
Notch Signaling and Cross-Talk in Hypoxia: A Candidate Pathway for High-Altitude Adaptation


Abstract
1. Introduction
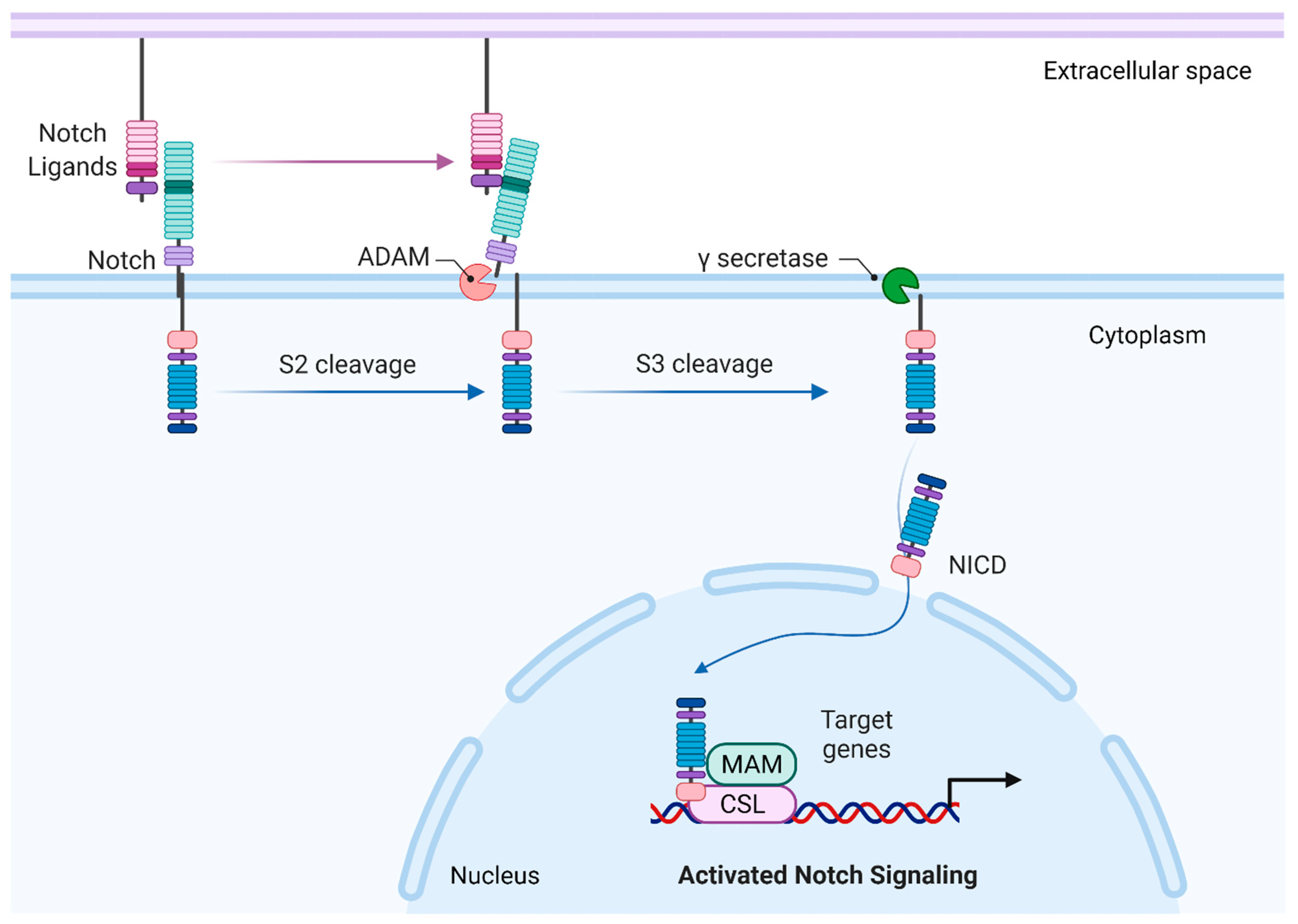
2. Canonical Notch Signaling
3. Notch under Positive Selection
4. Gene Transcription
4.1. Notch and HIF Cross-Talk
4.2. Epigenetic and Post-Transcriptional Modifications
4.2.1. Methylation
4.2.2. Alternative Splicing
4.2.3. MicroRNA
5. Hypoxic-Induced Cellular and Tissue Remodeling
5.1. O2 Delivery
5.1.1. Erythropoiesis
5.1.2. Angiogenesis
5.1.3. Vascular Tone
5.2. Cellular Metabolism
5.2.1. Glucose Homeostasis and Insulin Sensitivity
5.2.2. Glycolysis
5.2.3. Glutamine Metabolism
5.2.4. Mitochondrial Network and Respiration
5.2.5. β-Oxidation
5.3. Inflammation
5.4. Oxidative Stress
6. Future Directions
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Beall, C.M. Two Routes to Functional Adaptation: Tibetan and Andean High-Altitude Natives. Proc. Natl. Acad. Sci. USA 2007, 104 (Suppl. 1), 8655–8660. [Google Scholar] [CrossRef]
- Simonson, T.S. Altitude Adaptation: A Glimpse Through Various Lenses. High Alt. Med. Biol. 2015, 16, 125–137. [Google Scholar] [CrossRef]
- Simonson, T.S.; Yang, Y.; Huff, C.D.; Yun, H.; Qin, G.; Witherspoon, D.J.; Bai, Z.; Lorenzo, F.R.; Xing, J.; Jorde, L.B.; et al. Genetic Evidence for High-Altitude Adaptation in Tibet. Science 2010, 329, 72–75. [Google Scholar] [CrossRef]
- Beall, C.M. Andean, Tibetan, and Ethiopian Patterns of Adaptation to High-Altitude Hypoxia. Integr. Comp. Biol. 2006, 46, 18–24. [Google Scholar] [CrossRef]
- Pamenter, M.E.; Hall, J.E.; Tanabe, Y.; Simonson, T.S. Cross-Species Insights Into Genomic Adaptations to Hypoxia. Front. Genet. 2020, 11, 743. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-Inducible Factors in Physiology and Medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Barahona, A.; Villar, D.; Pescador, N.; Amigo, J.; del Peso, L. Genome-Wide Identification of Hypoxia-Inducible Factor Binding Sites and Target Genes by a Probabilistic Model Integrating Transcription-Profiling Data and in Silico Binding Site Prediction. Nucleic Acids Res. 2010, 38, 2332–2345. [Google Scholar] [CrossRef]
- Semenza, G.L. The Genomics and Genetics of Oxygen Homeostasis. Annu. Rev. Genom. Hum. Genet. 2020, 21, 183–204. [Google Scholar] [CrossRef] [PubMed]
- Iyer, N.V.; Kotch, L.E.; Agani, F.; Leung, S.W.; Laughner, E.; Wenger, R.H.; Gassmann, M.; Gearhart, J.D.; Lawler, A.M.; Yu, A.Y.; et al. Cellular and Developmental Control of O2 Homeostasis by Hypoxia-Inducible Factor 1α. Genes Dev. 1998, 12, 149–162. [Google Scholar] [CrossRef]
- Semenza, G.L.; Wang, G.L. A Nuclear Factor Induced by Hypoxia via de Novo Protein Synthesis Binds to the Human Erythropoietin Gene Enhancer at a Site Required for Transcriptional Activation. Mol. Cell. Biol. 1992, 12, 5447–5454. [Google Scholar]
- Semenza, G.L.; Roth, P.H.; Fang, H.M.; Wang, G.L. Transcriptional Regulation of Genes Encoding Glycolytic Enzymes by Hypoxia-Inducible Factor 1. J. Biol. Chem. 1994, 269, 23757–23763. [Google Scholar] [CrossRef]
- Horscroft, J.A.; Kotwica, A.O.; Laner, V.; West, J.A.; Hennis, P.J.; Levett, D.Z.H.; Howard, D.J.; Fernandez, B.O.; Burgess, S.L.; Ament, Z.; et al. Metabolic Basis to Sherpa Altitude Adaptation. Proc. Natl. Acad. Sci. USA 2017, 114, 6382–6387. [Google Scholar] [CrossRef]
- Ge, R.-L.; Simonson, T.S.; Gordeuk, V.; Prchal, J.T.; McClain, D.A. Metabolic Aspects of High-Altitude Adaptation in Tibetans. Exp. Physiol. 2015, 100, 1247–1255. [Google Scholar] [CrossRef]
- Bray, S.J. Notch Signalling in Context. Nat. Rev. Mol. Cell Biol. 2016, 17, 722–735. [Google Scholar] [CrossRef]
- Cordle, J.; Johnson, S.; Tay, J.Z.Y.; Roversi, P.; Wilkin, M.B.; de Madrid, B.H.; Shimizu, H.; Jensen, S.; Whiteman, P.; Jin, B.; et al. A Conserved Face of the Jagged/Serrate DSL Domain Is Involved in Notch Trans-Activation and Cis-Inhibition. Nat. Struct. Mol. Biol. 2008, 15, 849–857. [Google Scholar] [CrossRef]
- Rebay, I.; Fleming, R.J.; Fehon, R.G.; Cherbas, L.; Cherbas, P.; Artavanis-Tsakonas, S. Specific EGF Repeats of Notch Mediate Interactions with Delta and Serrate: Implications for Notch as a Multifunctional Receptor. Cell 1991, 67, 687–699. [Google Scholar] [CrossRef]
- Nam, Y.; Sliz, P.; Song, L.; Aster, J.C.; Blacklow, S.C. Structural Basis for Cooperativity in Recruitment of MAML Coactivators to Notch Transcription Complexes. Cell 2006, 124, 973–983. [Google Scholar] [CrossRef]
- Wilson, J.J.; Kovall, R.A. Crystal Structure of the CSL-Notch-Mastermind Ternary Complex Bound to DNA. Cell 2006, 124, 985–996. [Google Scholar] [CrossRef]
- Kopan, R.; Ilagan, M.X.G. The Canonical Notch Signaling Pathway: Unfolding the Activation Mechanism. Cell 2009, 137, 216–233. [Google Scholar] [CrossRef]
- Kitagawa, M. Notch Signalling in the Nucleus: Roles of Mastermind-like (MAML) Transcriptional Coactivators. J. Biochem. 2016, 159, 287–294. [Google Scholar] [CrossRef]
- Weng, A.P.; Millholland, J.M.; Yashiro-Ohtani, Y.; Arcangeli, M.L.; Lau, A.; Wai, C.; Del Bianco, C.; Rodriguez, C.G.; Sai, H.; Tobias, J.; et al. C-Myc Is an Important Direct Target of Notch1 in T-Cell Acute Lymphoblastic Leukemia/Lymphoma. Genes Dev. 2006, 20, 2096–2109. [Google Scholar] [CrossRef]
- Palomero, T.; Lim, W.K.; Odom, D.T.; Sulis, M.L.; Real, P.J.; Margolin, A.; Barnes, K.C.; O’Neil, J.; Neuberg, D.; Weng, A.P.; et al. NOTCH1 Directly Regulates C-MYC and Activates a Feed-Forward-Loop Transcriptional Network Promoting Leukemic Cell Growth. Proc. Natl. Acad. Sci. USA 2006, 103, 18261–18266. [Google Scholar] [CrossRef] [PubMed]
- Jarriault, S.; Le Bail, O.; Hirsinger, E.; Pourquié, O.; Logeat, F.; Strong, C.F.; Brou, C.; Seidah, N.G.; Isra l, A. Delta-1 Activation of Notch-1 Signaling Results in HES-1 Transactivation. Mol. Cell. Biol. 1998, 18, 7423–7431. [Google Scholar] [CrossRef]
- Maier, M.M.; Gessler, M. Comparative Analysis of the Human and Mouse Hey1 Promoter: Hey Genes Are New Notch Target Genes. Biochem. Biophys. Res. Commun. 2000, 275, 652–660. [Google Scholar] [CrossRef]
- Stoeck, A.; Lejnine, S.; Truong, A.; Pan, L.; Wang, H.; Zang, C.; Yuan, J.; Ware, C.; MacLean, J.; Garrett-Engele, P.W.; et al. Discovery of Biomarkers Predictive of GSI Response in Triple-Negative Breast Cancer and Adenoid Cystic Carcinoma. Cancer Discov. 2014, 4, 1154–1167. [Google Scholar] [CrossRef]
- Rangarajan, A.; Talora, C.; Okuyama, R.; Nicolas, M.; Mammucari, C.; Oh, H.; Aster, J.C.; Krishna, S.; Metzger, D.; Chambon, P.; et al. Notch Signaling Is a Direct Determinant of Keratinocyte Growth Arrest and Entry into Differentiation. EMBO J. 2001, 20, 3427–3436. [Google Scholar] [CrossRef]
- García-Peydró, M.; Fuentes, P.; Mosquera, M.; García-León, M.J.; Alcain, J.; Rodríguez, A.; de Miguel, P.G.; Menéndez, P.; Weijer, K.; Spits, H.; et al. The NOTCH1/CD44 Axis Drives Pathogenesis in a T Cell Acute Lymphoblastic Leukemia Model. J. Clin. Investig. 2018, 128, 2802–2818. [Google Scholar] [CrossRef]
- López-Arribillaga, E.; Rodilla, V.; Pellegrinet, L.; Guiu, J.; Iglesias, M.; Roman, A.C.; Gutarra, S.; González, S.; Muñoz-Cánoves, P.; Fernández-Salguero, P.; et al. Bmi1 Regulates Murine Intestinal Stem Cell Proliferation and Self-Renewal Downstream of Notch. Development 2015, 142, 41–50. [Google Scholar] [CrossRef]
- Haltiwanger, R.S.; Stanley, P. Modulation of Receptor Signaling by Glycosylation: Fringe Is an O-Fucose-Β1, 3-N-Acetylglucosaminyltransferase. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2002, 1573, 328–335. [Google Scholar] [CrossRef]
- Brückner, K.; Perez, L.; Clausen, H.; Cohen, S. Glycosyltransferase Activity of Fringe Modulates Notch–Delta Interactions. Nature 2000, 406, 411–415. [Google Scholar] [CrossRef]
