
Transposable Elements in Bats Show Differential Accumulation Patterns Determined by Class and Functionality

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
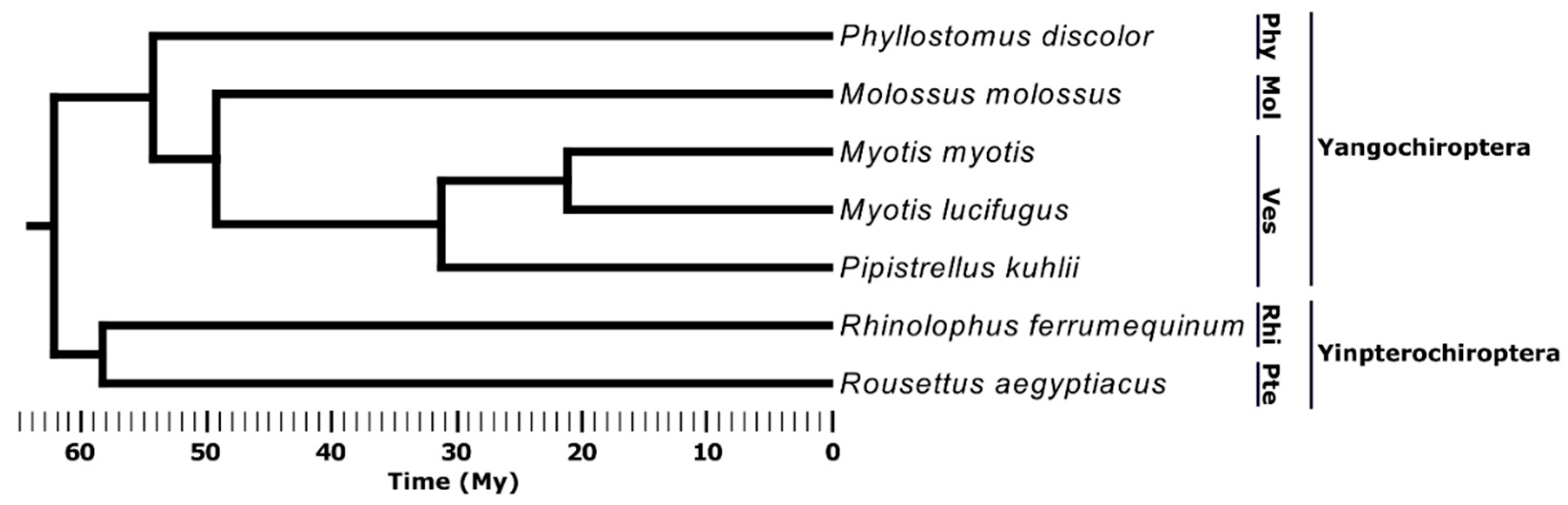
2.1. Species Selection and TE Annotation
2.2. TE Spatial Distribution Analysis
3. Results
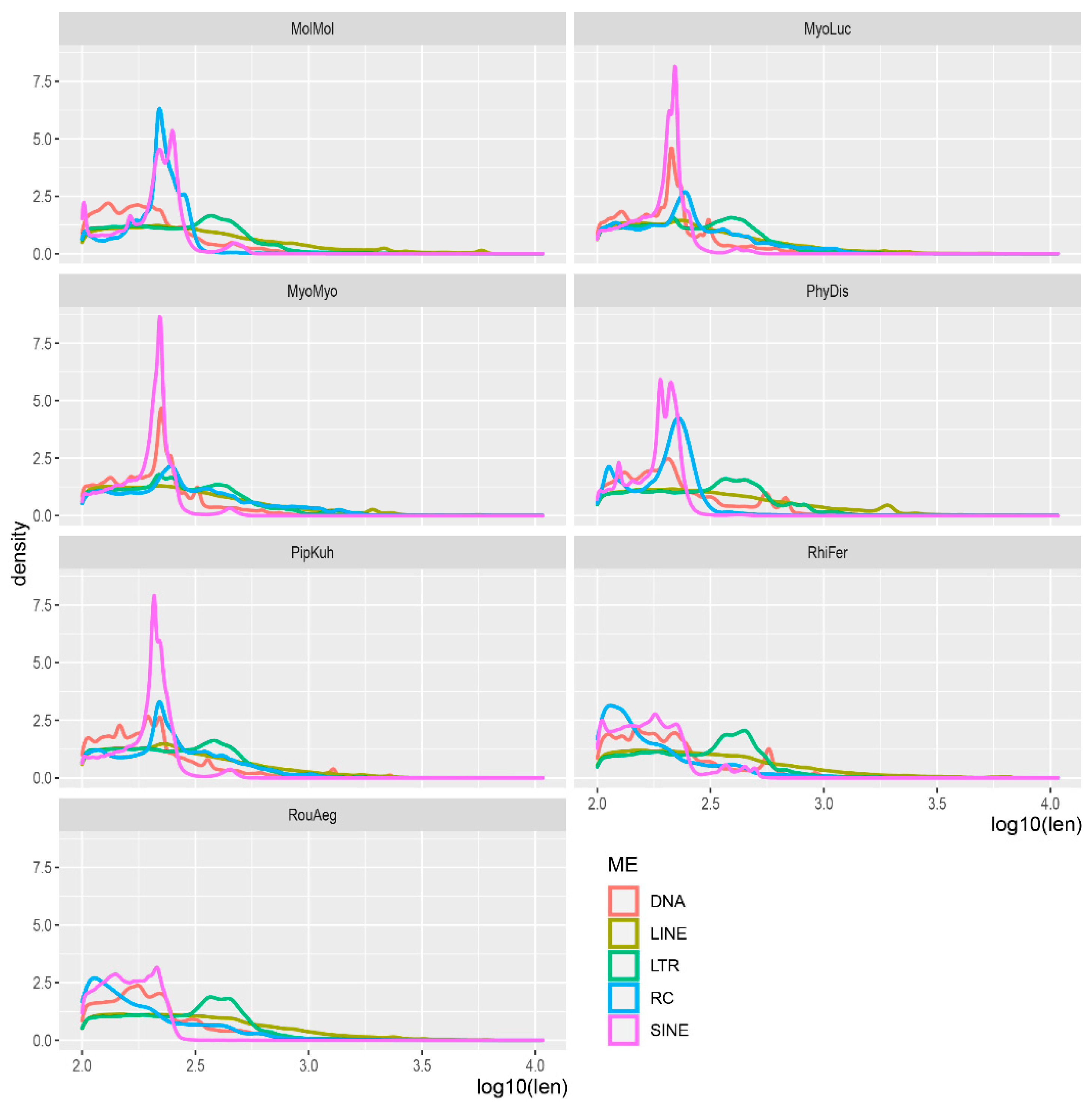
3.1. TE Composition in Seven Bat Species
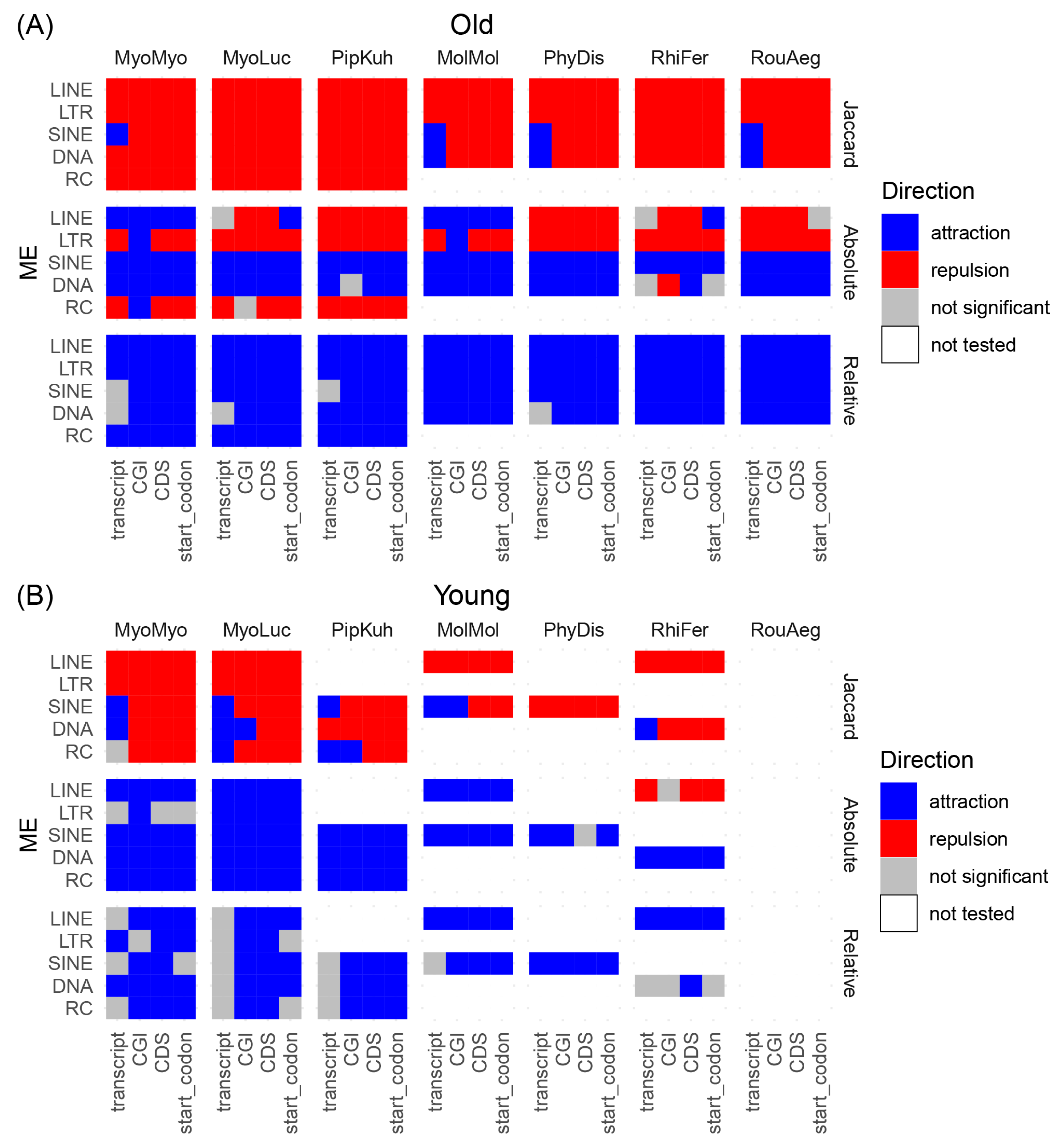
3.2. TE Distribution Relative to Genic Regions and CpG Islands
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McClintock, B. Chromosome organization and genic expression. Cold Spring Harb. Symp. Quant. Biol. 1951, 16, 13–47. [Google Scholar] [CrossRef] [PubMed]
- Plasterk, R.H. The origin of footprints of the Tc1 transposon of Caenorhabditis elegans. EMBO J. 1991, 10, 1919–1925. [Google Scholar] [CrossRef] [PubMed]
- Jiang, N.; Bao, Z.; Zhang, X.; Eddy, R.S.; Wessler, R.S. Pack-MULE transposable elements mediate gene evolution in plants. Nature 2004, 431, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Sen, S.K.; Han, K.; Wang, J.; Lee, J.; Wang, H.; Callinan, A.P.; Dyer, M.; Cordaux, R.; Liang, P.; Batzer, A.M. Human genomic deletions mediated by recombination between Alu elements. Am. J. Hum. Genet. 2006, 79, 41–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, J.; Zhang, Y.; Han, K.; Salem, A.H.; Sen, K.S.; Huff, D.C.; Zhou, Q.; Kirkness, F.E.; Levy, S.; Batzer, A.M.; et al. Mobile elements create structural variation: Analysis of a complete human genome. Genome Res. 2009, 19, 1516–1526. [Google Scholar] [CrossRef] [Green Version]
- Wells, J.N.; Feschotte, C. A Field Guide to Eukaryotic Transposable Elements. Annu Rev. Genet. 2020, 54, 539–561. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Zhang, Y.; Payer, L.M.; Lords, H.; Steranka, J.P.; Burns, K.H.; Xing, J. Polymorphic mobile element insertions contribute to gene expression and alternative splicing in human tissues. Genome Biol. 2020, 21, 185. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, T.; Preissl, S.; Amaral, M.L.; Grinstein, J.D.; Farah, E.N.; Destici, E.; Qiu, Y.; Hu, R.; Lee, A.Y.; et al. Transcriptionally active HERV-H retrotransposons demarcate topologically associating domains in human pluripotent stem cells. Nat. Genet. 2019, 51, 1380–1388. [Google Scholar] [CrossRef]
