
The Molecular Network behind Volatile Aroma Formation in Pear (Pyrus spp. Panguxiang) Revealed by Transcriptome Profiling via Fatty Acid Metabolic Pathways

 ,
,  ,
,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials and Sample Preparation
2.2. Determination of Volatile Substances
2.3. RNA Quantification and Qualification
2.4. Library Preparation and Sequencing
2.5. Data Analysis and Gene Annotation
2.6. Gene Co-Expression Network Analysis
2.7. Validation of DEGs through Quantitative Real-Time PCR (qRT–PCR)
3. Results
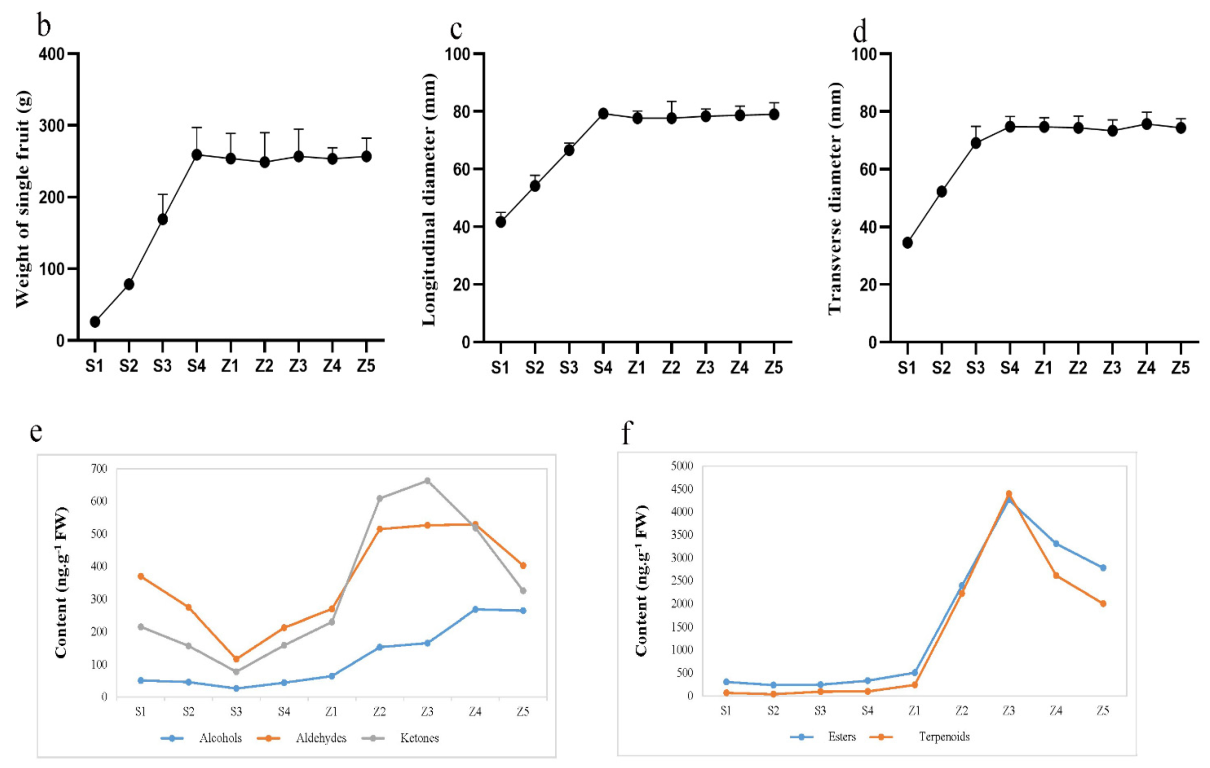
3.1. Sampling and Determination of Volatile Substance Content
3.2. Transcriptomic Analysis of Pyrus spp. Panguxiang Fruits
3.3. Functional Annotation and Classification of DEGs
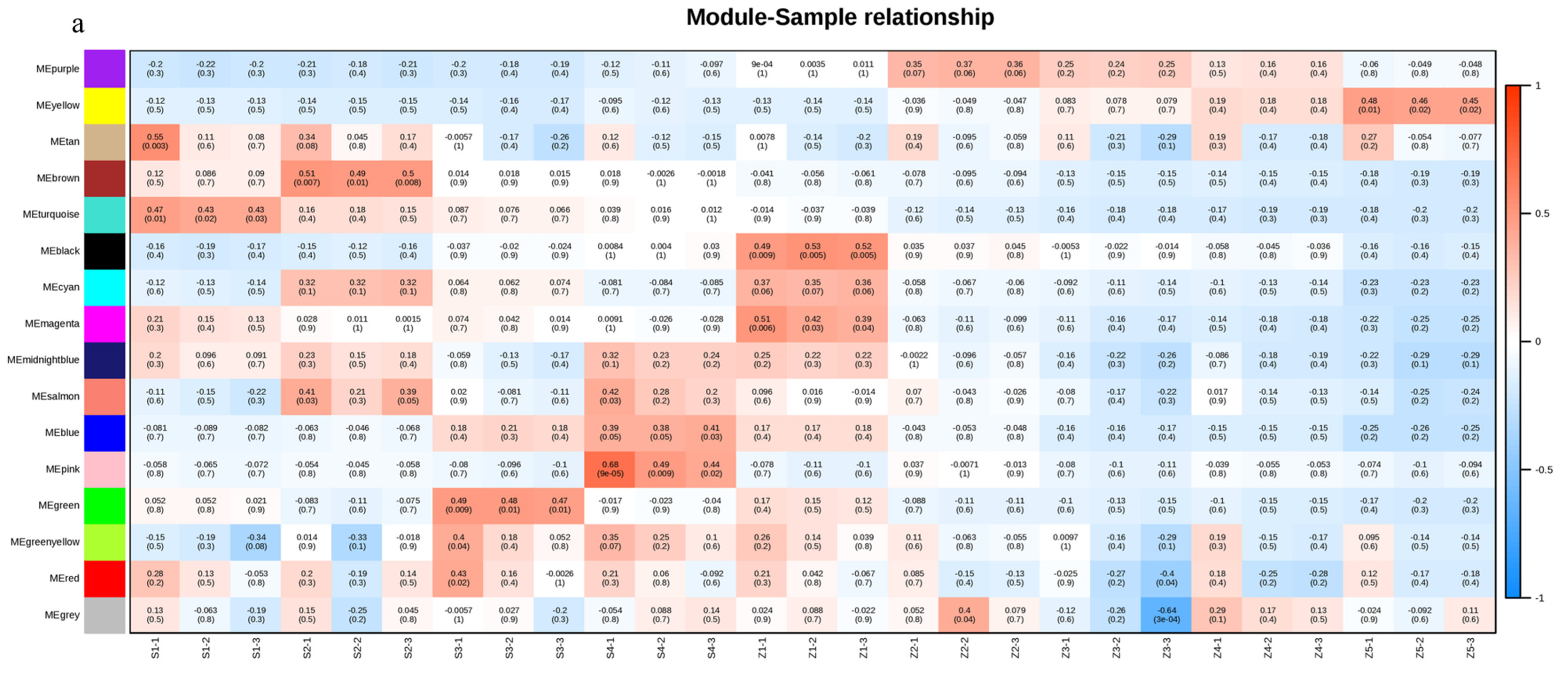
3.4. Co-Expression Analysis Identifies Key Gene Modules Involved in the Formation of Aroma Volatiles
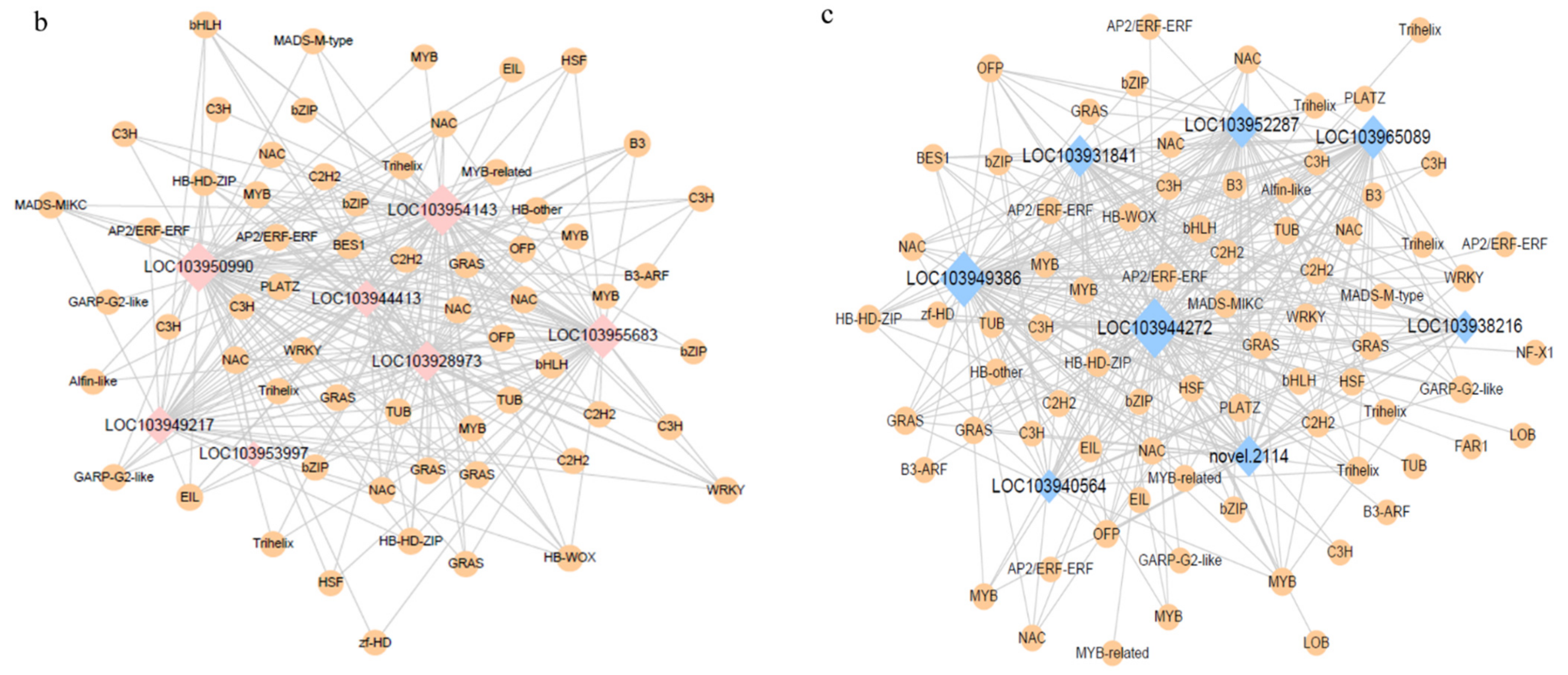
3.5. Co-Expression Network Analysis
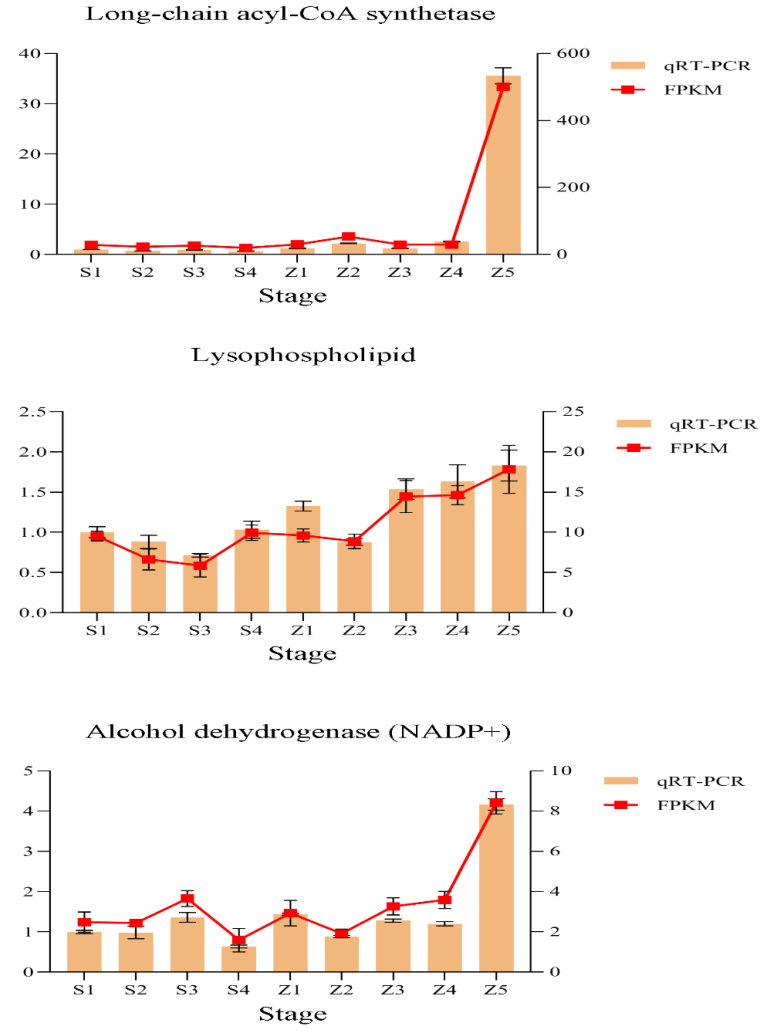