- Yang, L.-T.; Nichols, J.T.; Yao, C.; Manilay, J.O.; Robey, E.A.; Weinmaster, G. Fringe Glycosyltransferases Differentially Modulate Notch1 Proteolysis Induced by Delta1 and Jagged1. Mol. Biol. Cell 2005, 16, 927–942. [Google Scholar] [CrossRef]
- Landor, S.K.-J.; Lendahl, U. The Interplay between the Cellular Hypoxic Response and Notch Signaling. Exp. Cell Res. 2017, 356, 146–151. [Google Scholar] [CrossRef]
- Bigham, A.W.; Mao, X.; Mei, R.; Brutsaert, T.; Wilson, M.J.; Julian, C.G.; Parra, E.J.; Akey, J.M.; Moore, L.G.; Shriver, M.D. Identifying Positive Selection Candidate Loci for High-Altitude Adaptation in Andean Populations. Hum. Genom. 2009, 4, 79–90. [Google Scholar] [CrossRef]
- Xin, J.; Zhang, H.; He, Y.; Duren, Z.; Bai, C.; Chen, L.; Luo, X.; Yan, D.-S.; Zhang, C.; Zhu, X.; et al. Chromatin Accessibility Landscape and Regulatory Network of High-Altitude Hypoxia Adaptation. Nat. Commun. 2020, 11, 4928. [Google Scholar] [CrossRef]
- Zhang, Z.; Qiu, M.; Du, H.; Li, Q.; Yu, C.; Gan, W.; Peng, H.; Xia, B.; Xiong, X.; Song, X.; et al. Whole Genome Re-Sequencing Identifies Unique Adaption of Single Nucleotide Polymorphism, Insertion/Deletion and Structure Variation Related to Hypoxia in Tibetan Chickens. Gene Expr. Patterns 2021, 40, 119181. [Google Scholar] [CrossRef]
- Zhang, Y.; Zheng, X.; Zhang, Y.; Zhang, H.; Zhang, X.; Zhang, H. Comparative Transcriptomic and Proteomic Analyses Provide Insights into Functional Genes for Hypoxic Adaptation in Embryos of Tibetan Chickens. Sci. Rep. 2020, 10, 11213. [Google Scholar] [CrossRef]
- Jia, C.; Kong, X.; Koltes, J.E.; Gou, X.; Yang, S.; Yan, D.; Lu, S.; Wei, Z. Gene Co-Expression Network Analysis Unraveling Transcriptional Regulation of High-Altitude Adaptation of Tibetan Pig. PLoS ONE 2016, 11, e0168161. [Google Scholar] [CrossRef]
- Qiu, Q.; Zhang, G.; Ma, T.; Qian, W.; Wang, J.; Ye, Z.; Cao, C.; Hu, Q.; Kim, J.; Larkin, D.M.; et al. The Yak Genome and Adaptation to Life at High Altitude. Nat. Genet. 2012, 44, 946–949. [Google Scholar] [CrossRef]
- Bozkulak Esra Cagavi; Weinmaster Gerry Selective Use of ADAM10 and ADAM17 in Activation of Notch1 Signaling. Mol. Cell. Biol. 2009, 29, 5679–5695. [CrossRef]
- Wang, R.; Li, Y.; Tsung, A.; Huang, H.; Du, Q.; Yang, M.; Deng, M.; Xiong, S.; Wang, X.; Zhang, L.; et al. INOS Promotes CD24+ CD133+ Liver Cancer Stem Cell Phenotype through a TACE/ADAM17-Dependent Notch Signaling Pathway. Proc. Natl. Acad. Sci. USA 2018, 115, E10127–E10136. [Google Scholar] [CrossRef]
- Groot, A.J.; Cobzaru, C.; Weber, S.; Saftig, P.; Blobel, C.P.; Kopan, R.; Vooijs, M.; Franzke, C.-W. Epidermal ADAM17 Is Dispensable for Notch Activation. J. Investig. Dermatol. 2013, 133, 2286–2288. [Google Scholar] [CrossRef]
- Zhou, D.; Xue, J.; Lai, J.C.K.; Schork, N.J.; White, K.P.; Haddad, G.G. Mechanisms Underlying Hypoxia Tolerance in Drosophila Melanogaster: Hairy as a Metabolic Switch. PLoS Genet. 2008, 4, e1000221. [Google Scholar] [CrossRef]
- Azad, P.; Zhou, D.; Zarndt, R.; Haddad, G.G. Identification of Genes Underlying Hypoxia Tolerance in Drosophila by a P-Element Screen. G3 2012, 2, 1169–1178. [Google Scholar] [CrossRef]
- Zhou, D.; Udpa, N.; Gersten, M.; Visk, D.W.; Bashir, A.; Xue, J.; Frazer, K.A.; Posakony, J.W.; Subramaniam, S.; Bafna, V.; et al. Experimental Selection of Hypoxia-Tolerant Drosophila Melanogaster. Proc. Natl. Acad. Sci. USA 2011, 108, 2349–2354. [Google Scholar] [CrossRef]
- Zheng, X.; Linke, S.; Dias, J.M.; Zheng, X.; Gradin, K.; Wallis, T.P.; Hamilton, B.R.; Gustafsson, M.; Ruas, J.L.; Wilkins, S.; et al. Interaction with Factor Inhibiting HIF-1 Defines an Additional Mode of Cross-Coupling between the Notch and Hypoxia Signaling Pathways. Proc. Natl. Acad. Sci. USA 2008, 105, 3368–3373. [Google Scholar] [CrossRef]
- Gustafsson, M.V.; Zheng, X.; Pereira, T.; Gradin, K.; Jin, S.; Lundkvist, J.; Ruas, J.L.; Poellinger, L.; Lendahl, U.; Bondesson, M. Hypoxia Requires Notch Signaling to Maintain the Undifferentiated Cell State. Dev. Cell 2005, 9, 617–628. [Google Scholar] [CrossRef]
- Diez, H.; Fischer, A.; Winkler, A.; Hu, C.-J.; Hatzopoulos, A.K.; Breier, G.; Gessler, M. Hypoxia-Mediated Activation of Dll4-Notch-Hey2 Signaling in Endothelial Progenitor Cells and Adoption of Arterial Cell Fate. Exp. Cell Res. 2007, 313, 1–9. [Google Scholar] [CrossRef]
- Wang, R.; Zhang, Y.-W.; Zhang, X.; Liu, R.; Zhang, X.; Hong, S.; Xia, K.; Xia, J.; Zhang, Z.; Xu, H. Transcriptional Regulation of APH-1A and Increased g-Secretase Cleavage of APP and Notch by HIF-1 and Hypoxia. FASEB J.-Fed. Am. Soc. Exp. Biol. 2006, 20, 1275. [Google Scholar] [CrossRef]
- Villa, J.C.; Chiu, D.; Brandes, A.H.; Escorcia, F.E.; Villa, C.H.; Maguire, W.F.; Hu, C.-J.; de Stanchina, E.; Simon, M.C.; Sisodia, S.S.; et al. Nontranscriptional Role of Hif-1α in Activation of γ-Secretase and Notch Signaling in Breast Cancer. Cell Rep. 2014, 8, 1077–1092. [Google Scholar] [CrossRef]
- Lando, D.; Peet, D.J.; Gorman, J.J.; Whelan, D.A.; Whitelaw, M.L.; Bruick, R.K. FIH-1 Is an Asparaginyl Hydroxylase Enzyme That Regulates the Transcriptional Activity of Hypoxia-Inducible Factor. Genes Dev. 2002, 16, 1466–1471. [Google Scholar] [CrossRef]
- So, J.-H.; Kim, J.-D.; Yoo, K.-W.; Kim, H.-T.; Jung, S.-H.; Choi, J.-H.; Lee, M.-S.; Jin, S.-W.; Kim, C.-H. FIH-1, a Novel Interactor of Mindbomb, Functions as an Essential Anti-Angiogenic Factor during Zebrafish Vascular Development. PLoS ONE 2014, 9, e109517. [Google Scholar] [CrossRef]
- Childebayeva, A.; Jones, T.R.; Goodrich, J.M.; Leon-Velarde, F.; Rivera-Chira, M.; Kiyamu, M.; Brutsaert, T.D.; Dolinoy, D.C.; Bigham, A.W. LINE-1 and EPAS1 DNA Methylation Associations with High-Altitude Exposure. Epigenetics 2019, 14, 1–15. [Google Scholar] [CrossRef]
- Childebayeva, A.; Harman, T.; Weinstein, J.; Goodrich, J.M.; Dolinoy, D.C.; Day, T.A.; Bigham, A.W.; Brutsaert, T.D. DNA Methylation Changes Are Associated With an Incremental Ascent to High Altitude. Front. Genet. 2019, 10, 1062. [Google Scholar] [CrossRef]
- Reister, S.; Kordes, C.; Sawitza, I.; Häussinger, D. The Epigenetic Regulation of Stem Cell Factors in Hepatic Stellate Cells. Stem Cells Dev. 2011, 20, 1687–1699. [Google Scholar] [CrossRef]
- Piazzi, G.; Fini, L.; Selgrad, M.; Garcia, M.; Daoud, Y.; Wex, T.; Malfertheiner, P.; Gasbarrini, A.; Romano, M.; Meyer, R.L.; et al. Epigenetic Regulation of Delta-Like1 Controls Notch1 Activation in Gastric Cancer. Oncotarget 2011, 2, 1291–1301. [Google Scholar] [CrossRef][Green Version]
- Bartels, S.J.J.; Spruijt, C.G.; Brinkman, A.B.; Jansen, P.W.T.C.; Vermeulen, M.; Stunnenberg, H.G. A SILAC-Based Screen for Methyl-CpG Binding Proteins Identifies RBP-J as a DNA Methylation and Sequence-Specific Binding Protein. PLoS ONE 2011, 6, e25884. [Google Scholar] [CrossRef]
- Hirota, K.; Semenza, G.L. Regulation of Angiogenesis by Hypoxia-Inducible Factor 1. Crit. Rev. Oncol. Hematol. 2006, 59, 15–26. [Google Scholar] [CrossRef]
- Benedito, R.; Roca, C.; Sörensen, I.; Adams, S.; Gossler, A.; Fruttiger, M.; Adams, R.H. The Notch Ligands Dll4 and Jagged1 Have Opposing Effects on Angiogenesis. Cell 2009, 137, 1124–1135. [Google Scholar] [CrossRef]
- Jakobsson, L.; Franco, C.A.; Bentley, K.; Collins, R.T.; Ponsioen, B.; Aspalter, I.M.; Rosewell, I.; Busse, M.; Thurston, G.; Medvinsky, A.; et al. Endothelial Cells Dynamically Compete for the Tip Cell Position during Angiogenic Sprouting. Nat. Cell Biol. 2010, 12, 943–953. [Google Scholar] [CrossRef]
- Stenmark, K.R.; Fagan, K.A.; Frid, M.G. Hypoxia-Induced Pulmonary Vascular Remodeling: Cellular and Molecular Mechanisms. Circ. Res. 2006, 99, 675–691. [Google Scholar] [CrossRef]
