
Realistic Gene Transfer to Gene Duplication Ratios Identify Different Roots in the Bacterial Phylogeny Using a Tree Reconciliation Method


Abstract
:1. Introduction
2. Material and Methods
3. Results and Discussion
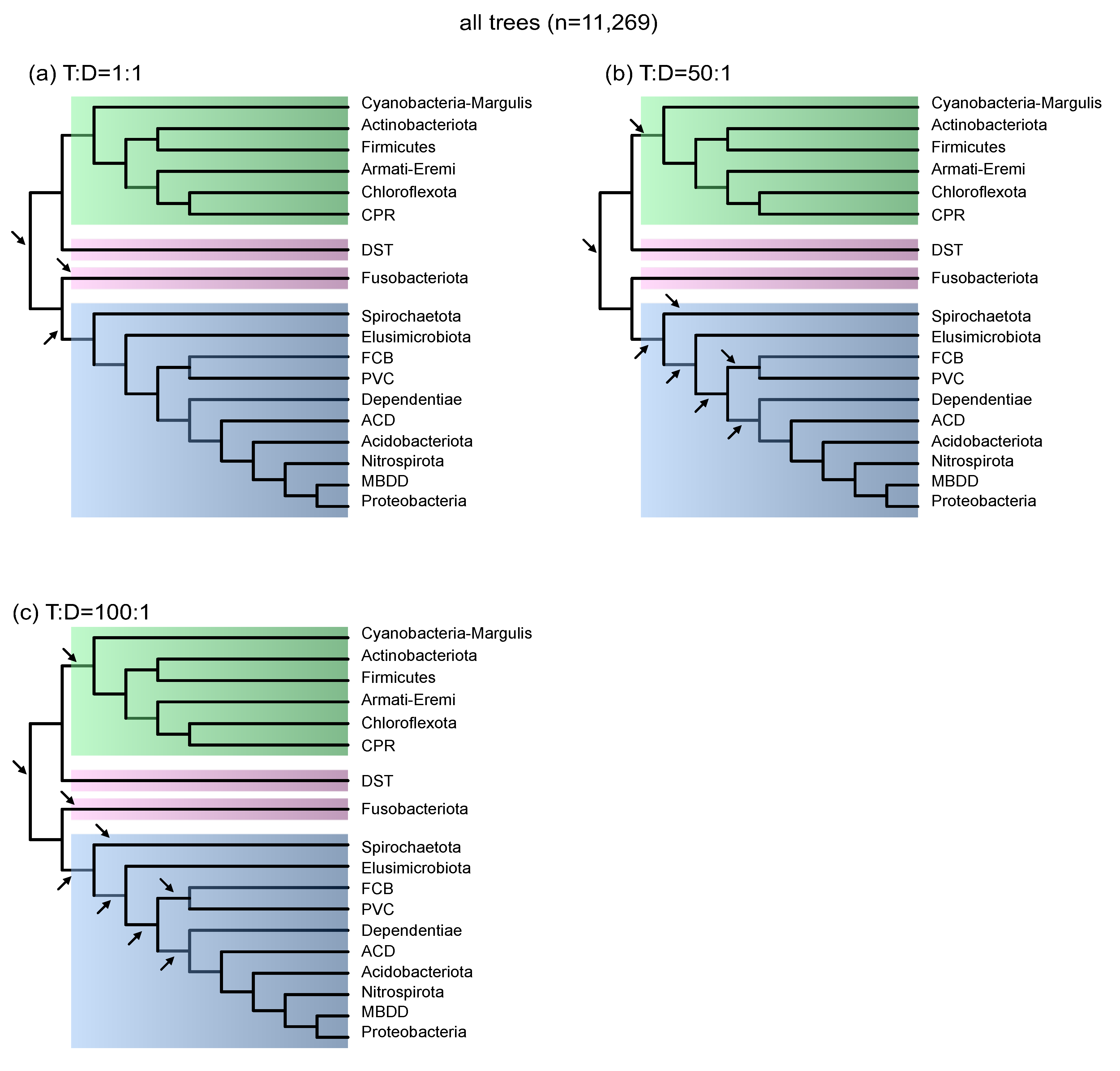
3.1. Gene Transfer to Gene Duplication Ratios Constrain the Number of Inferred Species Tree Roots
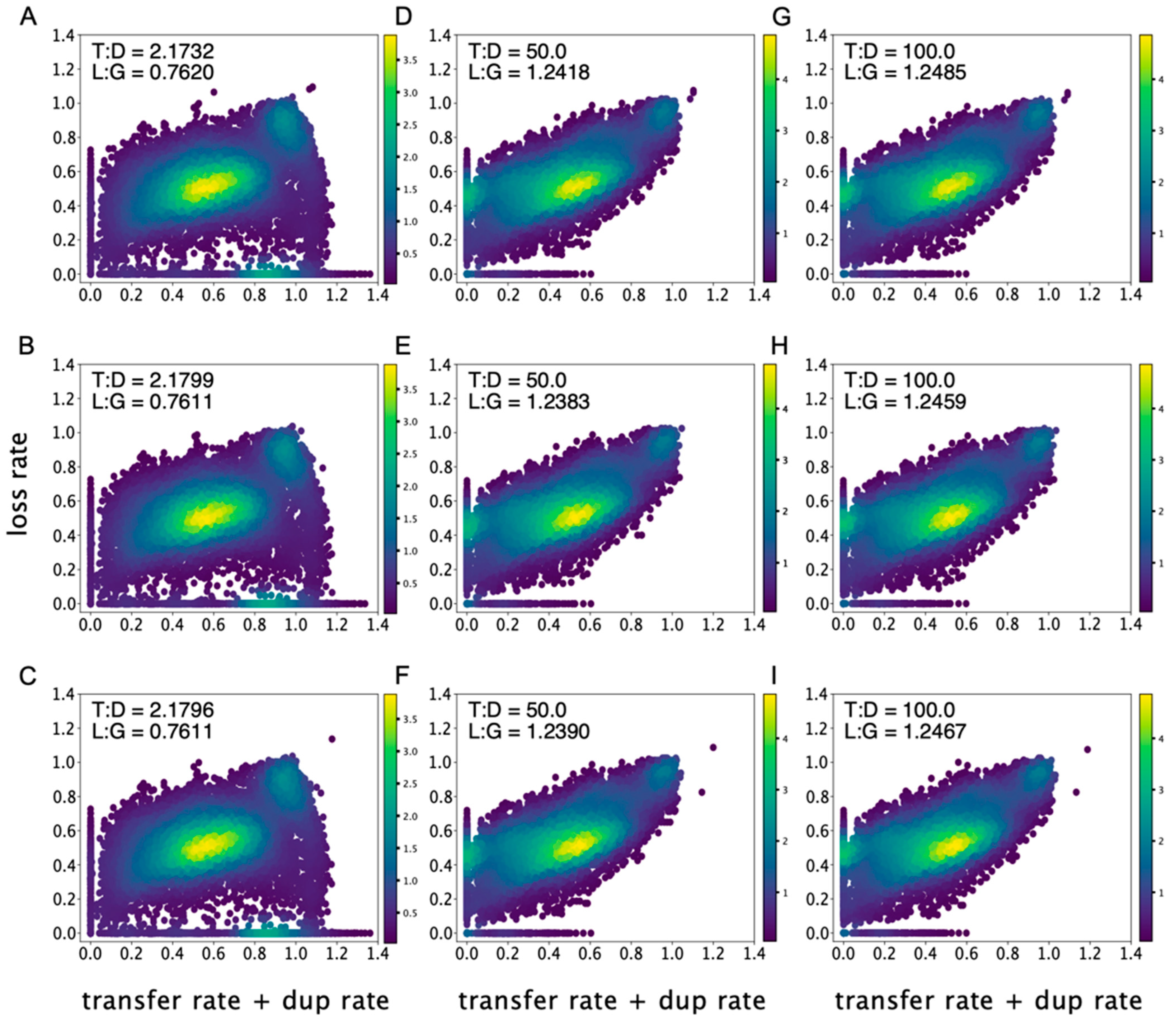
3.2. Imbalance of Gene Gains and Gene Losses
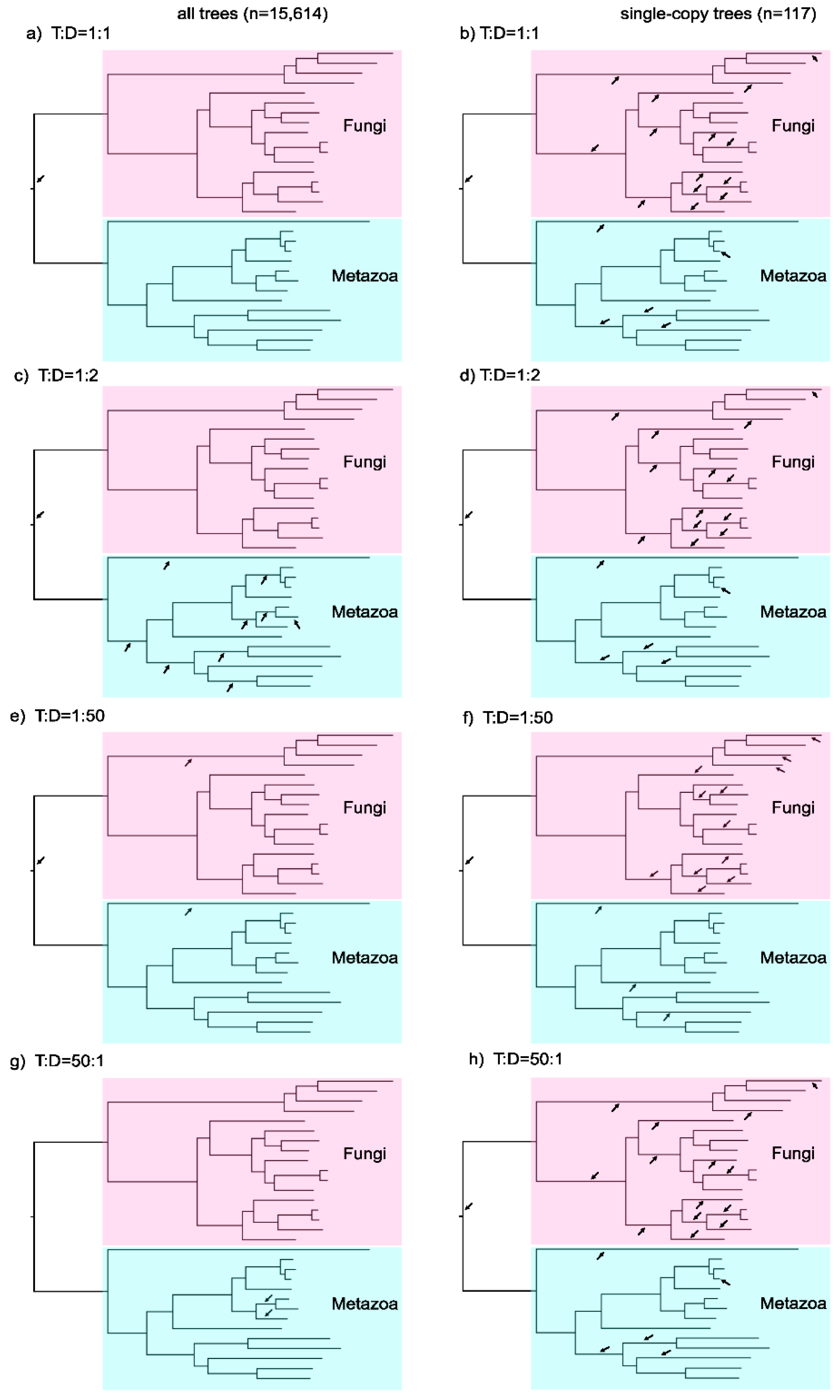
3.3. Testing ALE on a Simple Phylogenetic Case: The Fungal-Metazoan Root
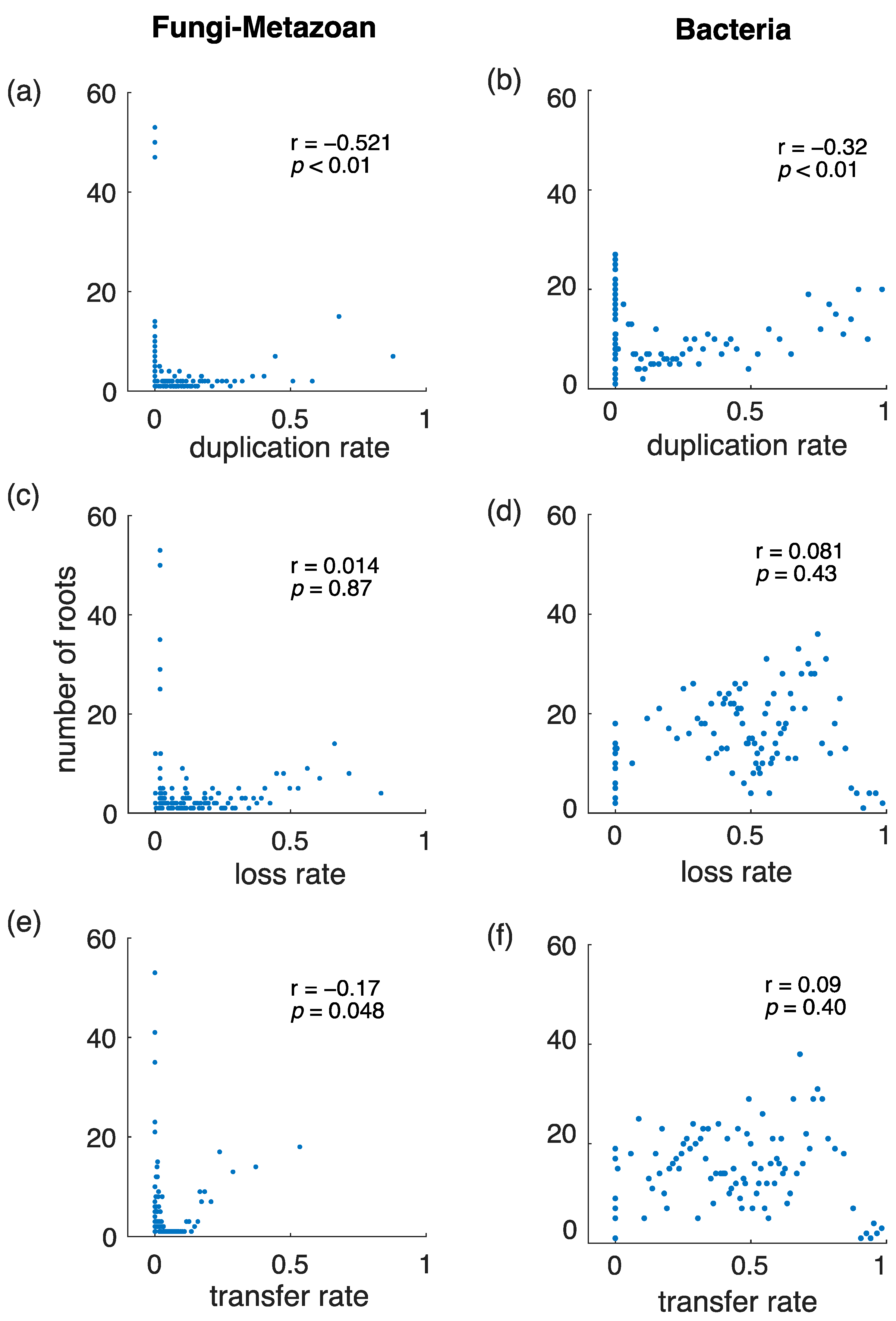
3.4. Gene Duplications Are More Informative Than Gene Transfers for Rooting Species Trees
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kluge, A.G.; Farris, J.S. Quantitative phyletics and the evolution of anurans. Syst. Zool. 1969, 18, 1–32. [Google Scholar] [CrossRef]
- Brown, J.R.; Doolittle, W.F. Root of the universal tree of life based on ancient aminoacyl-tRNA synthetase gene duplications. Proc. Natl. Acad. Sci. USA 1995, 92, 2441–2445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farris, J.S. Estimating phylogenetic trees from distance matrices. Am. Nat. 1972, 106, 645–668. [Google Scholar] [CrossRef]
- Tria, F.D.K.; Landan, G.; Dagan, T. Phylogenetic rooting using minimal ancestor deviation. Nat. Ecol. Evol. 2017, 1, 0193. [Google Scholar] [CrossRef] [PubMed]
- Lepage, T.; Bryant, D.; Philippe, H.; Lartillot, N. A general comparison of relaxed molecular clock models. Mol. Biol. Evol. 2007, 24, 2669–2680. [Google Scholar] [CrossRef] [Green Version]
- Williams, T.A.; Heaps, S.E.; Cherlin, S.; Nye, T.M.W.; Boys, R.J.; Embley, T.M. New substitution models for rooting phylogenetic trees. Philos. Trans. R. Soc. B Biol. Sci. 2015, 370, 20140336. [Google Scholar] [CrossRef] [Green Version]