3.6. Verification of the Expression Profiles of Key Genes
4. Discussion
4.1. Effect of Fruit Ripening and Storage on Aroma Content
4.2. WGCNA Explores the Formation of Volatile Aromas in Pear Fruits
4.3. Key Factors in the Formation of Volatile Aroma in Pears
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sun, Q.; Zhang, N.; Wang, J.; Zhang, H.; Li, D.; Shi, J.; Li, R.; Weeda, S.; Zhao, B.; Ren, S.; et al. Melatonin Promotes Ripening and Improves Quality of Tomato Fruit during Postharvest Life. J. Exp. Bot. 2015, 66, 657–668. [Google Scholar] [CrossRef] [PubMed]
- Spaho, N.; Gaši, F.; Leitner, E.; Blesić, M.; Akagić, A.; Žuljević, S.O.; Kurtović, M.; Ratković, D.Đ.; Murtić, M.S.; Akšić, M.F.; et al. Characterization of Volatile Compounds and Flavor in Spirits of Old Apple and Pear Cultivars from the Balkan Region. Foods 2021, 10, 1258. [Google Scholar] [CrossRef] [PubMed]
- Jennings, W.G.; Sevenants, M.R. Volatile Esters of Bartlett Pear. III.a. J. Food Sci. 1964, 29, 158–163. [Google Scholar] [CrossRef]
