
An Interplay between Viruses and Bacteria Associated with the White Sea Sponges Revealed by Metagenomics

Abstract
1. Introduction
2. Materials and Methods
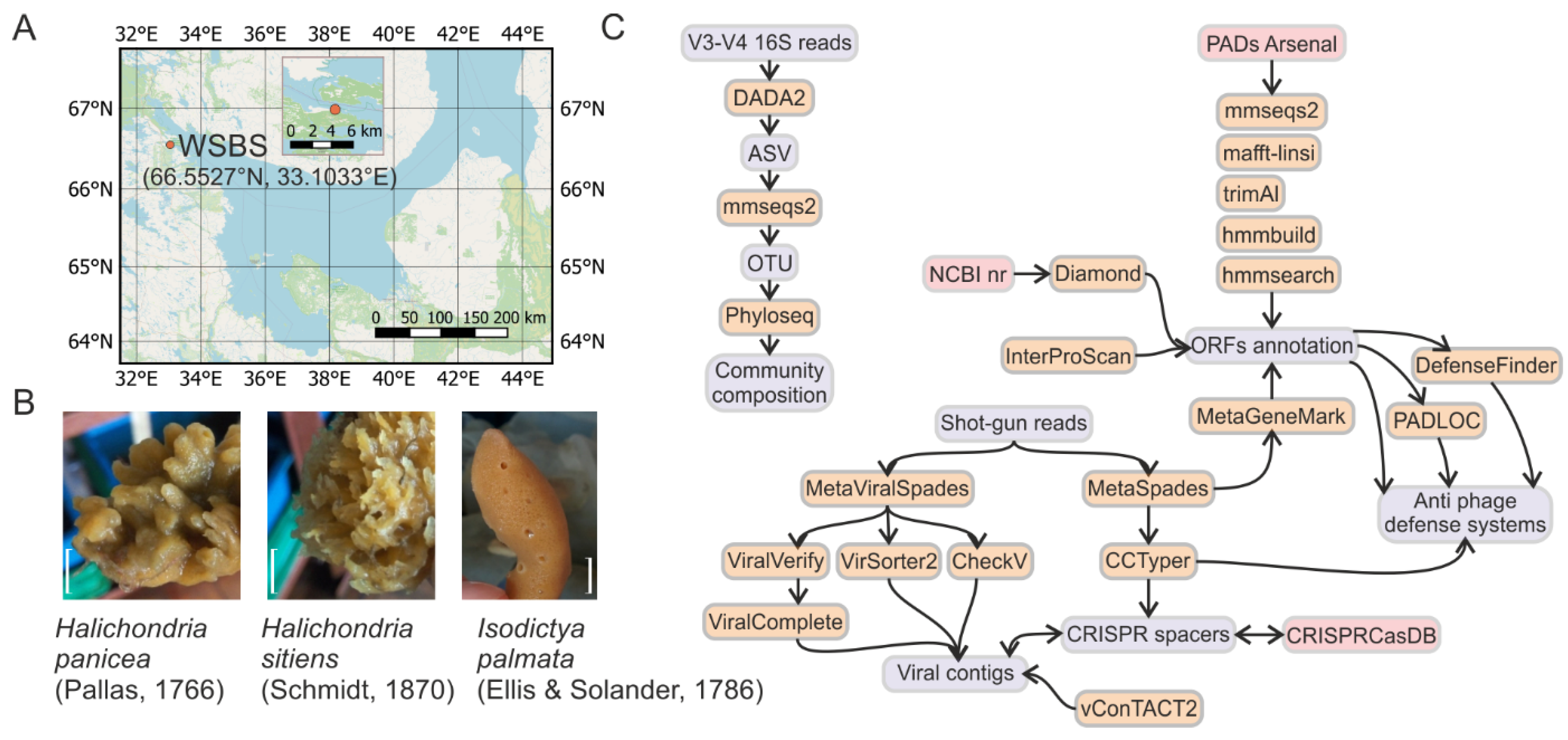
2.1. Sample Collection
2.2. Bacterial and Viral Fractions Isolation
2.3. DNA Extraction
2.4. High-Throughput Sequencing
2.5. 16S rRNA Data Analysis
2.6. Shot-Gun Metagenomes Assembly and Annotation
2.7. Identification of Viral Sequences and Taxonomic Assignment
2.8. Prediction and Quantification of Phage Defense Systems
2.9. CRISPR Spacer Detection and Analysis
3. Results
3.1. Samples Description and Processing
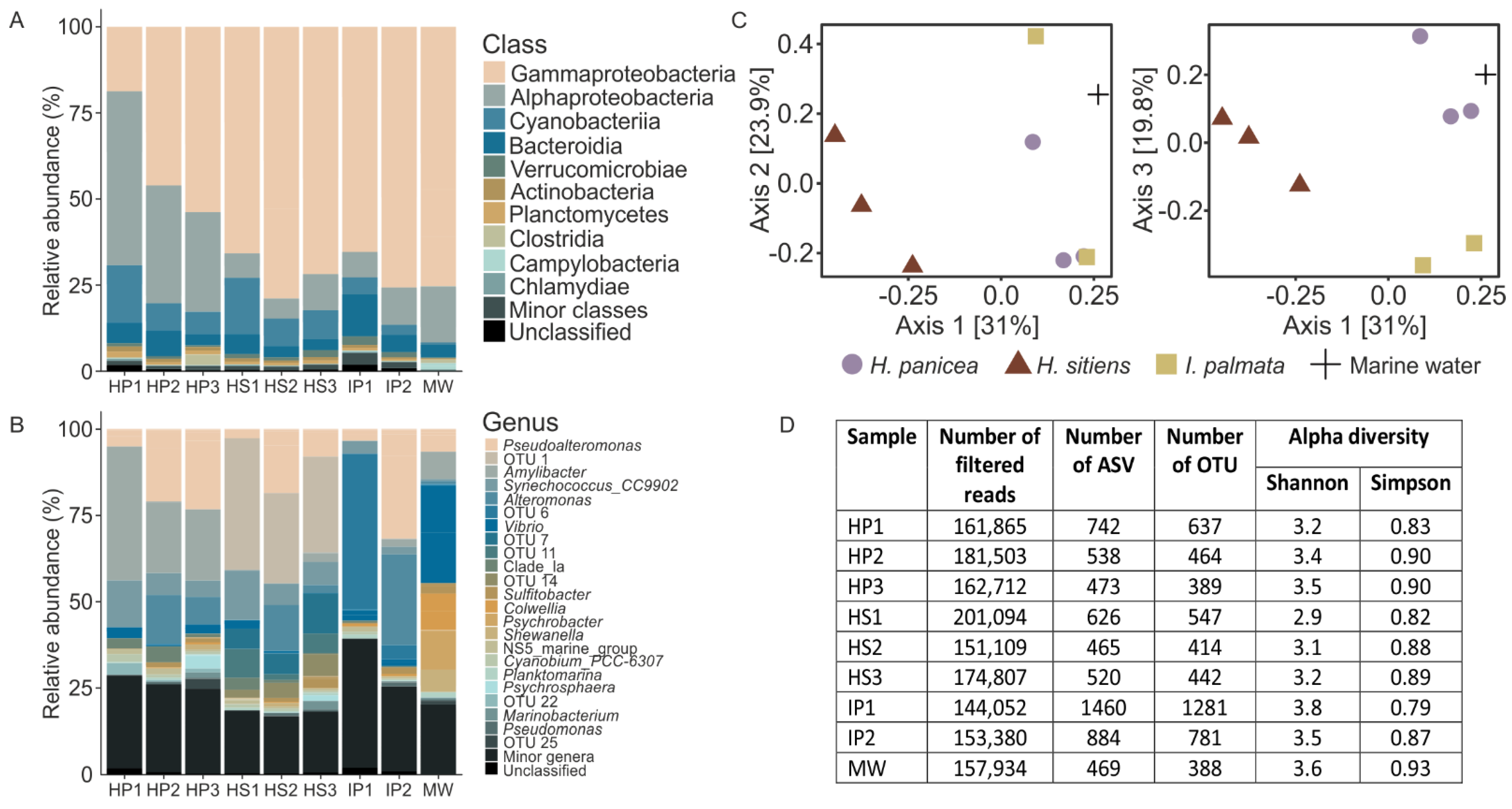
3.2. 16S Metagenomics Revealed Distinct Complex Bacterial Communities Associated with the White Sea Sponges and Marine Water
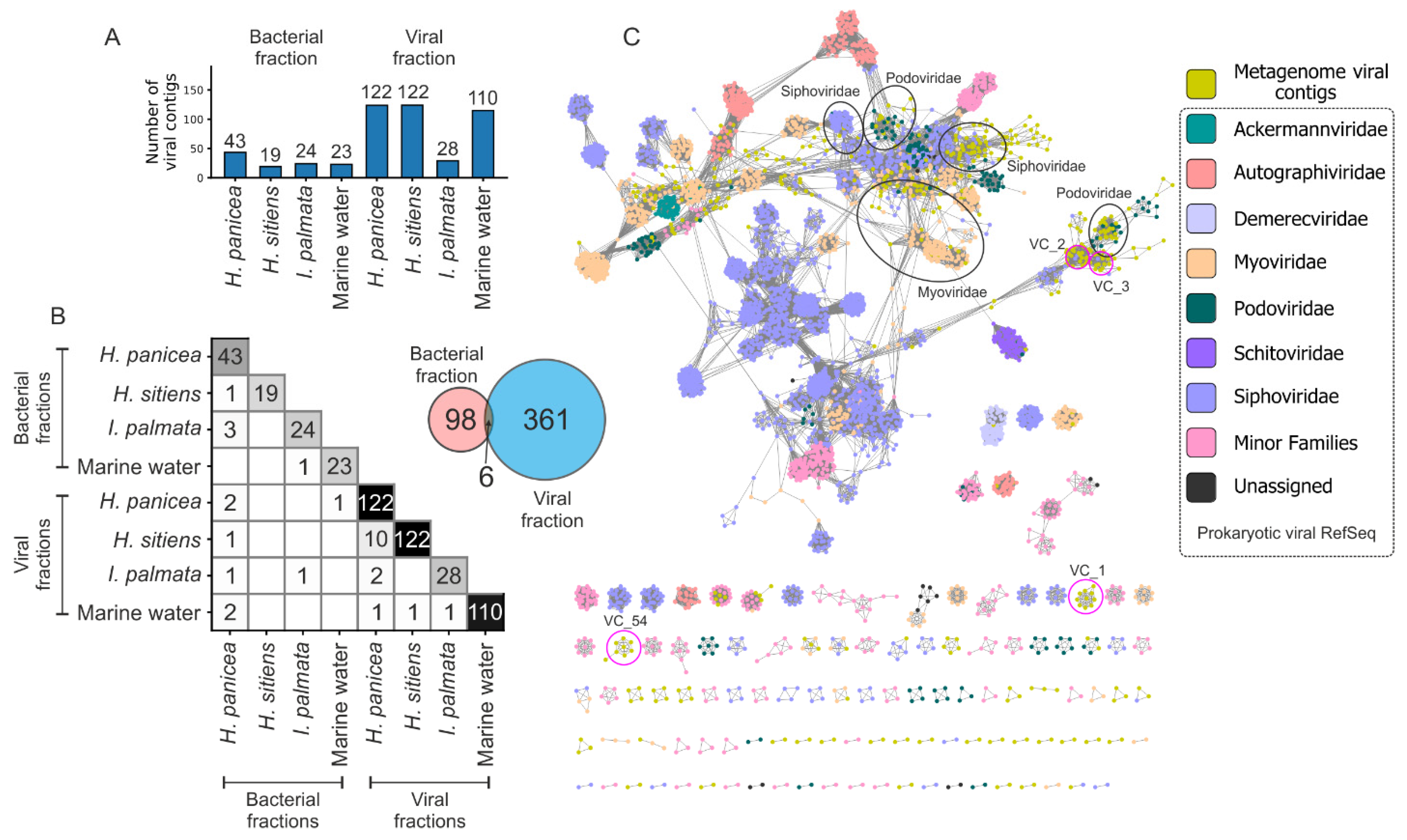
3.3. Enrichment and Diversity of Viromes from the White Sea
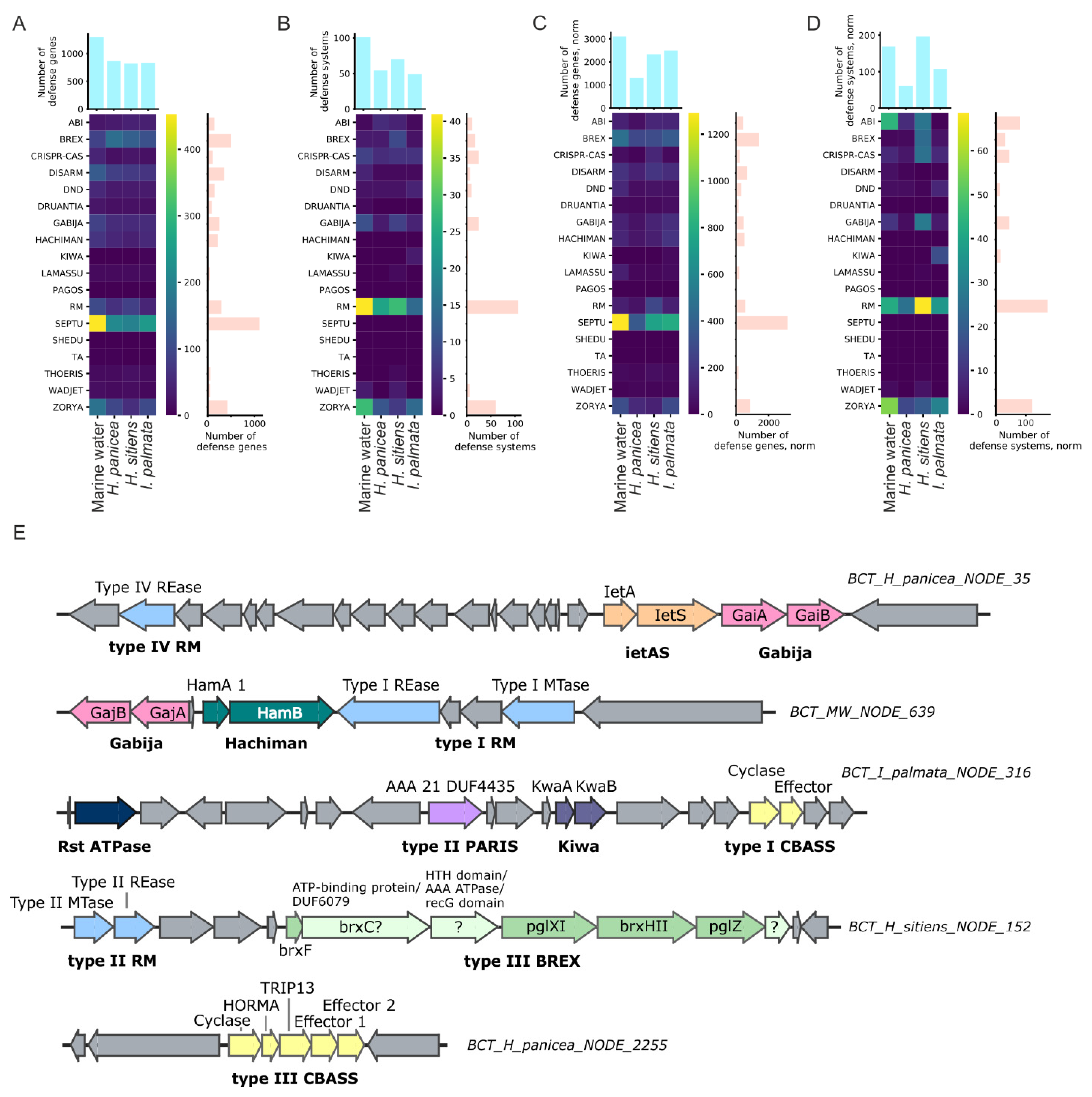
3.4. Antiphage Systems Detected in Metagenomes
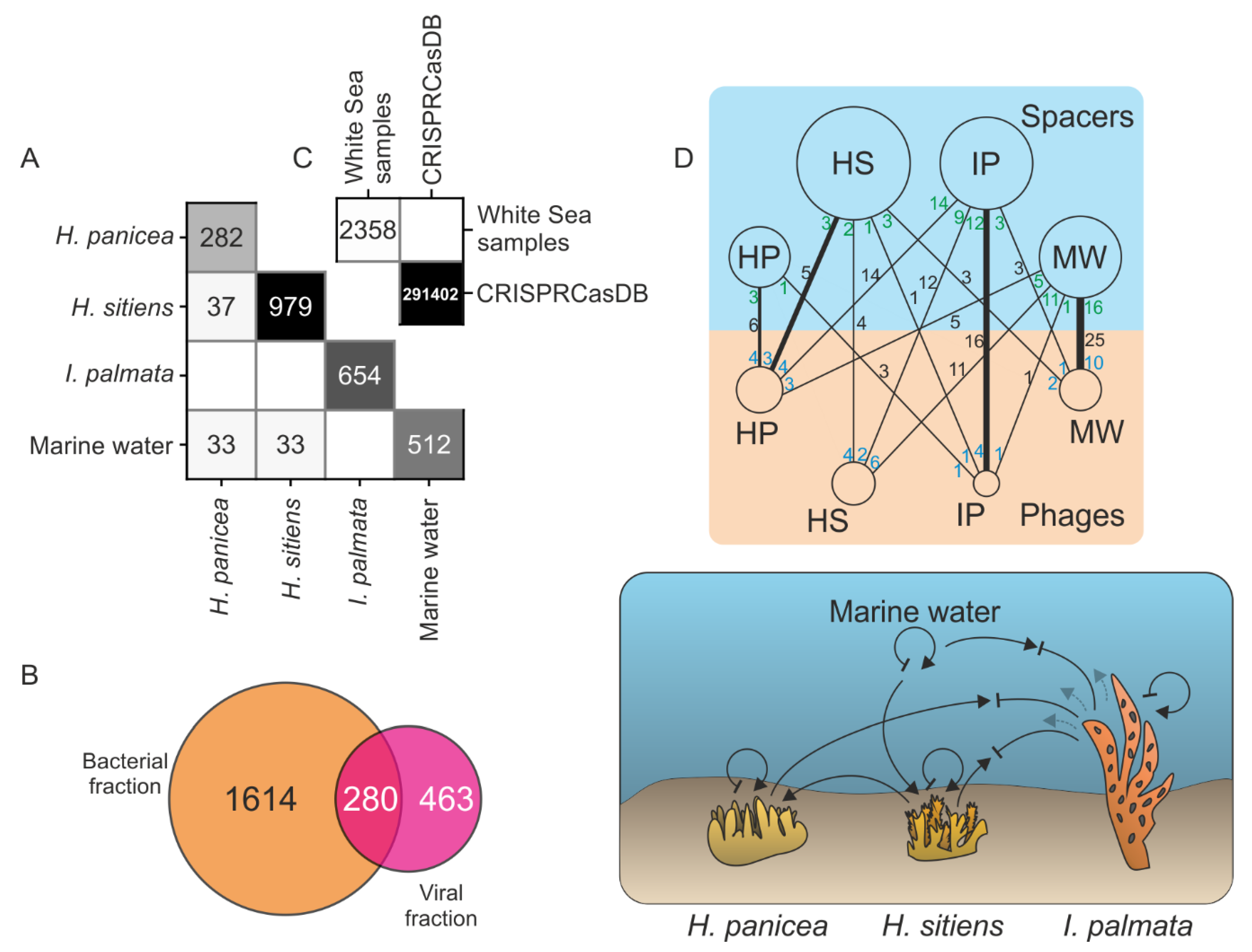
3.5. Analysis of CRISPR Spacers Show the Flow of Viruses between Marine Communities
4. Discussion
4.1. Sponge Species Specificity of Associated Bacterial and Viral Communities
4.2. Analysis of Defense Systems Repertoires of White Sea Sponges
4.3. Tracking the Viral Flow between Communities by a Spacer-Protospacer Network
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bordenstein, S.R.; Theis, K.R. Host Biology in Light of the Microbiome: Ten Principles of Holobionts and Hologenomes. PLoS Biol. 2015, 13, e1002226. [Google Scholar] [CrossRef]
