
Lipid Metabolism and Cancer


Abstract
1. Introduction
2. Function of Lipids in Tumor Cells
3. Synthesis and Storage of Lipids
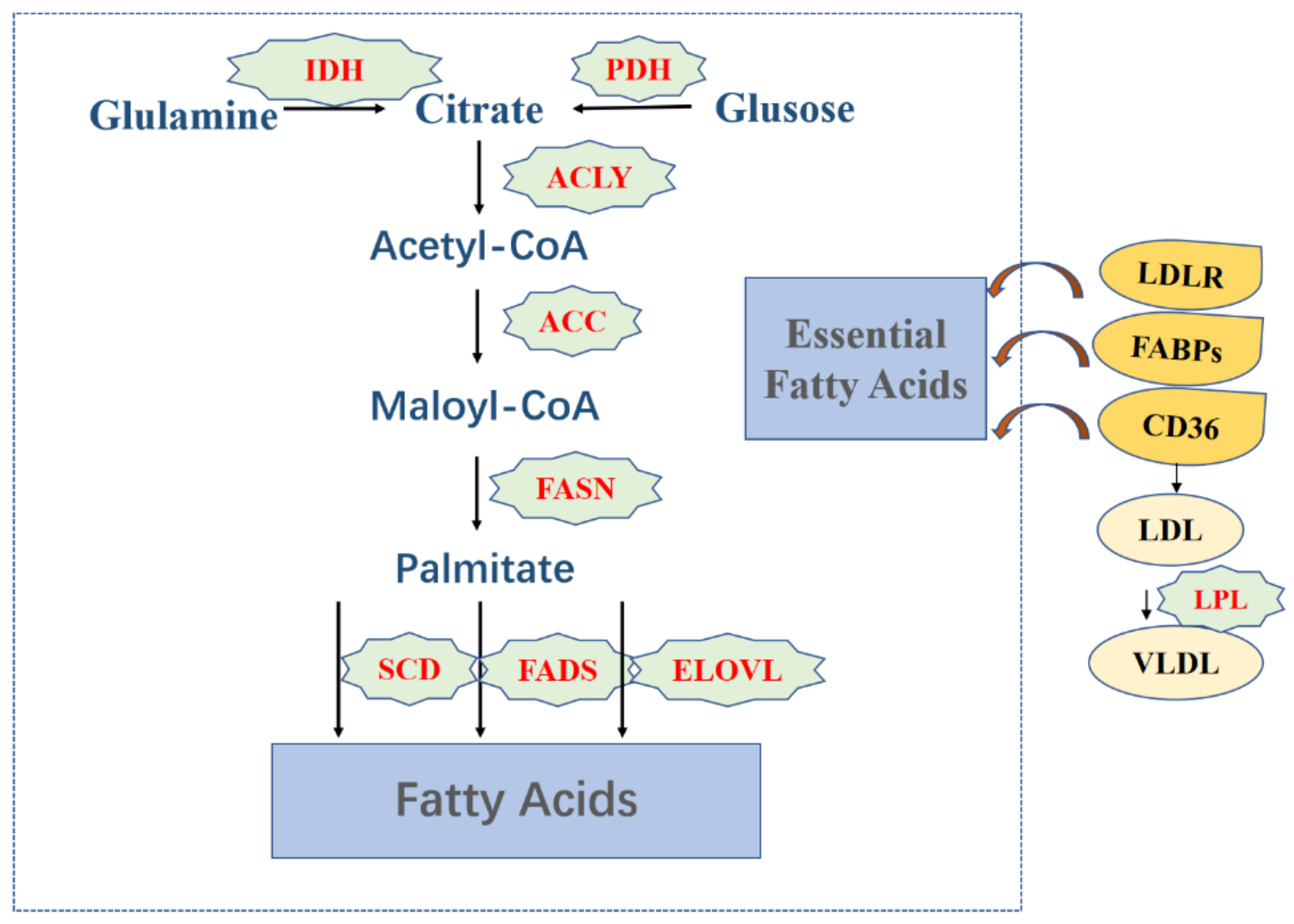
3.1. Fatty Acids
3.2. Phospholipids
3.3. Cholesterol
4. Lipid Metabolism and Tumors
4.1. Fatty Acids Metabolism Regulates Tumor Cells
4.2. Phospholipids Metabolism Regulates Tumor Cells
4.3. Cholesterol Metabolism Regulates Tumor Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Author, Year | Target Protein | Action Site | Inhibiter | Type of Cancer | Reference |
|---|---|---|---|---|---|
| Wei, J. et al., 2019 | ACLY | Catalyzes acetyl-CoA binding | SB-2049990 | Lung cancer, Breast cancer | [35] |
| Lally, J. et al., 2019 | ACC | Catalyzed acetyl-CoA carboxylation to Malonyl-CoA | ND-654 | Non-small cell lung cancer, liver cancer | [41] |
| VincentB, B.M. et al., 2018 | FASN | Catalytic synthesis of palmitate | TVB2640 | Pancreatic cancer | [46] |
| Tesfay, L. et al., 2019 | SCD1 | Elongate palmitate | BZ36 | Ovarian cancer | [49] |
| Jiang, M. et al., 2019 | CD36 | Exogenous intake of fatty acids | Anti-CD36 antibody | Stomach cancer | [51] |
| Bjarnadottir, O. et al., 2020 | HMGCR | HMG-CoA is converted to valproic acid | Statins | Breast cancer | [63] |
5. Regulation of Lipid Metabolism in the Tumor Microenvironment
5.1. Sterol Regulatory Element-Binding Proteins (SREBPs)
5.1.1. Fatty Acid Metabolism in the Tumor Microenvironment
5.1.2. Cholesterol Metabolism in the Tumor Microenvironment
5.2. Peroxisome Proliferator-Activated Receptors (PPARs)
5.2.1. Fatty Acid Metabolism in the Tumor Microenvironment
5.2.2. Lipogenesis
5.3. Liver X Receptors (LXRs)
5.4. AMP-Activated Protein Kinase (AMPK)
5.5. MicroRNA and LncRNA
5.5.1. microRNA
5.5.2. LncRNA
| Cancer Type | Gene | Signaling Pathway | Function | Model | Cell Lines | Author, Year | Reference |
|---|---|---|---|---|---|---|---|
| Hepatocellular carcinm | ACSL4 | c-Myc/SREBP1 | Regulate fatty acid metabolism | HCC tumor samples | Not mentioned | Chen et al., 2020 | [84] |
| ACADM | Cav1/SREBP-1 | Regulate fatty acid metabolism | FVB/N mice | Hep3B, PLC/PRF/5, LM3, MHCC97L150, Huh7, HLE | Ma et al., 2021 | [85] | |
| RIPK3 | ROS/caspase1/PPAR | Promote fatty acid metabolism | C57BL/6 wild type (WT) mice | H22 cells | Wu et al., 2020 | [96] | |
| SIRT6 | SIRT6/miR-122 | Regulation of fatty acid β oxidation | 6-month-old male mice | Huh7 | Elhanati et al., 2016 | [139] | |
| NEAT1 | miR-124-3p/ATGL/ DAG+FFA/ PPARα | Regulate triglyceride metabolism | Patients and tissue samples | L02, 293T, HepG2, Huh7, SK Hep-1 and HCCLM3 | Liu et al., 2018 | [153] | |
| SCD1 | ATP/AMPK/ mTOR/ SREBP1 | Regulate fatty acid metabolism | Not mentioned | Huh7.5, HepG2 and Bel-7402 | Liu et al., 2019 | [127] | |
| Breast cancer | KDM5B | AMPK/ KDM5B | Fatty acid metabolism | Not mentioned | MCF7 and MDA-MB-231 | Zhang et al., 2019 | [129] |
| ACAT2 | PI3K/AKT/ SREBP2 | Regulate cholesterol metabolism | breast tissues | MCF-7, T47D and BT474 | Huang et al., 2017 | [88] | |
| ABCA1 | MiR-33a/ABCA1 | Control cholesterol homeostasis | Not mentioned | SUM149 and SUM159 | Wolfe et al., 2016 | [142] | |
| FABP7 | PPARα/ FABP7 | Regulate fatty acid metabolism | Not mentioned | Hs578T, MCF7, MDA-MB-231, MDA-MB-435S, BT474 | Kwong et al., 2018 | [97] | |
| Pancreatic cancer | AMPK | AMPK/ACC | Regulate fatty acid metabolism | Not mentioned | AsPC-1 and PANC-1 | Gao et al., 2020 | [126] |
| SNHG16 | miR-195/SREBP2 | Regulaties lipogenesis | Not mentioned | HPDE6-C7, PANC-1, AsPC-1, BxPC-3, SW1990, HEK-293 | Yu, et al., 2019 | [148] | |
| Clear cell renal cell carcinoma | ECHS1 | AMPK/ GATA3/ ECHS1 | Regulate fatty acid oxidation | 6- to 8-month-old littermates | Human HEK293T, ACHN and 786-O cells | Qu et al., 2019 | [128] |
| Thyroid cancer | ACSL1 | SNHG7/miR449a/ACSL1 | Regulate fatty acid metabolism | Not mentioned | FTC133, TPC1, BCPAP, 8505C, Nthy-ori-3-1 cell lines | Guo et al., 2020 | [150] |
| Bladder cancer | SOX12 | miR-370/SOX12 | Regulate fatty acid | Not mentioned | the human BC cell lines 5637 and J82 | Huang et al., 2019 | [151] |
| Colorectal cancer | FASN | SREBP1/ FASN/CHOL | Regulate cholesterol synthesis | Not mentioned | HT-29 and HCT-8 | Jin et al., 2021 | [93] |
| Glioblastoma | ABCA1 | EGFR/AKT/ SREBP-1/LDLR | Regulates cholesterol metabolism | Not mentioned | U87, U87-EGFRvIII, U87-EGFR, U87-EGFR-PTEN, A431, LN229, T98 | Guo et al., 2011 | [119] |
| Gastric cancer | CDK6CCND1PIM-1 | miR33a/CDK6/CCND1 | Regulation of cholesterol homeostasis | Not mentioned | The human gastric carcinoma cell | Wang et al., 2015 | [148] |
| Prostate cancer | PCA3 | miR-132-3P/SREBP1 | Regulates triglyceride | prostate cancer | LNCaP cell | Guo et al., 2019 | [154] |
6. Lipid Metabolism and Programmed Cell Death
6.1. Lipid Metabolism and Tumor Cell Apoptosis
6.1.1. Fatty Acids
6.1.2. Cholesterol
6.1.3. Ceramide
6.1.4. Phospholipids
PI3K/AKT Signal Pathway
MAPK/ERK Signal Pathway
6.1.5. P53
6.2. Lipid Metabolism and Autophagy of Tumor Cells
6.3. Lipid Metabolism and Ferroptosis
6.3.1. MUFA and PUFA
ACSL4
LPCAT3
Lipoxygenase (LOX)
ALOXs
6.3.2. Phospholipid Metabolism
Glutathione Peroxidase 4 (GPX4)
Ferroptosis Inhibitory Protein 1 (FSP1)
COQ10 Antioxidant System
NADPH
6.3.3. Cholesterol
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- DeBose-Boyd, R.A. Significance and Regulation of Lipid Metabolism. Semin. Cell Dev. Biol. 2018, 81, 97. [Google Scholar] [CrossRef] [PubMed]
- Molendijk, J.; Robinson, H.; Djuric, Z.; Hill, M.M. Lipid Mechanisms in Hallmarks of Cancer. Mol. Omics 2020, 16, 6–18. [Google Scholar] [CrossRef] [PubMed]
- Skotland, T.; Kavaliauskiene, S.; Sandvig, K. The Role of Lipid Species in Membranes and Cancer-Related Changes. Cancer Metastasis Rev. 2020, 39, 343–360. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Geng, F.; Cheng, X.; Guo, D. Lipid Metabolism Reprogramming and Its Potential Targets in Cancer. Cancer Commun. 2018, 38, 27. [Google Scholar] [CrossRef] [PubMed]
- Bacci, M.; Lorito, N.; Smiriglia, A.; Morandi, A. Fat and Furious: Lipid Metabolism in Antitumoral Therapy Response and Resistance. Trends Cancer 2021, 7, 198–213. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Chen, H.; Tang, X.; Zhao, J.; Zhang, H.; Chen, Y.Q.; Chen, W. Δ6 Fatty Acid Desaturases in Polyunsaturated Fatty Acid Biosynthesis: Insights into the Evolution, Function with Substrate Specificities and Biotechnological Use. Appl. Microbiol. Biotechnol. 2020, 104, 9947–9963. [Google Scholar] [CrossRef]
- Dowhan, W. Understanding Phospholipid Function: Why Are There So Many Lipids? J. Biol. Chem. 2017, 292, 10755–10766. [Google Scholar] [CrossRef]
- Snaebjornsson, M.T.; Janaki-Raman, S.; Schulze, A. Greasing the Wheels of the Cancer Machine: The Role of Lipid Metabolism in Cancer. Cell Metab. 2020, 31, 62–76. [Google Scholar] [CrossRef]
- Guo, R.; Chen, Y.; Borgard, H.; Jijiwa, M.; Nasu, M.; He, M.; Deng, Y. The Function and Mechanism of Lipid Molecules and Their Roles in The Diagnosis and Prognosis of Breast Cancer. Molecules 2020, 25, 4864. [Google Scholar] [CrossRef]