- Qin, G.; Wang, Y.; Cao, B.; Wang, W.; Tian, S. Unraveling the Regulatory Network of the MADS Box Transcription Factor RIN in Fruit Ripening. Plant J. 2012, 70, 243–255. [Google Scholar] [CrossRef] [PubMed]
- Qin, G.; Tao, S.; Cao, Y.; Wu, J.; Zhang, H.; Huang, W.; Zhang, S. Evaluation of the Volatile Profile of 33 Pyrus Ussuriensis Cultivars by HS-SPME with GC-MS. Food Chem. 2012, 134, 2367–2382. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Jia, H.; Li, J.; Wang, Q.; Zhang, M.; Teng, Y. Emission of Volatile Esters and Transcription of Ethylene- and Aroma-Related Genes during Ripening of ‘Pingxiangli’ Pear Fruit (Pyrus Ussuriensis Maxim). Sci. Hortic. 2014, 170, 17–23. [Google Scholar] [CrossRef]
- Kou, X.; Liu, X.; Li, J.; Xiao, H.; Wang, J. Effects of Ripening, 1-Methylcyclopropene and Ultra-High-Pressure Pasteurisation on the Change of Volatiles in Chinese Pear Cultivars. J. Sci. Food Agric. 2012, 92, 177–183. [Google Scholar] [CrossRef]
