
Single-Cell Image-Based Analysis Reveals Chromatin Changes during the Acquisition of Tamoxifen Drug Resistance


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Culture of MCF7 and MCF7-Tamoxifen-Resistant Cell Line
2.2. Proliferation Assay
2.3. Invasiveness Assay
2.4. Immuno-Staining
2.5. Live Cell Exposure
2.6. Fluorescent Microscopy
2.7. Data Analysis and Statistics
3. Results
3.1. Characterize Phenotypic Changes between MCF7 and TamR
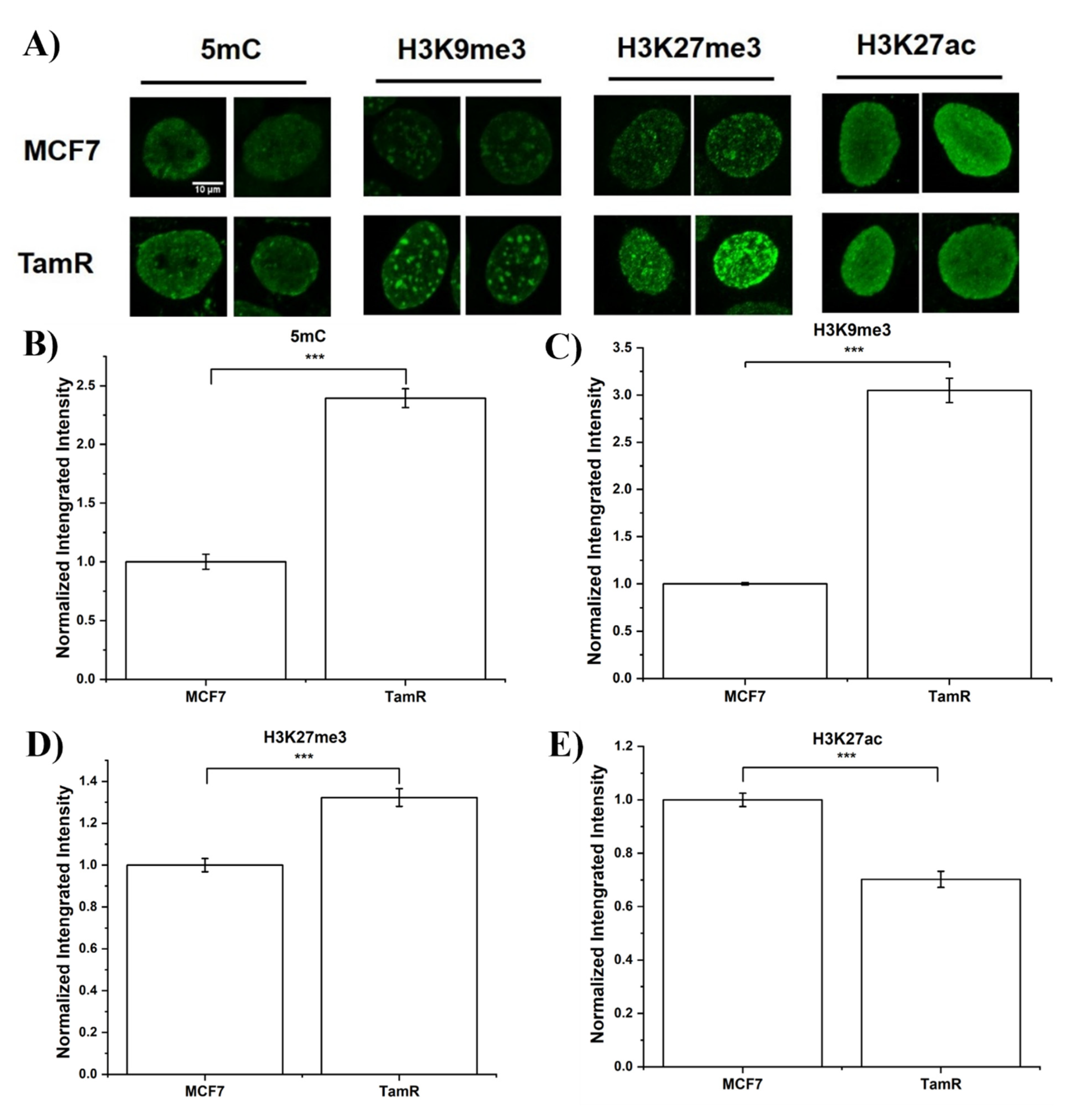
3.2. Assess Epigenome Difference between MCF7 and TamR
3.3. Tracking Epigenome Changes during Tam Treatment
4. Discussions
4.1. Epigenome and Morphological Changes Correlate with Drug Resistance in a Breast Cancer Cell Line
4.2. Dynamic Reprogramming of Epigenome during Tam Treatment
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vasan, N.; Baselga, J.; Hyman, D.M. A view on drug resistance in cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef] [Green Version]
- Si, W.; Shen, J.; Zheng, H.; Fan, W. The role and mechanisms of action of microRNAs in cancer drug resistance. Clin. Epigenet. 2019, 11, 25. [Google Scholar] [CrossRef] [PubMed]
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv. Pharm. Bull. 2017, 7, 339–348. [Google Scholar] [CrossRef]
- Mirzaei, S.A.; Reiisi, S.; Tabari, P.G.; Shekari, A.; Aliakbari, F.; Azadfallah, E.; Elahian, F. Broad blocking of MDR efflux pumps by acetylshikonin and acetoxyisovalerylshikonin to generate hypersensitive phenotype of malignant carcinoma cells. Sci. Rep. 2018, 8, 3446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef]
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug resistance in cancer: An overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szakacs, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 2006, 5, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.; Curry, E.; Magnani, L.; Wilhelm-Benartzi, C.; Borley, J. Poised epigenetic states and acquired drug resistance in cancer. Nat. Cancer 2014, 14, 747–753. [Google Scholar] [CrossRef]
- Glasspool, R.; Teodoridis, J.M.; Brown, R. Epigenetics as a mechanism driving polygenic clinical drug resistance. Br. J. Cancer 2006, 94, 1087–1092. [Google Scholar] [CrossRef] [Green Version]
- Bertolini, G.; Roz, L.; Perego, P.; Tortoreto, M.; Fontanella, E.; Gatti, L.; Pratesi, G.; Fabbri, A.; Andriani, F.; Tinelli, S.; et al. Highly tumorigenic lung cancer CD133+ cells display stem-like features and are spared by cisplatin treatment. Proc. Natl. Acad. Sci. USA 2009, 106, 16281–16286. [Google Scholar] [CrossRef] [Green Version]