- Graham, S.W.; Olmstead, R.G.; Barrett, S.C.H. Rooting phylogenetic trees with distant outgroups: A case study from the commelinoid monocots. Mol. Biol. Evol. 2002, 19, 1769–1781. [Google Scholar] [CrossRef] [Green Version]
- Huelsenbeck, J.P.; Bollback, J.P.; Levine, A.M. Inferring the root of a phylogenetic tree. Syst. Biol. 2002, 51, 32–43. [Google Scholar] [CrossRef] [Green Version]
- Wade, T.; Thiberio Rangel, L.; Kundu, S.; Fournier, G.P.; Bansal, M.S. Assessing the accuracy of phylogenetic rooting methods on prokaryotic gene families. PLoS ONE 2020, 15, e0232950. [Google Scholar] [CrossRef]
- Lamarca, A.P.; Mello, B.; Schrago, C.G. The performance of outgroup-free rooting under evolutionary radiations. Mol. Phylogenet. Evol. 2022, 169, 107434. [Google Scholar] [CrossRef]
- Goodman, M.; Czelusniak, J.; Moore, G.W.; Romero-Herrera, A.E.; Matsuda, G. Fitting the gene lineage into its species lineage, a parsimony strategy illustrated by cladograms constructed from globin sequences. Syst. Zool. 1979, 28, 132–163. [Google Scholar] [CrossRef]
- Page, R.D.M. Maps between trees and cladistic analysis of historical associations among genes, organisms, and areas. Syst. Biol. 1994, 43, 58–77. [Google Scholar]
- Bansal, M.S.; Alm, E.J.; Kellis, M. Efficient algorithms for the reconciliation problem with gene duplication, horizontal transfer and loss. Bioinformatics 2012, 28, i283–i291. [Google Scholar] [CrossRef]
- Szöllosi, G.J.; Rosikiewicz, W.; Boussau, B.; Tannier, E.; Daubin, V. Efficient exploration of the space of reconciled gene trees. Syst. Biol. 2013, 62, 901–912. [Google Scholar] [CrossRef] [Green Version]
