic Acid
2.6. Statistical Analyses
3. Results
3.1. Effect of Metformin and Glibenclamide on Kynurenic Acid Production In Vitro
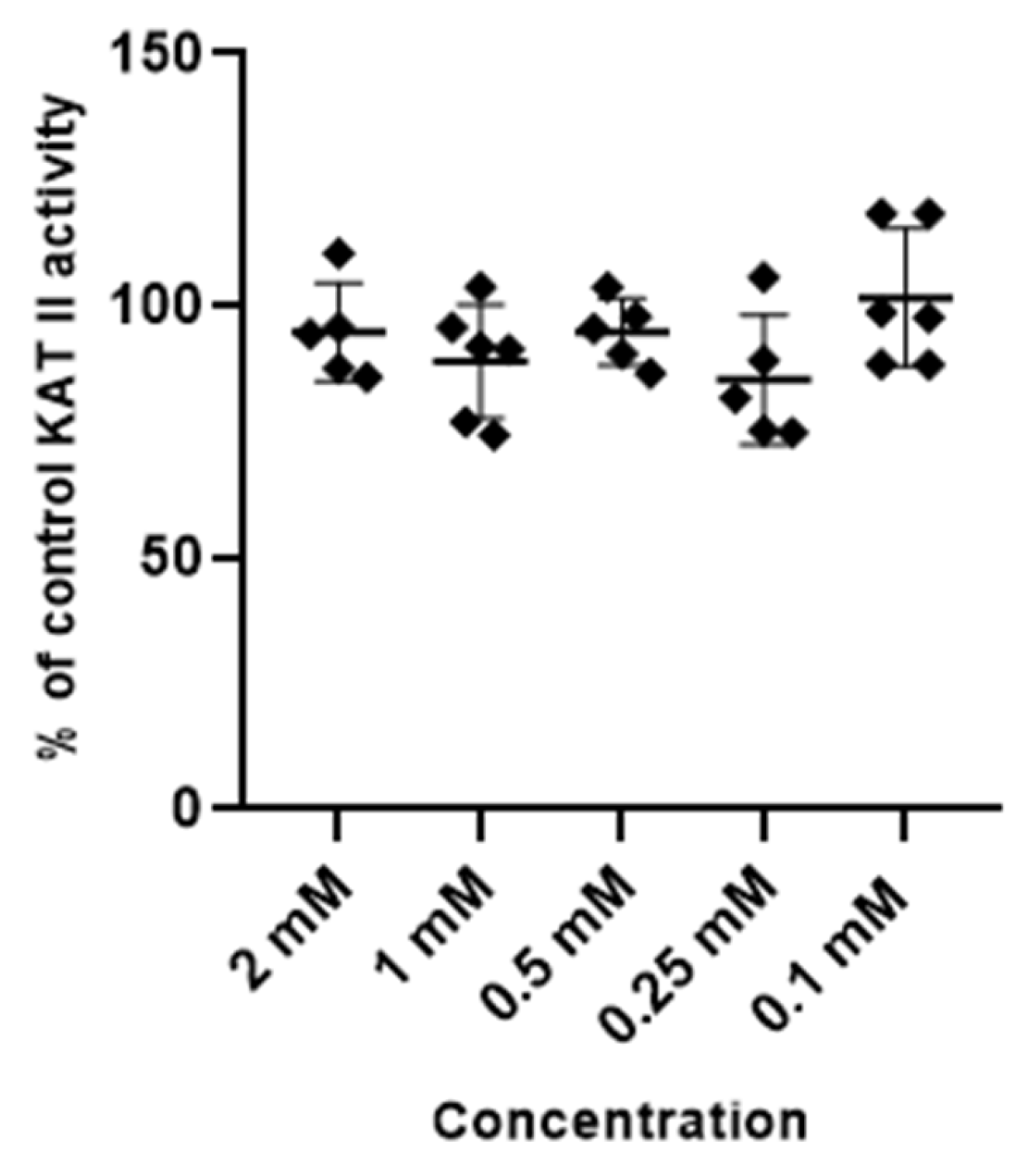
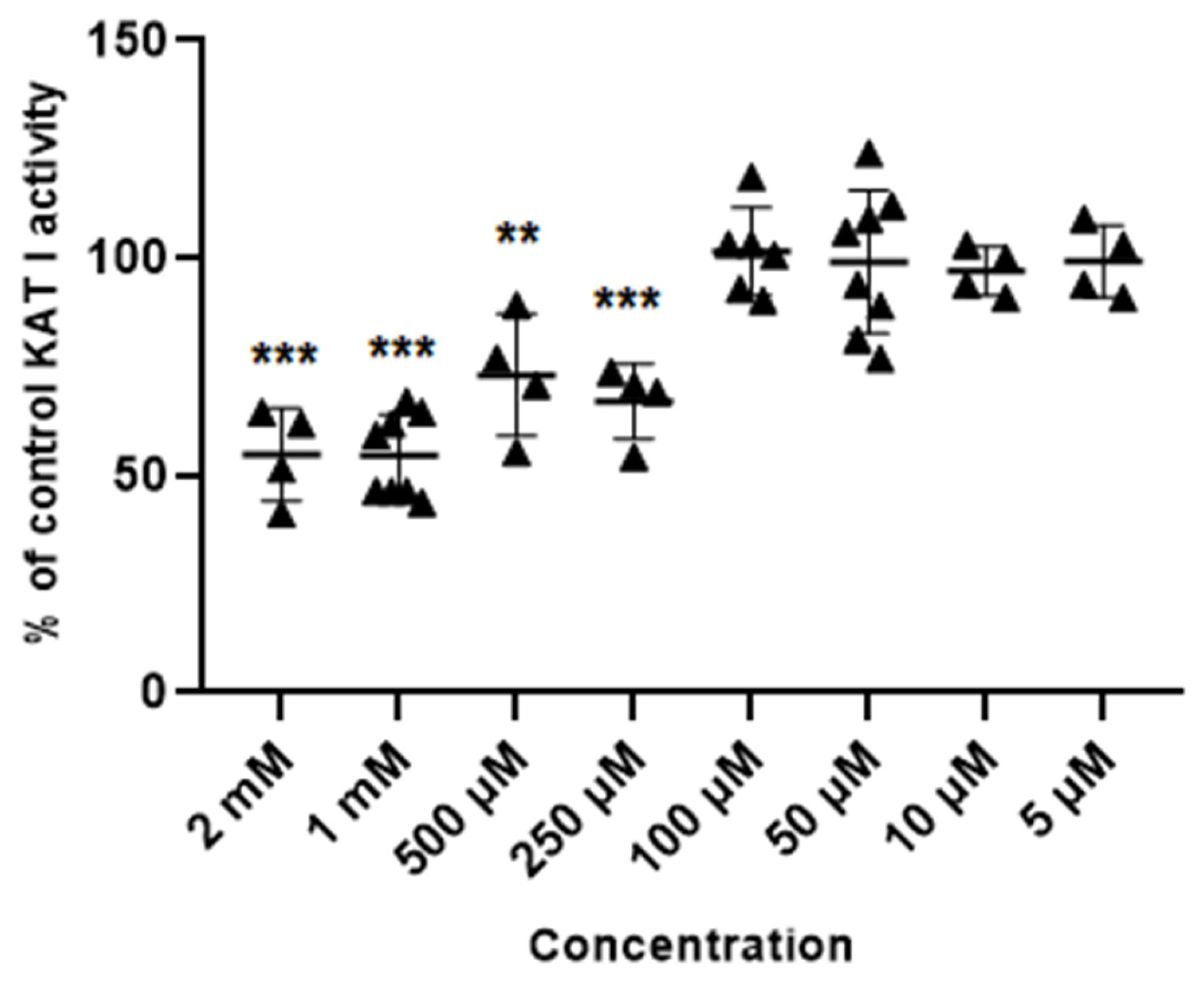
3.2. Effect of Metformin and Glibenclamide on the Activity of Kynurenine Aminotransferases (KATs) I and II
3.2.1. Metformin
3.2.2. Glibenclamide
4. Discussion
5. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Roth, W.; Zadeh, K.; Vekariya, R.; Ge, Y.; Mohamadzadeh, M. Tryptophan Metabolism and Gut-Brain Homeostasis. Int. J. Mol. Sci. 2021, 22, 2973. [Google Scholar] [CrossRef]
- Ostapiuk, A.; Urbanska, E.M. Kynurenic acid in neurodegenerative disorders-unique neuroprotection or double-edged sword? CNS Neurosci. Ther. 2022, 28, 19–35. [Google Scholar] [CrossRef]
- Liu, J.J.; Movassat, J.; Portha, B. Emerging role for kynurenines in metabolic pathologies. Curr. Opin. Clin. Nutr. Metab. Care 2019, 22, 82–90. [Google Scholar] [CrossRef]
- Savitz, J. The kynurenine pathway: A finger in every pie. Mol. Psychiatry 2020, 25, 131–147. [Google Scholar] [CrossRef] [PubMed]
- Stone, T.W.; Forrest, C.M.; Darlington, L.G. Kynurenine pathway inhibition as a therapeutic strategy for neuroprotection. FEBS J. 2012, 279, 1386–1397. [Google Scholar] [CrossRef] [PubMed]
- Agudelo, L.Z.; Ferreira, D.M.S.; Cervenka, I.; Bryzgalova, G.; Dadvar, S.; Jannig, P.R.; Pettersson-Klein, A.T.; Lakshmikanth, T.; Sustarsic, E.G.; Porsmyr-Palmertz, M.; et al. Kynurenic Acid and Gpr35 Regulate Adipose Tissue Energy Homeostasis and Inflammation. Cell Metab. 2018, 27, 378–392.e375. [Google Scholar] [CrossRef]
- Walczak, K.; Wnorowski, A.; Turski, W.A.; Plech, T. Kynurenic acid and cancer: Facts and controversies. Cell Mol. Life Sci. 2020, 77, 1531–1550. [Google Scholar] [CrossRef]
- Stone, T.W. Does kynurenic acid act on nicotinic receptors? An assessment of the evidence. J. Neurochem. 2020, 152, 627–649. [Google Scholar] [CrossRef] [PubMed]
- Lugo-Huitrón, R.; Blanco-Ayala, T.; Ugalde-Muñiz, P.; Carrillo-Mora, P.; Pedraza-Chaverrí, J.; Silva-Adaya, D.; Maldonado, P.D.; Torres, I.; Pinzón, E.; Ortiz-Islas, E.; et al. On the antioxidant properties of kynurenic acid: Free radical scavenging activity and inhibition of oxidative stress. Neurotoxicol. Teratol. 2011, 33, 538–547. [Google Scholar] [CrossRef] [PubMed]