- Diehl, A.G.; Ouyang, N.; Boyle, A.P. Transposable elements contribute to cell and species-specific chromatin looping and gene regulation in mammalian genomes. Nat. Commun. 2020, 11, 1796. [Google Scholar] [CrossRef] [Green Version]
- Oliver, K.R.; Greene, W.K. Mobile DNA and the TE-Thrust hypothesis: Supporting evidence from the primates. Mob. DNA 2011, 2, 8. [Google Scholar] [CrossRef] [Green Version]
- Oliver, K.R.; Greene, W.K. Transposable elements and viruses as factors in adaptation and evolution: An expansion and strengthening of the TE-Thrust hypothesis. Ecol. Evol. 2012, 2, 2912–2933. [Google Scholar] [CrossRef] [PubMed]
- Jurka, J.; Bao, W.; Kojima, K.K. Families of transposable elements, population structure and the origin of species. Biol. Direct 2011, 6, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chuong, E.B.; Elde, N.C.; Feschotte, C. Regulatory activities of transposable elements: From conflicts to benefits. Nat. Rev. Genet. 2017, 18, 71–86. [Google Scholar] [CrossRef] [Green Version]
- Gerdes, P.; Richardson, S.R.; Mager, D.L.; Faulkner, G.J. Transposable elements in the mammalian embryo: Pioneers surviving through stealth and service. Genome Biol. 2016, 17, 100. [Google Scholar] [CrossRef] [Green Version]
- Chuong, E.B.; Elde, N.C.; Feschotte, C. Regulatory evolution of innate immunity through co-option of endogenous retroviruses. Science 2016, 351, 1083–1087. [Google Scholar] [CrossRef] [Green Version]
- Lynch, V.J.; Nnamani, M.C.; Kapusta, A.; Brayer, K.; Plaza, S.L.; Mazur, E.C.; Emera, D.; Sheikh, S.Z.; Grutzner, F.; Bauersachs, S.; et al. Ancient transposable elements transformed the uterine regulatory landscape and transcriptome during the evolution of mammalian pregnancy. Cell Rep. 2015, 10, 551–561. [Google Scholar] [CrossRef] [Green Version]