- Dalerba, P.; Dylla, S.J.; Park, I.-K.; Liu, R.; Wang, X.; Cho, R.W.; Hoey, T.; Gurney, A.; Huang, E.H.; Simeone, D.M.; et al. Phenotypic characterization of human colorectal cancer stem cells. Proc. Natl. Acad. Sci. USA 2007, 104, 10158–10163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prieto-Vila, M.; Takahashi, R.-U.; Usuba, W.; Kohama, I.; Ochiya, T. Drug Resistance Driven by Cancer Stem Cells and Their Niche. Int. J. Mol. Sci. 2017, 18, 2574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dallas, N.A.; Xia, L.; Fan, F.; Gray, M.J.; Gaur, P.; Van Buren, G.; Samuel, S.; Kim, M.P.; Lim, S.J.; Ellis, L.M. Chemoresistant colorectal cancer cells, the cancer stem cell phenotype, and increased sensitivity to insulin-like growth factor-i receptor inhibition. Cancer Res. 2009, 69, 1951–1957. [Google Scholar] [CrossRef] [Green Version]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Prieto-Vila, M.; Usuba, W.; Takahashi, R.-U.; Shimomura, I.; Sasaki, H.; Ochiya, T.; Yamamoto, Y. Single-Cell Analysis Reveals a Preexisting Drug-Resistant Subpopulation in the Luminal Breast Cancer Subtype. Cancer Res. 2019, 79, 4412–4425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Guldner, I.H.; Golomb, S.M.; Sun, L.; Harris, J.A.; Lu, X.; Zhang, S. Single-cell profiling guided combinatorial immunotherapy for fast-evolving CDK4/6 inhibitor-resistant HER2-positive breast cancer. Nat. Commun. 2019, 10, 3817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.-C.W.; Lopez-Diaz, F.J.; Khan, S.Y.; Tariq, M.A.; Dayn, Y.; Vaske, C.J.; Radenbaugh, A.J.; Kim, H.J.; Emerson, B.M.; Pourmand, N. Single-cell analyses of transcriptional heterogeneity during drug tolerance transition in cancer cells by RNA sequencing. Proc. Natl. Acad. Sci. USA 2014, 111, E4726–E4735. [Google Scholar] [CrossRef] [Green Version]
- Laughney, A.M.; Kim, E.; Sprachman, M.M.; Miller, M.A.; Kohler, R.H.; Yang, K.S.; Orth, J.D.; Mitchison, T.J.; Weissleder, R. Single-cell pharmacokinetic imaging reveals a therapeutic strategy to overcome drug resistance to the microtubule inhibitor eribulin. Sci. Transl. Med. 2014, 6, 261ra152. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Cao, Y.; Kumar, V.; Zhang, X.; Leong, H.S.; Wong, A.M.L.; Ramakrishnan, N.; Hakimullah, M.; Teo, H.M.V.; Chong, F.T.; et al. Longitudinal single-cell RNA sequencing of patient-derived primary cells reveals drug-induced infidelity in stem cell hierarchy. Nat. Commun. 2018, 9, 4931. [Google Scholar] [CrossRef] [Green Version]
- Cheow, L.F.; Courtois, E.; Tan, Y.; Viswanathan, R.; Xing, Q.; Tan, R.Z.; Tan, D.S.W.; Loh, Y.-H.; Quake, S.R. Single-cell multimodal profiling reveals cellular epigenetic heterogeneity. Nat. Methods 2016, 13, 833–836. [Google Scholar] [CrossRef]