- Li, X.; Zhang, X.; Leathers, R.; Makino, A.; Huang, C.; Parsa, P.; Macias, J.; Yuan, J.X.-J.; Jamieson, S.W.; Thistlethwaite, P.A. Notch3 Signaling Promotes the Development of Pulmonary Arterial Hypertension. Nat. Med. 2009, 15, 1289–1297. [Google Scholar] [CrossRef]
- Ward, J.P.T.; McMurtry, I.F. Mechanisms of Hypoxic Pulmonary Vasoconstriction and Their Roles in Pulmonary Hypertension: New Findings for an Old Problem. Curr. Opin. Pharmacol. 2009, 9, 287–296. [Google Scholar] [CrossRef]
- Smith, K.A.; Voiriot, G.; Tang, H.; Fraidenburg, D.R.; Song, S.; Yamamura, H.; Yamamura, A.; Guo, Q.; Wan, J.; Pohl, N.M.; et al. Notch Activation of Ca2+ Signaling in the Development of Hypoxic Pulmonary Vasoconstriction and Pulmonary Hypertension. Am. J. Respir. Cell Mol. Biol. 2015, 53, 355–367. [Google Scholar] [CrossRef]
- Guo, Q.; Xu, H.; Yang, X.; Zhao, D.; Liu, S.; Sun, X.; Huang, J.-A. Notch Activation of Ca 2+-Sensing Receptor Mediates Hypoxia-Induced Pulmonary Hypertension. Hypertens. Res. 2017, 40, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Siervo, M.; Riley, H.L.; Fernandez, B.O.; Leckstrom, C.A.; Martin, D.S.; Mitchell, K.; Levett, D.Z.; Montgomery, H.E.; Mythen, M.G.; Grocott, M.P.; et al. Caudwell Xtreme Everest Research Group Effects of Prolonged Exposure to Hypobaric Hypoxia on Oxidative Stress, Inflammation and Gluco-Insular Regulation: The Not-so-Sweet Price for Good Regulation. PLoS ONE 2014, 9, e94915. [Google Scholar] [CrossRef]
- Young, P.M.; Rose, M.S.; Sutton, J.R.; Green, H.J.; Cymerman, A.; Houston, C.S. Operation Everest II: Plasma Lipid and Hormonal Responses during a Simulated Ascent of Mt. Everest. J. Appl. Physiol. 1989, 66, 1430–1435. [Google Scholar] [CrossRef]
- Pajvani, U.B.; Shawber, C.J.; Samuel, V.T.; Birkenfeld, A.L.; Shulman, G.I.; Kitajewski, J.; Accili, D. Inhibition of Notch Signaling Ameliorates Insulin Resistance in a FoxO1-Dependent Manner. Nat. Med. 2011, 17, 961–967. [Google Scholar] [CrossRef]
- Kim, J.-W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-Mediated Expression of Pyruvate Dehydrogenase Kinase: A Metabolic Switch Required for Cellular Adaptation to Hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef]
- Landor, S.K.-J.; Mutvei, A.P.; Mamaeva, V.; Jin, S.; Busk, M.; Borra, R.; Grönroos, T.J.; Kronqvist, P.; Lendahl, U.; Sahlgren, C.M. Hypo- and Hyperactivated Notch Signaling Induce a Glycolytic Switch through Distinct Mechanisms. Proc. Natl. Acad. Sci. USA 2011, 108, 18814–18819. [Google Scholar] [CrossRef]
- Slaninova, V.; Krafcikova, M.; Perez-Gomez, R.; Steffal, P.; Trantirek, L.; Bray, S.J.; Krejci, A. Notch Stimulates Growth by Direct Regulation of Genes Involved in the Control of Glycolysis and the Tricarboxylic Acid Cycle. Open Biol. 2016, 6, 150155. [Google Scholar] [CrossRef]
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive Glutamine Metabolism by IDH1 Mediates Lipogenesis under Hypoxia. Nature 2011, 481, 380–384. [Google Scholar] [CrossRef]
- Basak, N.P.; Roy, A.; Banerjee, S. Alteration of Mitochondrial Proteome Due to Activation of Notch1 Signaling Pathway. J. Biol. Chem. 2014, 289, 7320–7334. [Google Scholar] [CrossRef]
- Zhang, H.; Gao, P.; Fukuda, R.; Kumar, G.; Krishnamachary, B.; Zeller, K.I.; Dang, C.V.; Semenza, G.L. HIF-1 Inhibits Mitochondrial Biogenesis and Cellular Respiration in VHL-Deficient Renal Cell Carcinoma by Repression of C-MYC Activity. Cancer Cell 2007, 11, 407–420. [Google Scholar] [CrossRef]
- Levett, D.Z.; Radford, E.J.; Menassa, D.A.; Graber, E.F.; Morash, A.J.; Hoppeler, H.; Clarke, K.; Martin, D.S.; Ferguson-Smith, A.C.; Montgomery, H.E.; et al. Acclimatization of Skeletal Muscle Mitochondria to High-Altitude Hypoxia during an Ascent of Everest. FASEB J. 2012, 26, 1431–1441. [Google Scholar] [CrossRef]
- Bi, P.; Shan, T.; Liu, W.; Yue, F.; Yang, X.; Liang, X.-R.; Wang, J.; Li, J.; Carlesso, N.; Liu, X.; et al. Inhibition of Notch Signaling Promotes Browning of White Adipose Tissue and Ameliorates Obesity. Nat. Med. 2014, 20, 911–918. [Google Scholar] [CrossRef]
- Colleoni, F.; Padmanabhan, N.; Yung, H.-W.; Watson, E.D.; Cetin, I.; Tissot van Patot, M.C.; Burton, G.J.; Murray, A.J. Suppression of Mitochondrial Electron Transport Chain Function in the Hypoxic Human Placenta: A Role for MiRNA-210 and Protein Synthesis Inhibition. PLoS ONE 2013, 8, e55194. [Google Scholar] [CrossRef]
- Fukuda, R.; Zhang, H.; Kim, J.-W.; Shimoda, L.; Dang, C.V.; Semenza, G.L. HIF-1 Regulates Cytochrome Oxidase Subunits to Optimize Efficiency of Respiration in Hypoxic Cells. Cell 2007, 129, 111–122. [Google Scholar] [CrossRef]
- O’Brien, K.A.; Simonson, T.S.; Murray, A.J. Metabolic Adaptation to High Altitude. Curr. Opin. Endocr. Metab. Res. 2020, 11, 33–41. [Google Scholar] [CrossRef]
- Song, N.-J.; Yun, U.J.; Yang, S.; Wu, C.; Seo, C.-R.; Gwon, A.-R.; Baik, S.-H.; Choi, Y.; Choi, B.Y.; Bahn, G.; et al. Notch1 Deficiency Decreases Hepatic Lipid Accumulation by Induction of Fatty Acid Oxidation. Sci. Rep. 2016, 6, 19377. [Google Scholar] [CrossRef]
- Kuhlicke, J.; Frick, J.S.; Morote-Garcia, J.C.; Rosenberger, P.; Eltzschig, H.K. Hypoxia Inducible Factor (HIF)-1 Coordinates Induction of Toll-like Receptors TLR2 and TLR6 during Hypoxia. PLoS ONE 2007, 2, e1364. [Google Scholar] [CrossRef]
- Rius, J.; Guma, M.; Schachtrup, C.; Akassoglou, K.; Zinkernagel, A.S.; Nizet, V.; Johnson, R.S.; Haddad, G.G.; Karin, M. NF-KappaB Links Innate Immunity to the Hypoxic Response through Transcriptional Regulation of HIF-1alpha. Nature 2008, 453, 807–811. [Google Scholar] [CrossRef] [PubMed]
- Bonello, S.; Zähringer, C.; BelAiba, R.S.; Djordjevic, T.; Hess, J.; Michiels, C.; Kietzmann, T.; Görlach, A. Reactive Oxygen Species Activate the HIF-1α Promoter Via a Functional NFκB Site. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Shelly, L.; Miele, L.; Boykins, R.; Norcross, M.A.; Guan, E. Human Notch-1 Inhibits NF-ΚB Activity in the Nucleus Through a Direct Interaction Involving a Novel Domain. J. Immunol. 2001, 167, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Pajvani, U.B.; Qiang, L.; Kangsamaksin, T.; Kitajewski, J.; Ginsberg, H.N.; Accili, D. Inhibition of Notch Uncouples Akt Activation from Hepatic Lipid Accumulation by Decreasing MTorc1 Stability. Nat. Med. 2013, 19, 1054–1060. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Nioi, P.; Pickett, C.B. The Nrf2-Antioxidant Response Element Signaling Pathway and Its Activation by Oxidative Stress. J. Biol. Chem. 2009, 284, 13291–13295. [Google Scholar] [CrossRef] [PubMed]
- Kamata, H.; Manabe, T.; Oka, S.-I.; Kamata, K.; Hirata, H. Hydrogen Peroxide Activates IκB Kinases through Phosphorylation of Serine Residues in the Activation Loops. FEBS Lett. 2002, 519, 231–237. [Google Scholar] [CrossRef]
- Zhou, X.-L.; Wu, X.; Zhu, R.-R.; Xu, H.; Li, Y.-Y.; Xu, Q.-R.; Liu, S.; Lai, S.-Q.; Xu, X.; Wan, L.; et al. Notch1-Nrf2 Signaling Crosstalk Provides Myocardial Protection by Reducing ROS Formation. Biochem. Cell Biol. 2020, 98, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Yang, X.; Han, S.; Guo, H.; Zheng, Z.; Wang, H.; Guan, H.; Jia, Y.; Gao, J.; Yang, T.; et al. Notch1 Pathway Protects against Burn-Induced Myocardial Injury by Repressing Reactive Oxygen Species Production through JAK2/STAT3 Signaling. Oxid. Med. Cell. Longev. 2016, 2016, 5638943. [Google Scholar] [CrossRef] [PubMed]
- Pietras, A.; von Stedingk, K.; Lindgren, D.; Påhlman, S.; Axelson, H. JAG2 Induction in Hypoxic Tumor Cells Alters Notch Signaling and Enhances Endothelial Cell Tube Formation. Mol. Cancer Res. 2011, 9, 626–636. [Google Scholar] [CrossRef]
- Xing, F.; Okuda, H.; Watabe, M.; Kobayashi, A.; Pai, S.K.; Liu, W.; Pandey, P.R.; Fukuda, K.; Hirota, S.; Sugai, T.; et al. Hypoxia-Induced Jagged2 Promotes Breast Cancer Metastasis and Self-Renewal of Cancer Stem-like Cells. Oncogene 2011, 30, 4075–4086. [Google Scholar] [CrossRef] [PubMed]