- Friedli, M.; Trono, D. The developmental control of transposable elements and the evolution of higher species. Annu. Rev. Cell Dev. Biol. 2015, 31, 429–451. [Google Scholar] [CrossRef]
- Hancks, D.C.; Kazazian, H.H., Jr. Active human retrotransposons: Variation and disease. Curr. Opin. Genet. Dev. 2012, 22, 191–203. [Google Scholar] [CrossRef] [Green Version]
- Xing, J.; Wang, H.; Belancio, V.P.; Cordaux, R.; Deininger, P.L.; Batzer, M.A. Emergence of primate genes by retrotransposon-mediated sequence transduction. Proc. Natl. Acad. Sci. USA 2006, 103, 17608–17613. [Google Scholar] [CrossRef] [Green Version]
- Xing, J.; Witherspoon, D.J.; Jorde, L.B. Mobile element biology: New possibilities with high-throughput sequencing. Trends Genet. 2013, 29, 280–289. [Google Scholar] [CrossRef] [Green Version]
- Zoonomia, C. A comparative genomics multitool for scientific discovery and conservation. Nature 2020, 587, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Medstrand, P.; van de Lagemaat, L.N.; Mager, D.L. Retroelement distributions in the human genome: Variations associated with age and proximity to genes. Genome Res. 2002, 12, 1483–1495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennetzen, J.L. Transposable element contributions to plant gene and genome evolution. Plant Mol. Biol. 2000, 42, 251–269. [Google Scholar] [CrossRef]
- Sotero-Caio, C.G.; Platt, R.N.; Suh, A.; Ray, D.A. Evolution and Diversity of Transposable Elements in Vertebrate Genomes. Genome Biol. Evol. 2017, 9, 161–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wicker, T.; Sabot, F.; Hua-Van, A.; Bennetzen, J.L.; Capy, P.; Chalhoub, B.; Flavell, A.; Leroy, P.; Morgante, M.; Panaud, O.; et al. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 2007, 8, 973–982. [Google Scholar] [CrossRef]
- Cost, G.J.; Feng, Q.; Jacquier, A.; Boeke, J.D. Human L1 element target-primed reverse transcription in vitro. EMBO J. 2002, 21, 5899–5910. [Google Scholar] [CrossRef] [PubMed]