- Bell, C.C.; Fennell, K.A.; Chan, Y.C.; Rambow, F.; Yeung, M.M.; Vassiliadis, D.; Dawson, M.A. Targeting enhancer switching overcomes non-genetic drug resistance in acute myeloid leukaemia. Nat. Commun. 2019, 10, 2723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinhold, B. Epigenetics: The science of change. Environ. Health Perspect. 2006, 114, A160–A167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knowlden, J.M.; Hutcheson, I.R.; Jones, H.E.; Madden, T.; Gee, J.M.; Harper, M.E.; Nicholson, R.I. Elevated levels of epidermal growth factor receptor/c-erbB2 heterodimers mediate an autocrine growth regulatory pathway in tamoxifen-resistant MCF-7 cells. Endocrinology 2003, 144, 1032–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.Y.; Kim, Y.C.; Oh, J.H.; Kim, M.H. HOXA5 confers tamoxifen resistance via the PI3K/AKT signaling pathway in ER-positive breast cancer. J. Cancer 2021, 12, 4626–4637. [Google Scholar] [CrossRef] [PubMed]
- Ritch, S.J.; Brandhagen, B.N.; Goyeneche, A.A.; Telleria, C.M. Advanced assessment of migration and invasion of cancer cells in response to mifepristone therapy using double fluorescence cytochemical labeling. BMC Cancer 2019, 19, 376. [Google Scholar] [CrossRef] [PubMed]
- Justus, C.R.; Leffler, N.; Ruiz-Echevarria, M.; Yang, L.V. In vitro Cell Migration and Invasion Assays. J. Vis. Exp. 2014, e51046. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.F.; Xie, J.; Sánchez, O.F.; Bryan, C.; Freeman, J.L.; Yuan, C. Low dose lead exposure induces alterations on heterochromatin hallmarks persisting through SH-SY5Y cell differentiation. Chemosphere 2021, 264, 128486. [Google Scholar] [CrossRef]
- Xie, J.; Lin, L.; Sánchez, O.F.; Bryan, C.; Freeman, J.L.; Yuan, C. Pre-differentiation exposure to low-dose of atrazine results in persistent phenotypic changes in human neuronal cell lines. Environ. Pollut. 2020, 271, 116379. [Google Scholar] [CrossRef]
- Fasching, P.A.; Heusinger, K.; Haeberle, L.; Niklos, M.; Hein, A.; Bayer, C.M.; Rauh, C.; Schulz-Wendtland, R.; Bani, M.R.; Schrauder, M.; et al. Ki67, chemotherapy response, and prognosis in breast cancer patients receiving neoadjuvant treatment. BMC Cancer 2011, 11, 486. [Google Scholar] [CrossRef] [Green Version]
- Tan, P.-H.; Bay, B.H.; Yip, G.; Selvarajan, S.; Tan, P.; Wu, J.; Lee, C.H.; Li, K.B. Immunohistochemical detection of Ki67 in breast cancer correlates with transcriptional regulation of genes related to apoptosis and cell death. Mod. Pathol. 2005, 18, 374–381. [Google Scholar] [CrossRef]
- Smith, I.; Robertson, J.; Kilburn, L.; Wilcox, M.; Evans, A.; Holcombe, C.; Horgan, K.; Kirwan, C.; Mallon, E.; Sibbering, M.; et al. Long-term outcome and prognostic value of Ki67 after perioperative endocrine therapy in postmenopausal women with hormone-sensitive early breast cancer (POETIC): An open-label, multicentre, parallel-group, randomised, phase 3 trial. Lancet Oncol. 2020, 21, 1443–1454. [Google Scholar] [CrossRef]