- Lanner, F.; Lee, K.L.; Ortega, G.C.; Sohl, M.; Li, X.; Jin, S.; Hansson, E.M.; Claesson-Welsh, L.; Poellinger, L.; Lendahl, U.; et al. Hypoxia-Induced Arterial Differentiation Requires Adrenomedullin and Notch Signaling. Stem Cells Dev. 2013, 22, 1360–1369. [Google Scholar] [CrossRef]
- Jögi, A.; Øra, I.; Nilsson, H.; Lindeheim, A.; Makino, Y.; Poellinger, L.; Axelson, H.; Påhlman, S. Hypoxia Alters Gene Expression in Human Neuroblastoma Cells toward an Immature and Neural Crest-like Phenotype. Proc. Natl. Acad. Sci. USA 2002, 99, 7021–7026. [Google Scholar] [CrossRef]
- Mutvei, A.P.; Landor, S.K.-J.; Fox, R.; Braune, E.-B.; Tsoi, Y.L.; Phoon, Y.P.; Sahlgren, C.; Hartman, J.; Bergh, J.; Jin, S.; et al. Notch Signaling Promotes a HIF2α-Driven Hypoxic Response in Multiple Tumor Cell Types. Oncogene 2018, 37, 6083–6095. [Google Scholar] [CrossRef]
- Coleman, M.L.; McDonough, M.A.; Hewitson, K.S.; Coles, C.; Mecinovic, J.; Edelmann, M.; Cook, K.M.; Cockman, M.E.; Lancaster, D.E.; Kessler, B.M.; et al. Asparaginyl Hydroxylation of the Notch Ankyrin Repeat Domain by Factor Inhibiting Hypoxia-Inducible Factor. J. Biol. Chem. 2007, 282, 24027–24038. [Google Scholar] [CrossRef] [PubMed]
- Tseng, L.-C.; Zhang, C.; Cheng, C.-M.; Xu, H.; Hsu, C.-H.; Jiang, Y.-J. New Classes of Mind Bomb-Interacting Proteins Identified from Yeast Two-Hybrid Screens. PLoS ONE 2014, 9, e93394. [Google Scholar] [CrossRef] [PubMed]
- Shareef, M.M.; Udayakumar, T.S.; Sinha, V.K.; Saleem, S.M.; Griggs, W.W. Interaction of HIF-1 and Notch3 Is Required for the Expression of Carbonic Anhydrase 9 in Breast Carcinoma Cells. Genes Cancer 2013, 4, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Julian, C.G. Epigenomics and Human Adaptation to High Altitude. J. Appl. Physiol. 2017, 123, 1362–1370. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Liang, G. Rethinking How DNA Methylation Patterns Are Maintained. Nat. Rev. Genet. 2009, 10, 805–811. [Google Scholar] [CrossRef] [PubMed]
- Miranda, T.B.; Jones, P.A. DNA Methylation: The Nuts and Bolts of Repression. J. Cell. Physiol. 2007, 213, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Ogino, S.; Nosho, K.; Kirkner, G.J.; Kawasaki, T.; Chan, A.T.; Schernhammer, E.S.; Giovannucci, E.L.; Fuchs, C.S. A Cohort Study of Tumoral LINE-1 Hypomethylation and Prognosis in Colon Cancer. J. Natl. Cancer Inst. 2008, 100, 1734–1738. [Google Scholar] [CrossRef]
- James, S.J.; Cutler, P.; Melnyk, S.; Jernigan, S.; Janak, L.; Gaylor, D.W.; Neubrander, J.A. Metabolic Biomarkers of Increased Oxidative Stress and Impaired Methylation Capacity in Children with Autism. Am. J. Clin. Nutr. 2004, 80, 1611–1617. [Google Scholar] [CrossRef]
- Lachance, G.; Uniacke, J.; Audas, T.E.; Holterman, C.E.; Franovic, A.; Payette, J.; Lee, S. DNMT3a Epigenetic Program Regulates the HIF-2α Oxygen-Sensing Pathway and the Cellular Response to Hypoxia. Proc. Natl. Acad. Sci. USA 2014, 111, 7783–7788. [Google Scholar] [CrossRef] [PubMed]
- Herman, J.G.; Latif, F.; Weng, Y.; Lerman, M.I.; Zbar, B.; Liu, S.; Samid, D.; Duan, D.S.; Gnarra, J.R.; Linehan, W.M. Silencing of the VHL Tumor-Suppressor Gene by DNA Methylation in Renal Carcinoma. Proc. Natl. Acad. Sci. USA 1994, 91, 9700–9704. [Google Scholar] [CrossRef] [PubMed]
- Wenger, R.H.; Kvietikova, I.; Rolfs, A.; Camenisch, G.; Gassmann, M. Oxygen-Regulated Erythropoietin Gene Expression Is Dependent on a CpG Methylation-Free Hypoxia-Inducible Factor-1 DNA-Binding Site. Eur. J. Biochem. 1998, 253, 771–777. [Google Scholar] [CrossRef] [PubMed]
- Rössler, J.; Stolze, I.; Frede, S.; Freitag, P.; Schweigerer, L.; Havers, W.; Fandrey, J. Hypoxia-Induced Erythropoietin Expression in Human Neuroblastoma Requires a Methylation Free HIF-1 Binding Site. J. Cell. Biochem. 2004, 93, 153–161. [Google Scholar] [CrossRef]
- Yin, H.; Blanchard, K.L. DNA Methylation Represses the Expression of the Human Erythropoietin Gene by Two Different Mechanisms. Blood 2000, 95, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Schwanbeck, R. The Role of Epigenetic Mechanisms in Notch Signaling during Development. J. Cell. Physiol. 2015, 230, 969–981. [Google Scholar] [CrossRef] [PubMed]
- Ghoshal, P.; Nganga, A.J.; Moran-Giuati, J.; Szafranek, A.; Johnson, T.R.; Bigelow, A.J.; Houde, C.M.; Avet-Loiseau, H.; Smiraglia, D.J.; Ersing, N.; et al. Loss of the SMRT/NCoR2 Corepressor Correlates with JAG2 Overexpression in Multiple Myeloma. Cancer Res. 2009, 69, 4380–4387. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Li, J.; Ho, J.C.; Chia, G.S.; Kato, H.; Jha, S.; Yang, H.; Poellinger, L.; Lee, K.L. Hypoxia Is a Key Driver of Alternative Splicing in Human Breast Cancer Cells. Sci. Rep. 2017, 7, 4108. [Google Scholar] [CrossRef] [PubMed]
- Green, L.; Cookson, A.; Bruce, I.N.; Donn, R.P.; Ray, D.W. Identification of Multiple, Oxygen-Stable HIF1 Alpha Isoforms, and Augmented Expression of Adrenomedullin in Rheumatoid Arthritis. Clin. Exp. Rheumatol. 2013, 31, 672–682. [Google Scholar] [PubMed]
- Gothié, E.; Richard, D.E.; Berra, E.; Pagès, G. Identification of Alternative Spliced Variants of Human Hypoxia-Inducible Factor-1α. J. Biol. 2000, 275, 6922–6927. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-H.; Park, J.-W.; Chun, Y.-S. Non-Hypoxic Transcriptional Activation of the Aryl Hydrocarbon Receptor Nuclear Translocator in Concert with a Novel Hypoxia-Inducible Factor-1alpha Isoform. Nucleic Acids Res. 2004, 32, 5499–5511. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bowler, E.; Oltean, S. Alternative Splicing in Angiogenesis. Int. J. Mol. Sci. 2019, 20, 2067. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.-J.; Kim, I.; Lee, J.E.; Park, J.-W. PIN1 Transcript Variant 2 Acts as a Long Non-Coding RNA That Controls the HIF-1-Driven Hypoxic Response. Sci. Rep. 2019, 9, 10599. [Google Scholar] [CrossRef] [PubMed]
- Mirtschink, P.; Krishnan, J.; Grimm, F.; Sarre, A.; Hörl, M.; Kayikci, M.; Fankhauser, N.; Christinat, Y.; Cortijo, C.; Feehan, O.; et al. HIF-Driven SF3B1 Induces KHK-C to Enforce Fructolysis and Heart Disease. Nature 2015, 522, 444–449. [Google Scholar] [CrossRef]
- David, C.J.; Chen, M.; Assanah, M.; Canoll, P.; Manley, J.L. HnRNP Proteins Controlled by C-Myc Deregulate Pyruvate Kinase MRNA Splicing in Cancer. Nature 2010, 463, 364–368. [Google Scholar] [CrossRef]
- Williams, A.L.; Khadka, V.; Tang, M.; Avelar, A.; Schunke, K.J.; Menor, M.; Shohet, R.V. HIF1 Mediates a Switch in Pyruvate Kinase Isoforms after Myocardial Infarction. Physiol. Genom. 2018, 50, 479–494. [Google Scholar] [CrossRef]
- Luo, W.; Hu, H.; Chang, R.; Zhong, J.; Knabel, M.; O’Meally, R.; Cole, R.N.; Pandey, A.; Semenza, G.L. Pyruvate Kinase M2 Is a PHD3-Stimulated Coactivator for Hypoxia-Inducible Factor 1. Cell 2011, 145, 732–744. [Google Scholar] [CrossRef]
- Rosati, E.; Sabatini, R.; Rampino, G.; Tabilio, A.; Di Ianni, M.; Fettucciari, K.; Bartoli, A.; Coaccioli, S.; Screpanti, I.; Marconi, P. Constitutively Activated Notch Signaling Is Involved in Survival and Apoptosis Resistance of B-CLL Cells. Blood 2009, 113, 856–865. [Google Scholar] [CrossRef]
- Puente, X.S.; Pinyol, M.; Quesada, V.; Conde, L.; Ordóñez, G.R.; Villamor, N.; Escaramis, G.; Jares, P.; Beà, S.; González-Díaz, M.; et al. Whole-Genome Sequencing Identifies Recurrent Mutations in Chronic Lymphocytic Leukaemia. Nature 2011, 475, 101–105. [Google Scholar] [CrossRef]
- Larrayoz, M.; Rose-Zerilli, M.J.J.; Kadalayil, L.; Parker, H.; Blakemore, S.; Forster, J.; Davis, Z.; Steele, A.J.; Collins, A.; Else, M.; et al. Non-Coding NOTCH1 Mutations in Chronic Lymphocytic Leukemia; Their Clinical Impact in the UK CLL4 Trial. Leukemia 2017, 31, 510–514. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Brooks, A.N.; Fan, J.; Wan, Y.; Gambe, R.; Li, S.; Hergert, S.; Yin, S.; Freeman, S.S.; Levin, J.Z.; et al. Transcriptomic Characterization of SF3B1 Mutation Reveals Its Pleiotropic Effects in Chronic Lymphocytic Leukemia. Cancer Cell 2016, 30, 750–763. [Google Scholar] [CrossRef] [PubMed]