- Doyon, J.; Ranwez, V.; Daubin, V.; Berry, V. Models, algorithms and programs for phylogeny reconciliation. Brief. Bioinform. 2011, 12, 392–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimodaira, H. An approximately unbiased test of phylogenetic tree selection. Syst. Biol. 2002, 51, 492–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coleman, G.A.; Davín, A.A.; Mahendrarajah, T.A.; Szánthó, L.L.; Spang, A.; Hugenholtz, P.; Szöllősi, G.J.; Williams, T.A. A rooted phylogeny resolves early bacterial evolution. Science 2021, 372, eabe0511. [Google Scholar] [CrossRef]
- Xavier, J.C.; Gerhards, R.E.; Wimmer, J.L.E.; Brueckner, J.; Tria, F.D.K.; Martin, W.F. The metabolic network of the last bacterial common ancestor. Commun. Biol. 2021, 4, 413. [Google Scholar] [CrossRef]
- Pál, C.; Papp, B.; Lercher, M.J. Adaptive evolution of bacterial metabolic networks by horizontal gene transfer. Nat. Genet. 2005, 37, 1372–1375. [Google Scholar] [CrossRef]
- Dagan, T.; Artzy-Randrup, Y.; Martin, W.F. Modular networks and cumulative impact of lateral transfer in prokaryote genome evolution. Proc. Natl. Acad. Sci. USA 2008, 105, 10039–10044. [Google Scholar] [CrossRef] [Green Version]
- Treangen, T.J.; Rocha, E.P.C. Horizontal transfer, not duplication, drives the expansion of protein families in prokaryotes. PLoS Genet. 2011, 7, e1001284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tria, F.D.K.; Martin, W.F. Gene duplications are at least 50 times less frequent than gene transfers in prokaryotic genomes. Genome Biol. Evol. 2021, 13, evab224. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; Szklarczyk, D.; Forslund, K.; Cook, H.; Heller, D.; Walter, M.C.; Rattei, T.; Mende, D.R.; Sunagawa, S.; Kuhn, M.; et al. EGGNOG 4.5: A hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 2016, 44, D286–D293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Shimodaira, H. Testing regions with nonsmooth boundaries via multiscale bootstrap. J. Stat. Plan Inference 2008, 138, 1227–1241. [Google Scholar] [CrossRef]