- Berger, J.; Moller, D.E. The Mechanisms of Action of PPARs. Annu. Rev. Med. 2002, 53, 409–435. [Google Scholar] [CrossRef]
- de Carvalho, C.; Caramujo, M. The Various Roles of Fatty Acids. Molecules 2018, 23, 2583. [Google Scholar] [CrossRef] [PubMed]
- Olivecrona, G. Role of Lipoprotein Lipase in Lipid Metabolism. Curr. Opin. Lipidol. 2016, 27, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Park, J.K.; Coffey, N.J.; Limoges, A.; Le, A. The Heterogeneity of Lipid Metabolism in Cancer. In The Heterogeneity of Cancer Metabolism; Advances in Experimental Medicine and Biology; Le, A., Ed.; Springer International Publishing: Cham, Switzerland, 2021; Volume 1311, pp. 39–56. ISBN 978-3-030-65767-3. [Google Scholar]
- Menendez, J.A.; Lupu, R. Fatty Acid Synthase and the Lipogenic Phenotype in Cancer Pathogenesis. Nat. Rev. Cancer 2007, 7, 763–777. [Google Scholar] [CrossRef] [PubMed]
- Kalish, B.T.; Fell, G.L.; Nandivada, P.; Puder, M. Clinically Relevant Mechanisms of Lipid Synthesis, Transport, and Storage. JPEN J. Parenter Enter. Nutr. 2015, 39, 8S–17S. [Google Scholar] [CrossRef] [PubMed]
- Nickels, J.T. New Links between Lipid Accumulation and Cancer Progression. J. Biol. Chem. 2018, 293, 6635–6636. [Google Scholar] [CrossRef] [PubMed]
- Vance, J.E. Phospholipid Synthesis and Transport in Mammalian Cells. Traffic 2015, 16, 1–18. [Google Scholar] [CrossRef]
- Horibata, Y.; Mitsuhashi, S.; Shimizu, H.; Maejima, S.; Sakamoto, H.; Aoyama, C.; Ando, H.; Sugimoto, H. The Phosphatidylcholine Transfer Protein StarD7 Is Important for Myogenic Differentiation in Mouse Myoblast C2C12 Cells and Human Primary Skeletal Myoblasts. Sci. Rep. 2020, 10, 2845. [Google Scholar] [CrossRef]
- Madni, Z.K.; Tripathi, S.K.; Salunke, D.M. Structural Insights into the Lipid Transfer Mechanism of a Non-specific Lipid Transfer Protein. Plant J. 2020, 102, 340–352. [Google Scholar] [CrossRef]
- Sha, B.; Luo, M. PI Transfer Protein: The Specific Recognition of Phospholipids and Its Functions. Biochim. Biophys. Acta 1999, 1441, 268–277. [Google Scholar] [CrossRef]
- Jansen, M.; Treutner, K.-H.; Lynen Jansen, P.; Otto, J.; Schmitz, B.; Mueller, S.; Weiss, C.; Tietze, L.; Schumpelick, V. Phospholipids Reduce the Intraperitoneal Adhesion of Colonic Tumor Cells in Rats and Adhesion on Extracellular Matrix in Vitro. Int. J. Colorectal Dis. 2004, 19, 525–532. [Google Scholar] [CrossRef]
- Baxter, A.A.; Hulett, M.D.; Poon, I.K. The Phospholipid Code: A Key Component of Dying Cell Recognition, Tumor Progression and Host–Microbe Interactions. Cell Death Differ. 2015, 22, 1893–1905. [Google Scholar] [CrossRef] [PubMed]
- Kloska, A.; Węsierska, M.; Malinowska, M.; Gabig-Cimińska, M.; Jakóbkiewicz-Banecka, J. Lipophagy and Lipolysis Status in Lipid Storage and Lipid Metabolism Diseases. Int. J. Mol. Sci. 2020, 21, 6113. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Yang, H.; Song, B.-L. Mechanisms and Regulation of Cholesterol Homeostasis. Nat. Rev. Mol. Cell. Biol. 2020, 21, 225–245. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-S.; Yu, X.; Fordstrom, P.; Choi, K.; Chung, B.C.; Roh, S.-H.; Chiu, W.; Zhou, M.; Min, X.; Wang, Z. Cryo-EM Structures of NPC1L1 Reveal Mechanisms of Cholesterol Transport and Ezetimibe Inhibition. Sci. Adv. 2020, 6, eabb1989. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Song, B.; Xu, C. Cholesterol Metabolism in Cancer: Mechanisms and Therapeutic Opportunities. Nat. Metab. 2020, 2, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Krause, B.R.; Hartman, A.D. Adipose Tissue and Cholesterol Metabolism. J. Lipid Res. 1984, 25, 97–110. [Google Scholar] [CrossRef]
- Ouimet, M.; Marcel, Y.L. Regulation of Lipid Droplet Cholesterol Efflux From Macrophage Foam Cells. ATVB 2012, 32, 575–581. [Google Scholar] [CrossRef]
- Ronellenfitsch, M.W.; Luger, A.-L.; Steinbach, J.P. EGFR and MTOR as Therapeutic Targets in Glioblastoma. Oncotarget 2019, 10, 4721–4723. [Google Scholar] [CrossRef]
- Santos, C.R.; Schulze, A. Lipid Metabolism in Cancer: Lipid Metabolism in Cancer. FEBS J. 2012, 279, 2610–2623. [Google Scholar] [CrossRef]
- Maan, M.; Peters, J.M.; Dutta, M.; Patterson, A.D. Lipid Metabolism and Lipophagy in Cancer. Biochem. Biophys. Res. Commun. 2018, 504, 582–589. [Google Scholar] [CrossRef]
- Bogie, J.F.J.; Haidar, M.; Kooij, G.; Hendriks, J.J.A. Fatty Acid Metabolism in the Progression and Resolution of CNS Disorders. Adv. Drug Deliv. Rev. 2020, 159, 198–213. [Google Scholar] [CrossRef] [PubMed]
- Carracedo, A.; Cantley, L.C.; Pandolfi, P.P. Cancer Metabolism: Fatty Acid Oxidation in the Limelight. Nat. Rev. Cancer 2013, 13, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Koundouros, N.; Poulogiannis, G. Reprogramming of Fatty Acid Metabolism in Cancer. Br. J. Cancer 2020, 122, 4–22. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Leit, S.; Kuai, J.; Therrien, E.; Rafi, S.; Harwood, H.J.; DeLaBarre, B.; Tong, L. An Allosteric Mechanism for Potent Inhibition of Human ATP-Citrate Lyase. Nature 2019, 568, 566–570. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.; Zhou, F.; Wang, J.; Cao, H.; Chen, Y.; Liu, X.; Zhang, Z.; Dai, J.; He, X. Functional Polymorphisms of ATP Citrate Lyase Gene Predicts Clinical Outcome of Patients with Advanced Colorectal Cancer. World J. Surg. Oncol. 2015, 13, 42. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Min, X.; Shen, M.; Hua, Q.; Han, Y.; Zhao, L.; Liu, L.; Huang, G.; Liu, J.; Zhao, X. ACLY Facilitates Colon Cancer Cell Metastasis by CTNNB1. J. Exp. Clin. Cancer Res. 2019, 38, 401. [Google Scholar] [CrossRef]
- Bergers, G.; Fendt, S.-M. The Metabolism of Cancer Cells during Metastasis. Nat. Rev. Cancer 2021, 21, 162–180. [Google Scholar] [CrossRef]
- Nishi, K.; Suzuki, M.; Yamamoto, N.; Matsumoto, A.; Iwase, Y.; Yamasaki, K.; Otagiri, M.; Yumita, N. Glutamine Deprivation Enhances Acetyl-CoA Carboxylase Inhibitor-Induced Death of Human Pancreatic Cancer Cells. Anticancer Res. 2018, 38, 6683–6689. [Google Scholar] [CrossRef]
- Li, E.-Q.; Zhao, W.; Zhang, C.; Qin, L.-Z.; Liu, S.-J.; Feng, Z.-Q.; Wen, X.; Chen, C.-P. Synthesis and Anti-Cancer Activity of ND-646 and Its Derivatives as Acetyl-CoA Carboxylase 1 Inhibitors. Eur. J. Pharm. Sci. 2019, 137, 105010. [Google Scholar] [CrossRef]
- Lally, J.S.V.; Ghoshal, S.; DePeralta, D.K.; Moaven, O.; Wei, L.; Masia, R.; Erstad, D.J.; Fujiwara, N.; Leong, V.; Houde, V.P.; et al. Inhibition of Acetyl-CoA Carboxylase by Phosphorylation or the Inhibitor ND-654 Suppresses Lipogenesis and Hepatocellular Carcinoma. Cell Metab. 2019, 29, 174–182.e5. [Google Scholar] [CrossRef]