- Urbańska, E.M.C.-P.I.; Perzyński, A.; Derkacz, M.; Owe-Larsson, B. Endogenous Kynurenic Acid and Neurotoxicity. In Handbook of Neurotoxicity, 2nd ed.; Kostrzewa, R.M., Ed.; Springer: Berlin/Heidelberg, Germany, 2022; pp. 1035–1065. [Google Scholar]
- Kocki, T.; Kocki, J.; Wielosz, M.; Turski, W.A.; Urbanska, E.M. Carbamazepine enhances brain production of kynurenic acid in vitro. Eur. J. Pharmacol. 2004, 498, 325–326. [Google Scholar] [CrossRef]
- Kloc, R.; Luchowska, E.; Wielosz, M.; Owe-Larsson, B.; Urbanska, E.M. Memantine increases brain production of kynurenic acid via protein kinase A-dependent mechanism. Neurosci. Lett. 2008, 435, 169–173. [Google Scholar] [CrossRef]
- Kiluk, M.; Lewkowicz, J.; Kowalska, I.; Pawlak, D.; Łagoda, K.; Tankiewicz-Kwedlo, A. Alterations of the kynurenine pathway in patients with type 1 diabetes are associated with metabolic control of diabetes. Pol. Arch. Intern. Med. 2023, 133, 16581. [Google Scholar] [CrossRef]
- Kozieł, K.; Urbanska, E.M. Kynurenine Pathway in Diabetes Mellitus-Novel Pharmacological Target? Cells 2023, 12, 460. [Google Scholar] [CrossRef]
- Rohm, T.V.; Meier, D.T.; Olefsky, J.M.; Donath, M.Y. Inflammation in obesity, diabetes, and related disorders. Immunity 2022, 55, 31–55. [Google Scholar] [CrossRef]
- Chmiel-Perzyńska, I.; Perzyński, A.; Wielosz, M.; Urbańska, E.M. Hyperglycemia enhances the inhibitory effect of mitochondrial toxins and D,L-homocysteine on the brain production of kynurenic acid. Pharmacol. Rep. 2007, 59, 268–273. [Google Scholar] [PubMed]
- Chmiel-Perzyńska, I.; Perzyński, A.; Urbańska, E.M. Experimental diabetes mellitus type 1 increases hippocampal content of kynurenic acid in rats. Pharmacol. Rep. 2014, 66, 1134–1139. [Google Scholar] [CrossRef]
- da Silva Dias, I.C.; Carabelli, B.; Ishii, D.K.; de Morais, H.; de Carvalho, M.C.; Rizzo de Souza, L.E.; Zanata, S.M.; Brandão, M.L.; Cunha, T.M.; Ferraz, A.C.; et al. Indoleamine-2,3-Dioxygenase/Kynurenine Pathway as a Potential Pharmacological Target to Treat Depression Associated with Diabetes. Mol. Neurobiol. 2016, 53, 6997–7009. [Google Scholar] [CrossRef]