- Hickman, A.B.; Perez, Z.N.; Zhou, L.; Musingarimi, P.; Ghirlando, R.; Hinshaw, J.E.; Craig, N.L.; Dyda, F. Molecular architecture of a eukaryotic DNA transposase. Nat. Struct. Mol. Biol. 2005, 12, 715–721. [Google Scholar] [CrossRef] [PubMed]
- Jurka, J.; Kohany, O.; Pavlicek, A.; Kapitonov, V.V.; Jurka, M.V. Duplication, coclustering, and selection of human Alu retrotransposons. Proc. Natl. Acad. Sci. USA 2004, 101, 1268–1272. [Google Scholar] [CrossRef] [Green Version]
- Jurka, J. Sequence patterns indicate an enzymatic involvement in integration of mammalian retroposons. Proc. Natl. Acad. Sci. USA 1997, 94, 1872–1877. [Google Scholar] [CrossRef] [Green Version]
- Jurka, J.; Kohany, O.; Pavlicek, A.; Kapitonov, V.; Jurka, M. Clustering, duplication and chromosomal distribution of mouse SINE retrotransposons. Cytogenet. Genome Res. 2005, 110, 117–123. [Google Scholar] [CrossRef]
- Wu, H.; Caffo, B.; Jaffee, H.A.; Irizarry, R.A.; Feinberg, A.P. Redefining CpG islands using hidden Markov models. Biostatistics 2010, 11, 499–514. [Google Scholar] [CrossRef] [PubMed]
- Jebb, D.; Huang, Z.; Pippel, M.; Hughes, G.M.; Lavrichenko, K.; Devanna, P.; Winkler, S.; Jermiin, L.S.; Skirmuntt, E.C.; Katzourakis, A.; et al. Six reference-quality genomes reveal evolution of bat adaptations. Nature 2020, 583, 578–584. [Google Scholar] [CrossRef]
- Hubley, R.; Finn, R.D.; Clements, J.; Eddy, S.R.; Jones, T.A.; Bao, W.; Smit, A.F.; Wheeler, T.J. The Dfam database of repetitive DNA families. Nucleic Acids Res. 2016, 44, D81–D89. [Google Scholar] [CrossRef] [Green Version]
- Osmanski, A.B.; Paulat, N.S.; Korstian, J.; Grimshaw, J.R.; Halsey, M.; Sullivan, K.A.M.; Moreno-Santillán, D.D.; Crookshanks, C.; Roberts, J.; Garcia, C.; et al. Insights into mammalian TE diversity via the curation of 200+ mammalian genome assemblies. Science 2022. submitted. [Google Scholar]
- Foley, N.M.; Mason, V.C.; Harris, A.J.; Bredemeyer, K.R.; Damas, J.; Lewin, H.A.; Eizirik, E.; Gatesy, J.; Zoonomia, C.; Springer, M.S.; et al. A genomic timescale for placental mammal evolution. Science 2022, in press. [Google Scholar]