- Kim, S.-E.; Chang, M.; Yuan, C. One-pot approach for examining the DNA methylation patterns using an engineered methyl-probe. Biosens. Bioelectron. 2014, 58, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, O.F.; Mendonca, A.; Min, A.; Liu, J.; Yuan, C. Monitoring Histone Methylation (H3K9me3) Changes in Live Cells. ACS Omega 2019, 4, 13250–13259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez, O.F.; Mendonca, A.; Carneiro, A.D.; Yuan, C. Engineering Recombinant Protein Sensors for Quantifying Histone Acetylation. ACS Sens. 2017, 2, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Hiscox, S.; Morgan, L.; Barrow, D.; Dutkowski, C.; Wakeling, A.; Nicholson, R.I. Tamoxifen resistance in breast cancer cells is accompanied by an enhanced motile and invasive phenotype: Inhibition by gefitinib (`Iressa’, ZD1839). Clin. Exp. Metastasis 2004, 21, 201–212. [Google Scholar] [CrossRef]
- Zhu, Y.; Liu, Y.; Zhang, C.; Chu, J.; Wu, Y.; Li, Y.; Liu, J.; Li, Q.; Li, S.; Shi, Q.; et al. Tamoxifen-resistant breast cancer cells are resistant to DNA-damaging chemotherapy because of upregulated BARD1 and BRCA1. Nat. Commun. 2018, 9, 1595. [Google Scholar] [CrossRef]
- McQuin, C.; Goodman, A.; Chernyshev, V.; Kamentsky, L.; Cimini, B.A.; Karhohs, K.W.; Doan, M.; Ding, L.; Rafelski, S.M.; Thirstrup, D.; et al. CellProfiler 3.0: Next-generation image processing for biology. PLoS Biol. 2018, 16, e2005970. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.-J.; Evans, J.; Kim, K.; Chae, H.; Kim, S. Determining the Effect of DNA Methylation on Gene Expression in Cancer Cells, in Gene Function Analysis; Humana Press: Totowa, NJ, USA, 2014; pp. 161–178. [Google Scholar] [CrossRef]
- Baylin, S.B. DNA methylation and gene silencing in cancer. Nat. Clin. Pract. Oncol. 2005, 2, S4–S11. [Google Scholar] [CrossRef]
- Nicetto, D.; Zaret, K.S. Role of H3K9me3 heterochromatin in cell identity establishment and maintenance. Curr. Opin. Genet. Dev. 2019, 55, 1–10. [Google Scholar] [CrossRef]
- Creyghton, M.P.; Cheng, A.W.; Welstead, G.G.; Kooistra, T.; Carey, B.W.; Steine, E.J.; Hanna, J.; Lodato, M.A.; Frampton, G.M.; Sharp, P.A.; et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA 2010, 107, 21931–21936. [Google Scholar] [CrossRef] [Green Version]
- De Carvalho, D.; You, J.S.; Jones, P.A. DNA methylation and cellular reprogramming. Trends Cell Biol. 2010, 20, 609–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnakumar, R.; Blelloch, R.H. Epigenetics of cellular reprogramming. Curr. Opin. Genet. Dev. 2013, 23, 548–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flavahan, W.A.; Gaskell, E.; Bernstein, B.E. Epigenetic plasticity and the hallmarks of cancer. Science 2017, 357, eaal2380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feinberg, A.; Koldobskiy, A.P.F.M.A.; Göndör, A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat. Rev. Genet. 2016, 17, 284–299. [Google Scholar] [CrossRef]