- Nagies, F.S.P.; Brueckner, J.; Tria, F.D.K.; Martin, W.F. A spectrum of verticality across genes. PLoS Genet. 2020, 16, e1009200. [Google Scholar] [CrossRef]
- Cohen, O.; Pupko, T. Inference and characterization of horizontally transferred gene families using stochastic mapping. Mol. Biol. Evol. 2010, 27, 703–713. [Google Scholar] [CrossRef]
- Dagan, T.; Martin, W.F. Ancestral genome sizes specify the minimum rate of lateral gene transfer during prokaryote evolution. Proc. Natl. Acad. Sci. USA 2007, 104, 870–875. [Google Scholar] [CrossRef] [Green Version]
- Ochman, H.; Lawrence, J.G.; Groisman, E.A. Lateral gene transfer and the nature of bacterial innovation. Nature 2000, 405, 299–304. [Google Scholar] [CrossRef]
- Sela, I.; Wolf, Y.I.; Koonin, E.V. Theory of prokaryotic genome evolution. Proc. Natl. Acad. Sci. USA 2016, 113, 11399–11407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lane, N.; Martin, W.F. The energetics of genome complexity. Nature 2010, 467, 929–934. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Makarova, K.S.; Aravind, L. Horizontal Gene Transfer in Prokaryotes: Quantification and Classification. Dict. Genom. Transcr. Proteom. 2001, 55, 709–742. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Wolf, Y.I. Genomics of bacteria and archaea: The emerging dynamic view of the prokaryotic world. Nucleic Acids Res. 2008, 36, 6688–6719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snel, B.; Bork, P.; Huynen, M.A. Genomes in flux: The evolution of Archaeal and Proteobacterial gene content. Genome Res. 2002, 12, 17–25. [Google Scholar] [CrossRef] [Green Version]