- Inzucchi, S.E.; Bergenstal, R.M.; Buse, J.B.; Diamant, M.; Ferrannini, E.; Nauck, M.; Peters, A.L.; Tsapas, A.; Wender, R.; Matthews, D.R. Management of hyperglycaemia in type 2 diabetes: A patient-centered approach. Position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetologia 2012, 55, 1577–1596. [Google Scholar] [CrossRef] [PubMed]
- Dutta, S.; Shah, R.B.; Singhal, S.; Dutta, S.B.; Bansal, S.; Sinha, S.; Haque, M. Metformin: A Review of Potential Mechanism and Therapeutic Utility Beyond Diabetes. Drug Des. Devel. Ther. 2023, 17, 1907–1932. [Google Scholar] [CrossRef] [PubMed]
- Demaré, S.; Kothari, A.; Calcutt, N.A.; Fernyhough, P. Metformin as a potential therapeutic for neurological disease: Mobilizing AMPK to repair the nervous system. Expert Rev. Neurother. 2021, 21, 45–63. [Google Scholar] [CrossRef]
- Li, N.; Zhou, T.; Fei, E. Actions of Metformin in the Brain: A New Perspective of Metformin Treatments in Related Neurological Disorders. Int. J. Mol. Sci. 2022, 23, 8281. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, S.C.; Ferguson, E.L.; Choudhary, V.; Ranatunga, D.K.; Oni-Orisan, A.; Hayes-Larson, E.; Duarte Folle, A.; Mayeda, E.R.; Whitmer, R.A.; Gilsanz, P.; et al. Metformin Cessation and Dementia Incidence. JAMA Netw. Open 2023, 6, e2339723. [Google Scholar] [CrossRef] [PubMed]
- DiBona, V.L.; Shah, M.K.; Krause, K.J.; Zhu, W.; Voglewede, M.M.; Smith, D.M.; Crockett, D.P.; Zhang, H. Metformin reduces neuroinflammation and improves cognitive functions after traumatic brain injury. Neurosci. Res. 2021, 172, 99–109. [Google Scholar] [CrossRef]
- Woo, S.K.; Tsymbalyuk, N.; Tsymbalyuk, O.; Ivanova, S.; Gerzanich, V.; Simard, J.M. SUR1-TRPM4 channels, not K(ATP), mediate brain swelling following cerebral ischemia. Neurosci. Lett. 2020, 718, 134729. [Google Scholar] [CrossRef]
- Griepp, D.W.; Lee, J.; Moawad, C.M.; Davati, C.; Runnels, J.; Fiani, B. BIIB093 (intravenous glibenclamide) for the prevention of severe cerebral edema. Surg. Neurol. Int. 2021, 12, 80. [Google Scholar] [CrossRef] [PubMed]
- Costa, B.; Windlin, I.C.; Koterba, E.; Yamaki, V.N.; Rabelo, N.N.; Solla, D.J.F.; Samaia da Silva Coelho, A.C.; Telles, J.P.M.; Teixeira, M.J.; Figueiredo, E.G. Glibenclamide in aneurysmal subarachnoid hemorrhage: A randomized controlled clinical trial. J. Neurosurg. 2021, 137, 121–128. [Google Scholar] [CrossRef]