- Bosch, T.; McFall-Ngai, M. Metaorganisms as the New Frontier. Zoology 2011, 114, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Pita, L.; Rix, L.; Slaby, B.M.; Franke, A.; Hentschel, U. The Sponge Holobiont in a Changing Ocean: From Microbes to Ecosystems. Microbiome 2018, 6, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Freeman, C.J.; Easson, C.G.; Matterson, K.O.; Thacker, R.W.; Baker, D.M.; Paul, V.J. Microbial Symbionts and Ecological Divergence of Caribbean Sponges: A New Perspective on an Ancient Association. ISME J. 2020, 14, 1571–1583. [Google Scholar] [CrossRef]
- Tianero, M.D.; Balaich, J.N.; Donia, M.S. Localized Production of Defence Chemicals by Intracellular Symbionts of Haliclona Sponges. Nat. Microbiol. 2019, 4, 1149–1159. [Google Scholar] [CrossRef]
- Cleary, D.F.R.; de Voogd, N.J.; Polónia, A.R.M.; Freitas, R.; Gomes, N.C.M. Composition and Predictive Functional Analysis of Bacterial Communities in Seawater, Sediment and Sponges in the Spermonde Archipelago, Indonesia. Microb. Ecol. 2015, 70, 889–903. [Google Scholar] [CrossRef] [PubMed]
- Thomas, T.; Moitinho-Silva, L.; Lurgi, M.; Björk, J.R.; Easson, C.; Astudillo-García, C.; Olson, J.B.; Erwin, P.M.; López-Legentil, S.; Luter, H.; et al. Diversity, Structure and Convergent Evolution of the Global Sponge Microbiome. Nat. Commun. 2016, 7, 11870. [Google Scholar] [CrossRef]
- Souza, D.T.; Genuário, D.B.; Silva, F.S.P.; Pansa, C.C.; Kavamura, V.N.; Moraes, F.C.; Taketani, R.G.; Melo, I.S. Analysis of Bacterial Composition in Marine Sponges Reveals the Influence of Host Phylogeny and Environment. FEMS Microbiol. Ecol. 2017, 93, 204. [Google Scholar] [CrossRef]
- de Voogd, N.J.; Cleary, D.F.R.; Polónia, A.R.M.; Gomes, N.C.M. Bacterial Community Composition and Predicted Functional Ecology of Sponges, Sediment and Seawater from the Thousand Islands Reef Complex, West Java, Indonesia. FEMS Microbiol. Ecol. 2015, 91, 19. [Google Scholar] [CrossRef]