- Adams, B.D.; Kasinski, A.L.; Slack, F.J. Aberrant Regulation and Function of MicroRNAs in Cancer. Curr. Biol. 2014, 24, R762–R776. [Google Scholar] [CrossRef] [PubMed]
- Devlin, C.; Greco, S.; Martelli, F.; Ivan, M. MiR-210: More than a Silent Player in Hypoxia. IUBMB Life 2011, 63, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Lou, Y.-L.; Guo, F.; Liu, F.; Gao, F.-L.; Zhang, P.-Q.; Niu, X.; Guo, S.-C.; Yin, J.-H.; Wang, Y.; Deng, Z.-F. MiR-210 Activates Notch Signaling Pathway in Angiogenesis Induced by Cerebral Ischemia. Mol. Cell. Biochem. 2012, 370, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhao, Y.; Luo, R.; Bian, X.; Wang, Y. A positive feedback self-regulatory loop between miR-210 and HIF-1α mediated by CPEB2 is involved in trophoblast syncytialization: Implication of trophoblast malfunction in preeclampsia. Biol. Reprod. 2020, 102, 560–570. [Google Scholar] [PubMed]
- Dökümcü, K.; Simonian, M.; Farahani, R.M. MiR4673 Improves Fitness Profile of Neoplastic Cells by Induction of Autophagy. Cell Death Dis. 2018, 9, 1068. [Google Scholar] [CrossRef] [PubMed]
- Farahani, R.; Rezaei-Lotfi, S.; Simonian, M.; Hunter, N. Bi-Modal Reprogramming of Cell Cycle by MiRNA-4673 Amplifies Human Neurogenic Capacity. Cell Cycle 2019, 18, 848–868. [Google Scholar] [CrossRef]
- Huang, H.-L.; Shi, Y.-P.; He, H.-J.; Wang, Y.-H.; Chen, T.; Yang, L.-W.; Yang, T.; Chen, J.; Cao, J.; Yao, W.-M.; et al. MiR-4673 Modulates Paclitaxel-Induced Oxidative Stress and Loss of Mitochondrial Membrane Potential by Targeting 8-Oxoguanine-DNA Glycosylase-1. Cell. Physiol. Biochem. 2017, 42, 889–900. [Google Scholar] [CrossRef] [PubMed]
- Icli, B.; Li, H.; Pérez-Cremades, D.; Wu, W.; Ozdemir, D.; Haemmig, S.; Guimaraes, R.B.; Manica, A.; Marchini, J.F.; Orgill, D.P.; et al. MiR-4674 Regulates Angiogenesis in Tissue Injury by Targeting P38K Signaling in Endothelial Cells. Am. J. Physiol. Cell Physiol. 2020, 318, C524–C535. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.; Song, K.; Han, C.; Zhang, J.; Lu, L.; Chen, W.; Wu, T. Epigenetic Silencing of MiRNA-34a in Human Cholangiocarcinoma via EZH2 and DNA Methylation: Impact on Regulation of Notch Pathway. Am. J. Pathol. 2017, 187, 2288–2299. [Google Scholar] [CrossRef] [PubMed]
- Shayevitch, R.; Askayo, D.; Keydar, I.; Ast, G. The Importance of DNA Methylation of Exons on Alternative Splicing. RNA 2018, 24, 1351–1362. [Google Scholar] [CrossRef]
- Monge, C. LIFE IN THE ANDES AND CHRONIC MOUNTAIN SICKNESS. Science 1942, 95, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Beall, C.M.; Brittenham, G.M.; Strohl, K.P.; Blangero, J.; Williams-Blangero, S.; Goldstein, M.C.; Decker, M.J.; Vargas, E.; Villena, M.; Soria, R.; et al. Hemoglobin Concentration of High-Altitude Tibetans and Bolivian Aymara. Am. J. Phys. Anthropol. 1998, 106, 385–400. [Google Scholar] [CrossRef]
- Moore, L.G.; Niermeyer, S.; Zamudio, S. Human Adaptation to High Altitude: Regional and Life-Cycle Perspectives. Am. J. Phys. Anthropol. 1998, 107, 25–64. [Google Scholar] [CrossRef]
- Winslow, R.M.; Chapman, K.W.; Gibson, C.C.; Samaja, M.; Monge, C.C.; Goldwasser, E.; Sherpa, M.; Blume, F.D.; Santolaya, R. Different Hematologic Responses to Hypoxia in Sherpas and Quechua Indians. J. Appl. Physiol. 1989, 66, 1561–1569. [Google Scholar] [CrossRef]
- Semenza, G.L. Involvement of Oxygen-Sensing Pathways in Physiologic and Pathologic Erythropoiesis. Blood 2009, 114, 2015–2019. [Google Scholar] [CrossRef]
- Haase, V.H. Hypoxic Regulation of Erythropoiesis and Iron Metabolism. Am. J. Physiol. Renal Physiol. 2010, 299, F1–F13. [Google Scholar] [CrossRef]
- Lorenzo, F.R.; Huff, C.; Myllymäki, M.; Olenchock, B.; Swierczek, S.; Tashi, T.; Gordeuk, V.; Wuren, T.; Ri-Li, G.; McClain, D.A.; et al. A Genetic Mechanism for Tibetan High-Altitude Adaptation. Nat. Genet. 2014, 46, 951–956. [Google Scholar] [CrossRef] [PubMed]
- Hattangadi, S.M.; Wong, P.; Zhang, L.; Flygare, J.; Lodish, H.F. From Stem Cell to Red Cell: Regulation of Erythropoiesis at Multiple Levels by Multiple Proteins, RNAs, and Chromatin Modifications. Blood 2011, 118, 6258–6268. [Google Scholar] [CrossRef] [PubMed]
- Azad, P.; Stobdan, T.; Zhou, D.; Hartley, I.; Akbari, A.; Bafna, V.; Haddad, G.G. High-Altitude Adaptation in Humans: From Genomics to Integrative Physiology. J. Mol. Med. 2017, 95, 1269–1282. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Udpa, N.; Ronen, R.; Stobdan, T.; Liang, J.; Appenzeller, O.; Zhao, H.W.; Yin, Y.; Du, Y.; Guo, L.; et al. Whole-Genome Sequencing Uncovers the Genetic Basis of Chronic Mountain Sickness in Andean Highlanders. Am. J. Hum. Genet. 2013, 93, 452–462. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, M.M.; Callacondo, D.; Rojas-Camayo, J.; Quesada-Olarte, J.; Wang, X.; Uchida, N.; Maric, I.; Remaley, A.T.; Leon-Velarde, F.; Villafuerte, F.C.; et al. SENP1, but Not Fetal Hemoglobin, Differentiates Andean Highlanders with Chronic Mountain Sickness from Healthy Individuals among Andean Highlanders. Exp. Hematol. 2016, 44, 483–490e2. [Google Scholar] [CrossRef] [PubMed]
- Chiba, S. Concise Review: Notch Signaling in Stem Cell Systems. Stem Cells 2006, 24, 2437–2447. [Google Scholar] [CrossRef]
- Bigas, A.; Espinosa, L. Hematopoietic Stem Cells: To Be or Notch to Be. Blood J. Am. Soc. Hematol. 2012, 119, 3226–3235. [Google Scholar] [CrossRef] [PubMed]
- Robert-Moreno, A.; Espinosa, L.; de la Pompa, J.L.; Bigas, A. RBPjkappa-Dependent Notch Function Regulates Gata2 and Is Essential for the Formation of Intra-Embryonic Hematopoietic Cells. Development 2005, 132, 1117–1126. [Google Scholar] [CrossRef] [PubMed]
- Kumano, K.; Chiba, S.; Kunisato, A.; Sata, M.; Saito, T.; Nakagami-Yamaguchi, E.; Yamaguchi, T.; Masuda, S.; Shimizu, K.; Takahashi, T.; et al. Notch1 but Not Notch2 Is Essential for Generating Hematopoietic Stem Cells from Endothelial Cells. Immunity 2003, 18, 699–711. [Google Scholar] [CrossRef]
- Pajcini, K.V.; Speck, N.A.; Pear, W.S. Notch Signaling in Mammalian Hematopoietic Stem Cells. Leukemia 2011, 25, 1525–1532. [Google Scholar] [CrossRef]
- Milner, L.A.; Kopan, R.; Martin, D.I.; Bernstein, I.D. A Human Homologue of the Drosophila Developmental Gene, Notch, Is Expressed in CD34 Hematopoietic Precursors. Blood 1994, 83, 2057–2062. [Google Scholar] [CrossRef]
- Calvi, L.M.; Adams, G.B.; Weibrecht, K.W.; Weber, J.M.; Olson, D.P.; Knight, M.C.; Martin, R.P.; Schipani, E.; Divieti, P.; Bringhurst, F.R.; et al. Osteoblastic Cells Regulate the Haematopoietic Stem Cell Niche. Nature 2003, 425, 841–846. [Google Scholar] [CrossRef]
- Zeuner, A.; Francescangeli, F.; Signore, M.; Venneri, M.A.; Pedini, F.; Felli, N.; Pagliuca, A.; Conticello, C.; De Maria, R. The Notch2–Jagged1 Interaction Mediates Stem Cell Factor Signaling in Erythropoiesis. Cell Death Differ. 2010, 18, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Maillard, I.; Koch, U.; Dumortier, A.; Shestova, O.; Xu, L.; Sai, H.; Pross, S.E.; Aster, J.C.; Bhandoola, A.; Radtke, F.; et al. Canonical Notch Signaling Is Dispensable for the Maintenance of Adult Hematopoietic Stem Cells. Cell Stem Cell 2008, 2, 356–366. [Google Scholar] [CrossRef]
- Heng, T.S.P.; The Immunological Genome Project Consortium; Painter, M.W.; Elpek, K.; Lukacs-Kornek, V.; Mauermann, N.; Turley, S.J.; Koller, D.; Kim, F.S.; Wagers, A.J.; et al. The Immunological Genome Project: Networks of Gene Expression in Immune Cells. Nat. Immunol. 2008, 9, 1091–1094. [Google Scholar] [CrossRef] [PubMed]