- Turski, W.A.; Gramsbergen, J.B.; Traitler, H.; Schwarcz, R. Rat brain slices produce and liberate kynurenic acid upon exposure to L-kynurenine. J. Neurochem. 1989, 52, 1629–1636. [Google Scholar] [CrossRef]
- Kocki, T.; Urbańska, E.M.; Kocki, J.; Kloc, R.; Kocka, K.; Olajossy, M.; Owe-Larsson, B. Prolonged therapy with antidepressants increases hippocampal level of kynurenic acid and expression of Kat1 and Kat2 genes. Pharmacol. Rep. 2018, 70, 737–745. [Google Scholar] [CrossRef] [PubMed]
- Dudzińska, E.; Szymona, K.; Kloc, R.; Gil-Kulik, P.; Kocki, T.; Świstowska, M.; Bogucki, J.; Kocki, J.; Urbanska, E.M. Increased expression of kynurenine aminotransferases mRNA in lymphocytes of patients with inflammatory bowel disease. Therap. Adv. Gastroenterol. 2019, 12, 1756284819881304. [Google Scholar] [CrossRef] [PubMed]
- Zakrocka, I.; Turski, W.A.; Kocki, T. Angiotensin-converting enzyme inhibitors modulate kynurenic acid production in rat brain cortex in vitro. Eur. J. Pharmacol. 2016, 789, 308–312. [Google Scholar] [CrossRef]
- Luchowska, E.; Luchowski, P.; Paczek, R.; Ziembowicz, A.; Kocki, T.; Turski, W.A.; Wielosz, M.; Lazarewicz, J.; Urbanska, E.M. Dual effect of DL-homocysteine and S-adenosylhomocysteine on brain synthesis of the glutamate receptor antagonist, kynurenic acid. J. Neurosci. Res. 2005, 79, 375–382. [Google Scholar] [CrossRef]
- Zakrocka, I.; Targowska-Duda, K.M.; Wnorowski, A.; Kocki, T.; Jóźwiak, K.; Turski, W.A. Angiotensin II Type 1 Receptor Blockers Inhibit KAT II Activity in the Brain-Its Possible Clinical Applications. Neurotox. Res. 2017, 32, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Zakrocka, I.; Targowska-Duda, K.M.; Wnorowski, A.; Kocki, T.; Jóźwiak, K.; Turski, W.A. Influence of Cyclooxygenase-2 Inhibitors on Kynurenic Acid Production in Rat Brain in Vitro. Neurotox. Res. 2019, 35, 244–254. [Google Scholar] [CrossRef]
- Zhen, D.; Liu, J.; Zhang, X.D.; Song, Z. Kynurenic Acid Acts as a Signaling Molecule Regulating Energy Expenditure and Is Closely Associated With Metabolic Diseases. Front. Endocrinol. 2022, 13, 847611. [Google Scholar] [CrossRef]