- Jiang, L.; Fang, X.; Wang, H.; Li, D.; Wang, X. Ovarian Cancer-Intrinsic Fatty Acid Synthase Prevents Anti-Tumor Immunity by Disrupting Tumor-Infiltrating Dendritic Cells. Front. Immunol. 2018, 9, 2927. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.E.; Jung, B.H.; Park, J.; Kang, S.; Lee, H. Deciphering Fatty Acid Synthase Inhibition-Triggered Metabolic Flexibility in Prostate Cancer Cells through Untargeted Metabolomics. Cells 2020, 9, 2447. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Levantini, E.; Teo, J.T.; Goggi, J.; Clohessy, J.G.; Wu, C.S.; Chen, L.; Yang, H.; Krishnan, I.; Kocher, O.; et al. Fatty Acid Synthase Mediates EGFR Palmitoylation in EGFR Mutated Non-small Cell Lung Cancer. EMBO Mol. Med. 2018, 10, e8313. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.F.; Infante, J.R. Molecular Pathways: Fatty Acid Synthase. Clin. Cancer Res. 2015, 21, 5434–5438. [Google Scholar] [CrossRef] [PubMed]
- Vincent, B.M.; Tardiff, D.F.; Piotrowski, J.S.; Aron, R.; Lucas, M.C.; Chung, C.Y.; Bacherman, H.; Chen, Y.; Pires, M.; Subramaniam, R.; et al. Inhibiting Stearoyl-CoA Desaturase Ameliorates α-Synuclein Cytotoxicity. Cell Rep. 2018, 25, 2742–2754.e31. [Google Scholar] [CrossRef] [PubMed]
- Lingrand, M.; Lalonde, S.; Jutras-Carignan, A.; Bergeron, K.-F.; Rassart, E.; Mounier, C. SCD1 Activity Promotes Cell Migration via a PLD-MTOR Pathway in the MDA-MB-231 Triple-Negative Breast Cancer Cell Line. Breast Cancer 2020, 27, 594–606. [Google Scholar] [CrossRef] [PubMed]
- Carbone, M.; Melino, G. Stearoyl CoA Desaturase Regulates Ferroptosis in Ovarian Cancer Offering New Therapeutic Perspectives. Cancer Res. 2019, 79, 5149–5150. [Google Scholar] [CrossRef]
- Tesfay, L.; Paul, B.T.; Konstorum, A.; Deng, Z.; Cox, A.O.; Lee, J.; Furdui, C.M.; Hegde, P.; Torti, F.M.; Torti, S.V. Stearoyl-CoA Desaturase 1 Protects Ovarian Cancer Cells from Ferroptotic Cell Death. Cancer Res. 2019, 79, 5355–5366. [Google Scholar] [CrossRef]
- Feng, W.W.; Wilkins, O.; Bang, S.; Ung, M.; Li, J.; An, J.; del Genio, C.; Canfield, K.; DiRenzo, J.; Wells, W.; et al. CD36-Mediated Metabolic Rewiring of Breast Cancer Cells Promotes Resistance to HER2-Targeted Therapies. Cell Rep. 2019, 29, 3405–3420.e5. [Google Scholar] [CrossRef]
- Jiang, M.; Wu, N.; Xu, B.; Chu, Y.; Li, X.; Su, S.; Chen, D.; Li, W.; Shi, Y.; Gao, X.; et al. Fatty Acid-Induced CD36 Expression via O-GlcNAcylation Drives Gastric Cancer Metastasis. Theranostics 2019, 9, 5359–5373. [Google Scholar] [CrossRef]
- Ladanyi, A.; Mukherjee, A.; Kenny, H.A.; Johnson, A.; Mitra, A.K.; Sundaresan, S.; Nieman, K.M.; Pascual, G.; Benitah, S.A.; Montag, A.; et al. Adipocyte-Induced CD36 Expression Drives Ovarian Cancer Progression and Metastasis. Oncogene 2018, 37, 2285–2301. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Pych, E.; Corpron, C.; Harmon, C.M. Regulation of CD36 expression in human melanoma cells. Adv. Exp. Med. Biol. 2002, 507, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Urbanelli, L.; Buratta, S.; Logozzi, M.; Mitro, N.; Sagini, K.; Raimo, R.D.; Caruso, D.; Fais, S.; Emiliani, C. Lipidomic Analysis of Cancer Cells Cultivated at Acidic PH Reveals Phospholipid Fatty Acids Remodelling Associated with Transcriptional Reprogramming. J. Enzym. Inhib. Med. Chem. 2020, 35, 963–973. [Google Scholar] [CrossRef] [PubMed]
- Lope, V.; Guerrero-Zotano, Á.; Casas, A.; Baena-Cañada, J.M.; Bermejo, B.; Pérez-Gómez, B.; Criado-Navarro, I.; Antolín, S.; Sánchez-Rovira, P.; Ramos-Vázquez, M.; et al. Serum Phospholipids Fatty Acids and Breast Cancer Risk by Pathological Subtype. Nutrients 2020, 12, 3132. [Google Scholar] [CrossRef]
- Ngeow, J.; Eng, C. PTEN in Hereditary and Sporadic Cancer. Cold Spring Harb. Perspect. Med. 2020, 10, a036087. [Google Scholar] [CrossRef]
- Zhong, M.; Chen, Z.; Yan, Y.; Bahet, A.; Cai, X.; Chen, H.; Ran, H.; Qu, K.; Han, Z.; Zhuang, G.; et al. Expression of TIPE Family Members in Human Colorectal Cancer. Oncol. Lett. 2020, 21, 118. [Google Scholar] [CrossRef]
- Wise, H.M.; Hermida, M.A.; Leslie, N.R. Prostate Cancer, PI3K, PTEN and Prognosis. Clin. Sci. 2017, 131, 197–210. [Google Scholar] [CrossRef]
- Carnero, A.; Blanco-Aparicio, C.; Renner, O.; Link, W.; Leal, J. The PTEN/PI3K/AKT Signalling Pathway in Cancer, Therapeutic Implications. CCDT 2008, 8, 187–198. [Google Scholar] [CrossRef]
- Liu, H.Y.; Zhang, Y.Y.; Zhu, B.L.; Feng, F.Z.; Yan, H.; Zhang, H.Y.; Zhou, B. MiR-21 Regulates the Proliferation and Apoptosis of Ovarian Cancer Cells through PTEN/PI3K/AKT. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 4149–4155. [Google Scholar] [CrossRef]
- Yi, J.; Zhu, J.; Wu, J.; Thompson, C.B.; Jiang, X. Oncogenic Activation of PI3K-AKT-MTOR Signaling Suppresses Ferroptosis via SREBP-Mediated Lipogenesis. Proc. Natl. Acad. Sci. USA 2020, 117, 31189–31197. [Google Scholar] [CrossRef]
- Bilotta, M.T.; Petillo, S.; Santoni, A.; Cippitelli, M. Liver X Receptors: Regulators of Cholesterol Metabolism, Inflammation, Autoimmunity, and Cancer. Front. Immunol. 2020, 11, 584303. [Google Scholar] [CrossRef] [PubMed]
- Bjarnadottir, O.; Feldt, M.; Inasu, M.; Bendahl, P.-O.; Elebro, K.; Kimbung, S.; Borgquist, S. Statin Use, HMGCR Expression, and Breast Cancer Survival—The Malmö Diet and Cancer Study. Sci Rep. 2020, 10, 558. [Google Scholar] [CrossRef] [PubMed]
- Tsoi, H.; Chu, E.S.H.; Zhang, X.; Sheng, J.; Nakatsu, G.; Ng, S.C.; Chan, A.W.H.; Chan, F.K.L.; Sung, J.J.Y.; Yu, J. Peptostreptococcus Anaerobius Induces Intracellular Cholesterol Biosynthesis in Colon Cells to Induce Proliferation and Causes Dysplasia in Mice. Gastroenterology 2017, 152, 1419–1433.e5. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Wang, X.; Song, D.; Liu, X.; Gu, Y.; Xu, Z.; Wang, X.; Zhang, X.; Ye, Q.; Tong, Z.; et al. Cholesterol Induces Epithelial-to-Mesenchymal Transition of Prostate Cancer Cells by Suppressing Degradation of EGFR through APMAP. Cancer Res. 2019, 79, 3063–3075. [Google Scholar] [CrossRef]
- Testa, U. Colon Cancer Stem Cells. In Advances in Cancer Stem Cell Biology; Scatena, R., Mordente, E., Giardina, B., Eds.; Springer: New York, NY, USA, 2012; pp. 155–179. [Google Scholar] [CrossRef]
- Borgquist, S.; Giobbie-Hurder, A.; Ahern, T.P.; Garber, J.E.; Colleoni, M.; Láng, I.; Debled, M.; Ejlertsen, B.; von Moos, R.; Smith, I.; et al. Cholesterol, Cholesterol-Lowering Medication Use, and Breast Cancer Outcome in the BIG 1-98 Study. JCO 2017, 35, 1179–1188. [Google Scholar] [CrossRef]
- Jamnagerwalla, J.; Howard, L.E.; Allott, E.H.; Vidal, A.C.; Moreira, D.M.; Castro-Santamaria, R.; Andriole, G.L.; Freeman, M.R.; Freedland, S.J. Serum Cholesterol and Risk of High-Grade Prostate Cancer: Results from the REDUCE Study. Prostate Cancer Prostatic Dis 2018, 21, 252–259. [Google Scholar] [CrossRef]
- Schoeler, M.; Caesar, R. Dietary Lipids, Gut Microbiota and Lipid Metabolism. Rev. Endocr. Metab. Disord. 2019, 20, 461–472. [Google Scholar] [CrossRef]
- Sam, P.N.; Avery, E.; Claypool, S.M. Proteolytic Control of Lipid Metabolism. ACS Chem. Biol. 2019, 14, 2406–2423. [Google Scholar] [CrossRef]
- Morselli, E.; de Souza, S.R.; Gao, S.; Ávalos, Y.; Criollo, A.; Palmer, B.F.; Clegg, D.J. Impact of Estrogens and Estrogen Receptor-α in Brain Lipid Metabolism. Am. J. Physiol.-Endocrinol. Metab. 2018, 315, E7–E14. [Google Scholar] [CrossRef]