- Zhang, L.P.; Shen, Y.X.; Bu, Q.Z.; Ji, S.J. Effects of 1-Methylcyclopropene on the Metabolic Pathways of Aroma-Related Compounds in Nanguo Pear. J. Food Process. Preserv. 2014, 38, 1749–1758. [Google Scholar] [CrossRef]
- Rapparini, F.; Predieri, S. Pear Fruit Volatiles. Hortic. Rev. 2010, 28, 237–324. [Google Scholar] [CrossRef]
- Ban, Y.; Oyama-Okubo, N.; Honda, C.; Nakayama, M.; Moriguchi, T. Emitted and Endogenous Volatiles in ‘Tsugaru’ Apple: The Mechanism of Ester and (E,E)-α-Farnesene Accumulation. Food Chem. 2010, 118, 272–277. [Google Scholar] [CrossRef]
- Dong, J.; Zhang, Y.; Tang, X.; Jin, W.; Han, Z. Differences in Volatile Ester Composition between Fragaria × Ananassa and F. Vesca and Implications for Strawberry Aroma Patterns. Sci. Hortic. 2013, 150, 47–53. [Google Scholar] [CrossRef]
- Imahori, Y.; Yamamoto, K.; Tanaka, H.; Bai, J. Residual Effects of Low Oxygen Storage of Mature Green Fruit on Ripening Processes and Ester Biosynthesis during Ripening in Bananas. Postharvest. Biol. Technol. 2013, 77, 19–27. [Google Scholar] [CrossRef]