- Noto, Y.; Okamoto, H. Inhibition by kynurenine metabolites of proinsulin synthesis in isolated pancreatic islets. Acta Diabetol. Lat. 1978, 15, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Lam, C.K.; Chari, M.; Su, B.B.; Cheung, G.W.; Kokorovic, A.; Yang, C.S.; Wang, P.Y.; Lai, T.Y.; Lam, T.K. Activation of N-methyl-D-aspartate (NMDA) receptors in the dorsal vagal complex lowers glucose production. J. Biol. Chem. 2010, 285, 21913–21921. [Google Scholar] [CrossRef]
- Yokoi, N.; Beppu, M.; Yoshida, E.; Hoshikawa, R.; Hidaka, S.; Matsubara, T.; Shinohara, M.; Irino, Y.; Hatano, N.; Seino, S. Identification of putative biomarkers for prediabetes by metabolome analysis of rat models of type 2 diabetes. Metabolomics 2015, 11, 1277–1286. [Google Scholar] [CrossRef]
- Patterson, A.D.; Bonzo, J.A.; Li, F.; Krausz, K.W.; Eichler, G.S.; Aslam, S.; Tigno, X.; Weinstein, J.N.; Hansen, B.C.; Idle, J.R.; et al. Metabolomics reveals attenuation of the SLC6A20 kidney transporter in nonhuman primate and mouse models of type 2 diabetes mellitus. J. Biol. Chem. 2011, 286, 19511–19522. [Google Scholar] [CrossRef]
- Oxenkrug, G.F. Increased Plasma Levels of Xanthurenic and Kynurenic Acids in Type 2 Diabetes. Mol. Neurobiol. 2015, 52, 805–810. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, E.R.; Tuseth, N.; Eussen, S.J.; Ueland, P.M.; Strand, E.; Svingen, G.F.; Midttun, Ø.; Meyer, K.; Mellgren, G.; Ulvik, A.; et al. Associations of plasma kynurenines with risk of acute myocardial infarction in patients with stable angina pectoris. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 455–462. [Google Scholar] [CrossRef]
- Muzik, O.; Burghardt, P.; Yi, Z.; Kumar, A.; Seyoum, B. Successful metformin treatment of insulin resistance is associated with down-regulation of the kynurenine pathway. Biochem. Biophys. Res. Commun. 2017, 488, 29–32. [Google Scholar] [CrossRef]
- Frid, A.; Sterner, G.N.; Löndahl, M.; Wiklander, C.; Cato, A.; Vinge, E.; Andersson, A. Novel assay of metformin levels in patients with type 2 diabetes and varying levels of renal function: Clinical recommendations. Diabetes Care 2010, 33, 1291–1293. [Google Scholar] [CrossRef] [PubMed]
- Pocivavsek, A.; Elmer, G.I.; Schwarcz, R. Inhibition of kynurenine aminotransferase II attenuates hippocampus-dependent memory deficit in adult rats treated prenatally with kynurenine. Hippocampus 2019, 29, 73–77. [Google Scholar] [CrossRef]