- Moitinho-Silva, L.; Nielsen, S.; Amir, A.; Gonzalez, A.; Ackermann, G.L.; Cerrano, C.; Astudillo-Garcia, C.; Easson, C.; Sipkema, D.; Liu, F.; et al. The Sponge Microbiome Project. Gigascience 2017, 6, gix077. [Google Scholar] [CrossRef]
- Astudillo-García, C.; Bell, J.J.; Montoya, J.M.; Moitinho-Silva, L.; Thomas, T.; Webster, N.S.; Taylor, M.W. Assessing the Strength and Sensitivity of the Core Microbiota Approach on a Highly Diverse Sponge Reef. Environ. Microbiol. 2020, 22, 3985–3999. [Google Scholar] [CrossRef] [PubMed]
- Podell, S.; Blanton, J.M.; Neu, A.; Agarwal, V.; Biggs, J.S.; Moore, B.S.; Allen, E.E. Pangenomic Comparison of Globally Distributed Poribacteria Associated with Sponge Hosts and Marine Particles. ISME J. 2018, 13, 468–481. [Google Scholar] [CrossRef] [PubMed]
- Van Soest, R.W.M.; Boury-Esnault, N.; Vacelet, J.; Dohrmann, M.; Erpenbeck, D.; De Voogd, N.J.; Santodomingo, N.; Vanhoorne, B.; Kelly, M.; Hooper, J.N.A. Global Diversity of Sponges (Porifera). PLoS ONE 2012, 7, e35105. [Google Scholar] [CrossRef]
- Weisz, J.B.; Lindquist, N.; Martens, C.S. Do Associated Microbial Abundances Impact Marine Demosponge Pumping Rates and Tissue Densities? Oecologia 2007, 155, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Jahn, M.T.; Arkhipova, K.; Markert, S.M.; Stigloher, C.; Lachnit, T.; Pita, L.; Kupczok, A.; Ribes, M.; Stengel, S.T.; Rosenstiel, P.; et al. A Phage Protein Aids Bacterial Symbionts in Eukaryote Immune Evasion. Cell Host Microbe 2019, 26, 542–550.e5. [Google Scholar] [CrossRef]
- Horn, H.; Slaby, B.M.; Jahn, M.T.; Bayer, K.; Moitinho-Silva, L.; Förster, F.; Abdelmohsen, U.R.; Hentschel, U. An Enrichment of CRISPR and Other Defense-Related Features in Marine Sponge-Associated Microbial Metagenomes. Front. Microbiol. 2016, 7, 1751. [Google Scholar] [CrossRef]