- Pasello, M.; Giudice, A.M.; Scotlandi, K. The ABC Subfamily A Transporters: Multifaceted Players with Incipient Potentialities in Cancer. Semin. Cancer Biol. 2020, 60, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, X. Targeting the Wnt/β-Catenin Signaling Pathway in Cancer. J. Hematol. Oncol. 2020, 13, 165. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Saarinen, A.M.; Hitosugi, T.; Wang, Z.; Wang, L.; Ho, T.H.; Liu, J. Inhibition of Intracellular Lipolysis Promotes Human Cancer Cell Adaptation to Hypoxia. eLife 2017, 6, e31132. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Li, J.; Guo, D. SCAP/SREBPs Are Central Players in Lipid Metabolism and Novel Metabolic Targets in Cancer Therapy. CTMC 2018, 18, 484–493. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.D.; Shah, N.A.; Warrington, J.A.; Anderson, N.N.; Park, S.W.; Brown, M.S.; Goldstein, J.L. Combined Analysis of Oligonucleotide Microarray Data from Transgenic and Knockout Mice Identifies Direct SREBP Target Genes. Proc. Natl. Acad. Sci. USA 2003, 100, 12027–12032. [Google Scholar] [CrossRef]
- Knebel, B.; Hartwig, S.; Jacob, S.; Kettel, U.; Schiller, M.; Passlack, W.; Koellmer, C.; Lehr, S.; Müller-Wieland, D.; Kotzka, J. Inactivation of SREBP-1a Phosphorylation Prevents Fatty Liver Disease in Mice: Identification of Related Signaling Pathways by Gene Expression Profiles in Liver and Proteomes of Peroxisomes. Int. J. Mol. Sci. 2018, 19, 980. [Google Scholar] [CrossRef]
- Shimano, H. SREBP-1c and TFE3, Energy Transcription Factors That Regulate Hepatic Insulin Signaling. J. Mol. Med. 2007, 85, 437–444. [Google Scholar] [CrossRef]
- Barger, J.F.; Plas, D.R. Balancing Biosynthesis and Bioenergetics: Metabolic Programs in Oncogenesis. Endocr.-Relat. Cancer 2010, 17, R287–R304. [Google Scholar] [CrossRef]
- Yang, J.; Stack, M.S. Lipid Regulatory Proteins as Potential Therapeutic Targets for Ovarian Cancer in Obese Women. Cancers 2020, 12, 3469. [Google Scholar] [CrossRef]
- Guo, D.; Bell, E.; Mischel, P.; Chakravarti, A. Targeting SREBP-1-Driven Lipid Metabolism to Treat Cancer. CPD 2014, 20, 2619–2626. [Google Scholar] [CrossRef]
- Röhrig, F.; Schulze, A. The Multifaceted Roles of Fatty Acid Synthesis in Cancer. Nat. Rev. Cancer 2016, 16, 732–749. [Google Scholar] [CrossRef] [PubMed]
- Gouw, A.M.; Margulis, K.; Liu, N.S.; Raman, S.J.; Mancuso, A.; Toal, G.G.; Tong, L.; Mosley, A.; Hsieh, A.L.; Sullivan, D.K.; et al. The MYC Oncogene Cooperates with Sterol-Regulated Element-Binding Protein to Regulate Lipogenesis Essential for Neoplastic Growth. Cell Metab. 2019, 30, 556–572.e5. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Ding, C.; Chen, Y.; Hu, W.; Yu, C.; Peng, C.; Feng, X.; Cheng, Q.; Wu, W.; Lu, Y.; et al. ACSL4 Reprograms Fatty Acid Metabolism in Hepatocellular Carcinoma via C-Myc/SREBP1 Pathway. Cancer Lett. 2021, 502, 154–165. [Google Scholar] [CrossRef] [PubMed]
- Ma, A.P.Y.; Yeung, C.L.S.; Tey, S.K.; Mao, X.; Wong, S.W.K.; Ng, T.H.; Ko, F.C.F.; Kwong, E.M.L.; Tang, A.H.N.; Ng, I.O.-L.; et al. Suppression of ACADM-Mediated Fatty Acid Oxidation Promotes Hepatocellular Carcinoma via Aberrant CAV1/SREBP1 Signaling. Cancer Res. 2021, 81, 3679–3692. [Google Scholar] [CrossRef]
- Xue, L.; Qi, H.; Zhang, H.; Ding, L.; Huang, Q.; Zhao, D.; Wu, B.J.; Li, X. Targeting SREBP-2-Regulated Mevalonate Metabolism for Cancer Therapy. Front. Oncol. 2020, 10, 1510. [Google Scholar] [CrossRef]
- Kaymak, I.; Maier, C.R.; Schmitz, W.; Campbell, A.D.; Dankworth, B.; Ade, C.P.; Walz, S.; Paauwe, M.; Kalogirou, C.; Marouf, H.; et al. Mevalonate Pathway Provides Ubiquinone to Maintain Pyrimidine Synthesis and Survival in P53-Deficient Cancer Cells Exposed to Metabolic Stress. Cancer Res. 2020, 80, 189–203. [Google Scholar] [CrossRef]
- Huang, Y.; Jin, Q.; Su, M.; Ji, F.; Wang, N.; Zhong, C.; Jiang, Y.; Liu, Y.; Zhang, Z.; Yang, J.; et al. Leptin Promotes the Migration and Invasion of Breast Cancer Cells by Upregulating ACAT2. Cell Oncol. 2017, 40, 537–547. [Google Scholar] [CrossRef]
- Jin, Y.; Chen, Z.; Dong, J.; Wang, B.; Fan, S.; Yang, X.; Cui, M. SREBP1/FASN/Cholesterol Axis Facilitates Radioresistance in Colorectal Cancer. FEBS Open Bio 2021, 11, 1343–1352. [Google Scholar] [CrossRef]
- Brunmeir, R.; Xu, F. Functional Regulation of PPARs through Post-Translational Modifications. Int. J. Mol. Sci. 2018, 19, 1738. [Google Scholar] [CrossRef]
- Laganà, A.; Vitale, S.; Nigro, A.; Sofo, V.; Salmeri, F.; Rossetti, P.; Rapisarda, A.; La Vignera, S.; Condorelli, R.; Rizzo, G.; et al. Pleiotropic Actions of Peroxisome Proliferator-Activated Receptors (PPARs) in Dysregulated Metabolic Homeostasis, Inflammation and Cancer: Current Evidence and Future Perspectives. Int. J. Mol. Sci. 2016, 17, 999. [Google Scholar] [CrossRef]
- Montagner, A.; Polizzi, A.; Fouché, E.; Ducheix, S.; Lippi, Y.; Lasserre, F.; Barquissau, V.; Régnier, M.; Lukowicz, C.; Benhamed, F.; et al. Liver PPARα Is Crucial for Whole-Body Fatty Acid Homeostasis and Is Protective against NAFLD. Gut 2016, 65, 1202–1214. [Google Scholar] [CrossRef] [PubMed]
- Bogacka, I.; Xie, H.; Bray, G.A.; Smith, S.R. The Effect of Pioglitazone on Peroxisome Proliferator–Activated Receptor-γ Target Genes Related to Lipid Storage In Vivo. Diabetes Care 2004, 27, 8. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Nakajima, T.; Gonzalez, F.J.; Tanaka, N. PPARs as Metabolic Regulators in the Liver: Lessons from Liver-Specific PPAR-Null Mice. Int. J. Mol. Sci. 2020, 21, 2061. [Google Scholar] [CrossRef] [PubMed]
- Hong, F.; Pan, S.; Guo, Y.; Xu, P.; Zhai, Y. PPARs as Nuclear Receptors for Nutrient and Energy Metabolism. Molecules 2019, 24, 2545. [Google Scholar] [CrossRef]
- Wu, L.; Zhang, X.; Zheng, L.; Zhao, H.; Yan, G.; Zhang, Q.; Zhou, Y.; Lei, J.; Zhang, J.; Wang, J.; et al. RIPK3 Orchestrates Fatty Acid Metabolism in Tumor-Associated Macrophages and Hepatocarcinogenesis. Cancer Immunol. Res. 2020, 8, 710–721. [Google Scholar] [CrossRef]
- Kwong, S.C.; Jamil, A.H.A.; Rhodes, A.; Taib, N.A.; Chung, I. Metabolic Role of Fatty Acid Binding Protein 7 in Mediating Triple-Negative Breast Cancer Cell Death via PPAR-α Signaling. J. Lipid Res. 2019, 60, 1807–1817. [Google Scholar] [CrossRef]
- Yousefnia, S.; Momenzadeh, S.; Seyed Forootan, F.; Ghaedi, K.; Nasr Esfahani, M.H. The Influence of Peroxisome Proliferator-Activated Receptor γ (PPARγ) Ligands on Cancer Cell Tumorigenicity. Gene 2018, 649, 14–22. [Google Scholar] [CrossRef]
- Cheng, S.; Wang, G.; Wang, Y.; Cai, L.; Qian, K.; Ju, L.; Liu, X.; Xiao, Y.; Wang, X. Fatty Acid Oxidation Inhibitor Etomoxir Suppresses Tumor Progression and Induces Cell Cycle Arrest via PPARγ-Mediated Pathway in Bladder Cancer. Clin. Sci. 2019, 133, 1745–1758. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, X.; Wang, J.; Shen, Y.; Tang, X.; Yu, F.; Wang, R. Expression and Function of PPARs in Cancer Stem Cells. CSCR 2016, 11, 226–234. [Google Scholar] [CrossRef]