- Marsboom, G.; Rehman, J. Hypoxia Signaling in Vascular Homeostasis. Physiology 2018, 33, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Gnecchi-Ruscone, G.A.; Abondio, P.; De Fanti, S.; Sarno, S.; Sherpa, M.G.; Sherpa, P.T.; Marinelli, G.; Natali, L.; Di Marcello, M.; Peluzzi, D.; et al. Evidence of Polygenic Adaptation to High Altitude from Tibetan and Sherpa Genomes. Genome Biol. Evol. 2018, 10, 2919–2930. [Google Scholar] [CrossRef]
- Wei, C.; Wang, H.; Liu, G.; Zhao, F.; Kijas, J.W.; Ma, Y.; Lu, J.; Zhang, L.; Cao, J.; Wu, M.; et al. Genome-Wide Analysis Reveals Adaptation to High Altitudes in Tibetan Sheep. Sci. Rep. 2016, 6, 26770. [Google Scholar] [CrossRef] [PubMed]
- Dávila, R.D.; Julian, C.G.; Wilson, M.J.; Browne, V.A.; Rodriguez, C.; Bigham, A.W.; Shriver, M.D.; Vargas, E.; Moore, L.G. Do Anti-Angiogenic or Angiogenic Factors Contribute to the Protection of Birth Weight at High Altitude Afforded by Andean Ancestry? Reprod. Sci. 2010, 17, 861–870. [Google Scholar] [CrossRef]
- Soria, R.; Julian, C.G.; Vargas, E.; Moore, L.G.; Giussani, D.A. Graduated Effects of High-Altitude Hypoxia and Highland Ancestry on Birth Size. Pediatr. Res. 2013, 74, 633–638. [Google Scholar] [CrossRef]
- Yu, N.; Wu, J.-L.; Xiao, J.; Fan, L.; Chen, S.-H.; Li, W. HIF-1α Regulates Angiogenesis via Notch1/STAT3/ETBR Pathway in Trophoblastic Cells. Cell Cycle 2019, 18, 3502–3512. [Google Scholar] [CrossRef]
- Limbourg, F.P.; Takeshita, K.; Radtke, F.; Bronson, R.T.; Chin, M.T.; Liao, J.K. Essential Role of Endothelial Notch1 in Angiogenesis. Circulation 2005, 111, 1826–1832. [Google Scholar] [CrossRef]
- Dill, M.T.; Rothweiler, S.; Djonov, V.; Hlushchuk, R.; Tornillo, L.; Terracciano, L.; Meili-Butz, S.; Radtke, F.; Heim, M.H.; Semela, D. Disruption of Notch1 Induces Vascular Remodeling, Intussusceptive Angiogenesis, and Angiosarcomas in Livers of Mice. Gastroenterology 2012, 142, 967–977.e2. [Google Scholar] [CrossRef]
- Benedito, R.; Hellström, M. Notch as a Hub for Signaling in Angiogenesis. Exp. Cell Res. 2013, 319, 1281–1288. [Google Scholar] [CrossRef] [PubMed]
- Harrington, L.S.; Sainson, R.C.A.; Williams, C.K.; Taylor, J.M.; Shi, W.; Li, J.-L.; Harris, A.L. Regulation of Multiple Angiogenic Pathways by Dll4 and Notch in Human Umbilical Vein Endothelial Cells. Microvasc. Res. 2008, 75, 144–154. [Google Scholar] [CrossRef] [PubMed]
- Hellström, M.; Phng, L.-K.; Hofmann, J.J.; Wallgard, E.; Coultas, L.; Lindblom, P.; Alva, J.; Nilsson, A.-K.; Karlsson, L.; Gaiano, N.; et al. Dll4 Signalling through Notch1 Regulates Formation of Tip Cells during Angiogenesis. Nature 2007, 445, 776–780. [Google Scholar] [CrossRef] [PubMed]
- Gale, N.W.; Dominguez, M.G.; Noguera, I.; Pan, L.; Hughes, V.; Valenzuela, D.M.; Murphy, A.J.; Adams, N.C.; Lin, H.C.; Holash, J.; et al. Haploinsufficiency of Delta-like 4 Ligand Results in Embryonic Lethality Due to Major Defects in Arterial and Vascular Development. Proc. Natl. Acad. Sci. USA 2004, 101, 15949–15954. [Google Scholar] [CrossRef]
- Katoh, M.; Katoh, M. Precision Medicine for Human Cancers with Notch Signaling Dysregulation (Review). Int. J. Mol. Med. 2020, 45, 279–297. [Google Scholar] [CrossRef]
- Noguera-Troise, I.; Daly, C.; Papadopoulos, N.J.; Coetzee, S.; Boland, P.; Gale, N.W.; Lin, H.C.; Yancopoulos, G.D.; Thurston, G. Blockade of Dll4 Inhibits Tumour Growth by Promoting Non-Productive Angiogenesis. Nature 2006, 444, 1032–1037. [Google Scholar] [CrossRef]
- Ridgway, J.; Zhang, G.; Wu, Y.; Stawicki, S.; Liang, W.-C.; Chanthery, Y.; Kowalski, J.; Watts, R.J.; Callahan, C.; Kasman, I.; et al. Inhibition of Dll4 Signalling Inhibits Tumour Growth by Deregulating Angiogenesis. Nature 2006, 444, 1083–1087. [Google Scholar] [CrossRef] [PubMed]
- Wieland, E.; Rodriguez-Vita, J.; Liebler, S.S.; Mogler, C.; Moll, I.; Herberich, S.E.; Espinet, E.; Herpel, E.; Menuchin, A.; Chang-Claude, J.; et al. Endothelial Notch1 Activity Facilitates Metastasis. Cancer Cell 2017, 31, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Wohlfeil, S.A.; Häfele, V.; Dietsch, B.; Schledzewski, K.; Winkler, M.; Zierow, J.; Leibing, T.; Mohammadi, M.M.; Heineke, J.; Sticht, C.; et al. Hepatic Endothelial Notch Activation Protects against Liver Metastasis by Regulating Endothelial-Tumor Cell Adhesion Independent of Angiocrine Signaling. Cancer Res. 2019, 79, 598–610. [Google Scholar] [CrossRef] [PubMed]
- Woolard, J.; Bevan, H.S.; Harper, S.J.; Bates, D.O. Molecular Diversity of VEGF-A as a Regulator of Its Biological Activity. Microcirculation 2009, 16, 572–592. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Chathery, Y.; Wu, Y.; Rathore, N.; Tong, R.K.; Peale, F.; Bagri, A.; Tessier-Lavigne, M.; Koch, A.W.; Watts, R.J. Neuropilin-1 Binds to VEGF121 and Regulates Endothelial Cell Migration and Sprouting. J. Biol. Chem. 2007, 282, 24049–24056. [Google Scholar] [CrossRef] [PubMed]
- Abou Faycal, C.; Gazzeri, S.; Eymin, B. A VEGF-A/SOX2/SRSF2 Network Controls VEGFR1 Pre-MRNA Alternative Splicing in Lung Carcinoma Cells. Sci. Rep. 2019, 9, 336. [Google Scholar] [CrossRef] [PubMed]
- Boeckel, J.-N.; Guarani, V.; Koyanagi, M.; Roexe, T.; Lengeling, A.; Schermuly, R.T.; Gellert, P.; Braun, T.; Zeiher, A.; Dimmeler, S. Jumonji Domain-Containing Protein 6 (Jmjd6) Is Required for Angiogenic Sprouting and Regulates Splicing of VEGF-Receptor 1. Proc. Natl. Acad. Sci. USA 2011, 108, 3276–3281. [Google Scholar] [CrossRef] [PubMed]
- Kangsamaksin, T.; Murtomaki, A.; Kofler, N.M.; Cuervo, H.; Chaudhri, R.A.; Tattersall, I.W.; Rosenstiel, P.E.; Shawber, C.J.; Kitajewski, J. NOTCH Decoys That Selectively Block DLL/NOTCH or JAG/NOTCH Disrupt Angiogenesis by Unique Mechanisms to Inhibit Tumor Growth. Cancer Discov. 2015, 5, 182–197. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.-Q.; Jing, Z.-C. High-Altitude Pulmonary Hypertension. Eur. Respir. Rev. 2009, 18, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Groves, B.M.; Droma, T.; Sutton, J.R.; McCullough, R.G.; McCullough, R.E.; Zhuang, J.; Rapmund, G.; Sun, S.; Janes, C.; Moore, L.G. Minimal Hypoxic Pulmonary Hypertension in Normal Tibetans at 3,658 m. J. Appl. Physiol. 1993, 74, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Mol, B.W.J.; Roberts, C.T.; Thangaratinam, S.; Magee, L.A.; de Groot, C.J.M.; Hofmeyr, G.J. Pre-Eclampsia. Lancet 2016, 387, 999–1011. [Google Scholar] [CrossRef]
- Keyes, L.E.; Armaza, J.F.; Niermeyer, S.; Vargas, E.; Young, D.A.; Moore, L.G. Intrauterine Growth Restriction, Preeclampsia, and Intrauterine Mortality at High Altitude in Bolivia. Pediatr. Res. 2003, 54, 20–25. [Google Scholar] [CrossRef]
- Palmer, S.K.; Moore, L.G.; Young, D.; Cregger, B.; Berman, J.C.; Zamudio, S. Altered Blood Pressure Course during Normal Pregnancy and Increased Preeclampsia at High Altitude (3100 Meters) in Colorado. Am. J. Obstet. Gynecol. 1999, 180, 1161–1168. [Google Scholar] [CrossRef]
- Esteve-Valverde, E.; Ferrer-Oliveras, R.; Gil-Aliberas, N.; Baraldès-Farré, A.; Llurba, E.; Alijotas-Reig, J. Pravastatin for Preventing and Treating Preeclampsia: A Systematic Review. Obstet. Gynecol. Surv. 2018, 73, 40–55. [Google Scholar] [CrossRef] [PubMed]
- Murray, A.J. Energy Metabolism and the High-altitude Environment. Exp. Physiol. 2016. [CrossRef] [PubMed]
- Chicco, A.J.; Le, C.H.; Gnaiger, E.; Dreyer, H.C.; Muyskens, J.B.; D’Alessandro, A.; Nemkov, T.; Hocker, A.D.; Prenni, J.E.; Wolfe, L.M.; et al. Adaptive Remodeling of Skeletal Muscle Energy Metabolism in High-Altitude Hypoxia: Lessons from AltitudeOmics. J. Biol. Chem. 2018, 293, 6659–6671. [Google Scholar] [CrossRef] [PubMed]