- Ngollo, M.; Lebert, A.; Daures, M.; Judes, G.; Rifai, K.; Dubois, L.; Kemeny, J.-L.; Penault-Llorca, F.; Bignon, Y.-J.; Guy, L.; et al. Global analysis of H3K27me3 as an epigenetic marker in prostate cancer progression. BMC Cancer 2017, 17, 261. [Google Scholar] [CrossRef]
- Desplats, P.; Spencer, B.; Coffee, E.; Patel, P.; Michael, S.; Patrick, C.; Adame, A.; Rockenstein, E.; Masliah, E. α-Synuclein sequesters Dnmt1 from the nucleus a novel mechanism for epigenetic alterations in lewy body diseases. J. Biol. Chem. 2011, 286, 9031–9037. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Li, J.; Yin, G.; Zhao, Q.; Elias, D.; Lykkesfeldt, A.E.; Stenvang, J.; Brünner, N.; Wang, J.; Yang, H.; et al. Integrative analyses of gene expression and DNA methylation profiles in breast cancer cell line models of tamoxifen-resistance indicate a potential role of cells with stem-like properties. Breast Cancer Res. 2013, 15, R119. [Google Scholar] [CrossRef] [Green Version]
- Stone, A.; Zotenko, E.; Locke, W.J.; Korbie, D.; Millar, E.K.A.; Pidsley, R.; Stirzaker, C.; Graham, P.; Trau, M.; Musgrove, E.A.; et al. DNA methylation of oestrogen-regulated enhancers defines endocrine sensitivity in breast cancer. Nat. Commun. 2015, 6, 7758. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Zhang, Z.; Cenciarini, M.E.; Proietti, C.J.; Amasino, M.; Hong, T.; Yang, M.; Liao, Y.; Chiang, H.C.; Kaklamani, V.G.; et al. Tamoxifen Resistance in Breast Cancer Is Regulated by the EZH2–ERα–GREB1 Transcr. Axis. Cancer Res. 2018, 78, 671. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Chen, L.; Han, Y.; Wu, F.; Yang, W.-S.; Zhang, Z.; Huo, T.; Zhu, Y.; Yu, C.; Kim, H.; et al. Acetylation of histone H3K27 signals the transcriptional elongation for estrogen receptor alpha. Commun. Biol. 2020, 3, 165. [Google Scholar] [CrossRef] [Green Version]
- Waddell, A.; Mahmud, I.; Ding, H.; Huo, Z.; Liao, D. Pharmacological Inhibition of CBP/p300 Blocks Estrogen Receptor Alpha (ERα) Function through Suppressing Enhancer H3K27 Acetylation in Luminal Breast Cancer. Cancers 2021, 13, 2799. [Google Scholar] [CrossRef]
- Guo, J.U.; Su, Y.; Shin, J.H.; Shin, J.; Li, H.; Xie, B.; Zhong, C.; Hu, S.; Le, T.; Fan, G.; et al. Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain. Nat. Neurosci. 2014, 17, 215. [Google Scholar] [CrossRef] [Green Version]