- Mirkin, B.G.; Fenner, T.I.; Galperin, M.Y.; Koonin, E.V. Algorithms for computing parsimonious evolutionary scenarios for genome evolution, the last universal common ancestor and dominance of horizontal gene transfer in the evolution of prokaryotes. BMC Evol. Biol. 2003, 3, 1–34. [Google Scholar] [CrossRef]
- Puigbò, P.; Lobkovsky, A.E.; Kristensen, D.M.; Wolf, Y.I.; Koonin, E.V. Genomes in turmoil: Quantification of genome dynamics in prokaryote supergenomes. BMC Med. 2014, 12, 66. [Google Scholar] [CrossRef]
- Larsson, J.; Nylander, J.A.A.; Bergman, B. Genome fluctuations in cyanobacteria reflect evolutionary, developmental and adaptive traits. BMC Evol. Biol. 2011, 11, 187. [Google Scholar] [CrossRef] [Green Version]
- Stucken, K.; John, U.; Cembella, A.; Murillo, A.A.; Soto-Liebe, K.; Fuentes-Valdés, J.J.; Friedel, M.; Plominsky, A.M.; Vásquez, M.; Glöckner, G. The smallest known genomes of multicellular and toxic cyanobacteria: Comparison, minimal gene sets for linked traits and the evolutionary implications. PLoS ONE 2010, 5, e9235. [Google Scholar] [CrossRef] [Green Version]
- Herdman, M.; Janvier, M.; Rippka, R.; Stanier, R.Y. Genome size of cyanobacteria. J. Gen. Microbiol. 1979, 111, 73–85. [Google Scholar] [CrossRef] [Green Version]
- Hammerschmidt, K.; Landan, G.; Tria, F.D.K.; Alcorta, J.; Dagan, T. The order of trait emergence in the evolution of cyanobacterial multicellularity. Genome Biol. Evol. 2021, 13, evaa249. [Google Scholar] [CrossRef] [PubMed]
- Stechmann, A.; Cavalier-Smith, T. Rooting the eukaryote tree by using a derived gene fusion. Science 2002, 297, 89–91. [Google Scholar] [CrossRef] [PubMed]
- Katz, L.A.; Grant, J.R.; Parfrey, L.W.; Burleigh, J.G. Turning the crown upside down: Gene tree parsimony roots the eukaryotic tree of life. Syst. Biol. 2012, 61, 653–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tria, F.D.K.; Brueckner, J.; Skejo, J.; Xavier, J.C.; Kapust, N.; Knopp, M.; Wimmer, J.L.W.; Nagies, F.S.P.; Zimorski, V.; Gould, S.B.; et al. Gene duplications trace mitochondria to the onset of eukaryote complexity. Genome Biol. Evol. 2021, 13, evab055. [Google Scholar] [CrossRef]