- Levett, D.Z.H.; Viganò, A.; Capitanio, D.; Vasso, M.; De Palma, S.; Moriggi, M.; Martin, D.S.; Murray, A.J.; Cerretelli, P.; Grocott, M.P.W.; et al. Changes in Muscle Proteomics in the Course of the Caudwell Research Expedition to Mt. Everest. Proteomics 2015, 15, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Papandreou, I.; Cairns, R.A.; Fontana, L.; Lim, A.L.; Denko, N.C. HIF-1 Mediates Adaptation to Hypoxia by Actively Downregulating Mitochondrial Oxygen Consumption. Cell Metab. 2006, 3, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Bi, P.; Kuang, S. Notch Signaling as a Novel Regulator of Metabolism. Trends Endocrinol. Metab. 2015, 26, 248–255. [Google Scholar] [CrossRef]
- Kodama, K.; Tojjar, D.; Yamada, S.; Toda, K.; Patel, C.J.; Butte, A.J. Ethnic Differences in the Relationship Between Insulin Sensitivity and Insulin Response: A Systematic Review and Meta-Analysis. Diabetes Care 2013, 36, 1789–1796. [Google Scholar] [CrossRef]
- Schenk, S.; Saberi, M.; Olefsky, J.M. Insulin Sensitivity: Modulation by Nutrients and Inflammation. J. Clin. Investig. 2008, 118, 2992–3002. [Google Scholar] [CrossRef]
- Iiyori, N.; Alonso, L.C.; Li, J.; Sanders, M.H.; Garcia-Ocana, A.; O’Doherty, R.M.; Polotsky, V.Y.; O’Donnell, C.P. Intermittent Hypoxia Causes Insulin Resistance in Lean Mice Independent of Autonomic Activity. Am. J. Respir. Crit. Care Med. 2007, 175, 851–857. [Google Scholar] [CrossRef] [PubMed]
- Lecoultre, V.; Peterson, C.M.; Covington, J.D.; Ebenezer, P.J.; Frost, E.A.; Schwarz, J.-M.; Ravussin, E. Ten Nights of Moderate Hypoxia Improves Insulin Sensitivity in Obese Humans. Diabetes Care 2013, 36, e197–e198. [Google Scholar] [CrossRef]
- Hasan, S.S.; Jabs, M.; Taylor, J.; Wiedmann, L.; Leibing, T.; Nordström, V.; Federico, G.; Roma, L.P.; Carlein, C.; Wolff, G.; et al. Endothelial Notch Signaling Controls Insulin Transport in Muscle. EMBO Mol. Med. 2020, 12, e09271. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, K.A.; Atkinson, R.A.; Richardson, L.; Koulman, A.; Murray, A.J.; Harridge, S.D.R.; Martin, D.S.; Levett, D.Z.H.; Mitchell, K.; Mythen, M.G.; et al. Metabolomic and Lipidomic Plasma Profile Changes in Human Participants Ascending to Everest Base Camp. Sci. Rep. 2019, 9, 2297. [Google Scholar] [CrossRef] [PubMed]
- Cole, M.A.; Abd Jamil, A.H.; Heather, L.C.; Murray, A.J.; Sutton, E.R.; Slingo, M.; Sebag-Montefiore, L.; Tan, S.C.; Aksentijević, D.; Gildea, O.S.; et al. On the Pivotal Role of PPARa in Adaptation of the Heart to Hypoxia and Why Fat in the Diet Increases Hypoxic Injury. FASEB J. 2016, 30, 2684–2697. [Google Scholar] [CrossRef] [PubMed]
- Ivashkiv, L.B. The Hypoxia–Lactate Axis Tempers Inflammation. Nat. Rev. Immunol. 2019, 20, 85–86. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Chi, F.; Guo, T.; Punj, V.; Lee, W.N.P.; French, S.W.; Tsukamoto, H. NOTCH Reprograms Mitochondrial Metabolism for Proinflammatory Macrophage Activation. J. Clin. Investig. 2015, 125, 1579–1590. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond Aerobic Glycolysis: Transformed Cells Can Engage in Glutamine Metabolism That Exceeds the Requirement for Protein and Nucleotide Synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Bai, C.; Ruan, Y.; Liu, M.; Chu, Q.; Qiu, L.; Yang, C.; Li, B. Coordinative Metabolism of Glutamine Carbon and Nitrogen in Proliferating Cancer Cells under Hypoxia. Nat. Commun. 2019, 10, 201. [Google Scholar] [CrossRef] [PubMed]
- Hoppeler, H.; Howald, H.; Cerretelli, P. Human Muscle Structure after Exposure to Extreme Altitude. Experientia 1990, 46, 1185–1187. [Google Scholar] [CrossRef]
- Ashmore, T.; Fernandez, B.O.; Branco-Price, C.; West, J.A.; Cowburn, A.S.; Heather, L.C.; Griffin, J.L.; Johnson, R.S.; Feelisch, M.; Murray, A.J. Dietary Nitrate Increases Arginine Availability and Protects Mitochondrial Complex I and Energetics in the Hypoxic Rat Heart. J. Physiol. 2014, 592, 4715–4731. [Google Scholar] [CrossRef]
- Valenti, L.; Mendoza, R.M.; Rametta, R.; Maggioni, M.; Kitajewski, C.; Shawber, C.J.; Pajvani, U.B. Hepatic Notch Signaling Correlates with Insulin Resistance and Nonalcoholic Fatty Liver Disease. Diabetes 2013, 62, 4052–4062. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Lee, J.; He, C.; Zou, M.-H.; Xie, Z. Suppression of the MTORC1/STAT3/Notch1 Pathway by Activated AMPK Prevents Hepatic Insulin Resistance Induced by Excess Amino Acids. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E197–E209. [Google Scholar] [CrossRef] [PubMed]
- Eltzschig, H.K.; Carmeliet, P. Hypoxia and Inflammation. N. Engl. J. Med. 2011, 364, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Korbecki, J.; Simińska, D.; Gąssowska-Dobrowolska, M.; Listos, J.; Gutowska, I.; Chlubek, D.; Baranowska-Bosiacka, I. Chronic and Cycling Hypoxia: Drivers of Cancer Chronic Inflammation through HIF-1 and NF-ΚB Activation: A Review of the Molecular Mechanisms. Int. J. Mol. Sci. 2021, 22, 10701. [Google Scholar] [CrossRef] [PubMed]
- Osipo, C.; Golde, T.E.; Osborne, B.A.; Miele, L.A. Off the Beaten Pathway: The Complex Cross Talk between Notch and NF-ΚB. Lab. Investig. 2007, 88, 11–17. [Google Scholar] [CrossRef]
- Poellinger, L.; Lendahl, U. Modulating Notch Signaling by Pathway-Intrinsic and Pathway-Extrinsic Mechanisms. Curr. Opin. Genet. Dev. 2008, 18, 449–454. [Google Scholar] [CrossRef]
- Samon, J.B.; Champhekar, A.; Minter, L.M.; Telfer, J.C.; Miele, L.; Fauq, A.; Das, P.; Golde, T.E.; Osborne, B.A. Notch1 and TGFβ1 Cooperatively Regulate Foxp3 Expression and the Maintenance of Peripheral Regulatory T Cells. Blood J. Am. Soc. Hematol. 2008, 112, 1813–1821. [Google Scholar] [CrossRef] [PubMed]
- Radtke, F.; MacDonald, H.R.; Tacchini-Cottier, F. Regulation of Innate and Adaptive Immunity by Notch. Nat. Rev. Immunol. 2013, 13, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Radtke, F.; Wilson, A.; Stark, G.; Bauer, M.; van Meerwijk, J.; MacDonald, H.R.; Aguet, M. Deficient T Cell Fate Specification in Mice with an Induced Inactivation of Notch1. Immunity 1999, 10, 547–558. [Google Scholar] [CrossRef]
- Liu, Z.-J.; Tan, Y.; Beecham, G.W.; Seo, D.M.; Tian, R.; Li, Y.; Vazquez-Padron, R.I.; Pericak-Vance, M.; Vance, J.M.; Goldschmidt-Clermont, P.J.; et al. Notch Activation Induces Endothelial Cell Senescence and Pro-Inflammatory Response: Implication of Notch Signaling in Atherosclerosis. Atherosclerosis 2012, 225, 296–303. [Google Scholar] [CrossRef]
- Görlach, A.; Dimova, E.Y.; Petry, A.; Martínez-Ruiz, A.; Hernansanz-Agustín, P.; Rolo, A.P.; Palmeira, C.M.; Kietzmann, T. Reactive Oxygen Species, Nutrition, Hypoxia and Diseases: Problems Solved? Redox Biol 2015, 6, 372–385. [Google Scholar] [CrossRef]
- Lando, D.; Pongratz, I.; Poellinger, L.; Whitelaw, M.L. A Redox Mechanism Controls Differential DNA Binding Activities of Hypoxia-Inducible Factor (HIF) 1α and the HIF-like Factor. J. Biol. Chem. 2000, 275, 4618–4627. [Google Scholar] [CrossRef] [PubMed]
- Bailey, D.M.; Rimoldi, S.F.; Rexhaj, E.; Pratali, L.; Salinas Salmòn, C.; Villena, M.; McEneny, J.; Young, I.S.; Nicod, P.; Allemann, Y.; et al. Oxidative-Nitrosative Stress and Systemic Vascular Function in Highlanders with and without Exaggerated Hypoxemia. Chest 2013, 143, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Bailey, D.M.; Brugniaux, J.V.; Filipponi, T.; Marley, C.J.; Stacey, B.; Soria, R.; Rimoldi, S.F.; Cerny, D.; Rexhaj, E.; Pratali, L.; et al. Exaggerated Systemic Oxidative-Inflammatory-Nitrosative Stress in Chronic Mountain Sickness Is Associated with Cognitive Decline and Depression. J. Physiol. 2019, 597, 611–629. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.-C.; Qin, H.-Y.; He, F.; Wang, L.; Fu, W.; Liu, D.; Guo, F.-C.; Liang, L.; Dou, K.-F.; Han, H. Canonical Notch Pathway Protects Hepatocytes from Ischemia/Reperfusion Injury in Mice by Repressing Reactive Oxygen Species Production through JAK2/STAT3 Signaling. Hepatology 2011, 54, 979–988. [Google Scholar] [CrossRef] [PubMed]