- Karimi, E.; Slaby, B.M.; Soares, A.R.; Blom, J.; Hentschel, U.; Costa, R. Metagenomic Binning Reveals Versatile Nutrient Cycling and Distinct Adaptive Features in Alphaproteobacterial Symbionts of Marine Sponges. FEMS Microbiol. Ecol. 2018, 94, fiy074. [Google Scholar] [CrossRef]
- Fan, L.; Reynolds, D.; Liu, M.; Stark, M.; Kjelleberg, S.; Webster, N.S.; Thomas, T. Functional Equivalence and Evolutionary Convergence in Complex Communities of Microbial Sponge Symbionts. Proc. Natl. Acad. Sci. USA 2012, 109, E1878–E1887. [Google Scholar] [CrossRef]
- Rua, C.P.J.; Gregoracci, G.B.; Santos, E.O.; Soares, A.C.; Francini-Filho, R.B.; Thompson, F. Potential Metabolic Strategies of Widely Distributed Holobionts in the Oceanic Archipelago of St Peter and St Paul (Brazil). FEMS Microbiol. Ecol. 2015, 91, 43. [Google Scholar] [CrossRef][Green Version]
- Moreno-Pino, M.; Cristi, A.; Gillooly, J.F.; Trefault, N. Characterizing the Microbiomes of Antarctic Sponges: A Functional Metagenomic Approach. Sci. Rep. 2020, 10, 645. [Google Scholar] [CrossRef]
- Karimi, E.; Ramos, M.; Gonçalves, J.M.S.; Xavier, J.R.; Reis, M.P.; Costa, R. Comparative Metagenomics Reveals the Distinctive Adaptive Features of the Spongia Officinalis Endosymbiotic Consortium. Front. Microbiol. 2017, 8, 2499. [Google Scholar] [CrossRef]
- Pascelli, C.; Laffy, P.W.; Kupresanin, M.; Ravasi, T.; Webster, N.S. Morphological Characterization of Virus-like Particles in Coral Reef Sponges. PeerJ 2018, 6, e5625. [Google Scholar] [CrossRef]
- Laffy, P.W.; Wood-Charlson, E.M.; Turaev, D.; Jutz, S.; Pascelli, C.; Botté, E.S.; Bell, S.C.; Peirce, T.E.; Weynberg, K.D.; van Oppen, M.J.H.; et al. Reef Invertebrate Viromics: Diversity, Host Specificity and Functional Capacity. Environ. Microbiol. 2018, 20, 2125–2141. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.; Wemheuer, B.; Laffy, P.W.; Webster, N.S.; Thomas, T. Taxonomic, Functional and Expression Analysis of Viral Communities Associated with Marine Sponges. PeerJ 2021, 9, e10715. [Google Scholar] [CrossRef]