- Hahn, M.W. Bias in phylogenetic tree reconciliation methods: Implications for vertebrate genome evolution. Genome Biol. 2007, 8, R141. [Google Scholar] [CrossRef] [Green Version]
- Rannala, B. Identifiability of parameters in MCMC Bayesian inference of phylogeny. Syst. Biol. 2002, 51, 754–760. [Google Scholar] [CrossRef] [Green Version]
- Emms, D.M.; Kelly, S. STRIDE: Species tree root inference from gene duplication events. Mol. Biol. Evol. 2017, 34, 3267–3278. [Google Scholar] [CrossRef]
- Doolittle, W.F.; Bapteste, E. Pattern pluralism and the Tree of Life hypothesis. Proc. Natl. Acad. Sci. USA 2007, 104, 2043–2049. [Google Scholar] [CrossRef] [Green Version]
- Bapteste, E.; O’Malley, M.A.; Beiko, R.G.; Ereshefsky, M.; Gogarten, J.P.; Franklin-Hall, L.; Lapointe, F.; Dupré, J.; Dagan, T.; Boucher, Y.; et al. Prokaryotic evolution and the tree of life are two different things. Biol. Direct. 2009, 4, 34. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Transfer Rate | Duplication Rate | Loss Rate | Gain Rate | Loss/Gain Ratio | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Dataset | Mean | Median | Std | Mean | Median | Std | Mean | Median | Std | Mean | Median | Std | |
| 1:1 | 4.18 × 10−1 | 4.30 × 10−1 | 2.72 × 10−1 | 1.93 × 10−1 | 5.53 × 10−2 | 2.82 × 10−1 | 4.65 × 10−1 | 4.96 × 10−1 | 2.59 × 10−1 | 6.12 × 10−1 | 6.14 × 10−1 | 2.85 × 10−1 | 7.59 × 10−1 |
| 50:1 | 4.18 × 10−1 | 4.30 × 10−1 | 2.72 × 10−1 | 8.4 × 10−3 | 8.6 × 10−3 | 5.4 × 10−3 | 5.29 × 10−1 | 5.25 × 10−1 | 2.08 × 10−1 | 4.27 × 10−1 | 4.39 × 10−1 | 2.77 × 10−1 | 1.23 × 100 |