- Qin, G.; Tao, S.; Zhang, H.; Huang, W.; Wu, J.; Xu, Y.; Zhang, S. Evolution of the Aroma Volatiles of Pear Fruits Supplemented with Fatty Acid Metabolic Precursors. Molecules 2014, 19, 20183–20196. [Google Scholar] [CrossRef]
- Dudareva, N.; Klempien, A.; Muhlemann, J.K.; Kaplan, I. Biosynthesis, Function and Metabolic Engineering of Plant Volatile Organic Compounds. New Phytol. 2013, 198, 16–32. [Google Scholar] [CrossRef]
- Defilippi, B.G.; Manríquez, D.; Luengwilai, K.; González-Agüero, M. Chapter 1 Aroma Volatiles: Biosynthesis and Mechanisms of Modulation During Fruit Ripening. Adv. Bot. Res. 2009, 50, 1–37. [Google Scholar] [CrossRef]
- Gigot, C.; Ongena, M.; Fauconnier, M.-A.; Wathelet, J.-P.; Du Jardin, P.; Thonart, P. The Lipoxygenase Metabolic Pathway in Plants: Potential for Industrial Production of Natural Green Leaf Volatiles. 2010. Available online: https://popups.uliege.be/1780-4507/index.php?id=5669 (accessed on 20 September 2022).