- Zhou, K.; Qian, P.-Y.; Zhang, T.; Xu, Y.; Zhang, R. Unique Phage–Bacterium Interplay in Sponge Holobionts from the Southern Okinawa Trough Hydrothermal Vent. Environ. Microbiol. Rep. 2021, 13, 675–683. [Google Scholar] [CrossRef]
- Butina, T.V.; Khanaev, I.V.; Kravtsova, L.S.; Maikova, O.O.; Bukin, Y.S. Metavirome Datasets from Two Endemic Baikal Sponges Baikalospongia bacillifera. Data Br. 2020, 29, 105260. [Google Scholar] [CrossRef]
- Zhou, K.; Zhang, R.; Sun, J.; Zhang, W.; Tian, R.-M.; Chen, C.; Kawagucci, S.; Xu, Y. Potential Interactions between Clade SUP05 Sulfur-Oxidizing Bacteria and Phages in Hydrothermal Vent Sponges. Appl. Environ. Microbiol. 2019, 85, e00992-19. [Google Scholar] [CrossRef]
- Sun, G.; Xiao, J.; Wang, H.; Gong, C.; Pan, Y.; Yan, S.; Wang, Y. Efficient purification and concentration of viruses from a large body of high turbidity seawater. MethodsX 2014, 1, e197–e206. [Google Scholar] [CrossRef] [PubMed]
- Sambrook, J.; Fritsch, E.; Maniatis, T. Bacteriophage λ growth, purification, and DNA extraction. In Molecular Cloning, 2nd ed.; Nolan, C., Ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1989; Volume 1. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Steinegger, M.; Söding, J. MMseqs2 Enables Sensitive Protein Sequence Searching for the Analysis of Massive Data Sets. Nat. Biotechnol. 2017, 35, 1026–1028. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- FastQC: A Quality Control Tool for High Throughput Sequence Data. 2015. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 30 November 2021).