- Venkatesh, D.; O’Brien, N.A.; Zandkarimi, F.; Tong, D.R.; Stokes, M.E.; Dunn, D.E.; Kengmana, E.S.; Aron, A.T.; Klein, A.M.; Csuka, J.M.; et al. MDM2 and MDMX Promote Ferroptosis by PPARα-Mediated Lipid Remodeling. Genes Dev. 2020, 34, 526–543. [Google Scholar] [CrossRef]
- Luo, Y.; Xie, C.; Brocker, C.N.; Fan, J.; Wu, X.; Feng, L.; Wang, Q.; Zhao, J.; Lu, D.; Tandon, M.; et al. Intestinal PPARα Protects Against Colon Carcinogenesis via Regulation of Methyltransferases DNMT1 and PRMT6. Gastroenterology 2019, 157, 744–759.e4. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; He, K.; Tian, C.; Sun, H.; Zhu, C.; Bai, S.; Liu, J.; Wu, Q.; Xie, D.; Yue, T.; et al. Impaired Lipid Biosynthesis Hinders Anti-Tumor Efficacy of Intratumoral INKT Cells. Nat. Commun. 2020, 11, 438. [Google Scholar] [CrossRef] [PubMed]
- Wagner, N.; Wagner, K.-D. PPAR Beta/Delta and the Hallmarks of Cancer. Cells 2020, 9, 1133. [Google Scholar] [CrossRef] [PubMed]
- Calkin, A.C.; Tontonoz, P. Liver X Receptor Signaling Pathways and Atherosclerosis. ATVB 2010, 30, 1513–1518. [Google Scholar] [CrossRef]
- Guo, S.; Li, L.; Yin, H. Cholesterol Homeostasis and Liver X Receptor (LXR) in Atherosclerosis. CHDDT 2018, 18, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Endo-Umeda, K.; Makishima, M. Liver X Receptors Regulate Cholesterol Metabolism and Immunity in Hepatic Nonparenchymal Cells. Int. J. Mol. Sci. 2019, 20, 5045. [Google Scholar] [CrossRef]
- Schulman, I.G. Liver X Receptors Link Lipid Metabolism and Inflammation. FEBS Lett. 2017, 591, 2978–2991. [Google Scholar] [CrossRef]
- Wang, B.; Tontonoz, P. Liver X Receptors in Lipid Signalling and Membrane Homeostasis. Nat. Rev. Endocrinol. 2018, 14, 452–463. [Google Scholar] [CrossRef]
- Long, H.; Guo, X.; Qiao, S.; Huang, Q. Tumor LXR Expression Is a Prognostic Marker for Patients with Hepatocellular Carcinoma. Pathol. Oncol. Res. 2018, 24, 339–344. [Google Scholar] [CrossRef]
- Bovenga, F.; Sabbà, C.; Moschetta, A. Uncoupling Nuclear Receptor LXR and Cholesterol Metabolism in Cancer. Cell Metab. 2015, 21, 517–526. [Google Scholar] [CrossRef]
- Nazih, H.; Bard, J.M. Cholesterol, Oxysterols and LXRs in Breast Cancer Pathophysiology. Int. J. Mol. Sci. 2020, 21, 1356. [Google Scholar] [CrossRef] [PubMed]
- Munir, M.T.; Ponce, C.; Powell, C.A.; Tarafdar, K.; Yanagita, T.; Choudhury, M.; Gollahon, L.S.; Rahman, S.M. The Contribution of Cholesterol and Epigenetic Changes to the Pathophysiology of Breast Cancer. J. Steroid Biochem. Mol. Biol. 2018, 183, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Reinitz, F.; Youssef, M.; Hong, C.; Nathanson, D.; Akhavan, D.; Kuga, D.; Amzajerdi, A.N.; Soto, H.; Zhu, S.; et al. An LXR Agonist Promotes Glioblastoma Cell Death through Inhibition of an EGFR/AKT/SREBP-1/LDLR–Dependent Pathway. Cancer Discov. 2011, 1, 442–456. [Google Scholar] [CrossRef] [PubMed]
- Munir, M.T.; Ponce, C.; Santos, J.M.; Sufian, H.B.; Al-Harrasi, A.; Gollahon, L.S.; Hussain, F.; Rahman, S.M. VD3 and LXR Agonist (T0901317) Combination Demonstrated Greater Potency in Inhibiting Cholesterol Accumulation and Inducing Apoptosis via ABCA1-CHOP-BCL-2 Cascade in MCF-7 Breast Cancer Cells. Mol. Biol. Rep. 2020, 47, 7771–7782. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Alessi, D.R. LKB1 and AMPK and the Cancer-Metabolism Link—Ten Years After. BMC Biol. 2013, 11, 36. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Zhou, X.; Xu, H.; Melcher, K. Structure and Physiological Regulation of AMPK. Int. J. Mol. Sci. 2018, 19, 3534. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Li, J.; Miao, X.; Cui, W.; Miao, L.; Cai, L. A Minireview: Role of AMP-Activated Protein Kinase (AMPK) Signaling in Obesity-Related Renal Injury. Life Sci. 2021, 265, 118828. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of Metabolism and Mitochondrial Homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef]
- Angin, Y.; Beauloye, C.; Horman, S.; Bertrand, L. Regulation of Carbohydrate Metabolism, Lipid Metabolism, and Protein Metabolism by AMPK. Exp. Suppl. 2016, 107, 23–43. [Google Scholar] [CrossRef]
- Zang, Y.; Fan, L.; Chen, J.; Huang, R.; Qin, H. Improvement of Lipid and Glucose Metabolism by Capsiate in Palmitic Acid-Treated HepG2 Cells via Activation of the AMPK/SIRT1 Signaling Pathway. J. Agric. Food Chem. 2018, 66, 6772–6781. [Google Scholar] [CrossRef]
- He, Q.; Wang, L.; Zhao, R.; Yan, F.; Sha, S.; Cui, C.; Song, J.; Hu, H.; Guo, X.; Yang, M.; et al. Mesenchymal Stem Cell-Derived Exosomes Exert Ameliorative Effects in Type 2 Diabetes by Improving Hepatic Glucose and Lipid Metabolism via Enhancing Autophagy. Stem Cell Res. 2020, 11, 223. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhou, S.; Guo, H.; Zhang, J.; Ma, T.; Zheng, Y.; Zhang, Z.; Cai, L. Protective Effects of Sulforaphane on Type 2 Diabetes-Induced Cardiomyopathy via AMPK-Mediated Activation of Lipid Metabolic Pathways and NRF2 Function. Metabolism 2020, 102, 154002. [Google Scholar] [CrossRef] [PubMed]
- Ke, R.; Xu, Q.; Li, C.; Luo, L.; Huang, D. Mechanisms of AMPK in the Maintenance of ATP Balance during Energy Metabolism: AMPK and ATP Balance. Cell Biol. Int. 2018, 42, 384–392. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Liu, S.; Zhai, A.; Zhang, B.; Tian, G. AMPK-Mediated Regulation of Lipid Metabolism by Phosphorylation. Biol. Pharm. Bull. 2018, 41, 985–993. [Google Scholar] [CrossRef]
- Gao, L.; Xu, Z.; Huang, Z.; Tang, Y.; Yang, D.; Huang, J.; He, L.; Liu, M.; Chen, Z.; Teng, Y. CPI-613 Rewires Lipid Metabolism to Enhance Pancreatic Cancer Apoptosis via the AMPK-ACC Signaling. J. Exp. Clin. Cancer Res. 2020, 39, 73. [Google Scholar] [CrossRef]
- Liu, G.; Kuang, S.; Cao, R.; Wang, J.; Peng, Q.; Sun, C. Sorafenib Kills Liver Cancer Cells by Disrupting SCD1-mediated Synthesis of Monounsaturated Fatty Acids via the ATP-AMPK-mTOR-SREBP1 Signaling Pathway. FASEB J. 2019, 33, 10089–10103. [Google Scholar] [CrossRef]
- Qu, Y.-Y.; Zhao, R.; Zhang, H.-L.; Zhou, Q.; Xu, F.-J.; Zhang, X.; Xu, W.-H.; Shao, N.; Zhou, S.-X.; Dai, B.; et al. Inactivation of the AMPK-GATA3-ECHS1 Pathway Induces Fatty Acid Synthesis That Promotes Clear Cell Renal Cell Carcinoma Growth. Cancer Res. 2019, 80, 319–333. [Google Scholar] [CrossRef]
- Zhang, Z.-G.; Zhang, H.-S.; Sun, H.-L.; Liu, H.-Y.; Liu, M.-Y.; Zhou, Z. KDM5B Promotes Breast Cancer Cell Proliferation and Migration via AMPK-Mediated Lipid Metabolism Reprogramming. Exp. Cell Res. 2019, 379, 182–190. [Google Scholar] [CrossRef]
- Huang, C.; Freter, C. Lipid Metabolism, Apoptosis and Cancer Therapy. Int. J. Mol. Sci. 2015, 16, 924–949. [Google Scholar] [CrossRef]
- Zhou, L.; Hussain, M.M. Human MicroRNA-548p Decreases Hepatic Apolipoprotein B Secretion and Lipid Synthesis. ATVB 2017, 37, 786–793. [Google Scholar] [CrossRef]