- Kagohara, L.T.; Stein-O’Brien, G.L.; Kelley, D.; Flam, E.; Wick, H.C.; Danilova, L.V.; Easwaran, H.; Favorov, A.V.; Qian, J.; A Gaykalova, D.; et al. Epigenetic regulation of gene expression in cancer: Techniques, resources and analysis. Briefings Funct. Genom. 2018, 17, 49–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, H.; Xie, W. The role of 3D genome organization in development and cell differentiation. Nat. Rev. Mol. Cell Biol. 2019, 20, 535–550. [Google Scholar] [CrossRef]
- Rowley, M.J.; Corces, V.G. Organizational principles of 3D genome architecture. Nat. Rev. Genet. 2018, 19, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Mendonca, A.; Sánchez, O.; Zhao, H.; Lin, L.; Min, A.; Yuan, C. Development and application of novel BiFC probes for cell sorting based on epigenetic modification. Cytom. Part A 2022, 1–12. [Google Scholar] [CrossRef]
- Lyons, S.M.; Alizadeh, E.; Mannheimer, J.; Schuamberg, K.; Castle, J.; Schroder, B.; Turk, P.; Thamm, D.; Prasad, A. Changes in cell shape are correlated with metastatic potential in murine and human osteosarcomas. Biol. Open 2016, 5, 289–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uhler, C.; Shivashankar, G. Nuclear Mechanopathology and Cancer Diagnosis. Trends Cancer 2018, 4, 320–331. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.-H.; Wirtz, D. Cytoskeletal tension induces the polarized architecture of the nucleus. Biomaterials 2015, 48, 161–172. [Google Scholar] [CrossRef] [Green Version]
- Al-Jubori, A.A.; Sulaiman, G.M.; Tawfeeq, A.T.; Mohammed, H.A.; Khan, R.A.; Mohammed, S.A. Layer-by-Layer Nanoparticles of Tamoxifen and Resveratrol for Dual Drug Delivery System and Potential Triple-Negative Breast Cancer Treatment. Pharmaceutics 2021, 13, 1098. [Google Scholar] [CrossRef]
- Kastl, L.; Brown, I.; Schofield, A.C. Altered DNA methylation is associated with docetaxel resistance in human breast cancer cells. Int. J. Oncol. 2010, 36, 1235–1241. [Google Scholar] [PubMed] [Green Version]
- Gómez-Miragaya, J.; Morán, S.; Calleja-Cervantes, M.E.; Solé, A.C.; Pare, L.; Gómez, A.; Serra, V.; Dobrolecki, L.E.; Lewis, M.T.; Diaz-Lagares, A.; et al. The Altered Transcriptome and DNA Methylation Profiles of Docetaxel Resistance in Breast Cancer PDX Models. Mol. Cancer Res. 2019, 17, 2063–2076. [Google Scholar] [CrossRef] [PubMed]
- Furrer, D.; Dragic, D.; Fournier, F.; Droit, A.; Jacob, S.; Diorio, C. Association between genome-wide DNA methylation pattern and response to trastuzumab in HER2-positive breast cancer patients. FASEB J. 2019, 33, 250.6. [Google Scholar] [CrossRef]