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality Assessment Tool for Genome Assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and Sensitive Protein Alignment Using DIAMOND. Nat. Methods 2014, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Beier, S.; Flade, I.; Górska, A.; El-Hadidi, M.; Mitra, S.; Ruscheweyh, H.-J.; Tappu, R. MEGAN Community Edition—Interactive Exploration and Analysis of Large-Scale Microbiome Sequencing Data. PLoS Comput. Biol. 2016, 12, e1004957. [Google Scholar] [CrossRef]
- Antipov, D.; Raiko, M.; Lapidus, A.; Pevzner, P.A. MetaviralSPAdes: Assembly of Viruses from Metagenomic Data. Bioinformatics 2020, 36, 4126–4129. [Google Scholar] [CrossRef]
- Zhu, W.; Lomsadze, A.; Borodovsky, M. Ab Initio Gene Identification in Metagenomic Sequences. Nucleic Acids Res. 2010, 38, e132. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Guo, J.; Bolduc, B.; Zayed, A.A.; Varsani, A.; Dominguez-Huerta, G.; Delmont, T.O.; Pratama, A.A.; Gazitúa, M.C.; Vik, D.; Sullivan, M.B.; et al. VirSorter2: A Multi-Classifier, Expert-Guided Approach to Detect Diverse DNA and RNA Viruses. Microbiome 2021, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Nayfach, S.; Camargo, A.P.; Schulz, F.; Eloe-Fadrosh, E.; Roux, S.; Kyrpides, N.C. CheckV Assesses the Quality and Completeness of Metagenome-Assembled Viral Genomes. Nat. Biotechnol. 2020, 39, 578–585. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-Hit: A Fast Program for Clustering and Comparing Large Sets of Protein or Nucleotide Sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed]
- Bin Jang, H.; Bolduc, B.; Zablocki, O.; Kuhn, J.H.; Roux, S.; Adriaenssens, E.M.; Brister, J.R.; Kropinski, A.M.; Krupovic, M.; Lavigne, R.; et al. Taxonomic Assignment of Uncultivated Prokaryotic Virus Genomes Is Enabled by Gene-Sharing Networks. Nat. Biotechnol. 2019, 37, 632–639. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.; Wang, J.; Ramage, D.; Schwikowski, B. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Nishimura, Y.; Yoshida, T.; Kuronishi, M.; Uehara, H.; Ogata, H.; Goto, S. ViPTree: The Viral Proteomic Tree Server. Bioinformatics 2017, 33, 2379–2380. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Z.; Zhang, H.; Zhao, Y.; Zhang, Z.; Xiao, J. PADS Arsenal: A Database of Prokaryotic Defense Systems Related Genes. Nucleic Acids Res. 2020, 48, D590–D598. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. TrimAl: A Tool for Automated Alignment Trimming in Large-Scale Phylogenetic Analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.; Eddy, S.; Portugaly, E. Hidden Markov model speed heuristic and iterative HMM search procedure. BMC Bioinformatics 2010, 11, 431. [Google Scholar] [CrossRef]
- Payne, L.J.; Todeschini, T.C.; Wu, Y.; Perry, B.J.; Ronson, C.W.; Fineran, P.C.; Nobrega, F.L.; Jackson, S.A. Identification and Classification of Antiviral Defence Systems in Bacteria and Archaea with PADLOC Reveals New System Types. Nucleic Acids Res. 2021, 49, 10868–10878. [Google Scholar] [CrossRef] [PubMed]
- Tesson, F.; Hervé, A.; Touchon, M.; d’Humières, C.; Cury, J.; Bernheim, A. Systematic and Quantitative View of the Antiviral Arsenal of Prokaryotes. bioRxiv 2021, 2021.09.02.458658. [Google Scholar] [CrossRef]
- Russel, J.; Pinilla-Redondo, R.; Mayo-Muñoz, D.; Shah, S.A.; Sørensen, S.J. CRISPRCasTyper: Automated Identification, Annotation, and Classification of CRISPR-Cas Loci. CRISPR J. 2020, 3, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Pourcel, C.; Touchon, M.; Villeriot, N.; Vernadet, J.-P.; Couvin, D.; Toffano-Nioche, C.; Vergnaud, G. CRISPRCasdb a Successor of CRISPRdb Containing CRISPR Arrays and Cas Genes from Complete Genome Sequences, and Tools to Download and Query Lists of Repeats and Spacers. Nucleic Acids Res. 2020, 48, D535–D544. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A New Generation of Protein Database Search Programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Knobloch, S.; Jóhannsson, R.; Marteinsson, V. Bacterial Diversity in the Marine Sponge Halichondria Panicea from Icelandic Waters and Host-Specificity of Its Dominant Symbiont “Candidatus Halichondribacter Symbioticus”. FEMS Microbiol. Ecol. 2018, 95, fiy220. [Google Scholar] [CrossRef]