- Wang, M. Kidney MiR-33 Controls Fatty Acid Oxidation. Nat. Rev. Nephrol. 2020, 16, 66. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Xu, Y.; Zhu, Y.; Sun, H.; Juguilon, C.; Li, F.; Fan, D.; Yin, L.; Zhang, Y. Macrophage MiR-34a Is a Key Regulator of Cholesterol Efflux and Atherosclerosis. Mol. Ther. 2020, 28, 202–216. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Shi, Z.-M.; Chang, Y.-N.; Hu, Z.-M.; Qi, H.-X.; Hong, W. The Ways of Action of Long Non-Coding RNAs in Cytoplasm and Nucleus. Gene 2014, 547, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Hernando, C.; Suárez, Y.; Rayner, K.J.; Moore, K.J. MicroRNAs in Lipid Metabolism. Curr. Opin. Lipidol. 2011, 22, 86–92. [Google Scholar] [CrossRef]
- Thomas, M.; Deiters, A. MicroRNA MiR-122 as a Therapeutic Target for Oligonucleotides and Small Molecules. CMC 2013, 20, 3629–3640. [Google Scholar] [CrossRef]
- Simon, J.; Nuñez-García, M.; Fernández-Tussy, P.; Barbier-Torres, L.; Fernández-Ramos, D.; Gómez-Santos, B.; Buqué, X.; Lopitz-Otsoa, F.; Goikoetxea-Usandizaga, N.; Serrano-Macia, M.; et al. Targeting Hepatic Glutaminase 1 Ameliorates Non-Alcoholic Steatohepatitis by Restoring Very-Low-Density Lipoprotein Triglyceride Assembly. Cell Metab. 2020, 31, 605–622.e10. [Google Scholar] [CrossRef]
- Sengupta, D.; Cassel, T.; Teng, K.; Aljuhani, M.; Chowdhary, V.K.; Hu, P.; Zhang, X.; Fan, T.W.-M.; Ghoshal, K. Regulation of Hepatic Glutamine Metabolism by MiR-122. Mol. Metab. 2020, 34, 174–186. [Google Scholar] [CrossRef]
- Elhanati, S.; Ben-Hamo, R.; Kanfi, Y.; Varvak, A.; Glazz, R.; Lerrer, B.; Efroni, S.; Cohen, H.Y. Reciprocal Regulation between SIRT6 and MiR-122 Controls Liver Metabolism and Predicts Hepatocarcinoma Prognosis. Cell Rep. 2016, 14, 234–242. [Google Scholar] [CrossRef]
- Tao, Y.; Xu, S.; Wang, J.; Xu, L.; Zhang, C.; Chen, K.; Lian, Z.; Zhou, J.; Xie, H.; Zheng, S.; et al. Delivery of MicroRNA-33 Antagomirs by Mesoporous Silica Nanoparticles to Ameliorate Lipid Metabolic Disorders. Front. Pharmacol. 2020, 11, 921. [Google Scholar] [CrossRef]
- Najafi-Shoushtari, S.H.; Kristo, F.; Li, Y.; Shioda, T.; Cohen, D.E.; Gerszten, R.E.; Näär, A.M. MicroRNA-33 and the SREBP Host Genes Cooperate to Control Cholesterol Homeostasis. Science 2010, 328, 1566–1569. [Google Scholar] [CrossRef]
- Wolfe, A.R.; Bambhroliya, A.; Reddy, J.P.; Debeb, B.G.; Huo, L.; Larson, R.; Li, L.; Ueno, N.T.; Woodward, W.A. MiR-33a Decreases High-Density Lipoprotein-Induced Radiation Sensitivity in Breast Cancer. Int. J. Radiat. Oncol. Biol. Phys. 2016, 95, 791–799. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhou, X.; Shan, B.; Han, J.; Wang, F.; Fan, X.; Lv, Y.; Chang, L.; Liu, W. Downregulation of MicroRNA-33a Promotes Cyclin-Dependent Kinase 6, Cyclin D1 and PIM1 Expression and Gastric Cancer Cell Proliferation. Mol. Med. Rep. 2015, 12, 6491–6500. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulos, D.; Drosatos, K.; Hiyama, Y.; Goldberg, I.J.; Zannis, V.I. MicroRNA-370 Controls the Expression of MicroRNA-122 and Cpt1α and Affects Lipid Metabolism. J. Lipid Res. 2010, 51, 1513–1523. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y. MicroRNA-370 Suppresses SOX12 Transcription and Acts as a Tumor Suppressor in Bladder Cancer. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 2303–2312. [Google Scholar] [CrossRef]
- Huang, L.; Liu, X. MicroRNA-370 Promotes Cell Growth by Targeting WNK2 in Breast Cancer. DNA Cell Biol. 2019, 38, 501–509. [Google Scholar] [CrossRef]
- Lu, W.; Cao, F.; Wang, S.; Sheng, X.; Ma, J. LncRNAs: The Regulator of Glucose and Lipid Metabolism in Tumor Cells. Front. Oncol. 2019, 9, 1099. [Google Scholar] [CrossRef]
- Yu, Y.; Dong, J.-T.; He, B.; Zou, Y.-F.; Li, X.-S.; Xi, C.-H.; Yu, Y. LncRNA SNHG16 Induces the SREBP2 to Promote Lipogenesis and Enhance the Progression of Pancreatic Cancer. Future Oncol. 2019, 15, 3831–3844. [Google Scholar] [CrossRef]
- Guo, S.; Zhang, Y.; Wang, S.; Yang, T.; Ma, B.; Li, X.; Zhang, Y.; Jiang, X. LncRNA PCA3 Promotes Antimony-Induced Lipid Metabolic Disorder in Prostate Cancer by Targeting MIR-132-3 P/SREBP1 Signaling. Toxicol. Lett. 2021, 348, 50–58. [Google Scholar] [CrossRef]
- Guo, L.; Lu, J.; Gao, J.; Li, M.; Wang, H.; Zhan, X. The Function of SNHG7/MiR-449a/ACSL1 Axis in Thyroid Cancer. J. Cell Biochem. 2020, 121, 4034–4042. [Google Scholar] [CrossRef]
- Jiang, X.; Guo, S.; Zhang, Y.; Zhao, Y.; Li, X.; Jia, Y.; Xu, Y.; Ma, B. LncRNA NEAT1 Promotes Docetaxel Resistance in Prostate Cancer by Regulating ACSL4 via Sponging MiR-34a-5p and MiR-204-5p. Cell. Signal. 2020, 65, 109422. [Google Scholar] [CrossRef]
- Currie, E.; Schulze, A.; Zechner, R.; Walther, T.C.; Farese, R.V. Cellular Fatty Acid Metabolism and Cancer. Cell Metab. 2013, 18, 153–161. [Google Scholar] [CrossRef]
- Liu, X.; Liang, Y.; Song, R.; Yang, G.; Han, J.; Lan, Y.; Pan, S.; Zhu, M.; Liu, Y.; Wang, Y.; et al. Long Non-Coding RNA NEAT1-Modulated Abnormal Lipolysis via ATGL Drives Hepatocellular Carcinoma Proliferation. Mol. Cancer 2018, 17, 90. [Google Scholar] [CrossRef] [PubMed]
- Wójcik, P.; Žarković, N.; Gęgotek, A.; Skrzydlewska, E. Involvement of Metabolic Lipid Mediators in the Regulation of Apoptosis. Biomolecules 2020, 10, 402. [Google Scholar] [CrossRef] [PubMed]
- UN, D. Essential Fatty Acids, Lipid Peroxidation and Apoptosis. Prostaglandins Leukot. Essent. Fat. Acids 1999, 61, 157–163. [Google Scholar] [CrossRef]
- Zhang, C.; Yu, H.; Shen, Y.; Ni, X.; Shen, S.; Das, U.N. Polyunsaturated Fatty Acids Trigger Apoptosis of Colon Cancer Cells through a Mitochondrial Pathway. Arch. Med. Sci 2015, 11, 1081–1094. [Google Scholar] [CrossRef]
- Yang, J.; Zhu, S.; Lin, G.; Song, C.; He, Z. Vitamin D Enhances Omega-3 Polyunsaturated Fatty Acids-Induced Apoptosis in Breast Cancer Cells: ω-3 FFAs+VD 3 Enhances Apoptosis in Breast Cancer Cells. Cell Biol. Int. 2017, 41, 890–897. [Google Scholar] [CrossRef]
- Kim, S.; Jing, K.; Shin, S.; Jeong, S.; Han, S.-H.; Oh, H.; Yoo, Y.-S.; Han, J.; Jeon, Y.-J.; Heo, J.-Y.; et al. Ω3-Polyunsaturated Fatty Acids Induce Cell Death through Apoptosis and Autophagy in Glioblastoma Cells: In Vitro and in Vivo. Oncol. Rep. 2017, 39, 239–246. [Google Scholar] [CrossRef]
- Narayanan, B.A.; Narayanan, N.K.; Desai, D.; Pittman, B.; Reddy, B.S. Effects of a Combination of Docosahexaenoic Acid and 1,4-Phenylene Bis(Methylene) Selenocyanate on Cyclooxygenase 2, Inducible Nitric Oxide Synthase and β-Catenin Pathways in Colon Cancer Cells. Carcinogenesis 2004, 25, 2443–2449. [Google Scholar] [CrossRef]
- Giros, A.; Grzybowski, M.; Sohn, V.R.; Pons, E.; Fernandez-Morales, J.; Xicola, R.M.; Sethi, P.; Grzybowski, J.; Goel, A.; Boland, C.R.; et al. Regulation of Colorectal Cancer Cell Apoptosis by the N-3 Polyunsaturated Fatty Acids Docosahexaenoic and Eicosapentaenoic. Cancer Prev. Res. 2009, 2, 732–742. [Google Scholar] [CrossRef]
- Calder, P.C. The Role of Marine Omega-3 (n-3) Fatty Acids in Inflammatory Processes, Atherosclerosis and Plaque Stability. Mol. Nutr. Food Res. 2012, 56, 1073–1080. [Google Scholar] [CrossRef]
- Vona-Davis, L.; Rose, D.P. The Obesity-Inflammation-Eicosanoid Axis in Breast Cancer. J. Mammary Gland Biol. Neoplasia 2013, 18, 291–307. [Google Scholar] [CrossRef] [PubMed]