- Kumar, S.; Stecher, G.; Suleski, M.; Hedges, S.B. TimeTree: A Resource for Timelines, Timetrees, and Divergence Times. Mol. Biol. Evol. 2017, 34, 1812–1819. [Google Scholar] [CrossRef] [PubMed]
- Favorov, A.V.; Mularoni, L.; Cope, L.M.; Medvedeva, Y.; Mironov, A.A.; Makeev, V.; Wheelan, S.J. Exploring Massive, Genome Scale Datasets with the GenometriCorr Package. PLoS Comput. Biol. 2012, 8, e1002529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Platt, R.N., 2nd; Mangum, S.F.; Ray, D.A. Pinpointing the vesper bat transposon revolution using the Miniopterus natalensis genome. Mob. DNA 2016, 7, 12. [Google Scholar] [CrossRef] [Green Version]
- Ray, D.A.; Feschotte, C.; Pagan, H.J.; Smith, J.D.; Pritham, E.J.; Arensburger, P.; Atkinson, P.W.; Craig, N.L. Multiple waves of recent DNA transposon activity in the bat, Myotis lucifugus. Genome Res. 2008, 18, 717–728. [Google Scholar] [CrossRef] [Green Version]
- Ray, D.A.; Pagan, H.J.; Thompson, M.L.; Stevens, R.D. Bats with hATs: Evidence for recent DNA transposon activity in genus Myotis. Mol. Biol. Evol. 2007, 24, 632–639. [Google Scholar] [CrossRef]
- Pritham, E.J.; Feschotte, C. Massive amplification of rolling-circle transposons in the lineage of the bat Myotis lucifugus. Proc. Natl. Acad. Sci. USA 2007, 104, 1895–1900. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Scott, L.; Wichman, H.A. Tracing the history of LINE and SINE extinction in sigmodontine rodents. Mob. DNA 2019, 10, 22. [Google Scholar] [CrossRef] [Green Version]
- Wichman, H.A.; Scott, L.; Howell, E.K.; Martinez, A.R.; Yang, L.; Baker, R.J. Flying Around in the Genome: Characterization of LINE-1 in Chiroptera. Spec. Publ. Tex. Tech Univ. Mus. 2019, 71, 379–392. [Google Scholar]
- Hughes, J.F.; Coffin, J.M. Human endogenous retrovirus K solo-LTR formation and insertional polymorphisms: Implications for human and viral evolution. Proc. Natl. Acad. Sci. USA 2004, 101, 1668–1672. [Google Scholar] [CrossRef] [Green Version]