- Chen, J.L.; Yan, S.; Feng, Z.; Xiao, L.; Hu, X.S. Changes in the Volatile Compounds and Chemical and Physical Properties of Yali Pear (Pyrus Bertschneideri Reld) during Storage. Food Chem. 2006, 97, 248–255. [Google Scholar] [CrossRef]
- Lara, I.; Miró, R.M.; Fuentes, T.; Sayez, G.; Graell, J.; López, M.L. Biosynthesis of Volatile Aroma Compounds in Pear Fruit Stored under Long-Term Controlled-Atmosphere Conditions. Postharvest Biol. Technol. 2003, 29, 29–39. [Google Scholar] [CrossRef]
- Suwanagul, A.; Richardson, D.G. Identification of Headspace Volatile Compounds from Different Pear (Pyrus Communis L.) Varieties. Acta Hortic. 1998, 475, 605–623. [Google Scholar] [CrossRef]
- Takeoka, G.R.; Buttery, R.G.; Flath, R.A. Volatile Constituents of Asian Pear (Pyrus Serotina). J. Agric. Food Chem. 1992, 40, 1925–1929. [Google Scholar] [CrossRef]
- Fan, Z.; Hasing, T.; Johnson, T.S.; Garner, D.M.; Barbey, C.R.; Colquhoun, T.A.; Sims, C.A.; Resende, M.F.R.; Whitaker, V.M. Strawberry Sweetness and Consumer Preference Are Enhanced by Specific Volatile Compounds. Hortic. Res. 2021, 8, 1–15. [Google Scholar] [CrossRef]
- Chang, S.; Puryear, J.; Cairney, J. A Simple and Efficient Method for Isolating RNA from Pine Trees. Plant Mol. Biol. Report. 1993, 11, 113–116. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A Fast Spliced Aligner with Low Memory Requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. FeatureCounts: An Efficient General Purpose Program for Assigning Sequence Reads to Genomic Features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Varet, H.; Brillet-Guéguen, L.; Coppée, J.Y.; Dillies, M.A. SARTools: A DESeq2- and EdgeR-Based R Pipeline for Comprehensive Differential Analysis of RNA-Seq Data. PLoS ONE 2016, 11, e0157022. [Google Scholar] [CrossRef]
- Ali, M.; Hussain, R.M.; Rehman, N.U.; She, G.; Li, P.; Wan, X.; Guo, L.; Zhao, J.; Nishimura, M. De Novo Transcriptome Sequencing and Metabolite Profiling Analyses Reveal the Complex Metabolic Genes Involved in the Terpenoid Biosynthesis in Blue Anise Sage (Salvia guaranitica L.). DNA Res. 2018, 25, 597–617. [Google Scholar] [CrossRef]
- Apweiler, R.; Bairoch, A.; Wu, C.H.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; Magrane, M.; et al. UniProt: The Universal Protein Knowledgebase. Nucleic Acids Res. 2004, 32, D115–D119. [Google Scholar] [CrossRef]
- Bateman, A.; Martin, M.J.; O’Donovan, C.; Magrane, M.; Alpi, E.; Antunes, R.; Bely, B.; Bingley, M.; Bonilla, C.; Britto, R.; et al. UniProt: The Universal Protein Knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the Unification of Biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Koonin, E.V.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Krylov, D.M.; Makarova, K.S.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; Rao, B.S.; et al. A Comprehensive Evolutionary Classification of Proteins Encoded in Complete Eukaryotic Genomes. Genome Biol. 2004, 5, 1–28. [Google Scholar] [CrossRef]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The Protein Families Database. Nucleic Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG Resource for Deciphering the Genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef] [PubMed]