| 100:1 | 4.18 × 10−1 | 4.30 × 10−1 | 2.72 × 10−1 | 4.2 × 10−3 | 4.3 × 10−3 | 2.7 × 10−3 | 5.27 × 10−1 | 5.25 × 10−1 | 2.07 × 10−1 | 4.23 × 10−1 | 4.35 × 10−1 | 2.75 × 10−1 | 1.24 × 100 |
| 50:1 or more | 9.91 × 100 | 2.75 × 100 | 1.39 × 101 | 1.93 × 10−1 | 5.53 × 10−2 | 2.82 × 10−1 | 5.51 × 100 | 1.15 × 100 | 8.02 × 100 | 1.01 × 101 | 2.81 × 100 | 1.42 × 101 | 5.45 × 10−1 |
| Transfer Rate | Duplication Rate | Loss Rate | Gain Rate | Loss/Gain Ratio | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Dataset | Mean | Median | Std | Mean | Median | Std | Mean | Median | Std | Mean | Median | Std | |
| 1:1 | 3.56 × 10−2 | 1.00 × 10−6 | 7.73 × 10−2 | 7.30 × 10−2 | 5.67 × 10−6 | 1.29 × 10−1 | 1.40 × 10−1 | 8.81 × 10−2 | 1.67 × 10−1 | 1.08 × 10−1 | 5.54 × 10−2 | 1.62 × 10−1 | 1.29 × 101 |
| 1:2 | 3.56 × 10−2 | 1.00 × 10−6 | 7.73 × 10−2 | 7.21 × 10−2 | 2.00 × 10−6 | 1.54 × 10−1 | 1.54 × 10−1 | 8.59 × 10−2 | 2.22 × 10−1 | 1.06 × 10−1 | 3.00 × 10−6 | 2.31 × 10−1 | 1.44 × 101 |
| 1:50 | 3.56 × 10−2 | 1.00 × 10−6 | 7.73 × 10−2 | 1.77 × 101 | 5.00 × 10−5 | 3.86 × 101 | 1.15 × 101 | 1.71 × 10−1 | 2.42 × 101 | 1.81 × 101 | 5.1 × 10−5 | 3.94 × 101 | 6.36 × 10−1 |
| 50:1 | 3.56 × 10−2 | 1.00 × 10−6 | 7.73 × 10−2 | 7.0 × 10−4 | 2.00 × 10−8 | 1.5 × 10−3 | 1.27 × 10−1 | 8.52 × 10−2 | 1.42 × 10−1 | 3.63 × 10−2 | 1.02 × 10−6 | 7.89 × 10−2 | 3.52 × 101 |
| 100:1 | 3.56 × 10−2 | 1.00 × 10−6 | 7.73 × 10−2 | 4.0 × 10−4 | 1.00 × 10−8 | 8.0 × 10−4 | 1.27 × 10−1 | 8.44 × 10−2 | 1.41 × 10−1 | 3.59 × 10−2 | 1.00 × 10−6 | 7.81 × 10−2 | 3.54 × 101 |
| 1:1 (sc) | 4.8 × 10−3 | 1.00 × 10−10 | 9.3 × 10−3 | 2.26 × 10−7 | 1.0 × 10−10 | 6.34 × 10−7 | 4.6 × 10−3 | 1.0 × 10−10 | 9.4 × 10−3 | 4.8 × 10−3 | 2.0 × 10−10 | 9.3 × 10−3 | 9.58 × 10−1 |