- Meng, H.; Shen, Y.; Shen, J.; Zhou, F.; Shen, S.; Das, U.N. Effect of N-3 and n-6 Unsaturated Fatty Acids on Prostate Cancer (PC-3) and Prostate Epithelial (RWPE-1) Cells in Vitro. Lipids Health Dis. 2013, 12, 160. [Google Scholar] [CrossRef] [PubMed]
- Araujo, P.; Belghit, I.; Aarsæther, N.; Espe, M.; Lucena, E.; Holen, E. The Effect of Omega-3 and Omega-6 Polyunsaturated Fatty Acids on the Production of Cyclooxygenase and Lipoxygenase Metabolites by Human Umbilical Vein Endothelial Cells. Nutrients 2019, 11, 966. [Google Scholar] [CrossRef] [PubMed]
- Yan, G.; Zhao, H.; Zhang, Q.; Zhou, Y.; Wu, L.; Lei, J.; Wang, X.; Zhang, J.; Zhang, X.; Zheng, L.; et al. A RIPK3-PGE 2 Circuit Mediates Myeloid-Derived Suppressor Cell–Potentiated Colorectal Carcinogenesis. Cancer Res. 2018, 78, 5586–5599. [Google Scholar] [CrossRef]
- Saha, J.; Sarkar, D.; Pramanik, A.; Mahanti, K.; Adhikary, A.; Bhattacharyya, S. PGE2-HIF1α Reciprocal Induction Regulates Migration, Phenotypic Alteration and Immunosuppressive Capacity of Macrophages in Tumor Microenvironment. Life Sci. 2020, 253, 117731. [Google Scholar] [CrossRef]
- Bandyopadhyay, S.; Zhan, R.; Wang, Y.; Pai, S.K.; Hirota, S.; Hosobe, S.; Takano, Y.; Saito, K.; Furuta, E.; Iiizumi, M.; et al. Mechanism of Apoptosis Induced by the Inhibition of Fatty Acid Synthase in Breast Cancer Cells. Cancer Res. 2006, 66, 5934–5940. [Google Scholar] [CrossRef]
- Ventura, R.; Mordec, K.; Waszczuk, J.; Wang, Z.; Lai, J.; Fridlib, M.; Buckley, D.; Kemble, G.; Heuer, T.S. Inhibition of de Novo Palmitate Synthesis by Fatty Acid Synthase Induces Apoptosis in Tumor Cells by Remodeling Cell Membranes, Inhibiting Signaling Pathways, and Reprogramming Gene Expression. EBioMedicine 2015, 2, 808–824. [Google Scholar] [CrossRef]
- Luu, W.; Sharpe, L.J.; Gelissen, I.C.; Brown, A.J. The Role of Signalling in Cellular Cholesterol Homeostasis: Signalling in Cholesterol Homeostasis. IUBMB Life 2013, 65, 675–684. [Google Scholar] [CrossRef]
- Adlakha, Y.K.; Saini, N. MicroRNA: A Connecting Road between Apoptosis and Cholesterol Metabolism. Tumor Biol. 2016, 37, 8529–8554. [Google Scholar] [CrossRef]
- Hager, M.H.; Solomon, K.R.; Freeman, M.R. The Role of Cholesterol in Prostate Cancer. Curr. Opin. Clin. Nutr. Metab. Care 2006, 9, 379–385. [Google Scholar] [CrossRef]
- Mullen, T.D.; Obeid, L.M. Ceramide and Apoptosis: Exploring the Enigmatic Connections between Sphingolipid Metabolism and Programmed Cell Death. ACAMC 2012, 12, 340–363. [Google Scholar] [CrossRef] [PubMed]
- Verheij, M.; van Blitterswijk, W.J.; Bartelink, H. Radiation-Induced Apoptosis: The Ceramide-SAPK Signaling Pathway and Clinical Aspects. Acta Oncol. 1998, 37, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Kanto, T.; Kalinski, P.; Hunter, O.C.; Lotze, M.T.; Amoscato, A.A. Ceramide Mediates Tumor-Induced Dendritic Cell Apoptosis. J. Immunol. 2001, 167, 3773–3784. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef]
- Shariati, M.; Meric-Bernstam, F. Targeting AKT for Cancer Therapy. Expert Opin. Investig. Drugs 2019, 28, 977–988. [Google Scholar] [CrossRef]
- Sun, Y.; Liu, W.-Z.; Liu, T.; Feng, X.; Yang, N.; Zhou, H.-F. Signaling Pathway of MAPK/ERK in Cell Proliferation, Differentiation, Migration, Senescence and Apoptosis. J. Recept. Signal Transduct. 2015, 35, 600–604. [Google Scholar] [CrossRef]
- Gnanapradeepan, K.; Basu, S.; Barnoud, T.; Budina-Kolomets, A.; Kung, C.-P.; Murphy, M.E. The P53 Tumor Suppressor in the Control of Metabolism and Ferroptosis. Front. Endocrinol. 2018, 9, 124. [Google Scholar] [CrossRef]
- Goldstein, I.; Marcel, V.; Olivier, M.; Oren, M.; Rotter, V.; Hainaut, P. Understanding Wild-Type and Mutant P53 Activities in Human Cancer: New Landmarks on the Way to Targeted Therapies. Cancer Gene 2011, 18, 2–11. [Google Scholar] [CrossRef]
- Xie, Y.; Li, J.; Kang, R.; Tang, D. Interplay Between Lipid Metabolism and Autophagy. Front. Cell Dev. Biol. 2020, 8, 431. [Google Scholar] [CrossRef]
- Kimmelman, A.C.; White, E. Autophagy and Tumor Metabolism. Cell Metab. 2017, 25, 1037–1043. [Google Scholar] [CrossRef]
- Thiele, C.; Spandl, J. Cell Biology of Lipid Droplets. Curr. Opin. Cell Biol. 2008, 20, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Pol, A.; Gross, S.P.; Parton, R.G. Biogenesis of the Multifunctional Lipid Droplet: Lipids, Proteins, and Sites. J. Cell Biol. 2014, 204, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Tirinato, L.; Liberale, C.; Di Franco, S.; Candeloro, P.; Benfante, A.; La Rocca, R.; Potze, L.; Marotta, R.; Ruffilli, R.; Rajamanickam, V.P.; et al. Lipid Droplets: A New Player in Colorectal Cancer Stem Cells Unveiled by Spectroscopic Imaging. Stem Cells 2015, 33, 35–44. [Google Scholar] [CrossRef]
- Menard, J.A.; Christianson, H.C.; Kucharzewska, P.; Bourseau-Guilmain, E.; Svensson, K.J.; Lindqvist, E.; Chandran, V.I.; Kjellén, L.; Welinder, C.; Bengzon, J.; et al. Metastasis Stimulation by Hypoxia and Acidosis-Induced Extracellular Lipid Uptake Is Mediated by Proteoglycan-Dependent Endocytosis. Cancer Res. 2016, 76, 4828–4840. [Google Scholar] [CrossRef]
- Lass, A.; Zimmermann, R.; Oberer, M.; Zechner, R. Lipolysis—A Highly Regulated Multi-Enzyme Complex Mediates the Catabolism of Cellular Fat Stores. Prog. Lipid Res. 2011, 50, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Sztalryd, C.; Londos, C. Degradation of Perilipin Is Mediated through Ubiquitination-Proteasome Pathway. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2006, 1761, 83–90. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. Degradation of Lipid Droplet-Associated Proteins by Chaperone-Mediated Autophagy Facilitates Lipolysis. Nat. Cell Biol. 2015, 17, 759–770. [Google Scholar] [CrossRef]
- Eisenberg-Lerner, A.; Kimchi, A. The Paradox of Autophagy and Its Implication in Cancer Etiology and Therapy. Apoptosis 2009, 14, 376–391. [Google Scholar] [CrossRef]
- Masiero, E.; Agatea, L.; Mammucari, C.; Blaauw, B.; Loro, E.; Komatsu, M.; Metzger, D.; Reggiani, C.; Schiaffino, S.; Sandri, M. Autophagy Is Required to Maintain Muscle Mass. Cell Metab. 2009, 10, 507–515. [Google Scholar] [CrossRef]
- Magtanong, L.; Ko, P.-J.; To, M.; Cao, J.Y.; Forcina, G.C.; Tarangelo, A.; Ward, C.C.; Cho, K.; Patti, G.J.; Nomura, D.K.; et al. Exogenous Monounsaturated Fatty Acids Promote a Ferroptosis-Resistant Cell State. Cell Chem. Biol. 2019, 26, 420–432.e9. [Google Scholar] [CrossRef]
- Tang, D.; Kroemer, G. Ferroptosis. Curr. Biol. 2020, 30, R1292–R1297. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of Polyunsaturated Fatty Acids by Lipoxygenases Drives Ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975. [Google Scholar] [CrossRef] [PubMed]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.F.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized Arachidonic and Adrenic PEs Navigate Cells to Ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Li, X.; Zhang, X.; Kang, R.; Tang, D. Identification of ACSL4 as a Biomarker and Contributor of Ferroptosis. Biochem. Biophys. Res. Commun. 2016, 478, 1338–1343. [Google Scholar] [CrossRef]