- Szak, S.T.; Pickeral, O.K.; Makalowski, W.; Boguski, M.S.; Landsman, D.; Boeke, J.D. Molecular archeology of L1 insertions in the human genome. Genome Biol. 2002, 3, research0052.1–0052.18. [Google Scholar] [CrossRef]
- Saxonov, S.; Berg, P.; Brutlag, D.L. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc. Natl. Acad. Sci. USA 2006, 103, 1412–1417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abrusán, G.; Krambeck, H.-J. The Distribution of L1 and Alu Retroelements in Relation to GC Content on Human Sex Chromosomes Is Consistent with the Ectopic Recombination Model. J. Mol. Evol. 2006, 63, 484. [Google Scholar] [CrossRef]
- Kvikstad, E.M.; Makova, K.D. The (r)evolution of SINE versus LINE distributions in primate genomes: Sex chromosomes are important. Genome Res. 2010, 20, 600–613. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paulat, N.S.; McGuire, E.; Subramanian, K.; Osmanski, A.B.; Moreno-Santillán, D.D.; Ray, D.A.; Xing, J. Transposable Elements in Bats Show Differential Accumulation Patterns Determined by Class and Functionality. Life 2022, 12, 1190. https://doi.org/10.3390/life12081190
Paulat NS, McGuire E, Subramanian K, Osmanski AB, Moreno-Santillán DD, Ray DA, Xing J. Transposable Elements in Bats Show Differential Accumulation Patterns Determined by Class and Functionality. Life. 2022; 12(8):1190. https://doi.org/10.3390/life12081190
Chicago/Turabian StylePaulat, Nicole S., Erin McGuire, Krishnamurthy Subramanian, Austin B. Osmanski, Diana D. Moreno-Santillán, David A. Ray, and Jinchuan Xing. 2022. "Transposable Elements in Bats Show Differential Accumulation Patterns Determined by Class and Functionality" Life 12, no. 8: 1190. https://doi.org/10.3390/life12081190
APA StylePaulat, N. S., McGuire, E., Subramanian, K., Osmanski, A. B., Moreno-Santillán, D. D., Ray, D. A., & Xing, J. (2022). Transposable Elements in Bats Show Differential Accumulation Patterns Determined by Class and Functionality. Life, 12(8), 1190. https://doi.org/10.3390/life12081190