| 1:2 (sc) | 4.8 × 10−3 | 1.00 × 10−10 | 9.3 × 10−3 | 9.7 × 10−3 | 2.0 × 10−10 | 1.87 × 10−2 | 5.4 × 10−3 | 1.0 × 10−10 | 1.18 × 10−2 | 1.45 × 10−2 | 3.0 × 10−10 | 2.80 × 10−2 | 3.72 × 10−1 |
| 1:50 (sc) | 4.8 × 10−3 | 1.00 × 10−10 | 9.3 × 10−3 | 2.42 × 10−1 | 5.00 × 10−9 | 4.66 × 10−1 | 9.69 × 10−2 | 1.0 × 10−10 | 2.27 × 10−1 | 2.47 × 10−1 | 5.1 × 10−9 | 4.75 × 10−1 | 3.92 × 10−1 |
| 50:1 (sc) | 4.8 × 10−3 | 1.00 × 10−10 | 9.3 × 10−3 | 9.69 × 10−5 | 2.0 × 10−12 | 2.0 × 10−4 | 4.6 × 10−3 | 1.0 × 10−10 | 9.4 × 10−3 | 4.9 × 10−3 | 1.0 × 10−10 | 9.5 × 10−3 | 9.38 × 10−1 |
| 100:1 (sc) | 4.8 × 10−3 | 1.00 × 10−10 | 9.3 × 10−3 | 4.84 × 10−5 | 1.0 × 10−12 | 9.33 × 10−5 | 4.6 × 10−3 | 1.0 × 10−10 | 9.4 × 10−3 | 4.9 × 10−3 | 1.0 × 10−10 | 9.4 × 10−3 | 9.38 × 10−1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bremer, N.; Knopp, M.; Martin, W.F.; Tria, F.D.K. Realistic Gene Transfer to Gene Duplication Ratios Identify Different Roots in the Bacterial Phylogeny Using a Tree Reconciliation Method. Life 2022, 12, 995. https://doi.org/10.3390/life12070995
Bremer N, Knopp M, Martin WF, Tria FDK. Realistic Gene Transfer to Gene Duplication Ratios Identify Different Roots in the Bacterial Phylogeny Using a Tree Reconciliation Method. Life. 2022; 12(7):995. https://doi.org/10.3390/life12070995
Chicago/Turabian StyleBremer, Nico, Michael Knopp, William F. Martin, and Fernando D. K. Tria. 2022. "Realistic Gene Transfer to Gene Duplication Ratios Identify Different Roots in the Bacterial Phylogeny Using a Tree Reconciliation Method" Life 12, no. 7: 995. https://doi.org/10.3390/life12070995
APA StyleBremer, N., Knopp, M., Martin, W. F., & Tria, F. D. K. (2022). Realistic Gene Transfer to Gene Duplication Ratios Identify Different Roots in the Bacterial Phylogeny Using a Tree Reconciliation Method. Life, 12(7), 995. https://doi.org/10.3390/life12070995