- Coant, N.; Ben Mkaddem, S.; Pedruzzi, E.; Guichard, C.; Tréton, X.; Ducroc, R.; Freund, J.-N.; Cazals-Hatem, D.; Bouhnik, Y.; Woerther, P.-L.; et al. NADPH Oxidase 1 Modulates WNT and NOTCH1 Signaling to Control the Fate of Proliferative Progenitor Cells in the Colon. Mol. Cell. Biol. 2010, 30, 2636–2650. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.E.; Lawrence, E.S.; Simonson, T.S.; Fox, K. Seq-Ing Higher Ground: Functional Investigation of Adaptive Variation Associated With High-Altitude Adaptation. Front. Genet. 2020, 11, 471. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, K.A.; Griffin, J.L.; Murray, A.J.; Edwards, L.M. Mitochondrial Responses to Extreme Environments: Insights from Metabolomics. Extrem. Physiol. Med. 2015, 4, 7. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Subjects/Species | Highland/Hypoxic Location | Altitude (m) | Data Format | Test for Natural Selection | Positively Selected Notch Gene/Region | Reference |
|---|---|---|---|---|---|---|
|
Human, Andeans (n = 50) | Cerro de Pasco, Peru (Quechua) La Paz, Bolivia (Aymara) | 4300 3600 | SNP genotype, Affymetrix, Inc. Gene Chip Human mapping 500 k array | Locus-specific branch length for SNP loci | NOTCH1 | [33] |
|
Human, Tibetans (n = 131) | Lhasa, Tibet | 3680 | Chromatin accessibility landscape through paired ATAC-seq and RNA-seq, obtained from primary HUVECs | Variant interpretation model by paired expression and chromatin accessibility methodology to identify active selected regulatory elements | NOTCH1 regulatory element | [34] |
| Chicken, Tibetan (n = 9) | Xiangcheng County, Tibet | 3500 | Whole genome resequencing | Aligned to reference genome using SOAP2, detected short InDel and structure variants | NOTCH2 | [35] |
|
Pig, Tibetan (n = 2) | Diqing Tibetan autonomous prefecture, Yunnan province | 3500 | Lung tissue whole-transcriptome microarrays | Differential gene expression, regulatory and phenotypic impact factor analysis | RBPJ (CSL) | [37] |
| Yak, Tibetan (n = 1) | Huangyuan County, Qinghai province | 3700 | Short oligonucleotide analysis | Whole-genome shotgun assembly | ADAM17 | [38] |
| Species | %O2 | Duration of Hypoxia Exposure | Data Format | Test for Natural Selection | Positively Selected Notch Gene/Region | Reference |
|---|---|---|---|---|---|---|
| Drosophila (n = 100) | 5% O2 | 1 week | P-element insertion line screen | Genomewide screening of P-elements related to eclosion rate, rtPCR on selected P-element targets | Dip1, CG14782, mRpS18B, Mys45A, CG6230, Drp1, Rep2, osa, CG8116, Atg1, CG33169, Chro, pzg, polo, lqf, Scrib, Alh, tna, CG14185, ci | [43] |
| Drosophila (n = 200, derived from 27 parental strains) | 6% and 4% O2 | 3 weeks for Notch mutants | Whole genome re-sequencing | Aligned to reference genome using MAQ, identified loci with high-confidence allelic differences and regions with allelic frequency differences | Fixed SNPs/indels: Notch, Delta, Fringe, Sgg Hairless, HDAC4, Fur2, Bon, IP3K2, Nej, Pcaf, Change in gene regulation: E(spl) Cluster Genes, Aph-1, Nct, Ser. | [44] |
| Chicken, Tibetan (n = 9) | 13% O2 | 11 days | Transcriptomic and proteomic analysis of embryos | Differentially expressed protein. | NOTCH2 | [36] |
| Hypoxic Physiological Response | Related Signaling Factors | References | Notch Pathway Interplay | Effect of Notch Interplay | Model/Condition | References |
|---|---|---|---|---|---|---|
| Modification of gene transcription | HIF-1α, HIF-2α | Reviewed in [6] | Notch ICD | Recruitment of HIF to HREs | Mouse myogenic and embryonic teratocarcinoma cells, hypoxia (1% O2) | [45,46] |
| DLL4, HEY2 | Notch pathway activation | Mouse myogenic and embryonic teratocarcinoma cells and human epithelial and embryonic kidney cells, hypoxia (1% O2) | [46,47] | |||
| γ-Secretase complex | Enhanced cleavage of Notch ICD | Human epithelial and breast cancer cells, hypoxia (NiCl2 and 1% O2) | [48,49] | |||
| Factor inhibiting HIF (FIH) | [50] | Notch ICD | Cellular differentiation | Mouse myogenic and embryonic teratocarcinoma cells, hypoxia (1% O2) | [45,46] | |
| Mindbomb 1 and 2 | Angiogenesis | Zebrafish embryos | [51] | |||
| DNA methylation | [52,53] | NOTCH1, NOTCH3, DLL1 | Notch pathway expression | Rat hepatic stellate cells and human gastric cancer cells | [54,55] | |
| CSL (or RBP-J) | Methylation-dependent DNA binding | Human leukemia cells | [56] | |||
| Angiogenesis | HIF via VEGF | Reviewed in [57] | Notch via DLL4 and Jagged1 | Differential VEGFR expression for selection of tip and stalk cells | Mouse embryo | [58,59] |
| Increased vascular tone | Pulmonary vascular remodeling | [60] | Notch3 pathway | Smooth muscle cell proliferation in small pulmonary arteries | Human and rodent pulmonary hypertension | [61] |
| Increased intracellular Ca2+ | Reviewed in [62] | Notch3 pathway | Upregulation of TPRC channels and increased expression of the Ca2+-sending receptor | [63,64] | ||
| Loss of insulin sensitivity | Increased plasma glucose and insulin | [65,66] | Notch ICD via FoxO1 | Insulin resistance, increased glucose-6 phosphatase expression | Mouse liver, normoxia | [67] |
| Upregulated glycolysis | HIF via PDK | [68] | Notch via PI3K/AKT serine/threonine kinase | Increased glucose uptake, and upregulation of glycolytic genes | Human breast cancer cells and Drosophila | [69,70] |
| Notch via p53 | Glycolytic dependency, suppressed mitochondrial activity | Human breast cancer cells | [69] | |||
| Glutamine metabolism | HIF-2α | [71] | Notch1 pathway | Decreased glutamine consumption and expression of glutaminase, ornithine aminotransferase and glutamine dehydrogenase 1 | Human immortalized leukemia cells and T lymphocytes | [72] |
| Loss in mitochondrial density | HIF via PGC1α | [73,74] | HES1 | Suppressed PGC1α | Mouse adipocytes | [75] |
| Suppressed respiratory complex I | HIF via mir-210 | [76] | Notch1 pathway | Decreased complex I activity and subunit expression | Breast cancer and immortalized leukemia cells | [72] |
| Complex IV subunit switch | HIF | [77] | Notch1 via p53 | Downregulated CIV | Breast cancer | [32] |
| Suppression of β-oxidation | HIF, PPARα | [3,12,78] | Notch1 pathway | Notch1 pathway inhibition increased PPARA and CPT1 expression | Mouse models of Notch deficiency, liver and adipose | [75,79] |
| Inflammation | HIF, NF-kB | [65,80,81,82] | Notch ICD | Interaction with NF-kB subunit | Human T cells | [83] |
| Oxidative stress | ROS via HIF, NF-kB, Nrf2 | [84,85,86] | Notch1 via Nrf2 | Increased cell viability, reduced ROS formation, increased antioxidant activities | Neonate rat myocardial cells, hypoxia–reoxygenation | [87] |
| Notch1 via JAK2/STAT3 | Activated mitochondrial SOD expression and decreased ROS production | Rat myocardium, burn injury | [88] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
O’Brien, K.A.; Murray, A.J.; Simonson, T.S. Notch Signaling and Cross-Talk in Hypoxia: A Candidate Pathway for High-Altitude Adaptation. Life 2022, 12, 437. https://doi.org/10.3390/life12030437
O’Brien KA, Murray AJ, Simonson TS. Notch Signaling and Cross-Talk in Hypoxia: A Candidate Pathway for High-Altitude Adaptation. Life. 2022; 12(3):437. https://doi.org/10.3390/life12030437
Chicago/Turabian StyleO’Brien, Katie A., Andrew J. Murray, and Tatum S. Simonson. 2022. "Notch Signaling and Cross-Talk in Hypoxia: A Candidate Pathway for High-Altitude Adaptation" Life 12, no. 3: 437. https://doi.org/10.3390/life12030437
APA StyleO’Brien, K. A., Murray, A. J., & Simonson, T. S. (2022). Notch Signaling and Cross-Talk in Hypoxia: A Candidate Pathway for High-Altitude Adaptation. Life, 12(3), 437. https://doi.org/10.3390/life12030437