- O’Donovan, C.; Martin, M.J.; Glemet, E.; Codani, J.J.; Apweiler, R. Removing Redundancy in SWISS-PROT and TrEMBL. Bioinformatics 1999, 15, 258–259. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bairoch, A.; Apweiler, R. The SWISS-PROT Protein Sequence Database and Its Supplement TrEMBL in 2000. Nucleic Acids Res. 2000, 28, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Jiao, C.; Sun, H.; Rosli, H.G.; Pombo, M.A.; Zhang, P.; Banf, M.; Dai, X.; Martin, G.B.; Giovannoni, J.J.; et al. ITAK: A Program for Genome-Wide Prediction and Classification of Plant Transcription Factors, Transcriptional Regulators, and Protein Kinases. Mol. Plant 2016, 9, 1667–1670. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R Package for Weighted Correlation Network Analysis. BMC Bioinform. 2008, 9, 1–13. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE Guidelines: Minimum Information for Publication of Quantitative Real-Time PCR Experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef]
- Abdullah-Zawawi, M.R.; Ahmad-Nizammuddin, N.F.; Govender, N.; Harun, S.; Mohd-Assaad, N.; Mohamed-Hussein, Z.A. Comparative Genome-Wide Analysis of WRKY, MADS-Box and MYB Transcription Factor Families in Arabidopsis and Rice. Sci. Rep. 2021, 11, 1–18. [Google Scholar] [CrossRef]
- Saltveit, M.E. Effect of Ethylene on Quality of Fresh Fruits and Vegetables. Postharvest Biol. Technol. 1999, 15, 279–292. [Google Scholar] [CrossRef]
- Li, G.; Jia, H.; Wu, R.; Hussain, S.; Teng, Y. Characterization of Aromatic Volatile Constituents in 11 Asian Pear Cultivars Belonging to Different Species. Afr. J. Agric. Res. 2012, 7, 4761–4770. [Google Scholar] [CrossRef]
- López-Nicolás, J.M.; Andreu-Sevilla, A.J.; Carbonell-Barrachina, Á.A.; García-Carmona, F. Effects of Addition of α-Cyclodextrin on the Sensory Quality, Volatile Compounds, and Color Parameters of Fresh Pear Juice. J. Agric. Food Chem. 2009, 57, 9668–9675. [Google Scholar] [CrossRef]
- Moya-León, M.A.; Vergara, M.; Bravo, C.; Montes, M.E.; Moggia, C. 1-MCP Treatment Preserves Aroma Quality of ‘Packham’s Triumph’ Pears during Long-Term Storage. Postharvest Biol. Technol. 2006, 42, 185–197. [Google Scholar] [CrossRef]
- Yang, Y.; Han, L.; Yuan, Y.; Li, J.; Hei, N.; Liang, H. Gene Co-Expression Network Analysis Reveals Common System-Level Properties of Prognostic Genes across Cancer Types. Nat. Commun. 2014, 5, 1–9. [Google Scholar] [CrossRef]
- El-Sharkawy, I.; Liang, D.; Xu, K. Transcriptome Analysis of an Apple (Malus × Domestica) Yellow Fruit Somatic Mutation Identifies a Gene Network Module Highly Associated with Anthocyanin and Epigenetic Regulation. J. Exp. Bot. 2015, 66, 7359–7376. [Google Scholar] [CrossRef]
- Yuan, L.; Chen, L.; Qian, K.; Qian, G.; Wu, C.L.; Wang, X.; Xiao, Y. Co-Expression Network Analysis Identified Six Hub Genes in Association with Progression and Prognosis in Human Clear Cell Renal Cell Carcinoma (CcRCC). Genom. Data 2017, 14, 132–140. [Google Scholar] [CrossRef]
- Song, J.; Bangerth, F. Fatty Acids as Precursors for Aroma Volatile Biosynthesis in Pre-Climacteric and Climacteric Apple Fruit. Postharvest Biol. Technol. 2003, 30, 113–121. [Google Scholar] [CrossRef]