- Si, X.; Liu, Y.; Lv, J.; Ding, H.; Zhang, X.A.; Shao, L.; Yang, N.; Cheng, H.; Sun, L.; Zhu, D.; et al. ERα propelled aberrant global DNA hypermethylation by activating the DNMT1 gene to enhance anticancer drug resistance in human breast cancer cells. Oncotarget 2016, 7, 20966–20980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al Emran, A.; Marzese, D.M.; Menon, D.R.; Stark, M.; Torrano, J.; Hammerlindl, H.; Zhang, G.; Brafford, P.; Salomon, M.P.; Nelson, N.; et al. Distinct histone modifications denote early stress-induced drug tolerance in cancer. Oncotarget 2017, 9, 8206–8222. [Google Scholar] [CrossRef] [Green Version]
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Däbritz, J.H.M.; Zhao, Z.; Yu, Y.; Dörr, J.R.; Dimitrova, L.; Lenze, D.; Barbosa, I.A.M.; et al. Senescence-associated reprogramming promotes cancer stemness. Nature 2017, 553, 96–100. [Google Scholar] [CrossRef] [Green Version]
- Taube, J.; Sphyris, N.; Johnson, K.S.; Reisenauer, K.; Nesbit, T.A.; Joseph, R.; Vijay, G.V.; Sarkar, T.R.; Bhangre, N.A.; Song, J.J.; et al. The H3K27me3-demethylase KDM6A is suppressed in breast cancer stem-like cells, and enables the resolution of bivalency during the mesenchymal-epithelial transition. Oncotarget 2017, 8, 65548–65565. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.-Y.; Hung, M.-H.; Wang, D.-S.; Chu, P.-Y.; Su, J.-C.; Teng, T.-H.; Huang, C.-T.; Chao, T.-T.; Wang, C.-Y.; Shiau, C.-W.; et al. Tamoxifen induces apoptosis through cancerous inhibitor of protein phosphatase 2A–dependent phospho-Akt inactivation in estrogen receptor–negative human breast cancer cells. Breast Cancer Res. 2014, 16, 431. [Google Scholar] [CrossRef] [Green Version]
- Mandlekar, S.; Kong, A.-N.T. Mechanisms of tamoxifen-induced apoptosis. Apoptosis 2001, 6, 469–477. [Google Scholar] [CrossRef]
- Durso, A.; Brickner, J.H. Mechanisms of epigenetic memory. Trends Genet. 2014, 30, 230–236. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, H.; Lin, L.F.; Hahn, J.; Xie, J.; Holman, H.F.; Yuan, C. Single-Cell Image-Based Analysis Reveals Chromatin Changes during the Acquisition of Tamoxifen Drug Resistance. Life 2022, 12, 438. https://doi.org/10.3390/life12030438
Zhao H, Lin LF, Hahn J, Xie J, Holman HF, Yuan C. Single-Cell Image-Based Analysis Reveals Chromatin Changes during the Acquisition of Tamoxifen Drug Resistance. Life. 2022; 12(3):438. https://doi.org/10.3390/life12030438
Chicago/Turabian StyleZhao, Han, Li F. Lin, Joshua Hahn, Junkai Xie, Harvey F. Holman, and Chongli Yuan. 2022. "Single-Cell Image-Based Analysis Reveals Chromatin Changes during the Acquisition of Tamoxifen Drug Resistance" Life 12, no. 3: 438. https://doi.org/10.3390/life12030438
APA StyleZhao, H., Lin, L. F., Hahn, J., Xie, J., Holman, H. F., & Yuan, C. (2022). Single-Cell Image-Based Analysis Reveals Chromatin Changes during the Acquisition of Tamoxifen Drug Resistance. Life, 12(3), 438. https://doi.org/10.3390/life12030438