- Millman, A.; Melamed, S.; Amitai, G.; Sorek, R. Diversity and Classification of Cyclic-Oligonucleotide-Based Anti-Phage Signalling Systems. Nat. Microbiol. 2020, 5, 1608–1615. [Google Scholar] [CrossRef]
- Garrett, S.C. Pruning and Tending Immune Memories: Spacer Dynamics in the CRISPR Array. Front. Microbiol. 2021, 12, 739. [Google Scholar] [CrossRef]
- Vorontsova, D.; Datsenko, K.A.; Medvedeva, S.; Bondy-Denomy, J.; Savitskaya, E.E.; Pougach, K.; Logacheva, M.; Wiedenheft, B.; Davidson, A.R.; Severinov, K.; et al. Foreign DNA Acquisition by the I-F CRISPR–Cas System Requires All Components of the Interference Machinery. Nucleic Acids Res. 2015, 43, 10848–10860. [Google Scholar] [CrossRef] [PubMed]
- Wichels, A.; Würtz, S.; Döpke, H.; Schütt, C.; Gerdts, G. Bacterial Diversity in the Breadcrumb Sponge Halichondria Panicea (Pallas). FEMS Microbiol. Ecol. 2006, 56, 102–118. [Google Scholar] [CrossRef]
- Naim, M.A.; Morillo, J.A.; Sørensen, S.J.; Waleed, A.A.S.; Smidt, H.; Sipkema, D. Host-Specific Microbial Communities in Three Sympatric North Sea Sponges. FEMS Microbiol. Ecol. 2014, 90, 390–403. [Google Scholar] [CrossRef]
- Strehlow, B.; Schuster, A.; Francis, W.; Canfield, D. Metagenomic Data for Halichondria panicea from Illumina and Nanopore Sequencing and Preliminary Genome Assemblies for the Sponge and Two Microbial Symbionts. bioRxiv 2021, 2021.10.18.464794. [Google Scholar] [CrossRef]
- Ereskovsky, A.; Ozerov, D.A.; Pantyulin, A.N.; Tzetlin, A.B. Mass Mortality Event of White Sea Sponges as the Result of High Temperature in Summer 2018. Polar Biol. 2019, 42, 2313–2318. [Google Scholar] [CrossRef]
- Kennedy, J.; Codling, C.E.; Jones, B.V.; Dobson, A.D.W.; Marchesi, J.R. Diversity of Microbes Associated with the Marine Sponge, Haliclona simulans, Isolated from Irish Waters and Identification of Polyketide Synthase Genes from the Sponge Metagenome. Environ. Microbiol. 2008, 10, 1888–1902. [Google Scholar] [CrossRef]
- Croué, J.; West, N.J.; Escande, M.L.; Intertaglia, L.; Lebaron, P.; Suzuki, M.T. A Single Betaproteobacterium Dominates the Microbial Community of the Crambescidine-Containing Sponge Crambe Crambe. Sci. Reports 2013, 3, 2583. [Google Scholar] [CrossRef]
- Pascelli, C.; Laffy, P.W.; Botté, E.; Kupresanin, M.; Rattei, T.; Lurgi, M.; Ravasi, T.; Webster, N.S. Viral Ecogenomics across the Porifera. Microbiome 2020, 8. [Google Scholar] [CrossRef]
- Roux, S.; Hallam, S.J.; Woyke, T.; Sullivan, M.B. Viral Dark Matter and Virus-Host Interactions Resolved from Publicly Available Microbial Genomes. Elife 2015, 4, e08490. [Google Scholar] [CrossRef] [PubMed]
- Steinert, G.; Busch, K.; Bayer, K.; Kodami, S.; Arbizu, P.M.; Kelly, M.; Mills, S.; Erpenbeck, D.; Dohrmann, M.; Wörheide, G.; et al. Compositional and Quantitative Insights into Bacterial and Archaeal Communities of South Pacific Deep-Sea Sponges (Demospongiae and Hexactinellida). Front. Microbiol. 2020, 11, 716. [Google Scholar] [CrossRef] [PubMed]
- Welsh, J.E.; Steenhuis, P.; De Moraes, K.R.; Van Der Meer, J.; Thieltges, D.W.; Brussaard, C.P.D. Marine Virus Predation by Non-Host Organisms. Sci. Rep. 2020, 10, 5221. [Google Scholar] [CrossRef]
- Thomas, T.; Rusch, D.; DeMaere, M.Z.; Yung, P.Y.; Lewis, M.; Halpern, A.; Heidelberg, K.B.; Egan, S.; Steinberg, P.D.; Kjelleberg, S. Functional Genomic Signatures of Sponge Bacteria Reveal Unique and Shared Features of Symbiosis. ISME J. 2010, 4, 1557–1567. [Google Scholar] [CrossRef] [PubMed]
- Goldfarb, T.; Sberro, H.; Weinstock, E.; Cohen, O.; Doron, S.; Charpak-Amikam, Y.; Afik, S.; Ofir, G.; Sorek, R. BREX Is a Novel Phage Resistance System Widespread in Microbial Genomes. EMBO J. 2015, 34, 169–183. [Google Scholar] [CrossRef]