- Kodali, M.; Attaluri, S.; Madhu, L.N.; Shuai, B.; Upadhya, R.; Gonzalez, J.J.; Rao, X.; Shetty, A.K. Metformin treatment in late middle age improves cognitive function with alleviation of microglial activation and enhancement of autophagy in the hippocampus. Aging Cell 2021, 20, e13277. [Google Scholar] [CrossRef] [PubMed]
- Modzelewski, R.; Stefanowicz-Rutkowska, M.M.; Matuszewski, W.; Bandurska-Stankiewicz, E.M. Gestational Diabetes Mellitus-Recent Literature Review. J. Clin. Med. 2022, 11, 5736. [Google Scholar] [CrossRef]
- DeMarsilis, A.; Reddy, N.; Boutari, C.; Filippaios, A.; Sternthal, E.; Katsiki, N.; Mantzoros, C. Pharmacotherapy of type 2 diabetes: An update and future directions. Metabolism 2022, 137, 155332. [Google Scholar] [CrossRef] [PubMed]
- Mehta, R.I.; Tosun, C.; Ivanova, S.; Tsymbalyuk, N.; Famakin, B.M.; Kwon, M.S.; Castellani, R.J.; Gerzanich, V.; Simard, J.M. Sur1-Trpm4 Cation Channel Expression in Human Cerebral Infarcts. J. Neuropathol. Exp. Neurol. 2015, 74, 835–849. [Google Scholar] [CrossRef]
- Khanna, A.; Walcott, B.P.; Kahle, K.T.; Simard, J.M. Effect of glibenclamide on the prevention of secondary brain injury following ischemic stroke in humans. Neurosurg. Focus 2014, 36, E11. [Google Scholar] [CrossRef]
- Zubov, A.; Muruzheva, Z.; Tikhomirova, M.; Karpenko, M. Glibenclamide as a neuroprotective antidementia drug. Arch. Physiol. Biochem. 2022, 128, 1693–1696. [Google Scholar] [CrossRef]
- Tosun, C.; Kurland, D.B.; Mehta, R.; Castellani, R.J.; deJong, J.L.; Kwon, M.S.; Woo, S.K.; Gerzanich, V.; Simard, J.M. Inhibition of the Sur1-Trpm4 channel reduces neuroinflammation and cognitive impairment in subarachnoid hemorrhage. Stroke 2013, 44, 3522–3528. [Google Scholar] [CrossRef]
- Ortega, F.J.; Jolkkonen, J.; Mahy, N.; Rodríguez, M.J. Glibenclamide enhances neurogenesis and improves long-term functional recovery after transient focal cerebral ischemia. J. Cereb. Blood Flow Metab. 2013, 33, 356–364. [Google Scholar] [CrossRef]
- Zubov, A.S.; Ivleva, I.S.; Pestereva, N.S.; Tiutiunnik, T.V.; Traktirov, D.S.; Karpenko, M.N. Glibenclamide alters serotonin and dopamine levels in the rat striatum and hippocampus, reducing cognitive impairment. Psychopharmacology 2022, 239, 2787–2798. [Google Scholar] [CrossRef] [PubMed]