- Shintoku, R.; Takigawa, Y.; Yamada, K.; Kubota, C.; Yoshimoto, Y.; Takeuchi, T.; Koshiishi, I.; Torii, S. Lipoxygenase-mediated Generation of Lipid Peroxides Enhances Ferroptosis Induced by Erastin and RSL3. Cancer Sci. 2017, 108, 2187–2194. [Google Scholar] [CrossRef]
- Mashima, T.; Seimiya, H.; Tsuruo, T. De Novo Fatty-Acid Synthesis and Related Pathways as Molecular Targets for Cancer Therapy. Br. J. Cancer 2009, 100, 1369–1372. [Google Scholar] [CrossRef]
- Zou, Y.; Palte, M.J.; Deik, A.A.; Li, H.; Eaton, J.K.; Wang, W.; Tseng, Y.-Y.; Deasy, R.; Kost-Alimova, M.; Dančík, V.; et al. A GPX4-Dependent Cancer Cell State Underlies the Clear-Cell Morphology and Confers Sensitivity to Ferroptosis. Nat. Commun. 2019, 10, 1617. [Google Scholar] [CrossRef]
- Conrad, M.; Pratt, D.A. The Chemical Basis of Ferroptosis. Nat. Chem. Biol. 2019, 15, 1137–1147. [Google Scholar] [CrossRef]
- Wang, B.; Tontonoz, P. Phospholipid Remodeling in Physiology and Disease. Annu. Rev. Physiol. 2019, 81, 165–188. [Google Scholar] [CrossRef]
- Hashidate-Yoshida, T.; Harayama, T.; Hishikawa, D.; Morimoto, R.; Hamano, F.; Tokuoka, S.M.; Eto, M.; Tamura-Nakano, M.; Yanobu-Takanashi, R.; Mukumoto, Y.; et al. Fatty Acid Remodeling by LPCAT3 Enriches Arachidonate in Phospholipid Membranes and Regulates Triglyceride Transport. eLife 2015, 4, e06328. [Google Scholar] [CrossRef]
- Kuhn, H.; Banthiya, S.; van Leyen, K. Mammalian Lipoxygenases and Their Biological Relevance. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2015, 1851, 308–330. [Google Scholar] [CrossRef] [PubMed]
- Rådmark, O.; Werz, O.; Steinhilber, D.; Samuelsson, B. 5-Lipoxygenase, a Key Enzyme for Leukotriene Biosynthesis in Health and Disease. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2015, 1851, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Mashima, R.; Okuyama, T. The Role of Lipoxygenases in Pathophysiology; New Insights and Future Perspectives. Redox Biol. 2015, 6, 297–310. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.K.; Rao, G.N. Emerging Role of 12/15-Lipoxygenase (ALOX15) in Human Pathologies. Prog. Lipid Res. 2019, 73, 28–45. [Google Scholar] [CrossRef]
- Anthonymuthu, T.S.; Kenny, E.M.; Shrivastava, I.; Tyurina, Y.Y.; Hier, Z.E.; Ting, H.-C.; Dar, H.H.; Tyurin, V.A.; Nesterova, A.; Amoscato, A.A.; et al. Empowerment of 15-Lipoxygenase Catalytic Competence in Selective Oxidation of Membrane ETE-PE to Ferroptotic Death Signals, HpETE-PE. J. Am. Chem. Soc. 2018, 140, 17835–17839. [Google Scholar] [CrossRef]
- Probst, L.; Dächert, J.; Schenk, B.; Fulda, S. Lipoxygenase Inhibitors Protect Acute Lymphoblastic Leukemia Cells from Ferroptotic Cell Death. Biochem. Pharmacol. 2017, 140, 41–52. [Google Scholar] [CrossRef]
- Sato, H.; Tamba, M.; Ishii, T.; Bannai, S. Cloning and Expression of a Plasma Membrane Cystine/Glutamate Exchange Transporter Composed of Two Distinct Proteins. J. Biol. Chem. 1999, 274, 11455–11458. [Google Scholar] [CrossRef]
- Brigelius-Flohé, R.; Maiorino, M. Glutathione Peroxidases. Biochim. Biophys. Acta (BBA) Gen. Subj. 2013, 1830, 3289–3303. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Wu, M.; Xu, L.-G.; Li, X.; Zhai, Z.; Shu, H.-B. AMID, an Apoptosis-Inducing Factor-Homologous Mitochondrion-Associated Protein, Induces Caspase-Independent Apoptosis. J. Biol. Chem. 2002, 277, 25617–25623. [Google Scholar] [CrossRef] [PubMed]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ Oxidoreductase FSP1 Acts Parallel to GPX4 to Inhibit Ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Goya Grocin, A.; Xavier da Silva, T.N.; Panzilius, E.; Scheel, C.H.; et al. FSP1 Is a Glutathione-Independent Ferroptosis Suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef]
- Kuang, F.; Liu, J.; Tang, D.; Kang, R. Oxidative Damage and Antioxidant Defense in Ferroptosis. Front. Cell Dev. Biol. 2020, 8, 586578. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Hayano, M.; Pagano, N.C.; Stockwell, B.R. Cell-Line Selectivity Improves the Predictive Power of Pharmacogenomic Analyses and Helps Identify NADPH as Biomarker for Ferroptosis Sensitivity. Cell Chem. Biol. 2016, 23, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Ding, C.-K.C.; Rose, J.; Sun, T.; Wu, J.; Chen, P.-H.; Lin, C.-C.; Yang, W.-H.; Chen, K.-Y.; Lee, H.; Xu, E.; et al. MESH1 Is a Cytosolic NADPH Phosphatase That Regulates Ferroptosis. Nat. Metab. 2020, 2, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Lee, J.H.; Park, J.-W. Down-Regulation of IDH2 Sensitizes Cancer Cells to Erastin-Induced Ferroptosis. Biochem. Biophys. Res. Commun. 2020, 525, 366–371. [Google Scholar] [CrossRef]
- Girotti, A.W.; Korytowski, W. Cholesterol Hydroperoxide Generation, Translocation, and Reductive Turnover in Biological Systems. Cell Biochem. Biophys. 2017, 75, 413–419. [Google Scholar] [CrossRef]
- Viswanathan, V.S.; Ryan, M.J.; Dhruv, H.D.; Gill, S.; Eichhoff, O.M.; Seashore-Ludlow, B.; Kaffenberger, S.D.; Eaton, J.K.; Shimada, K.; Aguirre, A.J.; et al. Dependency of a Therapy-Resistant State of Cancer Cells on a Lipid Peroxidase Pathway. Nature 2017, 547, 453–457. [Google Scholar] [CrossRef]
- Ingold, I.; Berndt, C.; Schmitt, S.; Doll, S.; Poschmann, G.; Buday, K.; Roveri, A.; Peng, X.; Porto Freitas, F.; Seibt, T.; et al. Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell 2018, 172, 409–422.e21. [Google Scholar] [CrossRef]
- Forcina, G.C.; Dixon, S.J. GPX4 at the Crossroads of Lipid Homeostasis and Ferroptosis. Proteomics 2019, 19, 1800311. [Google Scholar] [CrossRef] [PubMed]







| Author Year | Function of Lipids | Type of Lipids | Reference |
|---|---|---|---|
| Cui, J. et al., 2020 Berger, J. et al., 2002 | Engergy torage and metabolism | Triglycerides, diacylglycerin, Monoacylglycerol, long-chain fatty acids, sterol esters, PPARβ, PPARγ. | [6,10] |
| Dowhan, W. 2017 De Carvalho, C. & Caramujo, M. 2018 | Signal transduction | Dicylglycero, arachidonic aicd, phosphatidic acid, ysophosphatidic acid, PI-4-phosphate. | [7,11] |
| Dowhan, W. 2017 Molendijk, J. et al., 2020 | Membrane structure construction | SREBPs, LXRs, PC, PE, PL, PS, sphingomyelin | [2,7] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, H.; Wang, M.; Su, J.; Li, Y.; Long, J.; Chu, J.; Wan, X.; Cao, Y.; Li, Q. Lipid Metabolism and Cancer. Life 2022, 12, 784. https://doi.org/10.3390/life12060784
Cheng H, Wang M, Su J, Li Y, Long J, Chu J, Wan X, Cao Y, Li Q. Lipid Metabolism and Cancer. Life. 2022; 12(6):784. https://doi.org/10.3390/life12060784
Chicago/Turabian StyleCheng, Hui, Meng Wang, Jingjing Su, Yueyue Li, Jiao Long, Jing Chu, Xinyu Wan, Yu Cao, and Qinglin Li. 2022. "Lipid Metabolism and Cancer" Life 12, no. 6: 784. https://doi.org/10.3390/life12060784
APA StyleCheng, H., Wang, M., Su, J., Li, Y., Long, J., Chu, J., Wan, X., Cao, Y., & Li, Q. (2022). Lipid Metabolism and Cancer. Life, 12(6), 784. https://doi.org/10.3390/life12060784