- Gordeeva, J.; Morozova, N.; Sierro, N.; Isaev, A.; Sinkunas, T.; Tsvetkova, K.; Matlashov, M.; Truncaite, L.; Morgan, R.D.; Ivanov, N.V.; et al. BREX System of Escherichia Coli Distinguishes Self from Non-Self by Methylation of a Specific DNA Site. Nucleic Acids Res. 2019, 47, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Deng, X.; Li, Y.; Ma, X.; Feng, J.; Yu, B.; Chen, Y.; Luo, Y.; Wang, X.; Chen, M.; et al. Structure of Schlafen13 Reveals a New Class of TRNA/RRNA- Targeting RNase Engaged in Translational Control. Nat. Commun. 2018, 9, 1165. [Google Scholar] [CrossRef] [PubMed]
- Garvie, C.W.; Wu, X.; Papanastasiou, M.; Lee, S.; Fuller, J.; Schnitzler, G.R.; Horner, S.W.; Baker, A.; Zhang, T.; Mullahoo, J.P.; et al. Structure of PDE3A-SLFN12 Complex Reveals Requirements for Activation of SLFN12 RNase. Nat. Commun. 2021, 12, 4375. [Google Scholar] [CrossRef] [PubMed]
- Picton, D.M.; Luyten, Y.A.; Morgan, R.D.; Nelson, A.; Smith, D.L.; Dryden, D.T.F.; Hinton, J.C.D.; Blower, T.R. The Phage Defence Island of a Multidrug Resistant Plasmid Uses Both BREX and Type IV Restriction for Complementary Protection from Viruses. Nucleic Acids Res. 2021, 49, 11257–11273. [Google Scholar] [CrossRef]
- Koonin, E.V.; Makarova, K.S.; Wolf, Y.I. Evolutionary Genomics of Defense Systems in Archaea and Bacteria. Annu. Rev. Microbiol. 2017, 71, 233. [Google Scholar] [CrossRef] [PubMed]
- Isaev, A.B.; Musharova, O.S.; Severinov, K.V. Microbial Arsenal of Antiviral Defenses—Part, I. Biochemistry 2021, 86, 319–337. [Google Scholar] [CrossRef]
- Dion, M.B.; Plante, P.L.; Zufferey, E.; Shah, S.A.; Corbeil, J.; Moineau, S. Streamlining CRISPR Spacer-Based Bacterial Host Predictions to Decipher the Viral Dark Matter. Nucleic Acids Res. 2021, 49, 3127–3138. [Google Scholar] [CrossRef]
- Yosef, I.; Goren, M.G.; Qimron, U. Proteins and DNA Elements Essential for the CRISPR Adaptation Process in Escherichia Coli. Nucleic Acids Res. 2012, 40, 5569–5576. [Google Scholar] [CrossRef]
- Pucciarelli, S.; Devaraj, R.R.; Mancini, A.; Ballarini, P.; Castelli, M.; Schrallhammer, M.; Petroni, G.; Miceli, C. Microbial Consortium Associated with the Antarctic Marine Ciliate Euplotes focardii: An Investigation from Genomic Sequences. Microb. Ecol. 2015, 70, 484–497. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Number of Reads after Filtering | Total Number of Contigs | Total Assembly Length, Mb | N50 | L50 | Number of Contigs > 5 kb | Total Assembly Length (Contigs > 5 kb), Mb |
|---|---|---|---|---|---|---|---|
| H.panicea (Bct) | 156,744,899 | 1,048,333 | 704.2 | 1678 | 62,534 | 10,147 | 102.6 |
| H. sitiens (Bct) | 151,978,145 | 660,860 | 515.4 | 2309 | 36,966 | 11,252 | 105.4 |
| I. palmata (Bct) | 151,705,427 | 984,734 | 626.6 | 1646 | 49,855 | 8239 | 87.0 |
| Marine water (Bct) | 139,611,995 | 1,409,157 | 783.8 | 1271 | 74,205 | 7222 | 82.2 |
| H.panicea (Vrs) | 105,797,172 | 773,423 | 477.2 | 1253 | 54,605 | 3723 | 50.6 |
| H. sitiens (Vrs) | 81,791,648 | 416,792 | 250.4 | 1313 | 21,446 | 2579 | 33.8 |
| I. palmata (Vrs) | 74,156,030 | 214,337 | 111.1 | 1227 | 8941 | 965 | 11.1 |
| Marine water (Vrs) | 107,464,178 | 1,078,397 | 691.0 | 1437 | 56,832 | 7208 | 100.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rusanova, A.; Fedorchuk, V.; Toshchakov, S.; Dubiley, S.; Sutormin, D. An Interplay between Viruses and Bacteria Associated with the White Sea Sponges Revealed by Metagenomics. Life 2022, 12, 25. https://doi.org/10.3390/life12010025
Rusanova A, Fedorchuk V, Toshchakov S, Dubiley S, Sutormin D. An Interplay between Viruses and Bacteria Associated with the White Sea Sponges Revealed by Metagenomics. Life. 2022; 12(1):25. https://doi.org/10.3390/life12010025
Chicago/Turabian StyleRusanova, Anastasiia, Victor Fedorchuk, Stepan Toshchakov, Svetlana Dubiley, and Dmitry Sutormin. 2022. "An Interplay between Viruses and Bacteria Associated with the White Sea Sponges Revealed by Metagenomics" Life 12, no. 1: 25. https://doi.org/10.3390/life12010025
APA StyleRusanova, A., Fedorchuk, V., Toshchakov, S., Dubiley, S., & Sutormin, D. (2022). An Interplay between Viruses and Bacteria Associated with the White Sea Sponges Revealed by Metagenomics. Life, 12(1), 25. https://doi.org/10.3390/life12010025