- de Sant’Anna, J.R.; Franco, C.C.; Mathias, P.C.; de Castro-Prado, M.A. Assessment of in vivo and in vitro genotoxicity of glibenclamide in eukaryotic cells. PLoS ONE 2015, 10, e0120675. [Google Scholar] [CrossRef]
- Shen, Z.; Xiang, M.; Chen, C.; Ding, F.; Wang, Y.; Shang, C.; Xin, L.; Zhang, Y.; Cui, X. Glutamate excitotoxicity: Potential therapeutic target for ischemic stroke. Biomed. Pharmacother. 2022, 151, 113125. [Google Scholar] [CrossRef]
- Wilkinson, C.M.; Brar, P.S.; Balay, C.J.; Colbourne, F. Glibenclamide, a Sur1-Trpm4 antagonist, does not improve outcome after collagenase-induced intracerebral hemorrhage. PLoS ONE 2019, 14, e0215952. [Google Scholar] [CrossRef] [PubMed]
- Kung, T.F.C.; Wilkinson, C.M.; Dirks, C.A.; Jickling, G.C.; Colbourne, F. Glibenclamide does not improve outcome following severe collagenase-induced intracerebral hemorrhage in rats. PLoS ONE 2021, 16, e0252584. [Google Scholar] [CrossRef] [PubMed]
- Wątroba, M.; Grabowska, A.D.; Szukiewicz, D. Effects of Diabetes Mellitus-Related Dysglycemia on the Functions of Blood-Brain Barrier and the Risk of Dementia. Int. J. Mol. Sci. 2023, 24, 69. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bednarz, K.; Kozieł, K.; Urbańska, E.M. Novel Activity of Oral Hypoglycemic Agents Linked with Decreased Formation of Tryptophan Metabolite, Kynurenic Acid. Life 2024, 14, 127. https://doi.org/10.3390/life14010127
Bednarz K, Kozieł K, Urbańska EM. Novel Activity of Oral Hypoglycemic Agents Linked with Decreased Formation of Tryptophan Metabolite, Kynurenic Acid. Life. 2024; 14(1):127. https://doi.org/10.3390/life14010127
Chicago/Turabian StyleBednarz, Kinga, Kamila Kozieł, and Ewa M. Urbańska. 2024. "Novel Activity of Oral Hypoglycemic Agents Linked with Decreased Formation of Tryptophan Metabolite, Kynurenic Acid" Life 14, no. 1: 127. https://doi.org/10.3390/life14010127
APA StyleBednarz, K., Kozieł, K., & Urbańska, E. M. (2024). Novel Activity of Oral Hypoglycemic Agents Linked with Decreased Formation of Tryptophan Metabolite, Kynurenic Acid. Life, 14(1), 127. https://doi.org/10.3390/life14010127