- Echeverría, G.; Fuentes, T.; Graell, J.; Lara, I.; López, M.L. Aroma Volatile Compounds of ‘Fuji’ Apples in Relation to Harvest Date and Cold Storage Technology: A Comparison of Two Seasons. Postharvest Biol. Technol. 2004, 32, 29–44. [Google Scholar] [CrossRef]
- Schaffer, R.J.; Friel, E.N.; Souleyre, E.J.F.; Bolitho, K.; Thodey, K.; Ledger, S.; Bowen, J.H.; Ma, J.H.; Nain, B.; Cohen, D.; et al. A Genomics Approach Reveals That Aroma Production in Apple Is Controlled by Ethylene Predominantly at the Final Step in Each Biosynthetic Pathway. Plant Physiol. 2007, 144, 1899–1912. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Gene ID | Primer Sequence (5′-3′) | The Size Products | |
|---|---|---|---|---|
| long-chain acyl-CoA synthetase | LOC103949217 | F:AACCGACTATCTTCTGTGCTGTTCC | R:AGCCGCCTGATGAAATCTTCTGTG | 81 bp |
| lysophospholipid acyltransferase | LOC103931841 | F:CCAATGCTACTCGGCTATGCTTCC | R:CGATTCCTCCTTCCTTCCATGCG | 139 bp |
| alcohol dehydrogenase (NADP+) | LOC103952287 | F:CACCAGCATTGGGCTTGAAATCG | R:CACCTCCATCCCTGCCTCCTTC | 121 bp |
| endogenous reference gene | AY338250 | F:CAGGCTGACTGTGCTGTCCT | R:TCACACCGAGGGTGAAAGCA | 115 bp |
| TF Family | Black | Blue | Brown | Cyan | Green | Green-Yellow | Grey | Magenta | Midnight Blue | Pink | Purple | Red | Salmon | Tan | Yellow | Turquoise |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AP2/ERF-ERF | 21 | 7 | 1 | 10 | 10 | 2 | 8 | 38 | ||||||||
| C2H2 | 1 | 4 | 8 | 5 | 2 | 3 | 2 | 6 | 1 | 5 | 32 | |||||
| C3H | 13 | 9 | 1 | 10 | 2 | 2 | 2 | 5 | 1 | 7 | 22 | |||||
| GRAS | 4 | 10 | 10 | 5 | 26 | |||||||||||
| MYB | 1 | 7 | 1 | 7 | 1 | 4 | 3 | 2 | 2 | 7 | 37 | |||||
| NAC | 2 | 4 | 11 | 8 | 1 | 4 | 2 | 6 | 36 | |||||||
| Trihelix | 2 | 1 | 4 | 1 | 2 | 6 | 22 | |||||||||
| Total | 4 | 48 | 53 | 3 | 54 | 0 | 3 | 9 | 1 | 24 | 6 | 13 | 2 | 2 | 44 | 213 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, H.; Quan, J.; Rana, S.; Wang, Y.; Li, Z.; Cai, Q.; Ma, S.; Geng, X.; Liu, Z. The Molecular Network behind Volatile Aroma Formation in Pear (Pyrus spp. Panguxiang) Revealed by Transcriptome Profiling via Fatty Acid Metabolic Pathways. Life 2022, 12, 1494. https://doi.org/10.3390/life12101494
Li H, Quan J, Rana S, Wang Y, Li Z, Cai Q, Ma S, Geng X, Liu Z. The Molecular Network behind Volatile Aroma Formation in Pear (Pyrus spp. Panguxiang) Revealed by Transcriptome Profiling via Fatty Acid Metabolic Pathways. Life. 2022; 12(10):1494. https://doi.org/10.3390/life12101494
Chicago/Turabian StyleLi, Huiyun, Jine Quan, Sohel Rana, Yanmei Wang, Zhi Li, Qifei Cai, Shuhong Ma, Xiaodong Geng, and Zhen Liu. 2022. "The Molecular Network behind Volatile Aroma Formation in Pear (Pyrus spp. Panguxiang) Revealed by Transcriptome Profiling via Fatty Acid Metabolic Pathways" Life 12, no. 10: 1494. https://doi.org/10.3390/life12101494
APA StyleLi, H., Quan, J., Rana, S., Wang, Y., Li, Z., Cai, Q., Ma, S., Geng, X., & Liu, Z. (2022). The Molecular Network behind Volatile Aroma Formation in Pear (Pyrus spp. Panguxiang) Revealed by Transcriptome Profiling via Fatty Acid Metabolic Pathways. Life, 12(10), 1494. https://doi.org/10.3390/life12101494